4.1. Synthesis of Muraymycin Analogues
General methods: All chemicals were purchased from standard suppliers. Reactions involving oxygen and/or moisture sensitive reagents were carried out under an atmosphere of nitrogen or argon using anhydrous solvents. Anhydrous solvents were obtained in the following manner: THF and MeCN were dried with a solvent purification system (MBRAUN MB SPS 800). Pyridine and
i-PrOH were dried over CaH
2 and distilled. MeOH was dried over activated molecular sieves (3 Å) and degassed. All of the thus obtained solvents were stored over molecular sieves (4 Å; in case of MeOH 3 Å). All other solvents were of technical quality and distilled prior to use, and deionized water was used throughout. Column chromatography was carried out on silica gel 60 (0.040–0.063 mm, 230–400 mesh ASTM, VWR) under flash conditions except where indicated. TLC was performed on aluminum plates precoated with silica gel 60 F
254 (VWR). Visualization of the spots was carried out using UV light (254 nm) and/or staining under heating (H
2SO
4 staining solution: 4 g vanillin, 25 mL conc. H
2SO
4, 80 mL AcOH and 680 mL MeOH; KMnO
4 staining solution: 1 g KMnO
4, 6 g K
2CO
3 and 1.5 mL 1.25 M NaOH solution, all dissolved in 100 mL H
2O; ninhydrin staining solution: 0.3 g ninhydrin, 3 mL AcOH and 100 mL 1-butanol; bromocresol green staining solution: 0.1 g bromocresol green, 500 mL EtOH, 5 mL 0.1 M aqueous NaOH solution). Semipreparative HPLC was performed on a VWR-Hitachi system equipped with an L-2300 pump, an L-2200 autosampler, an L-2300 column oven (24 °C), an L-2455 Diode Array Detector (DAD) and a LiChroCart
TM column (10 × 250 mm) containing reversed phase silica gel Nucleodur™ 100-5 C18ec (10 µm) purchased from Macherey-Nagel. Method 1: eluent A water (+0.1% TFA), eluent B MeCN (+0.1% TFA); 0–15 min gradient of B (10%–20%), 15–25 min gradient of B (20%–40%), 25–32 min gradient of B (40%–100%), 32–40 min 100% B, 40–41 min gradient of B (100%–30%), 41–45 min 30% B; flow 2 mL/min. Method 2: eluent A water (+0.1% TFA), eluent B MeCN (+0.1% TFA); 0–15 min gradient of B (20%–30%), 15–25 min gradient of B (30%–40%), 25-32 min gradient of B (40%–80%), 32–40 min 80% B, 40–41 min gradient of B (80%–30%), 41–45 min 30% B; flow 2 mL/min. Method 3: eluent A water, eluent B MeCN, 0–25 min gradient of B (5%–50%), 25–40 min gradient of B (50%–100%), 40–45 min 100% B, 45–50 min gradient of B (100%–5%), flow 5 mL/min. 500 MHz-
1H, 126 MHz-
13C, as well as 202 MHz-
31P NMR spectra were recorded on a Bruker UltraShield
TM-500 spectrometer. 376 MHz-
19F NMR spectra were recorded on a Bruker UltraShield
TM-400 spectrometer. 300 MHz-
1H and 76 MHz-
13C NMR spectra were recorded on a Bruker Fourier 300 spectrometer. All
13C and
19F NMR spectra are
1H-decoupled. All spectra were recorded at room temperature except where indicated and were referenced internally to solvent reference frequencies wherever possible. Chemical shifts (δ) are quoted in ppm and coupling constants (
J) are reported in Hz. Assignment of signals was carried out using H,H-COSY, HSQC, and HMBC spectra obtained on the spectrometers mentioned above. The numbering of atoms of muraymycin target structures is depicted in the
Supplementary Materials Figure S5. Mass spectra were measured on a Finnigan Surveyor MSQ Plus mass spectrometer. For ESI measurements in the positive mode, solutions in MeOH were used. High resolution mass spectra were measured on a Thermo Scientific Q Exactive Orbitrap mass spectrometer with ESI ionization mode and on a Finnigan sectorfield mass spectrometer type MAT 95S with chemical ionization (CI).
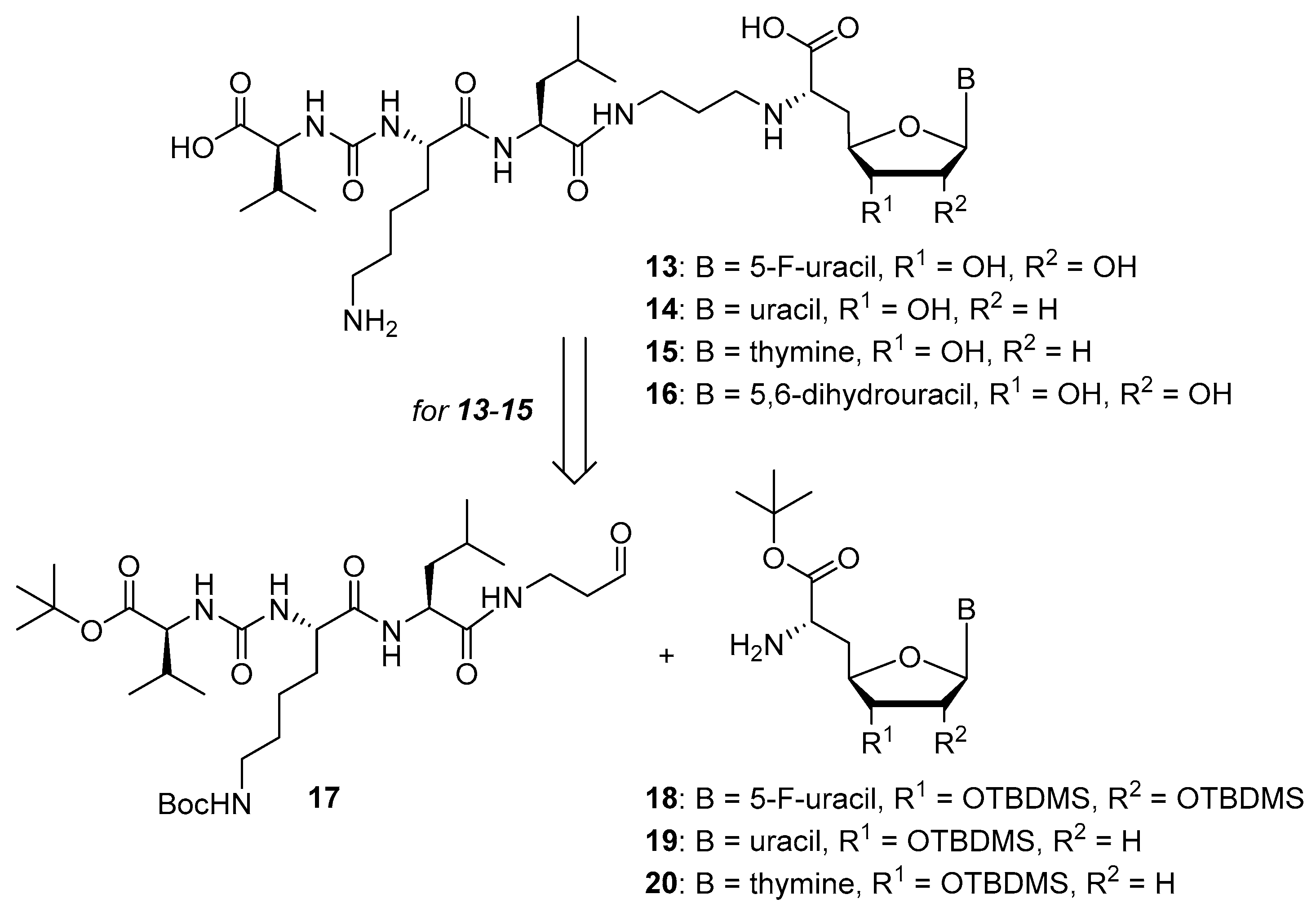
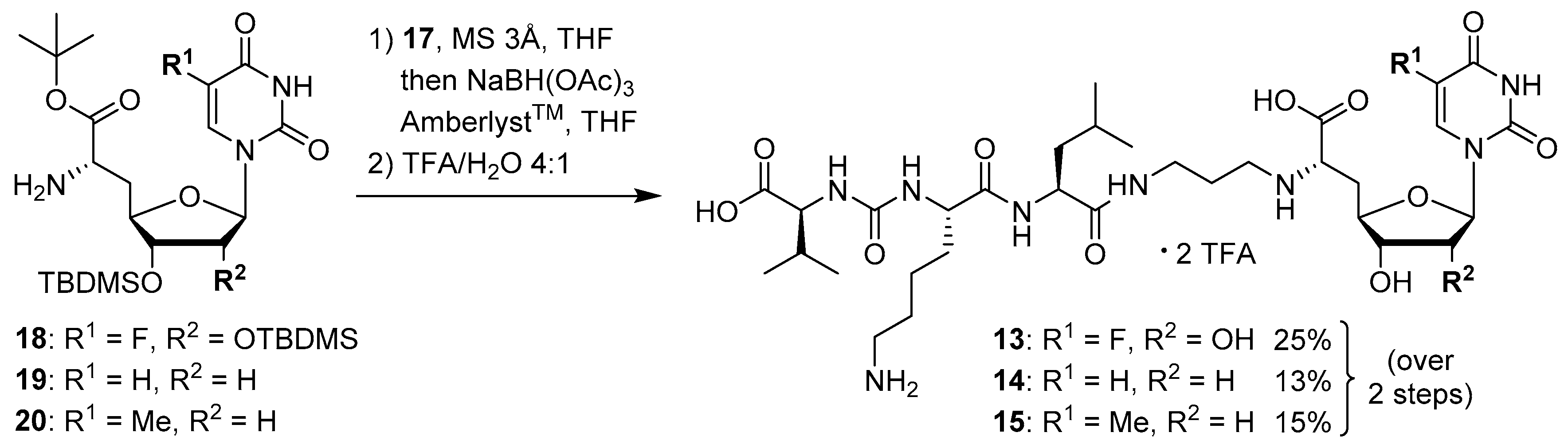
5-Fluorouridine-derived muraymycin analogue (
13): Nucleosyl amino acid ester
18 (35 mg, 58 μmol) was dissolved in THF (3 mL), and a solution of aldehyde
17 [
42] (46 mg, 75 μmol) in THF (3 mL) was added. The reaction mixture was stirred over molecular sieve (3 Å) at rt for 24 h. NaBH(OAc)
3 (33 mg, 0.16 mmol) and Amberlyst 15
TM (3.4 mg, 11 μmol) were added and stirring was continued at rt for 24 h. The reaction mixture was filtered and the residue was washed with EtOAc (150 mL). The combined filtrates were washed with sat. NaHCO
3 solution (100 mL) and brine (50 mL). The aqueous layer was extracted with EtOAc (3 × 100 mL). The combined organics were dried over Na
2SO
4 and the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (98:2→95:5, CH
2Cl
2-MeOH) to give the fully protected muraymycin analogue as a colorless solid. This was dissolved in aq. TFA (80%, 4.6 mL) and stirred at rt for 24 h. The reaction mixture was then diluted with water (12 mL) and the solvent was evaporated under reduced pressure. The resultant crude product was purified by semipreparative HPLC (method 1) to give
13 (bis-TFA salt) as a colorless foam (11 mg, 25% over 2 steps from
18).
1H NMR (500 MHz, D
2O): δ [ppm] = 0.83 (d,
J = 6.0 Hz, 3H, Leu-5-H), 0.88 (d,
J = 5.7 Hz, 3H, Leu-5-H), 0.89 (d,
J = 6.9 Hz, 3H, Val-4-H), 0.93 (d,
J = 6.9 Hz, 3H, Val-4-H), 1.33–1.48 (m, 2H, Lys-4-H), 1.50–1.70 (m, 6H, Lys-3-H
a, Lys-5-H, Leu-3-H, Leu-4-H), 1.73–1.87 (m, 1H, Lys-3-H
b), 1.85–1.92 (m, 2H, 2″-H), 2.10–2.16 (m, 1H, Val-3-H), 2.23–2.33 (m, 1H, 5′-H
a), 2.39–2.44 (m, 1H, 5′-H
b), 2.96 (t,
J = 7.6 Hz, 2H, Lys-6-H), 3.06 (t,
J = 7.6 Hz, 2H, 1″-H), 3.20–3.29 (m, 2H, 3″-H
a, 3″-H
b), 3.85–3.90 (m, 1H, 6′-H), 4.03 (dd,
J = 12.3, 6.3 Hz, 1H, 3′-H), 4.05 (d,
J = 5.4 Hz, 1H, Val-2-H), 4.09 (dd,
J = 8.5, 5.4 Hz, 1H, Lys-2-H), 4.14 (ddd,
J = 9.8, 6.3, 2.8 Hz, 1H, 4′-H), 4.23 (dd,
J = 10.1, 5.0 Hz, 1H, Leu-2-H), 4.35 (dd,
J = 5.7, 3.8 Hz, 1H, 2′-H), 5.73 (d,
J = 3.8 Hz, 1H, 1′-H), 7.81 (d,
J = 6.3 Hz, 1H, 6-H).
