Natural Products in Alzheimer’s Disease Therapy: Would Old Therapeutic Approaches Fix the Broken Promise of Modern Medicines?

Abstract
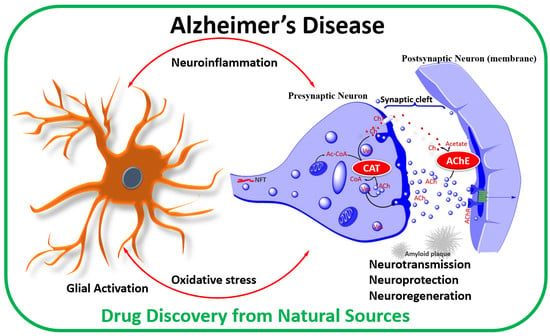
:
{kind=link}
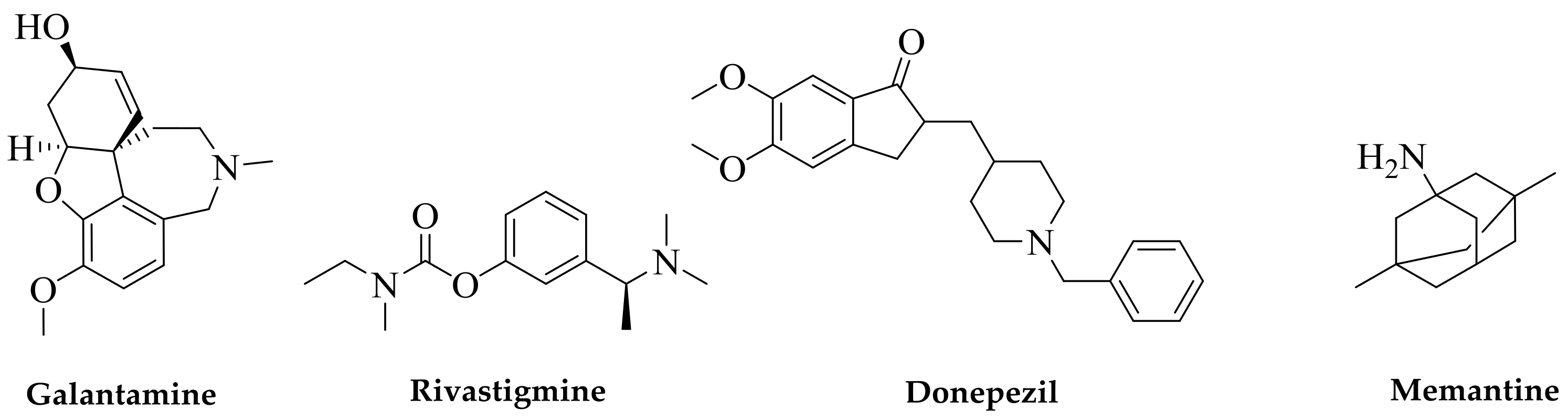{kind=link}
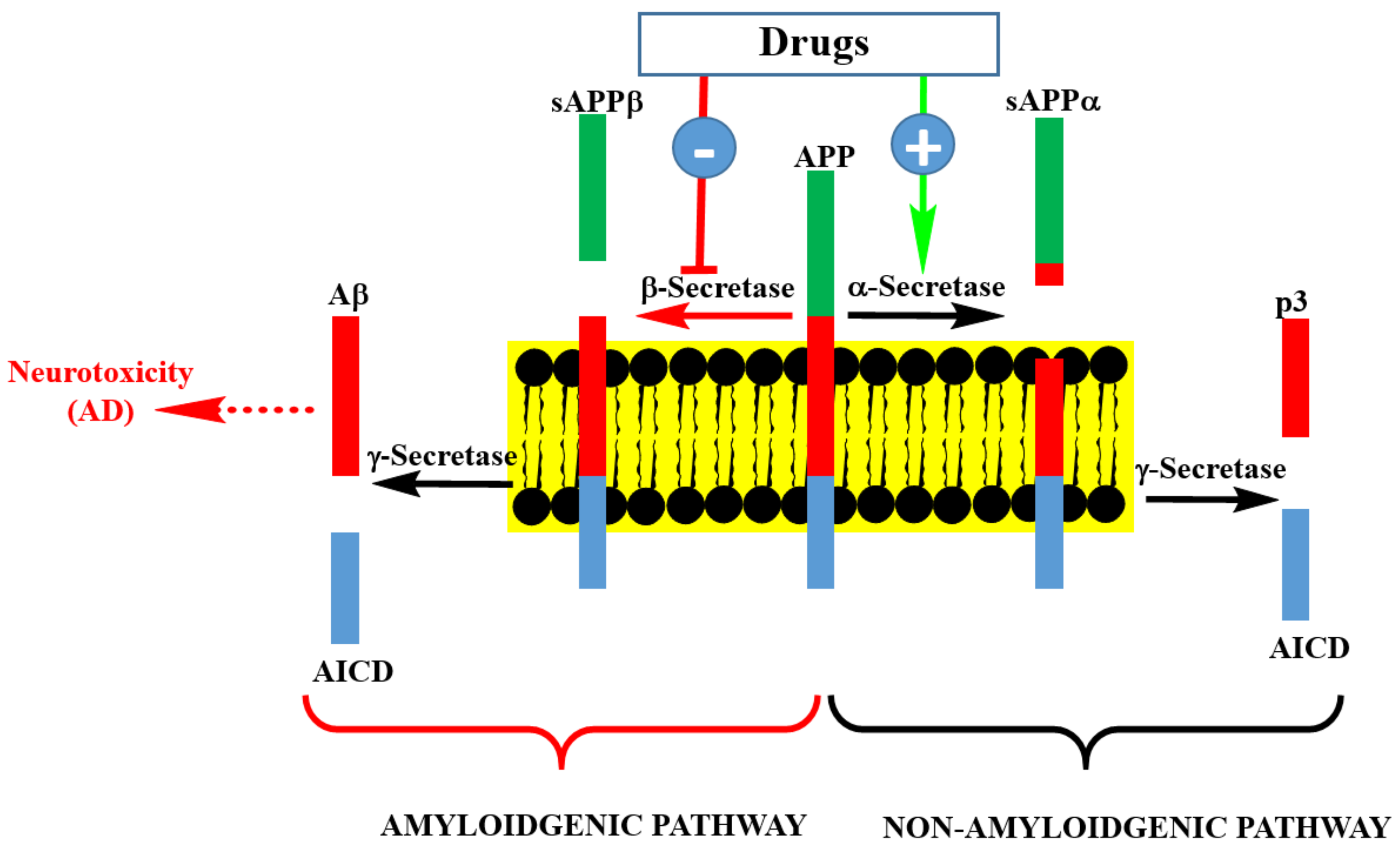{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. AD Pathology