13C NMR (126 MHz, D
2O): δ [ppm] = 17.02 (Leu-C-5), 18.55 (Leu-C-5), 20.70 (Val-C-4), 22.08 (Lys-C-4), 22.15 (Val-C-4), 24.44 (Lys-C-5), 25.70 (C-2″), 26.31 (Leu-C-4), 30.02 (Val-C-3), 30.83 (Lys-C-3), 32.91 (C-5′), 36.01 (C-3′), 39.28 (Lys-C-6), 39.55 (Leu-C-3), 44.36 (C-1″), 52.65 (Leu-C-2), 54.11 (Lys-C-2), 58.95 (Val-C-2), 59.70 (C-6′), 72.83 (C-2′), 72.89 (C-3′), 79.96 (C-4′), 91.38 (C-1′), 116.45 (d,
JCF = 291.4 Hz, TFA-CF
3), 126.45 (d,
JCF = 34.8 Hz, C-6), 140.76 (d,
JCF = 233.7 Hz, C-5), 150.02 (C-2), 159.36 (urea-C=O), 159.55 (C-4), 163.07 (d,
JCF = 35.8 Hz, TFA-COO), 172.02 (C-7′), 174.98 (Leu-C-1), 175.48 (Lys-C-1), 176.80 (Val-C-1).
19F NMR (376 MHz, D
2O): δ [ppm] = −166.24 (1F, 5-F), −75.59 (6F, TFA-CF
3). MS (ESI
+):
m/z = 761.5 [M + H]
+. HRMS (ESI
+): calcd.: 761.3840 [M + H]
+, 783.3659 [M + Na]
+, found: 761.3843, 783.3646. HPLC (semipreparative): t
R = 24.2 min (method 1).
2’-Deoxyuridine-derived muraymycin analogue (
14): Nucleosyl amino acid ester
19 (12.6 mg, 27.6 μmol) was dissolved in THF (1.7 mL), and a solution of aldehyde
17 [
42] (17 mg, 28 μmol) in THF (1.7 mL) was added. The reaction mixture was stirred over molecular sieve (3 Å) at rt for 24 h. NaBH(OAc)
3 (12.3 mg, 58.0 μmol) and Amberlyst 15
TM (1.9 mg, 5.5 μmol) were added and stirring was continued at rt for 24 h. The reaction mixture was filtered and the residue was washed with EtOAc (150 mL). The combined filtrates were washed with sat. NaHCO
3 solution (100 mL) and brine (50 mL). The aqueous layer was extracted with EtOAc (3 × 100 mL). The combined organics were dried over Na
2SO
4 and the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (98:2→96:4→95:5, CH
2Cl
2-MeOH) to give the fully protected muraymycin analogue as a colorless solid. This was dissolved in aq. TFA (80%, 1.3 mL) and stirred at rt for 24 h. The reaction mixture was then diluted with water (12 mL) and the solvent was evaporated under reduced pressure. The resultant crude product was purified by semipreparative HPLC (method 1) to give
14 (bis-TFA salt) as a colorless foam (3.5 mg, 13% over 2 steps from
19).
1H NMR (500 MHz, D
2O): δ [ppm] = 0.84 (d,
J = 6.0 Hz, 3H, Leu-5-H), 0.89 (d,
J = 6.0 Hz, 3H, Leu-5-H), 0.90 (d,
J = 6.6 Hz, 3H, Val-4-H), 0.93 (d,
J = 6.9 Hz, 3H, Val-4-H), 1.35–1.46 (m, 2H, Lys-4-H), 1.50–1.69 (m, 6H, Lys-3-H
a, Lys-5-H, Leu-3-H, Leu-4-H), 1.72–1.80 (m, 1H, Lys-3-H
b), 1.85–1.91 (m, 2H, 2″-H), 2.10–2.17 (m, 1H, Val-3-H), 2.20 (ddd,
J = 15.1, 10.4, 6.0 Hz, 1H, 5′-H
a), 2.33 (ddd,
J = 14.5, 7.3, 5.7 Hz, 1H, 2′-H
a), 2.38 (ddd,
J = 14.8, 6.9, 2.8 Hz 1H, 5′-H
b), 2.45–2.51 (m, 1H, 2′-H
b), 2.96 (t,
J = 7.6 Hz, 2H, Lys-6-H), 3.04 (t,
J = 7.6 Hz, 2H, 1″-H), 3.19–3.32 (m, 2H, 3″-H
a, 3″-H
b), 3.88 (t,
J = 6.3 Hz, 1H, 6′-H), 4.03 (ddd,
J = 10.4, 5.0, 2.8 Hz, 1H, 4′-H), 4.05 (d,
J = 5.4 Hz, 1H, Val-2-H), 4.09 (dd,
J = 8.8, 5.7 Hz, 1H, Lys-2-H), 4.23 (dd,
J = 9.8, 4.7 Hz, 1H, Leu-2-H), 4.28–4.32 (m, 1H, 3′-H), 5.85 (d,
J = 8.2 Hz, 1H, 5-H), 6.16 (dd,
J = 6.6, 6.6 Hz, 1H, 1′-H), 7.66 (d,
J = 8.2 Hz, 1H, 6-H).
13C NMR (126 MHz, D
2O): δ [ppm] = 17.03 (Val-C-4), 18.56 (Val-C-4), 20.71 (Leu-C-4), 22.10 (Leu-C-4), 22.16 (Lys-C-4), 24.46 (Lys-C-5), 25.69 (C-2″), 26.32 (Leu-C-4), 30.02 (Val-C-3), 30.84 (Lys-C-3), 32.95 (C-5′), 35.99 (C-3″), 37.47 (C-2′), 39.29 (Lys-C-6), 39.56 (Leu-C-6), 44.25 (C-1″), 52.65 (Leu-C-2), 54.13 (Lys-C-2), 58.95 (Val-C-2), 59.60 (C-6′), 73.53 (C-3′), 82.56 (C-4′), 86.33 (C-1′), 102.30 (C-5), 142.87 (C-6), 151.51 (C-2), 159.56 (urea-C=O), 166.27 (C-4), 171.90 (C-7′), 174.97 (Leu-C-1), 175.48 (Lys-C-1), 176.79 (Val-C-1).
19F NMR (376 MHz, D
2O): δ [ppm] = −75.59 (TFA-CF
3). HRMS (ESI
+): calcd.: 727.3985 [M + H]
+, found: 727.3966. HPLC (semipreparative): t
R = 25.9 min (method 1).
Thymidine-derived muraymycin analogue (
15): Nucleosyl amino acid ester
20 (25 mg, 53 μmol) was dissolved in THF (3 mL), and a solution of aldehyde
17 [
42] (42 mg, 68 μmol) in THF (3 mL) was added. The reaction mixture was stirred over molecular sieve (3 Å) at rt for 24 h. NaBH(OAc)
3 (23 mg, 0.11 mmol) and Amberlyst 15
TM (3.5 mg, 11 μmol) were added and stirring was continued at rt for 24 h. The reaction mixture was filtered and the residue was washed with EtOAc (150 mL). The combined filtrates were washed with sat. NaHCO
3 solution (100 mL) and brine (50 mL). The aqueous layer was extracted with EtOAc (3 × 100 mL). The combined organics were dried over Na
2SO
4 and the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (98:2→95:5, CH
2Cl
2-MeOH) to give the fully protected muraymycin analogue as a colorless solid. This was dissolved in aq. TFA (80%, 6.5 mL) and stirred at rt for 24 h. The reaction mixture was then diluted with water (12 mL) and the solvent was evaporated under reduced pressure. The resultant crude product was purified by semipreparative HPLC (method 2) to give
15 (bis-TFA salt) as a colorless foam (9.0 mg, 15% over 2 steps from
20).
1H NMR (500 MHz, D
2O): δ [ppm] = 0.83 (d,
J = 6.0 Hz, 3H, Leu-5-H), 0.88 (d,
J = 5.7 Hz, 3H, Leu-5-H), 0.89 (d,
J = 6.6 Hz, 3H, Val-4-H), 0.93 (d,
J = 6.9 Hz, 3H, Val-4-H), 1.31–1.45 (m, 2H, Lys-4-H), 1.50–1.68 (m, 6H, Lys-3-H
a, Lys-5-H, Leu-3-H, Leu-4-H), 1.73–1.76 (m, 1H, Lys-3-H
b), 1.85–1.90 (m, 2H, 2″-H), 1.86 (s, 3H, 7-H), 2.10–2.16 (m, 1H, Val-3-H), 2.20–2.25 (m, 1H, 5′-H
a), 2.30 (ddd,
J = 14.2, 6.9, 5.0 Hz, 1H, 2′-H
a), 2.39–2.44 (m, 1H, 5′-H
b), 2.46–2.51 (m, 1H, 2′-H
b), 2.96 (t,
J = 7.6 Hz, 2H, Lys-6-H), 3.06 (t,
J = 7.3 Hz, 2H, 1″-H), 3.20–3.29 (m, 2H, 3″-H), 3.94–3.97 (m, 1H, 6′-H), 4.02 (ddd,
J = 10.4, 4.7, 2.8 Hz, 1H, 4′-H), 4.05 (d,
J = 5.4 Hz, 1H, Val-2-H), 4.05–4.10 (dd,
J = 8.5, 5.4 Hz, 1H, Lys-2-H), 4.21 (dd,
J = 9.8, 4.7 Hz, 1H, Leu-2-H), 4.30–4.33 (m, 1H, 3′-H), 6.16 (dd,
J = 6.6, 6.6 Hz, 1H, 1′-H), 7.44 (s, 1H, 6-H).
13C NMR (126 MHz, D
2O): δ [ppm] = 11.57 (C-7), 17.02 (Val-C-4), 18.54 (Val-C-4), 20.70 (Leu-C-4), 22.09 (Leu-C-4), 22.16 (Lys-C-4), 24.45 (Lys-C-5), 25.67 (C-2″), 26.32 (Leu-C-4), 30.00 (Val-C-3), 30.82 (Lys-C-3), 32.73 (C-5′), 35.96 (C-3″), 37.24 (C-2′), 39.28 (Lys-C-6), 39.55 (Leu-C-6), 44.19 (C-1″), 52.62 (Leu-C-2), 54.15 (Lys-C-2), 58.91 (Val-C-2), 59.16 (C-6′), 73.16 (C-3′), 82.30 (C-4′), 85.96 (C-1′), 111.52 (C-5), 116.41 (d,
JCF = 291.4 Hz, TFA-CF
3), 138.34 (C-6), 151.65 (C-2), 159.56 (urea-C=O), 163.05 (d,
JCF = 35.7 Hz, TFA-COO), 166.51 (C-4), 171.57 (C-7′), 174.95 (Leu-C-1), 175.46 (Lys-C-1), 176.73 (Val-C-1).
19F NMR (376 MHz, D
2O): δ [ppm] = −75.59 (TFA-CF
3). MS (ESI
+):
m/z = 741.5 [M + H]
+, 763.5 [M + Na]
+. HRMS (ESI
+): calcd.: 741.4141 [M + H]
+, found: 741.4122. HPLC (semipreparative): t
R = 13.9 min (method 2).
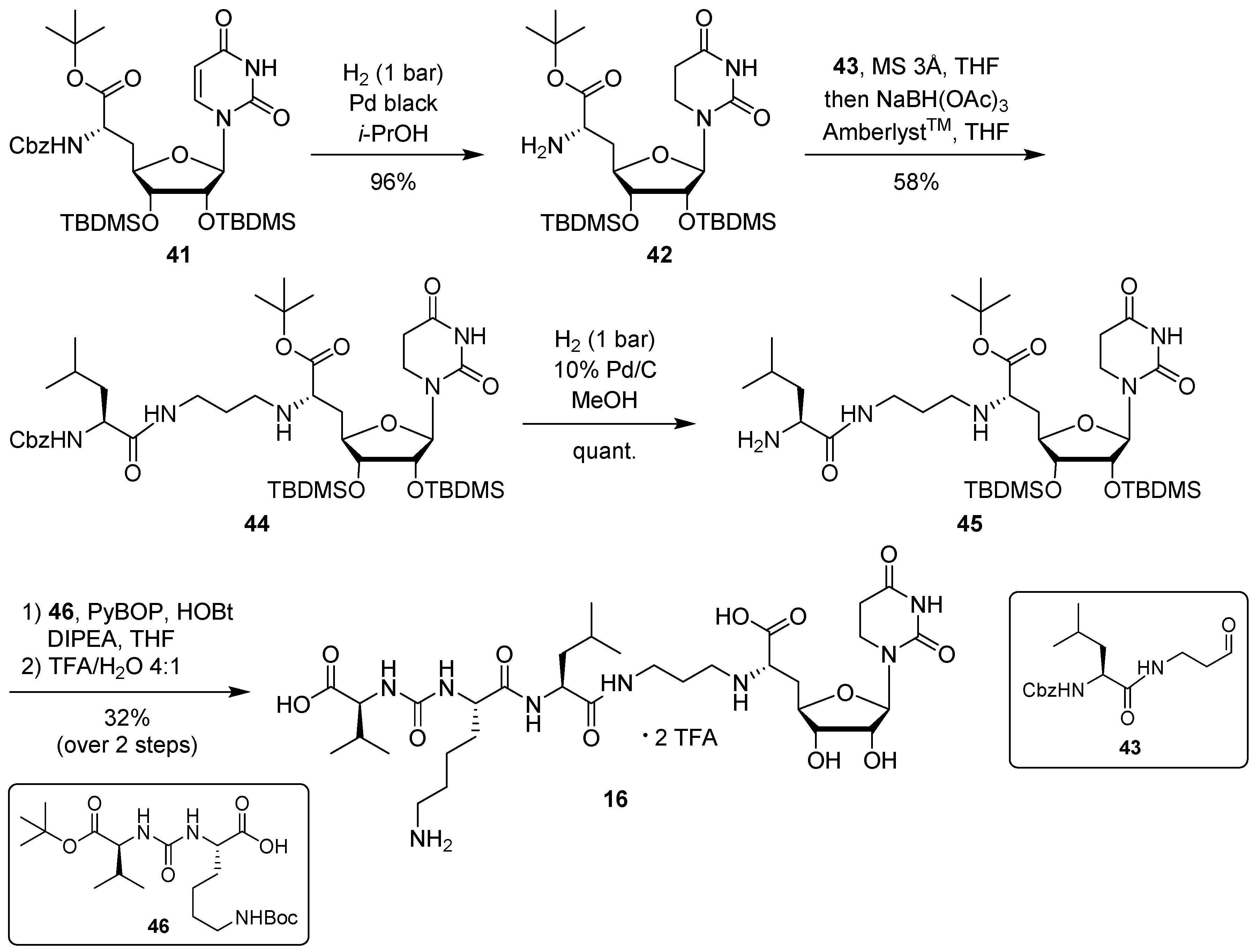
5,6-Dihydrouridine-derived muraymycin analogue (
16): Protected urea dipeptide
46 [
24,
41] (15.5 mg, 34.8 μmol) was dissolved in THF (5 mL), and 1-hydroxybenzotriazole (HOBt, 4.9 mg, 36 μmol), benzotriazole-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOB, 18.7 mg, 35.9 μmol) and DIPEA (12.2 μL, 71.8 μmol) were added. The reaction mixture was stirred at rt for 30 min. After cooling to 0 °C, a solution of 5,6-dihydrouridine derivative
45 (25.2 mg, 33.2 μmol) in THF (7 mL) was added. The reaction mixture was stirred at 0 °C for 30 min and at rt for 3 h. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (98:2→95:5, CH
2Cl
2-MeOH) to give the fully protected muraymycin analogue. This was dissolved in aq. TFA (80%, 5 mL) and stirred at rt for 24 h. The reaction mixture was then diluted with water (15 mL) and the solvent was evaporated under reduced pressure. The resultant crude product was purified by semipreparative HPLC (method 3) to give
16 (bis-TFA salt) as a colorless solid (7.9 mg, 32% over 2 steps from
45).
1H NMR (500 MHz, D
2O, 15 °C): δ [ppm] = 0.79 (d,
J = 5.0 Hz, 3H, Leu-5-H), 0.82–0.87 (m, 6H, Leu-5-H, Val-4-H), 0.89 (d,
J = 6.4 Hz, 3H, Val-4-H), 1.31–1.41 (m, 2H, Lys-4-H), 1.44–1.56 (m, 3H, Leu-3-H, Leu-4-H), 1.56–1.66 (m, 2H, Lys-5-H), 1.67–1.76 (m, 2H, Lys-3-H), 1.78–1.89 (m, 2H, 2″-H), 2.04–2.16 (m, 2H, Val-3-H, 5′-H
a), 2.29–2.37 (m, 1H, 5′-H
b), 2.68 (t,
J = 6.5 Hz, 2H, 5-H), 2.89–2.94 (m, 2H, Lys-6-H), 2.98–3.04 (m, 2H, 1″-H), 3.16–3.25 (m, 2H, 3″-H), 3.45 (t,
J = 6.5 Hz, 2H, 6-H), 3.84–3.88 (m, 1H, 6′-H), 3.89–3.95 (m, 1H, 3′-H), 3.96–4.09 (m, 3H, 4′-H, Lys-2-H, Val-2-H), 4.12–4.22 (m, 1H, Leu-2-H), 4.25 (dd,
J = 5.3, 4.9 Hz, 1H, 2′-H), 5.67 (d,
J = 4.9 Hz, 1H, 1′-H).
13C NMR (126 MHz, D
2O): δ [ppm] = 17.04 (Val-C-4), 18.56 (Val-C-4), 20.72 (Leu-C-5), 22.10 (Lys-C-4), 22.16 (Leu-C-5), 24.46 (Leu-C-4), 25.69 (C-2″), 26.32 (Lys-C-5), 30.01 (Val-C-3), 30.21 (C-5), 30.83 (Lys-C-3), 33.04 (C-5′), 36.00 (C-3″), 37.56 (C-6), 39.30 (Lys-C-6), 39.60 (Leu-C-3), 44.31 (C-1″), 52.65 (Leu-C-2), 54.11 (Lys-C-2), 58.95 (Val-C-2), 59.42 (C-6′), 70.50 (C-2′), 73.30 (C-3′), 79.07 (C-4′), 89.51 (C-1′), 116.47 (q,
JCF = 291.4 Hz, TFA-CF
3), 154.45 (C-2), 159.58 (urea-C=O), 163.10 (q,
JCF = 35.6 Hz, TFA-COO), 171.47 (C-7′), 173.75 (C-4), 174.99 (Lys-C-1), 175.46 (Leu-C-1), 176.78 (Val-C-1).
19F NMR (376 MHz, D
2O): δ [ppm] = −76.04 (TFA-CF
3). HRMS (ESI
+): calcd.: 745.4090 [M + H]
+, found: 745.4074. HPLC (semipreparative): t
R = 5.9 min (method 3).
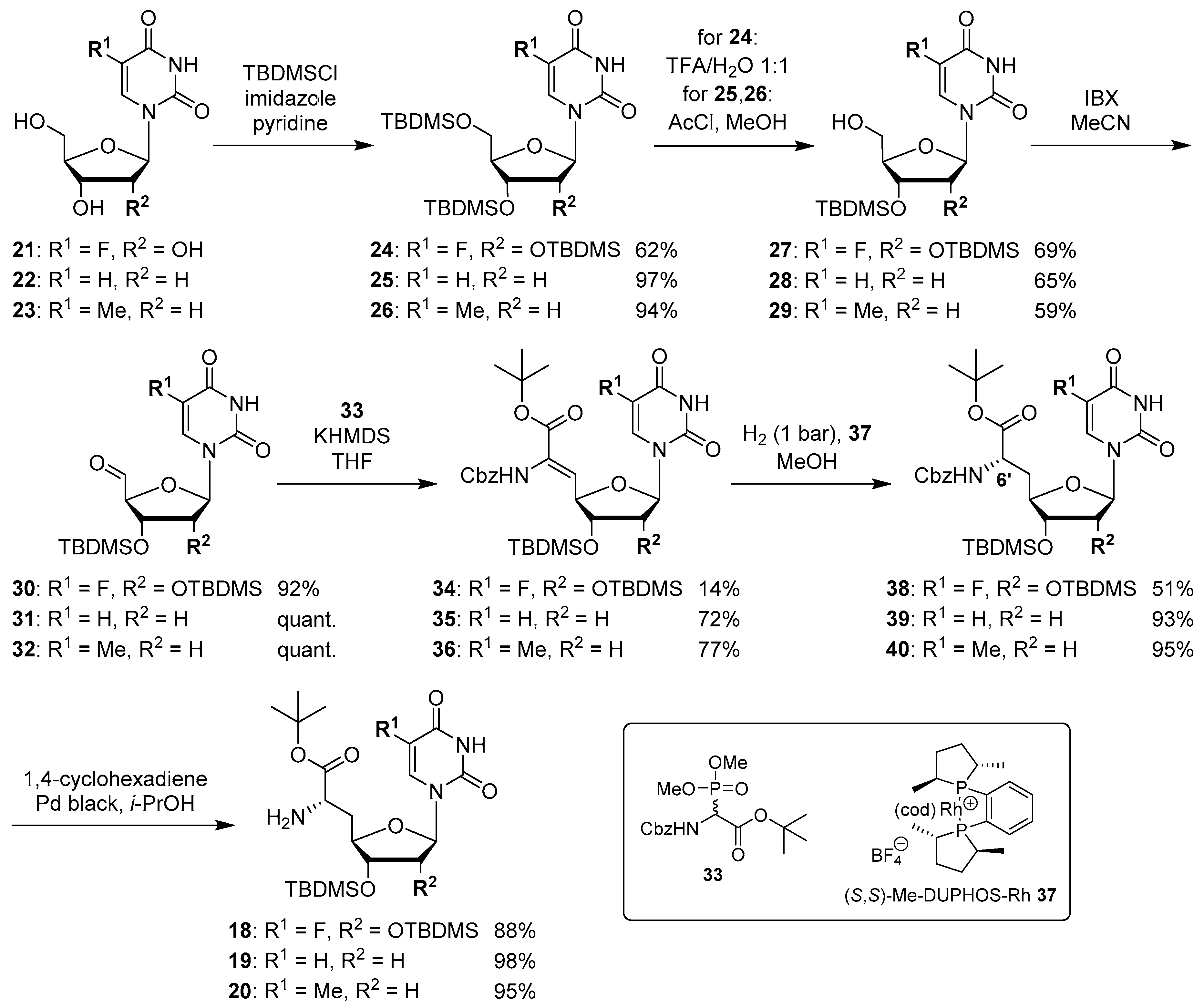
(6′S)-5-Fluorouridinyl amino acid tert-butyl ester (18): Cbz-protected nucleosyl amino acid ester 38 (25 mg, 34 μmol) was dissolved in i-PrOH (2 mL). 1,4-Cyclohexadiene (31 μL, 0.34 mmol) and Pd black (~4 mg, 38 μmol) were added and the reaction mixture was stirred at rt for 3 h. After filtration through a syringe filter and washing the filter with EtOAc (3 × 5 mL), the solvent of the combined filtrates was evaporated under reduced pressure to give 18 as a colorless foam (18 mg, 88%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.08 (s, 6H, Si(CH3)2C(CH3)3), 0.12 (s, 6H, Si(CH3)2C(CH3)3), 0.90 (s, 9H, Si(CH3)2C(CH3)3), 0.91 (s, 9H, Si(CH3)2C(CH3)3), 1.47 (s, 9H, COOC(CH3)3), 1.86 (ddd, J = 17.0, 10.4, 6.3 Hz, 1H, 5′-Ha), 2.18 (ddd, J = 14.2, 6.3, 2.8 Hz, 1H, 5′-Hb), 3.63 (dd, J = 6.3, 6.3 Hz, 1H, 6′-H), 3.67 (dd, J = 6.6, 4.1 Hz, 1H, 3′-H), 4.16 (dd, J = 4.1, 2.8 Hz, 1H, 2′-H), 4.21 (ddd, J = 10.7, 6.6, 2.9 Hz, 1H, 4′-H), 5.63 (d, J = 1.9 Hz, 1H, 1′-H), 7.58 (d, JHF = 6.0 Hz, 1H, 6-H). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.84 (Si(CH3)2C(CH3)3), −4.31 (Si(CH3)2C(CH3)3), 17.91 (Si(CH3)2C(CH3)3), 18.05 (Si(CH3)2C(CH3)3), 25.76 (Si(CH3)2C(CH3)3), 25.84 (Si(CH3)2C(CH3)3), 27.98 (COOC(CH3)3), 37.80 (C-5′), 53.18 (C-6′), 75.01 (C-2′), 75.19 (C-3′), 81.34 (C-4′), 81.74 (COOC(CH3)3), 91.56 (C-1′), 124.26 (d, JCF = 34.8 Hz, C-6), 140.4 (d, JCF = 237.4 Hz, C-5), 148.55 (C-2), 156.82 (d, JCF = 26.6 Hz, C-4). 19F NMR (376 MHz, CDCl3): −164.54. MS (ESI+): m/z = 604.4 [M + H]+, 626.4 [M + Na]+. HRMS (ESI+): calcd.: 604.3244 [M + H]+, 626.3064 [M + Na]+, found: 604.3232, 626.3051. TLC: Rf = 0.31 (95:5, CH2Cl2-MeOH).
(6′S)-2′-Deoxyuridinyl amino acid tert-butyl ester (19): Cbz-protected nucleosyl amino acid ester 39 (50 mg, 85 μmol) was dissolved in i-PrOH (5 mL). 1,4-Cyclohexadiene (80 μL, 0.85 mmol) and Pd black (~4 mg, 38 μmol) were added and the reaction mixture was stirred at rt for 1.5 h. After filtration through a syringe filter and washing the filter with EtOAc (3 × 10 mL), the solvent of the combined filtrates was evaporated under reduced pressure to give 19 as a colorless foam (38 mg, 98%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.08 (s, 6H, Si(CH3)2C(CH3)3), 0.89 (s, 9H, Si(CH3)2C(CH3)3), 1.47 (s, 9H, COOC(CH3)3), 1.84 (ddd, J = 14.2, 9.5, 6.9 Hz, 1H, 5′-Ha), 2.11–2.17 (m, 2H, 2′-Ha, 5′-Hb), 2.30 (ddd, J = 13.6, 6.6, 5.4 Hz, 1H, 2′-Hb), 3.57 (dd, J = 6.3, 6.3 Hz, 1H, 6′-H), 3.95 (ddd, J = 9.1, 5.0, 3.5 Hz, 1H, 4′-H), 4.11 (dd, J = 12.0, 5.0 Hz, 1H, 3′-H), 5.76 (d, J = 7.9 Hz, 1H, 5-H), 6.16 (dd, J = 6.0, 6.0 Hz, 1H, 1′-H), 7.44 (d, J = 8.2 Hz, 1H, 6-H). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.71 (Si(CH3)2C(CH3)3), 17.90 (Si(CH3)2C(CH3)3), 25.66 (Si(CH3)2C(CH3)3), 28.00 (COOC(CH3)3), 37.85 (C-5′), 40.58 (C-2′), 53.09 (C-6′), 74.89 (C-3′), 81.65 (COOC(CH3)3), 84.28 (C-4′), 85.25 (C-1′), 102.55 (C-5), 139.88 (C-6), 149.90 (C-2), 162.86 (C-4), 173.81 (C-7′). MS (ESI+): m/z = 456.1 [M + H]+, 478.2 [M + Na]+. HRMS (ESI+): calcd.: 456.2524 [M + H]+, found: 456.2498.
(6′S)-Thymidinyl amino acid tert-butyl ester (20): Cbz-protected nucleosyl amino acid ester 40 (50 mg, 83 μmol) was dissolved in i-PrOH (5 mL). 1,4-Cyclohexadiene (77 μL, 0.82 mmol) and Pd black (~4 mg, 38 μmol) were added and the reaction mixture was stirred at rt for 2.5 h. After filtration through a syringe filter and washing the filter with EtOAc (3 × 10 mL), the solvent of the combined filtrates was evaporated under reduced pressure to give 20 as a colorless foam (37 mg, 95%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.09 (s, 6H, Si(CH3)2C(CH3)3), 0.90 (s, 9H, Si(CH3)2C(CH3)3), 1.47 (s, 9H, COOC(CH3)3), 1.95 (d, J = 1.1 Hz, 3H, 7-H), 1.86 (ddd, J = 16.1, 9.3, 6.9 Hz, 1H, 5′-Ha), 2.13–2.18 (m, 2H, 2′-Ha, 5′-Hb), 2.26 (ddd, J = 13.6, 6.7, 5.0 Hz, 1H, 2′-Hb), 3.58 (dd, J = 6.4, 6.4 Hz, 1H, 6′-H), 3.93 (ddd, J = 9.2, 4.9, 4.0 Hz, 1H, 4′-H), 4.13 (dd, J = 12.1, 5.0 Hz, 1H, 3′-H), 6.17 (dd, J = 6.5, 6.5 Hz, 1H, 1′-H), 7.20 (d, J = 1.1 Hz, 1H, 6-H). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.71 (Si(CH3)2C(CH3)3), 12.58 (C-7), 17.90 (Si(CH3)2C(CH3)3), 25.67 (Si(CH3)2C(CH3)3), 27.98 (COOC(CH3)3), 37.86 (C-5′), 40.33 (C-2′), 53.10 (C-6′), 75.00 (C-3′), 81.57 (COOC(CH3)3), 84.04 (C-4′), 84.87 (C-1′), 111.07 (C-5), 135.63 (C-6), 149.97 (C-2), 163.43 (C-4), 173.88 (C-7′). MS (ESI+): m/z = 470.1 [M + H]+, 492.3 [M + Na]+. HRMS (ESI+): calcd.: 470.2681 [M + H]+, found: 470.2658.
2′,3′,5′-Tris-O-TBDMS-5-fluorouridine (24): 5-Fluorouridine 21 (2.00 g, 7.62 mmol) was coevaporated with dry pyridine (2 × 14 mL). Pyridine (26 mL), imidazole (1.56 g, 22.8 mmol) and TBDMSCl (5.74 g, 38.1 mmol) were added and the reaction mixture was stirred at rt for 4 d. Water (90 mL) was then added at 0 °C. The solvent was evaporated under reduced pressure, the resultant colorless residue was dissolved in EtOAc (100 mL) and washed with sat. NaHCO3 solution (3 × 100 mL) and brine (2 × 20 mL). The organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (7:3, petroleum ether-EtOAc) to give 24 as a colorless foam (2.87 g, 62%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.07 (s, 6H, Si(CH3)2C(CH3)3), 0.09 (s, 6H, Si(CH3)2C(CH3)3), 0.16 (s, 6H, Si(CH3)2C(CH3)3), 0.89 (s, 9H, Si(CH3)2C(CH3)3), 0.92 (s, 9H, Si(CH3)2C(CH3)3), 0.97 (s, 9H, Si(CH3)2C(CH3)3), 3.77 (dd, J = 11.7, 1.0 Hz, 1H, 5′-Ha), 4.01 (dd, J = 11.7, 1.2 Hz, 1H, 5′-Hb), 4.06–4.10 (m, 3H, 2′-H, 3′-H, 4′-H), 5.89 (dd, J = 4.1, 1.2 Hz, 1H, 1′-H), 8.18 (d, JHF = 6.3 Hz, 1H, 6-H), 8.73 (bs, 1H, 3-NH). 13C NMR (126 MHz, CDCl3): δ [ppm] = −5.54 (Si(CH3)2C(CH3)3), −4.81 (Si(CH3)2C(CH3)3), −3.94 (Si(CH3)2C(CH3)3), 17.92 (Si(CH3)2C(CH3)3), 18.05 (Si(CH3)2C(CH3)3), 18.61 (Si(CH3)2C(CH3)3), 25.70 (Si(CH3)2C(CH3)3), 25.79 (Si(CH3)2C(CH3)3), 26.06 (Si(CH3)2C(CH3)3), 62.15 (C-5′), 71.16 (C-3′), 76.12 (C-4′), 85.11 (C-2′), 88.78 (C-1′), 124.41 (d, JCF = 33.9 Hz, C-6), 140.37 (d, JCF = 236.4 Hz, C-5), 148.65 (C-2), 156.67 (d, JCF = 26.6 Hz, C-4). 19F NMR (376 MHz, CDCl3): δ [ppm] = −164.60. MS (ESI−): m/z = 605.3 [M + H]+, 627.3 [M + Na]+. HRMS (ESI)+: calcd.: 605.3268 [M + H]+, found: 605.3261. TLC: Rf = 0.85 (1:1, petroleum ether-EtOAc).
3′,5′-Bis-O-TBDMS-2′-deoxyuridine (25): 2′-Deoxyuridine 22 (5.09 g, 22.3 mmol) was coevaporated with dry pyridine (2 × 40 mL). Pyridine (75 mL), imidazole (4.58 g, 66.9 mmol) and TBDMSCl (10.1 g, 66.9 mmol) were added and the reaction mixture was stirred at rt for 31 h. Water (50 mL) was then added at 0 °C. The solvent was evaporated under reduced pressure, the resultant colorless residue was dissolved in EtOAc (250 mL) and washed with sat. NaHCO3 solution (3 × 125 mL) and brine (50 mL). The organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (3:2, petroleum ether-EtOAc) to give 25 as a colorless solid (9.90 g, 97%). 1H NMR (300 MHz, CDCl3): δ [ppm] = 0.08 (s, 3H, Si(CH3)2C(CH3)3), 0.09 (s, 3H, Si(CH3)2C(CH3)3), 0.11 (s, 3H, Si(CH3)2C(CH3)3), 0.11 (s, 3H, Si(CH3)2C(CH3)3), 0.90 (s, 9H, Si(CH3)2C(CH3)3), 0.98 (s, 9H, Si(CH3)2C(CH3)3), 2.07 (ddd, J = 13.2, 6.2, 6.2 Hz, 1H, 2′-Ha), 2.33 (ddd, J = 13.2, 6.2, 4.2 Hz, 1H, 2′-Hb), 3.77 (dd, J = 11.3, 2.5 Hz, 1H, 5′-Ha), 3.90 (dd, J = 11.3, 2.5 Hz, 1H, 5′-Hb), 3.92–3.94 (m, 1H, 4′-H), 4.42 (ddd, J = 6.0, 6.0, 3.8 Hz, 1H, 3′-H), 5.68 (d, J = 8.0 Hz, 1H, 5-H), 6.29 (dd, J = 6.2, 6.2 Hz, 1H, 1′-H), 7.47 (d, J = 8.0 Hz, 1H, 6-H), 8.16 (s, 1H, 3-NH). 13C NMR (126 MHz, CDCl3): δ [ppm] = −5.43 (Si(CH3)2C(CH3)3), −5.36 (Si(CH3)2C(CH3)3), −4.72 (Si(CH3)2C(CH3)3), −4.47 (Si(CH3)2C(CH3)3), 18.12 (Si(CH3)2C(CH3)3), 18.50 (Si(CH3)2C(CH3)3), 25.86 (Si(CH3)2C(CH3)3), 26.02 (Si(CH3)2C(CH3)3), 42.00 (C-2′), 62.60 (C-5′), 71.38 (C-3′), 85.35 (C-1′), 87.94 (C-4′), 102.25 (C-5), 140.34 (C-6), 150.10 (C-2), 162.95 (C-4). HRMS (CI): calcd.: 457.2549 [M + H]+, found: 457.2538. TLC: Rf = 0.40 (1:1, CH2Cl2-EtOAc).
3′,5′-Bis-O-TBDMS-thymidine (26): Thymidine 23 (5.40 g, 22.3 mmol) was coevaporated with dry pyridine (2 × 22 mL). Pyridine (50 mL), imidazole (3.88 g, 57.0 mmol) and TBDMSCl (8.43 g, 56.0 mmol) were added and the reaction mixture was stirred at rt for 48 h. Water (50 mL) was then added at 0 °C. The solvent was evaporated under reduced pressure, the resultant colorless residue was dissolved in EtOAc (250 mL) and washed with sat. NaHCO3 solution (3 × 125 mL) and brine (50 mL). The organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (3:1, petroleum ether-EtOAc) to give 26 as a colorless solid (9.81 g, 94%). 1H NMR (300 MHz, CDCl3): δ [ppm] = 0.08 (s, 3H, Si(CH3)2C(CH3)3), 0.08 (s, 3H, Si(CH3)2C(CH3)3), 0.11 (s, 3H, Si(CH3)2C(CH3)3), 0.11 (s, 3H, Si(CH3)2C(CH3)3), 0.90 (s, 9H, Si(CH3)2C(CH3)3), 0.93 (s, 9H, Si(CH3)2C(CH3)3), 1.92 (d, J = 0.8 Hz, 3H, 7-H), 1.95–2.05 (m, 1H, 2′-Ha), 2.25 (ddd, J = 13.1, 5.8, 2.5 Hz, 1H, 2′-Hb) 3.76 (dd, J = 11.4, 2.5 Hz, 1H, 5′-Ha), 3.87 (dd, J = 11.4, 2.5 Hz, 1H, 5′-Hb), 3.94 (dd, J = 2.5, 2.5 Hz, 1H, 4′-H), 4.41 (ddd, J = 5.8, 2.5, 2.5 Hz, 1H, 3′-H), 6.33 (dd, J = 8.0, 5.8 Hz, 1H, 1′-H), 7.47 (d, J = 0.8 Hz, 1H, 6-H), 8.03 (s, 1H, 3-NH). 13C NMR (126 MHz, CDCl3): δ [ppm] = −5.29 (Si(CH3)2C(CH3)3), −5.20 (Si(CH3)2C(CH3)3), −4.67 (Si(CH3)2C(CH3)3), −4.48 (Si(CH3)2C(CH3)3), 12.68 (C-7), 18.17 (Si(CH3)2C(CH3)3), 18.57 (Si(CH3)2C(CH3)3), 25.90 (Si(CH3)2C(CH3)3), 26.09 (Si(CH3)2C(CH3)3), 41.53 (C-2′), 63.16 (C-5′), 72.45 (C-3′), 85.00 (C-1′), 88.01 (C-4′), 110.93 (C-5), 135.64 (C-6), 150.17 (C-2), 163.52 (C-4). HRMS (CI): calcd.: 471.2705 [M + H]+, found: 471.2694. TLC: Rf = 0.29 (1:1, CH2Cl2-EtOAc).
2′,3′-Bis-O-TBDMS-5-fluorouridine (27): 2′,3′,5′-Tris-O-TBDMS-5-fluorouridine 24 (1.09 g, 1.80 mmol) was dissolved in THF (22 mL) and aq. TFA (50%, 5.4 mL) was added slowly at 0 °C over 40 min. The reaction mixture was stirred at 0 °C for 5.5 h. After conversion was sufficient according to TLC, sat. NaHCO3 solution (70 mL) was added until pH 8 was reached. The aqueous layer was extracted with EtOAc (3 × 70 mL) and the combined organics were dried over Na2SO4. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (7:3, petroleum ether-EtOAc) to give 27 as a colorless foam (610 mg, 69%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.09 (s, 6H, Si(CH3)2C(CH3)3), 0.10 (s, 6H, Si(CH3)2C(CH3)3), 0.90 (s, 9H, Si(CH3)2C(CH3)3), 0.92 (s, 9H, Si(CH3)2C(CH3)3), 2.45 (bs, 1H, 5′-OH), 3.80 (ddd, J = 11.9, 5.0, 1.9 Hz, 1H, 5′-Ha), 4.04 (ddd, J = 11.6, 2.2 Hz, 1H, 5′-Hb), 4.11 (d, J = 5.0 Hz, 1H, 4′-H), 4.16 (dd, J = 4.7, 4.4 Hz, 1H, 3′-H), 4.34 (dd, J = 4.7, 4.4 Hz, 1H, 2′-H), 5.60 (d, J = 6.0 Hz, 1H, 1′-H), 8.12 (d, JHF = 6.6 Hz, 1H, 6-H), 9.00 (bs, 1H, 3-NH). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.84 (Si(CH3)2C(CH3)3), −4.55 (Si(CH3)2C(CH3)3), 17.95 (Si(CH3)2C(CH3)3), 18.05 (Si(CH3)2C(CH3)3), 25.75 (Si(CH3)2C(CH3)3), 25.80 (Si(CH3)2C(CH3)3), 60.89 (C-5′), 70.76 (C-4′), 74.87 (C-2′), 84.88 (C-3′), 91.89 (C-1′), 126.12 (d, JCF = 34.8 Hz, C-6), 139.29 (d, JCF = 236.4 Hz, C-5), 148.76 (C-2), 156.78 (d, JCF = 26.6 Hz, C-4). 19F NMR (376 MHz, CDCl3): δ [ppm] = −165.47. MS (ESI+): m/z = 513.2 [M + Na]+. HRMS (ESI+): calcd.: 491.2403 [M + H]+, 513.2223 [M + Na]+, found: 491.2392, 513.2210. TLC: Rf = 0.19 (7:3, petroleum ether-EtOAc).
3′-O-TBDMS-2′-deoxyuridine (28): 3′,5′-Bis-O-TBDMS-2′-deoxyuridine 25 (7.09 g, 15.5 mmol) was dissolved in MeOH (200 mL) and cooled to −10 °C. Acetyl chloride (273 μL, 3.84 mmol) was added slowly at this temperature. The mixture was warmed to 0 °C and stirred at this temperature for 2.5 h. After conversion was sufficient according to TLC, sat. NaHCO3 solution (150 mL) was added and the mixture was stirred for 15 min. Then EtOAc (300 mL), water (100 mL) and brine (200 mL) were added. The organic layer was washed with sat. NaHCO3 solution (100 mL), brine (2 x 100 mL) and water (100 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (3:2, petroleum ether-EtOAc→7:3, petroleum ether-EtOAc→EtOAc) to give 28 as a colorless solid (3.47 g, 65%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.08 (s, 6H, Si(CH3)2C(CH3)3), 0.89 (s, 9H, Si(CH3)2C(CH3)3), 2.26–2.30 (m, 2H, 2′-H), 2.51 (dd, J = 4.6, 4.6 Hz, 1H, 5′-OH), 3.76 (ddd, J = 11.6, 5.8, 3.2 Hz, 1H, 5′-Ha), 3.89–3.91 (m, 1H, 5′-Hb), 3.92–3.94 (m, 1H, 4′-H), 4.48 (ddd, J = 5.9, 4.2, 4.2 Hz, 1H, 3′-H), 5.73 (d, J = 8.1 Hz, 1H, 5-H), 6.18 (dd, J = 6.6, 6.6 Hz, 1H, 1′-H), 7.68 (d, J = 8.1 Hz, 1H, 6-H), 9.33 (s, 1H, 3-NH). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.90 (Si(CH3)2C(CH3)3), −4.74 (Si(CH3)2C(CH3)3), 17.92 (Si(CH3)2C(CH3)3), 25.67 (Si(CH3)2C(CH3)3), 40.87 (C-2′), 61.73 (C-5′), 71.38 (C-3′), 86.60 (C-1′), 87.61 (C-4′), 102.40 (C-5), 141.15 (C-6), 150.30 (C-2), 163.56 (C-4). HRMS (CI): calcd.: 343.1684 [M + H]+, found: 343.1687. TLC: Rf = 0.44 (EtOAc).
3′-O-TBDMS-thymidine (29): 3′,5′-Bis-O-TBDMS-thymidine 26 (7.02 g, 14.9 mmol) was dissolved in MeOH (340 mL) and cooled to −10 °C. Acetyl chloride (265 μL, 3.73 mmol) was added slowly at this temperature. The mixture was warmed to 0 °C and stirred at this temperature for 3.5 h. After conversion was sufficient according to TLC, sat. NaHCO3 solution (150 mL) was added and the mixture was stirred for 15 min. Then EtOAc (300 mL), water (100 mL) and brine (400 mL) were added. The organic layer was washed with sat. NaHCO3 solution (2 × 100 mL), water (100 mL) and brine (100 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (1:1, petroleum ether-EtOAc) to give 29 as a colorless solid (3.21 g, 59%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.09 (s, 6H, Si(CH3)2C(CH3)3), 0.90 (s, 9H, Si(CH3)2C(CH3)3), 1.92 (d, J = 1.1 Hz, 3H, 7-H), 2.22 (ddd, J = 13.3, 6.4, 3.8 Hz, 1H, 2′-Ha), 2.33–2.42 (m, 1H, 2′-Hb), 2.60 (brs, 1H, 5′-OH), 3.76 (ddd, J = 12.4, 6.5, 3.5 Hz, 1H, 5′-Ha), 3.90–3.95 (m, 2H, 4′-H, 5′-Hb), 4.48–4.52 (m, 1H, 3′-H), 6.14 (dd, J = 6.8, 6.8 Hz, 1H, 1′-H), 7.36 (d, J = 1.1 Hz, 1H, 6-H), 8.63 (s, 1H, 3-NH). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.87 (Si(CH3)2C(CH3)3), −4.71 (Si(CH3)2C(CH3)3), 12.52 (C-7), 17.94 (Si(CH3)2C(CH3)3), 25.70 (Si(CH3)2C(CH3)3), 40.41 (C-2′), 62.05 (C-5′), 71.60 (C-3′), 87.09 (C-1′), 87.56 (C-4′), 111.00 (C-5), 137.05 (C-6), 150.24 (C-2), 163.59 (C-4). HRMS (CI): calcd.: 357.1840 [M + H]+, found: 357.1848. TLC: Rf = 0.35 (1:1, petroleum ether-EtOAc).
2′,3′-Bis-O-TBDMS-5-fluorouridine-5′-aldehyde (30): 2′,3′-Bis-O-TBDMS-5-fluorouridine 27 (992 mg, 2.02 mmol) was dissolved in MeCN (21 mL) and IBX (1.19 g, 4.26 mmol) was added. The reaction mixture was stirred under reflux for 1.5 h. After cooling to 0 °C, stirring was continued for 30 min. The reaction mixture was then filtered and the solid residue was washed with EtOAc (6 × 40 mL). The solvent of the combined filtrates was evaporated under reduced pressure to give 30 as a colorless foam (910 mg, 92%). This material was used in the following step without further purification and characterization with respect to its limited stability. 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.04 (s, 6H, Si(CH3)2C(CH3)3), 0.14 (s, 6H, Si(CH3)2C(CH3)3), 0.89 (s, 9H, Si(CH3)2C(CH3)3), 0.94 (s, 9H, Si(CH3)2C(CH3)3), 4.18 (dd, J = 5.4, 4.1 Hz, 1H, 4′-H), 4.21 (dd, J = 4.1, 4.1 Hz, 1H, 3′-H), 4.64 (d, J = 3.5 Hz, 1H, 2′-H), 5.82 (dd, J = 5.4, 1.0 Hz, 1H, 1′-H), 8.02 (d, JHF = 6.0 Hz, 1H, 6-H), 8.53 (brs, 1H, 3-NH), 9.83 (s, 1H, 5’-H).
3′-O-TBDMS-2′-deoxyuridine-5′-aldehyde (31): 3′-O-TBDMS-2′-deoxyuridine 28 (1.25 g, 3.66 mmol) was dissolved in MeCN (35 mL) and IBX (2.56 g, 9.15 mmol) was added. The reaction mixture was stirred under reflux for 2 h. After cooling to 0 °C, stirring was continued for 30 min. The reaction mixture was then filtered and the solid residue was washed with EtOAc (3 × 50 mL). The solvent of the combined filtrates was evaporated under reduced pressure to give 31 as a colorless foam (1.25 g, quant.). This material was used in the following step without further purification and characterization with respect to its limited stability. 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.13 (s, 3H, Si(CH3)2C(CH3)3), 0.14 (s, 3H, Si(CH3)2C(CH3)3), 0.91 (s, 9H, Si(CH3)2C(CH3)3), 1.97–2.03 (m, 1H, 2′-Ha), 2.37 (ddd, J = 13.6, 5.8, 2.2 Hz, 1H, 2′-Hb), 4.52 (d, J = 2.0 Hz, 1H, 4′-H), 4.64–4.68 (m, 1H, 3′-H), 5.81 (d, J = 8.2 Hz, 1H, 5-H), 6.31 (dd, J = 8.1, 5.8 Hz, 1H, 1′-H), 7.85 (d, J = 8.2 Hz, 1H, 6-H), 9.26 (brs, 1H, 3-NH), 9.75 (s, 1H, 5′-H).
3′-O-TBDMS-thymidine-5′-aldehyde (32): 3′-O-TBDMS-thymidine 29 (1.32 g, 3.70 mmol) was dissolved in MeCN (35 mL) and IBX (2.59 g, 9.25 mmol) was added. The reaction mixture was stirred under reflux for 1 h. After cooling to 0 °C, stirring was continued for 30 min. The reaction mixture was then filtered and the solid residue was washed with EtOAc (3 × 40 mL). The solvent of the combined filtrates was evaporated under reduced pressure to give 32 as a colorless foam (1.31 g, quant.). This material was used in the following step without further purification and characterization with respect to its limited stability. 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.15 (s, 3H, Si(CH3)2C(CH3)3), 0.16 (s, 3H, Si(CH3)2C(CH3)3), 0.94 (s, 9H, Si(CH3)2C(CH3)3), 1.98 (d, J = 1.1 Hz, 3H, 7-H), 2.03–2.12 (m, 1H, 2′-Ha), 2.34 (ddd, J = 13.4, 5.9, 1.9 Hz, 1H, 2′-Hb), 4.50 (d, J = 2.1 Hz, 1H, 4′-H), 4.69 (ddd, J = 5.2, 2.1, 1.9 Hz, 1H, 3′-H), 6.33 (dd, J = 8.1, 5.9 Hz, 1H, 1′-H), 7.59 (d, J = 1.1 Hz, 1H, 6-H), 8.76 (brs, 1H, 3-NH), 9.78 (s, 1H, 5′-H).
Z-6′-
N-Cbz-5′,6′-didehydro-5-fluorouridinyl amino acid
tert-butyl ester (
34): THF (8 mL) was added to a solution of KHMDS in toluene (0.5 M, 2.84 mL, 1.42 mmol). After cooling to –86 °C, a solution of phosphonate
33 [
31] (480 mg, 1.42 mmol) in THF (10 mL) was added slowly at this temperature. The mixture was stirred at –86 °C for 30 min. Subsequently, a solution of aldehyde
30 (900 mg, 1.85 mmol) in THF (12 mL) was added dropwise. Stirring was continued for 16 h while the solution was allowed to slowly warm to rt. After cooling to 10 °C, MeOH (3.2 mL) was added. The mixture was diluted with EtOAc (70 mL) and washed with half-sat. NaCl solution (2 × 65 mL). The organic layer was dried over Na
2SO
4 and the solvent was evaporated under reduced pressure. The resultant crude product was purified by threefold column chromatography (7:3, petroleum ether-EtOAc and 3:1, petroleum ether-EtOAc, respectively) to give
34 (9:1 mixture of
Z and
E isomers as determined by
1H NMR spectroscopy) as a colorless foam (184 mg, 14%).
Z-
34:
1H NMR (500 MHz, CDCl
3): δ [ppm] = 0.07 (s, 6H, Si(C
H3)
2C(CH
3)
3), 0.12 (s, 6H, Si(C
H3)
2C(CH
3)
3), 0.90 (s, 9H, Si(CH
3)
2C(C
H3)
3), 0.91 (s, 9H, Si(CH
3)
2C(C
H3)
3), 1.49 (s, 9H, COOC(C
H3)
3), 3.90 (dd,
J = 3.8, 3.8 Hz, 1H, 3′-H), 4.28 (dd,
J = 3.5, 3.5 Hz, 1H, 2′-H), 4.92 (dd,
J = 7.0, 7.0 Hz, 1H, 4′-H), 5.13–5.18 (m, 2H, 1″-H), 5.60 (d,
J = 2.9 Hz, 1H, 1′-H), 6.24 (d,
J = 7.8 Hz, 1H, 5′-H), 6.80 (brs, 1H, 6′-NH), 7.34–7.36 (m, 5H, aryl-H), 7.39 (d,
JHF = 6.0 Hz, 1H, 6-H), 8.48 (d,
JHF = 4.4 Hz, 1H, 3-NH).
13C NMR (126 MHz, CDCl
3): δ [ppm] = −4.81 (Si(
CH
3)
2C(CH
3)
3), −4.47 (Si(
CH
3)
2C(CH
3)
3), 18.02 (Si(CH
3)
2C(CH
3)
3), 18.10 (Si(CH
3)
2C(CH
3)
3), 25.75 (Si(CH
3)
2C(
CH
3)
3), 25.82 (Si(CH
3)
2C(
CH
3)
3), 27.84 (COOC(
CH
3)
3), 67.60 (C-1″), 74.90 (C-2′), 76.03 (C-3′), 79.44 (C-4′), 82.90 (COO
C(CH
3)
3), 92.21 (C-1′), 124.28 (C-5′), 124.55 (d,
JCF = 34.8 Hz, C-6), 128.20 (C-3″/C-4″), 128.52 (C-3″/C-4″), 131.28 (C-2″), 135.71 (C-5″), 140.26 (d,
JCF = 238.3 Hz, C-5), 148.09 (C-2), 156.19 (d,
JCF = 27.5 Hz, C-4).
19F NMR (376 MHz, CDCl
3): δ [ppm] = −164.45. MS (ESI
+):
m/z = 736.4 [M + H]
+, 758.5 [M + Na]
+. HRMS (ESI
+): calcd.: 736.3455 [M + H]
+, 758.3275 [M + Na]
+, found: 736.3433, 758.3256. TLC:
Rf = 0.60 (1:1, petroleum ether-EtOAc).
E-
29:
1H NMR (500 MHz, CDCl
3): δ [ppm] = 0.07 (s, 6H, Si(C
H3)
2C(CH
3)
3), 0.12 (s, 6H, Si(C
H3)
2C(CH
3)
3), 0.90 (s, 9H, Si(CH
3)
2C(C
H3)
3), 0.91 (s, 9H, Si(CH
3)
2C(C
H3)
3), 1.49 (s, 9H, COOC(C
H3)
3), 3.84 (dd,
J = 5.7, 4.1 Hz, 1H, 3′-H), 4.23 (dd,
J = 3.8, 3.5 Hz, 1H, 2′-H), 5.16 (d,
J = 1.3 Hz, 2H, 1″-H), 5.46 (dd,
J = 9.8, 6.0 Hz, 1H, 4′-H), 5.68 (d,
J = 4.1 Hz, 1H, 1′-H), 6.86 (d,
J = 9.5 Hz, 1H, 5′-H), 7.26–7.38 (m, 5H, aryl-H), 7.55 (d,
JHF = 6.3 Hz, 1H, 6-H).
19F NMR (376 MHz, CDCl
3): δ [ppm] = −164.09. MS (ESI
+):
m/z = 736.4 [M + H]
+, 758.5 [M + Na]
+. TLC:
Rf = 0.66 (1:1, petroleum ether-EtOAc).
Z-6′-
N-Cbz-5′,6′-didehydro-2′-deoxyuridinyl amino acid
tert-butyl ester (
35): KO
t-Bu (460 mg, 4.06 mmol) was dissolved in THF (15 mL). After cooling to −87 °C, a solution of phosphonate
33 [
31] (1.50 g, 4.02 mmol) in THF (30 mL) was added slowly at this temperature. The mixture was stirred at −86 °C for 30 min. Subsequently, a solution of aldehyde
31 (1.25 g, 3.66 mmol) in THF (12 mL) was added dropwise. Stirring was continued for 18.5 h while the solution was allowed to slowly warm to rt. After cooling to 0 °C, MeOH (5 mL) was added and the mixture was stirred for further 40 min. It was then diluted with EtOAc (150 mL) and washed with half-sat. NaCl solution (3 × 100 mL). The organic layer was dried over Na
2SO
4 and the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (1:1, petroleum ether-EtOAc) to give
35 as a colorless foam (1.54 g, 72%).
1H NMR (500 MHz, CDCl
3): δ [ppm] = 0.08 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.09 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.88 (s, 9H, Si(CH
3)
2C(C
H3)
3), 1.47 (s, 9H, COOC(C
H3)
3), 2.16 (ddd,
J = 13.6, 6.2, 6.2 Hz, 1H, 2′-H
a), 2.41 (ddd,
J = 13.6, 6.2, 4.7 Hz, 1H, 2′-H
b), 4.33 (ddd,
J = 6.2, 4.7, 4.6 Hz, 1H, 3′-H), 4.71 (dd,
J = 7.8, 4.6 Hz, 1H, 4′-H), 5.14 (d,
J = 4.0 Hz, 2H, 1″-H), 5.73 (dd,
J = 8.1, 2.2 Hz, 1H, 5-H), 6.13 (d,
J = 7.8 Hz, 1H, 5′-H), 6.14 (dd,
J = 6.2, 6.2 Hz, 1H, 1′-H), 6.78 (s, 1H, 6′-NH), 7.29-7.40 (m, 6H, aryl-H, 6-H), 8.75 (brs, 1H, 3-NH).
13C NMR (126 MHz, CDCl
3): δ [ppm] = −4.76 (Si(
CH
3)
2C(CH
3)
3), −4.64 (Si(
CH
3)
2C(CH
3)
3), 18.06 (Si(CH
3)
2C(CH
3)
3), 25.81 (Si(CH
3)
2C(
CH
3)
3), 28.03 (COOC(
CH
3)
3), 40.88 (C-2′), 67.75 (C-1″), 75.97 (C-3′), 82.85 (C-4′), 82.92 (COO
C(CH
3)
3), 86.69 (C-1′), 102.60 (C-5), 124.91 (C-5′), 128.40 (C-3″,C-7″), 128.51 (C-5″), 128.68 (C-4″, C-6″), 130.36 (C-6′), 135.85 (C-2″), 139.78 (C-6), 149.98 (C-2), 153.70 (Cbz-C=O), 162.83 (C-4), 171.13 (C-7′). HRMS (CI): calcd.: 588.2736 [M + H]
+, found: 588.2717. TLC:
Rf = 0.26 (EtOAc).
Z-6′-
N-Cbz-5′,6′-didehydro-thymidinyl amino acid
tert-butyl ester (
36): KO
t-Bu (466 mg, 4.06 mmol) was dissolved in THF (35 mL). After cooling to −80 °C, a solution of phosphonate
33 [
31] (1.52 g, 4.06 mmol) in THF (30 mL) was added slowly at this temperature. The mixture was stirred at –86 °C for 30 min. Subsequently, a solution of aldehyde
32 (1.31 g, 3.70 mmol) in THF (12 mL) was added dropwise. Stirring was continued for 21 h while the solution was allowed to slowly warm to rt. After cooling to 0 °C, MeOH (5 mL) was added. The mixture was diluted with EtOAc (150 mL) and washed with half-sat. NaCl solution (3 × 100 mL). The organic layer was dried over Na
2SO
4 and the solvent was removed under reduced pressure. The resultant crude product was purified by twofold column chromatography (3:2, petroleum ether-EtOAc) to give
36 (95:5 mixture of
Z and
E isomers as determined by
1H NMR spectroscopy) as a colorless foam (1.71 g, 77%).
1H NMR (300 MHz, CDCl
3): δ [ppm] = 0.09 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.09 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.89 (s, 9H, Si(CH
3)
2C(C
H3)
3), 1.47 (s, 9H, COOC(C
H3)
3), 1.92 (s, 3H, 7-H), 2.16 (ddd,
J = 13.5, 6.2, 6.2 Hz, 1H, 2′-H
a), 2.36 (ddd,
J = 13.5, 6.2, 4.7 Hz, 1H, 2′-H
b), 4.35 (ddd,
J = 6.2, 4.7, 4.6 Hz, 1H, 3′-H), 4.70 (dd,
J = 7.8, 4.6 Hz, 1H, 4′-H), 5.14 (d,
J = 4.0 Hz, 2H, 1″-H), 6.13 (d,
J = 7.8 Hz, 1 H, 5′-H), 6.14 (dd,
J = 6.2, 6.2 Hz, 1H, 1′-H), 6.81 (s, 1H, 6′-NH), 7.13 (s, 1H, 6-H), 7.29–7.40 (m, 5H, aryl-H), 8.71 (s, 1H, 3-NH).
13C NMR (126 MHz, CDCl
3): δ [ppm] = −4.76 (Si(
CH
3)
2C(CH
3)
3), −4.64 (Si(
CH
3)
2C(CH
3)
3), 12.78 (C-7), 18.05 (Si(CH
3)
2C(CH
3)
3), 25.81 (Si(CH
3)
2C(
CH
3)
3), 28.02 (COOC(
CH
3)
3), 40.57 (C-2′), 67.73 (C-1″), 76.13 (C-3′), 82.85 (COO
C(CH
3)
3), 83.34 (C-4′), 86.39 (C-1′), 111.20 (C-5), 124.95 (C-5′), 128.38 (C-3″, C-7″), 128.49 (C-5″), 128.68 (C-4″, C-6″), 130.67 (C-6′), 135.61 (C-6), 135.86 (C-2″), 150.12 (C-2), 153.71 (Cbz-C=O), 162.91 (C-4), 163.67 (C-7′). HRMS (CI): calcd.: 602.2892 [M + H]
+, found: 602.2888. TLC:
Rf = 0.31 (55:45, petroleum ether-EtOAc).
(6′S)-6′-N-Cbz-5-fluorouridinyl amino acid tert-butyl ester (38): Under strict exclusion of oxygen, didehydro amino acid 34 (9:1 mixture of Z and E isomers, 280 mg, 0.381 mmol) was dissolved in MeOH (13 mL). After addition of (S,S)-Me-DUPHOS-Rh 37 (~8 mg, 12 μmol), the solution was stirred under an H2 atmosphere (~1 bar) at rt for 6 d. The reaction was monitored by 1H NMR spectroscopy. Additional catalyst 37 (~4 mg, 6 μmol) was added after further 5 d and 2 d, respectively, until the conversion of starting material 34 was complete. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by threefold column chromatography (3:1, petroleum ether-EtOAc and 9:1, petroleum ether-EtOAc and CH2Cl2→95:5, CH2Cl2-EtOAc, respectively) to give 38 as a colorless foam (133 mg, 51%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.07 (s, 6H, Si(CH3)2C(CH3)3), 0.12 (s, 6H, Si(CH3)2C(CH3)3), 0.89 (s, 9H, Si(CH3)2C(CH3)3), 0.91 (s, 9H, Si(CH3)2C(CH3)3), 1.46 (s, 9H, COOC(CH3)3), 2.02 (ddd, J = 14.2, 8.2, 3.8 Hz, 1H, 5′-Ha), 2.19 (ddd, J = 14.2, 8.2, 2.2 Hz, 1H, 5′-Hb), 3.63 (dd, J = 6.0, 4.1 Hz, 1H, 3′-H), 4.14 (dd, J = 4.1, 3.2 Hz, 1H, 2′-H), 4.25 (ddd, J = 10.8, 6.3, 1.9 Hz, 1H, 4′-H), 4.45 (ddd, J = 7.6, 7.6, 3.8 Hz, 1H, 6′-H), 5.12 (s, 2H, 1″-H), 5.61 (d, J = 6.9 Hz, 1H, 6′-NH), 5.62 (d, 3J = 1.9 Hz, 1H, 1′-H), 7.31–7.38 (m, 5H, aryl-H), 7.84 (d, JHF = 6.0 Hz, 1H, 6-H), 8.95 (brs, 1H, 3-NH). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.85 (Si(CH3)2C(CH3)3), −4.33 (Si(CH3)2C(CH3)3), 17.96 (Si(CH3)2C(CH3)3), 17.99 (Si(CH3)2C(CH3)3), 25.75 (Si(CH3)2C(CH3)3), 25.79 (Si(CH3)2C(CH3)3), 27.92 (COOC(CH3)3), 36.35 (C-5′), 51.86 (C-6′), 66.97 (C-1″), 75.15 (C-2′), 75.24 (C-3′), 80.24 (C-4′), 82.98 (COOC(CH3)3), 91.20 (C-1′), 124.72 (d, JCF = 30.0 Hz, C-6), 128.22 (C-3″/C-4″), 128.26 (C-3″/C-4″), 128.51 (C-2″), 139.51 (C-5″), 140.65 (d, JCF = 238.3 Hz, C-5), 148.39 (C-2), 155.49 (d, JCF = 32.9 Hz, C-4). 19F NMR (376 MHz, CDCl3): δ [ppm] = −164.51. MS (ESI+): m/z = 738.5 [M + H]+, 760.4 [M + Na]+. HRMS (ESI+): calcd.: 738.3612 [M + H]+, 760.3431 [M + Na]+, found: 738.3594, 760.3413. TLC: Rf = 0.53 (1:1, petroleum ether-EtOAc).
(6′S)-6′-N-Cbz-2′-deoxyuridinyl amino acid tert-butyl ester (39): Under strict exclusion of oxygen, didehydro amino acid 35 (600 mg, 1.02 mmol) was dissolved in MeOH (35 mL). After addition of (S,S)-Me-DUPHOS-Rh 37 (~15 mg, 21 μmol), the solution was stirred under an H2 atmosphere (~1 bar) at rt for 4 d. The reaction was monitored by 1H NMR spectroscopy. Additional catalyst 37 (~4 mg, 6 μmol) was added and stirring was continued for 2 d until the conversion of starting material 35 was complete. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (3:2, petroleum ether-EtOAc) to give 39 as a colorless foam (562 mg, 93%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.05 (s, 6H, Si(CH3)2C(CH3)3), 0.89 (s, 9H, Si(CH3)2C(CH3)3), 1.43 (s, 9H, COOC(CH3)3), 1.93–2.02 (m, 1H, 5′-Ha), 2.03–2.11 (m, 1H, 2′-Ha), 2.20 (ddd, J = 14.2, 6.7, 2.9 Hz, 1H, 5′-Hb), 2.23–2.31 (m, 1H, 2′-Hb), 3.90–3.97 (m, 1H, 4′-H), 4.05 (dd, J = 10.8, 4.9 Hz, 1H, 3′-H), 4.41 (dd, J = 11.6, 6.7 Hz, 1H, 6′-H), 5.10 (s, 2H, 1″-H), 5.55 (d, J = 6.8 Hz, 1H, 6′-NH), 5.77 (d, J = 8.0 Hz, 1H, 5-H), 6.14 (dd, J = 6.1, 6.1 Hz, 1H, 1′-H), 7.27–7.39 (m, 5H, aryl-H), 7.54 (d, J = 8.0 Hz, 1H, 6-H), 8.92 (s, 1H, 3-NH). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.74 (Si(CH3)2C(CH3)3), −4.48 (Si(CH3)2C(CH3)3), 18.03 (Si(CH3)2C(CH3)3), 25.79 (Si(CH3)2C(CH3)3), 28.06 (COOC(CH3)3), 36.14 (C-5′), 40.64 (C-2′), 52.23 (C-6′), 67.09 (C-1″), 74.92 (C-3′), 82.92 (COOC(CH3)3), 83.31 (C-4′), 85.63 (C-1′), 102.68 (C-5), 128.30, 128.37, 128.67 (C-3″, C-4″, C-5″, C-6″, C-7″), 136.38 (C-2″), 140.35 (C-6), 150.15 (C-2), 155.69 (Cbz-C=O), 163.29 (C-4), 170.61 (C-7′). HRMS (CI): calcd.: 590.2892 [M + H]+, found: 590.2885. TLC: Rf = 0.25 (1:1, petroleum ether-EtOAc).
(6′S)-6′-N-Cbz-thymidinyl amino acid tert-butyl ester (40): Under strict exclusion of oxygen, didehydro amino acid 36 (600 mg, 0.997 mmol) was dissolved in MeOH (35 mL). After addition of (S,S)-Me-DUPHOS-Rh 37 (~20 mg, 28 μmol), the solution was stirred under an H2 atmosphere (~1 bar) at rt for 7 d. The reaction was monitored by 1H NMR spectroscopy. After complete conversion of starting material 36, the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (1:1, petroleum ether-EtOAc) to give 40 as a colorless foam (566 mg, 95%). 1H NMR (500 MHz, CDCl3): δ [ppm] = 0.06 (s, 6H, Si(CH3)2C(CH3)3), 0.88 (s, 9H, Si(CH3)2C(CH3)3), 1.43 (s, 9H, COOC(CH3)3), 1.97 (s, 3H, 7-H), 1.99–2.09 (m, 2H, 2′-Ha, 5′-Ha), 2.14–2.20 (m, 1H, 5′-Hb), 2.22 (ddd, J = 13.5, 6.5, 4.3 Hz, 1H, 2′-Hb), 3.92–3.97 (m, 1H, 4′-H), 4.07 (ddd, J = 6.5, 4.3, 4.3 Hz, 1H, 3′-H), 4.42 (dd, J = 11.8, 7.1 Hz, 1H, 6′-H), 5.10 (s, 2H, 1″-H), 5.54 (d, J = 6.7 Hz, 1H, 6′-NH), 6.18 (dd, J = 6.5, 6.5 Hz, 1H, 1′-H), 7.29–7.36 (m, 5H, aryl-H), 7.37 (s, 1H, 6-H), 8.53 (brs, 1H, 3-NH). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.71 (Si(CH3)2C(CH3)3), −4.47 (Si(CH3)2C(CH3)3), 12.46 (C-7), 18.05 (Si(CH3)2C(CH3)3), 25.18 (Si(CH3)2C(CH3)3), 28.06 (COOC(CH3)3), 36.26 (C-5′), 40.39 (C-2′), 52.16 (C-6′), 67.06 (C-1″), 75.11 (C-3′), 82.89 (COOC(CH3)3), 83.26 (C-4′), 85.27 (C-1′), 111.33 (C-5), 128.26, 128.37, 128.68 (C-3″, C-4″, C-5″, C-6″, C-7″), 136.00 (C-6), 136.41 (C-2″), 150.18 (C-2), 155.65 (Cbz-C=O), 163.74 (C-4), 170.65 (C-7′). HRMS (CI): calcd.: 604.3049 [M + H]+, found: 604.3029. TLC: Rf = 0.30 (1:1, petroleum ether-EtOAc).
(6′
S)-5,6-dihydrouridinyl amino acid
tert-butyl ester (
42): (6′
S)-6′-
N-Cbz-uridinyl amino acid
tert-butyl ester
41 [
31] (100 mg, 0.139 mmol) was dissolved in
i-PrOH (20 mL) and stirred over molecular sieve (3 Å) at rt for 15 min. Pd black (130 mg, 1.22 mmol) was added and the reaction mixture was stirred under an H
2 atmosphere (~1 bar) at rt for 4 h. After filtration through a syringe filter and washing the filter with
i-PrOH (50 °C, 45 mL), the solvent of the combined filtrates was evaporated under reduced pressure to give
42 as a colorless foam (82 mg, 96%).
1H NMR (300 MHz, CDCl
3): δ [ppm] = 0.06 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.07 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.08 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.09 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.90 (s, 9H, Si(CH
3)
2C(C
H3)
3), 0.91 (s, 9H, Si(CH
3)
2C(C
H3)
3), 1.46 (s, 9H, COOC(C
H3)
3), 1.68 (ddd,
J = 13.9, 9.4, 7.1 Hz, 1H, 5′-H
a), 2.12 (ddd,
J = 13.9, 6.0, 2.6 Hz, 1H, 5′-H
b), 2.63-–2.69 (m, 2H, 5-H), 3.29–3.40 (m, 1H, 6-H
a), 3.49 (dd,
J = 6.3, 6.3 Hz, 1H, 6-H
b), 3.56 (dd,
J = 7.1, 7.0 Hz, 1H, 6′-H), 3.74 (dd,
J = 5.7, 4.6 Hz, 1H, 3′-H), 4.01 (ddd,
J = 9.4, 5.7, 2.6 Hz, 1H, 4′-H), 4.14 (dd,
J = 4.6, 4.0 Hz, 1H, 2′-H), 5.59 (d,
J = 4.0 Hz, 1H, 1′-H), 7.61 (brs, 1H, 3-NH).
13C NMR (126 MHz, CDCl
3): δ [ppm] = −4.49 (Si(
CH
3)
2C(CH
3)
3), −4.40 (Si(
CH
3)
2C(CH
3)
3), −4.38 (Si(
CH
3)
2C(CH
3)
3), −4.00 (Si(
CH
3)
2C(CH
3)
3), 18.15 (Si(CH
3)
2C(CH
3)
3), 18.24 (Si(CH
3)
2C(CH
3)
3), 25.93 (Si(CH
3)
2C(
CH
3)
3), 26.02 (Si(CH
3)
2C(
CH
3)
3), 28.16 (COOC(
CH
3)
3), 31.29 (C-5), 38.47 (C-5′), 38.70 (C-6), 53.62 (C-6′), 73.52 (C-2′), 76.14 (C-3′), 80.81 (C-4′), 81.69 (COO
C(CH
3)
3), 91.64 (C-1′), 152.17 (C-2), 169.44 (C-4), 173.97 (C-7′). HRMS (CI): calcd.: 588.3495 [M + H]
+, found: 588.3488.
N-Cbz-protected 5,6-dihydrouridine derivative (
44): Nucleosyl amino acid ester
42 (100 mg, 0.170 mmol) was dissolved in THF (14 mL) and aldehyde
43 [
24,
34] (70 mg, 0.22 mmol) was added. The reaction mixture was stirred over molecular sieve (3 Å) at rt for 23 h. NaBH(OAc)
3 (80 mg, 0.39 mmol) and Amberlyst 15
TM (14 mg, 44 μmol) were added and stirring was continued at rt for 22 h. The reaction mixture was filtered and the residue was washed with EtOAc (150 mL). The combined filtrates were washed with sat. NaHCO
3 solution (100 mL) and brine (50 mL). The aqueous layer was extracted with EtOAc (3 × 100 mL). The combined organics were dried over Na
2SO
4 and the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (98:2, CH
2Cl
2-MeOH) to give
44 as a colorless solid (93 mg, 58%).
1H NMR (500 MHz, CD
3OD): δ [ppm] = 0.11 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.12 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.13 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.14 (s, 3H, Si(C
H3)
2C(CH
3)
3), 0.84–0.91 (m, 6H, Leu-5-H), 0.92 (s, 9H, Si(CH
3)
2C(C
H3)
3), 0.93 (s, 9H, Si(CH
3)
2C(C
H3)
3), 1.49 (s, 9H, COOC(C
H3)
3), 1.53 (dd,
J = 7.2, 7.2 Hz, 2H, Leu-3-H), 1.63–1.72 (m, 3H, 2″-H, Leu-4-H), 1.78 (ddd,
J = 13.4, 11.2, 4.9 Hz, 1H, 5′-H
a), 1.99 (ddd,
J = 13.4, 9.6, 2.7 Hz, 1H, 5′-H
b), 2.51–2.58 (m, 1H, 1″-H
a), 2.60–2.67 (m, 3H, 1″-H
b, 5-H), 3.18–3.36 (m, 2H, 6′-H, 3″-H
a), 3.39 (ddd,
J = 12.9, 6.6, 6.6 Hz, 1H, 6-H
a), 3.47 (ddd,
J = 12.9, 6.7, 6.7 Hz, 1H, 6-H
b), 3.56 (ddd,
J = 8.1, 4.4, 4.4 Hz, 1H, 3″-H
b), 3.85 (dd,
J = 4.8, 4.8 Hz, 1H, 3′-H), 3.88–3.94 (m, 1H, 4′-H), 4.04–4.14 (m, 1H, Leu-2-H), 4.25 (dd,
J = 4.8, 4.6 Hz, 1H, 2′-H), 5.09 (s, 2H, 1″′-H), 5.77 (d,
J = 4.6 Hz, 1H, 1′-H), 7.25–7.39 (m, 5H, aryl-H).
13C NMR (126 MHz, CD
3OD): δ [ppm] = −4.36 (Si(
CH
3)
2C(CH
3)
3), −4.34 (Si(
CH
3)
2C(CH
3)
3), −4.13 (Si(
CH
3)
2C(CH
3)
3), −3.93 (Si(
CH
3)
2C(CH
3)
3), 18.90 (Si(CH
3)
2C(CH
3)
3), 18.94 (Si(CH
3)
2C(CH
3)
3), 23.37 (Leu-C-5), 23.46 (Leu-C-5), 25.99 (Leu-C-4), 26.41 (Si(CH
3)
2C(
CH
3)
3), 26.48 (Si(CH
3)
2C(
CH
3)
3), 28.44 (COOC(
CH
3)
3), 29.97 (C-2″), 31.93 (C-5), 37.46 (C-5′), 38.09 (C-6), 39.00 (C-3″), 42.22 (Leu-C-3), 46.02 (C-1″), 55.23 (Leu-C-2), 60.41 (C-6′), 67.71 (C-1″′), 74.14 (C-2′), 77.15 (C-3′), 81.10 (C-4′), 83.24 (COO
C(CH
3)
3), 91.77 (C-1′), 128.85 (C-3″′, C-7″′), 129.09 (C-5″′), 129.50 (C-4″′, C-6″′), 138.20 (C-2″′), 154.93 (C-2), 158.39 (Cbz-C=O), 172.49 (C-4), 175.67 (C-7′), 181.44 (Leu-C-1). HRMS (ESI
+): calcd.: 892.5282 [M + H]
+, found: 892.5261.
5,6-Dihydrouridine derivative (45): N-Cbz-protected 5,6-dihydrouridine derivative 44 (51 mg, 57 μmol) was dissolved in MeOH (10 mL) and Pd/C (10%, 3.6 mg, 3.4 μmol Pd) was added. The reaction mixture was stirred under an H2 atmosphere (~1 bar) at rt for 2 h. After filtration through a syringe filter and washing the filter with EtOAc (50 °C, 20 mL), the solvent of the combined filtrates was evaporated under reduced pressure to give 45 as a colorless solid (42 mg, quant.). 1H NMR (500 MHz, CD3OD): δ [ppm] = 0.11 (s, 3H, Si(CH3)2C(CH3)3), 0.12 (s, 3H, Si(CH3)2C(CH3)3), 0.13 (s, 3H, Si(CH3)2C(CH3)3), 0.14 (s, 3H, Si(CH3)2C(CH3)3), 0.89–0.96 (m, 6H, Leu-5-H), 0.92 (s, 9H, Si(CH3)2C(CH3)3), 0.94 (s, 9H, Si(CH3)2C(CH3)3), 1.41–1.55 (m, 2H, Leu-3-H), 1.49 (s, 9H, COOC(CH3)3), 1.64–1.72 (m, 3H, 2″-H, Leu-4-H), 1.76 (ddd, J = 13.9, 10.8, 4.9 Hz, 1H, 5′-Ha), 1.96–2.02 (m, 1H, 5′-Hb), 2.52 (ddd, J = 11.6, 7.0, 7.0 Hz, 1H, 1″-Ha), 2.59–2.68 (m, 3H, 5-H, 1″-Hb), 3.19–3.30 (m, 2H, 6′-H, 3″-Ha), 3.41 (ddd, J = 13.6, 6.8, 6.8 Hz, 1H, 6-Ha), 3.49 (ddd, J = 13.6, 6.8, 6.8 Hz, 1H, 6-Hb), 3.85 (dd, J = 4.8, 4.8 Hz, 1H, 3′-H), 3.88–3.93 (m, 1H, 4′-H), 4.04–4.14 (m, 1H, Leu-2-H), 4.24 (dd, J = 4.8, 4.5 Hz, 1H, 2′-H), 5.76 (d, J = 4.5 Hz, 1H, 1′-H). 13C NMR (126 MHz, CD3OD): δ [ppm] = −4.36 (Si(CH3)2C(CH3)3), −4.35 (Si(CH3)2C(CH3)3), −4.13 (Si(CH3)2C(CH3)3), −3.92 (Si(CH3)2C(CH3)3), 18.90 (Si(CH3)2C(CH3)3), 18.93 (Si(CH3)2C(CH3)3), 22.58 (Leu-C-5), 23.41 (Leu-C-5), 25.92 (Leu-C-4), 26.41 (Si(CH3)2C(CH3)3), 26.48 (Si(CH3)2C(CH3)3), 28.46 (COOC(CH3)3), 30.43 (C-2″), 31.95 (C-5), 38.17 (C-5′), 38.25 (C-6), 38.85 (C-3″), 45.71 (Leu-C-3), 46.16 (C-1″), 54.68 (Leu-C-2), 60.94 (C-6′), 74.24 (C-2′), 77.16 (C-3′), 81.13 (C-4′), 82.77 (COOC(CH3)3), 91.61 (C-1′), 154.93 (C-2), 172.49 (C-4), 174.97 (C-7′), 178.16 (Leu-C-1). MS (ESI+): calcd.: 758.49 [M + H]+, found: 758.57.



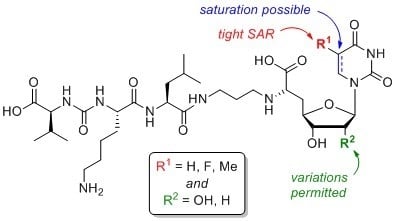









{kind=link}
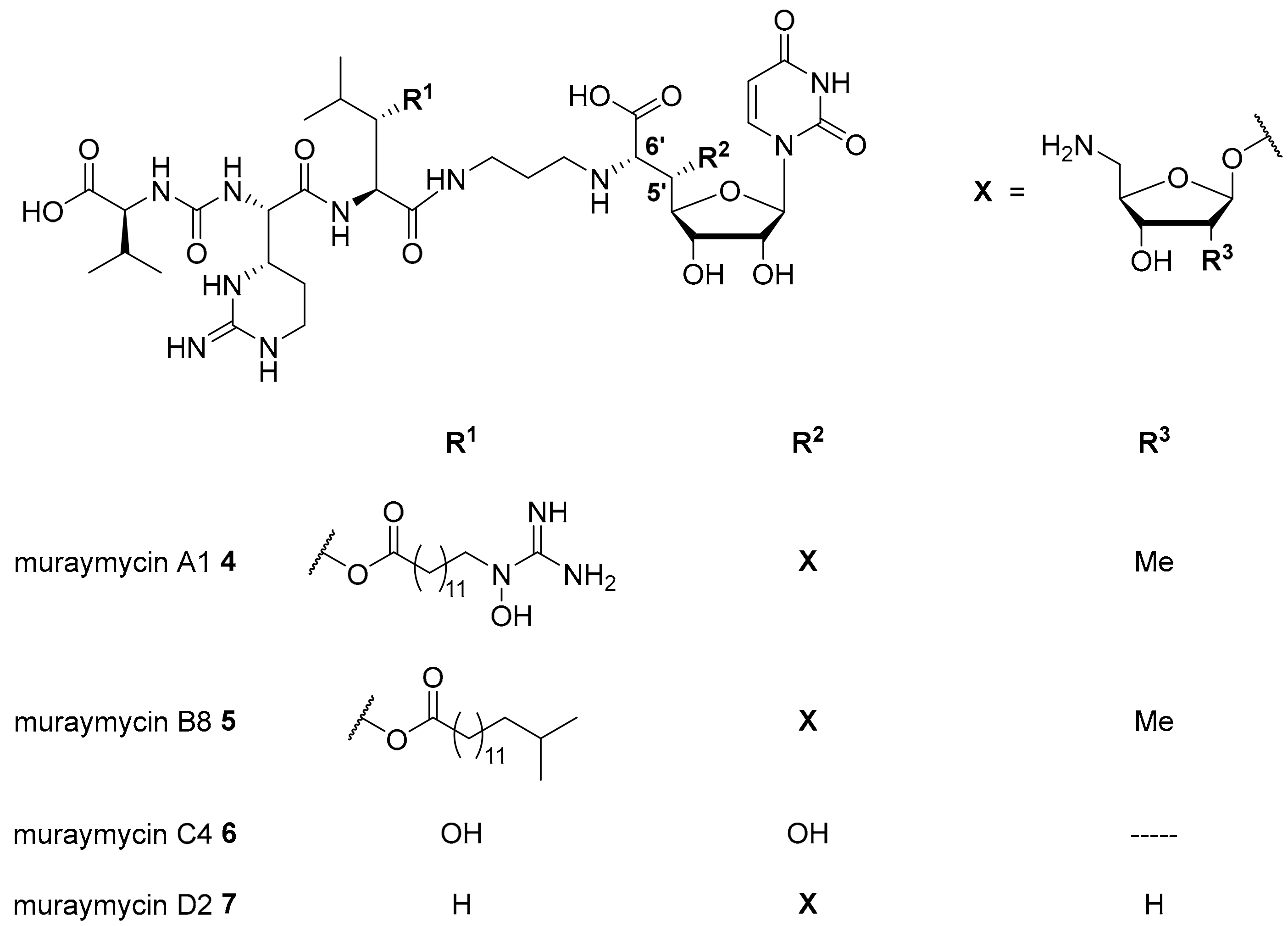{kind=link}
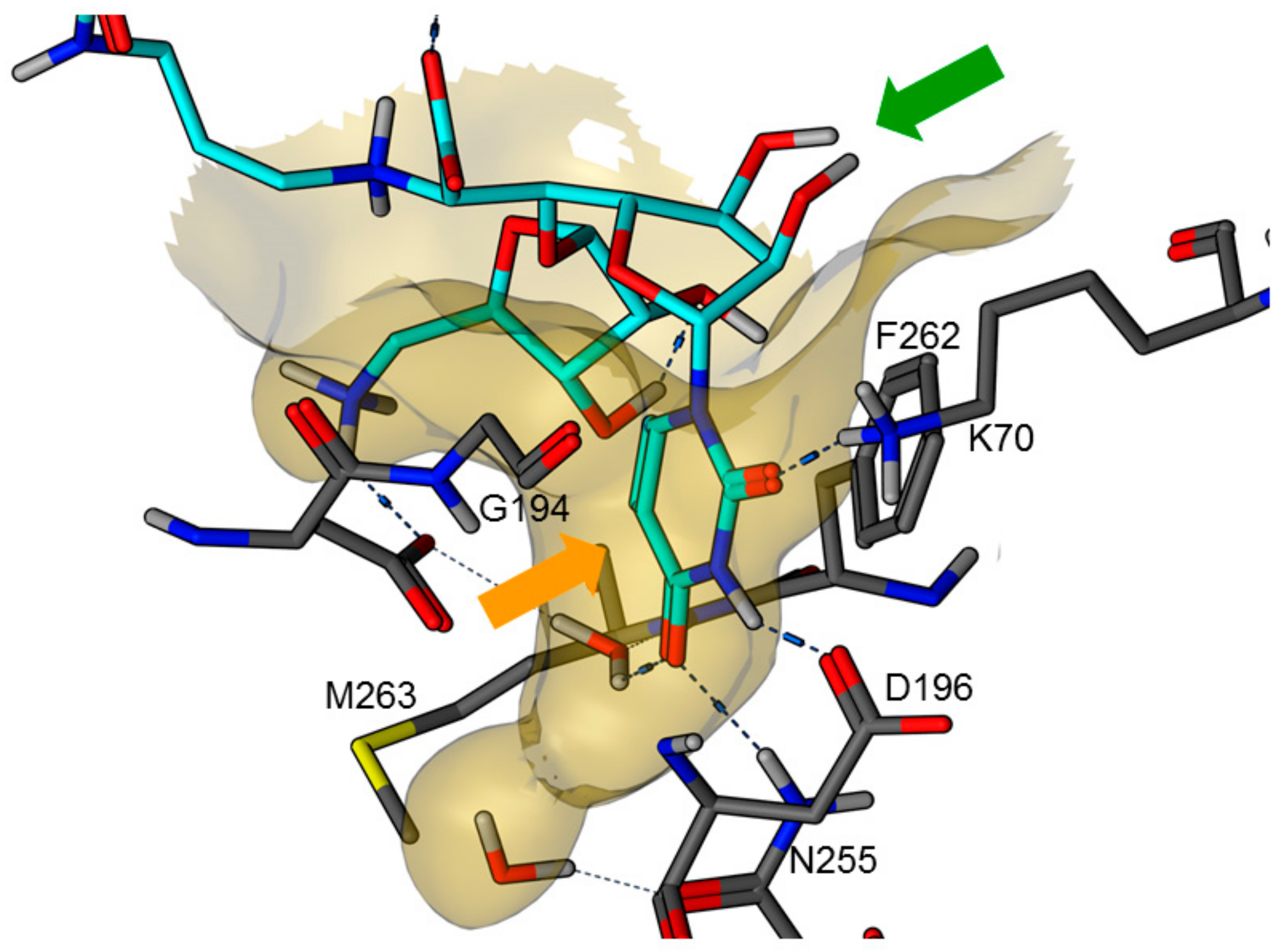{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





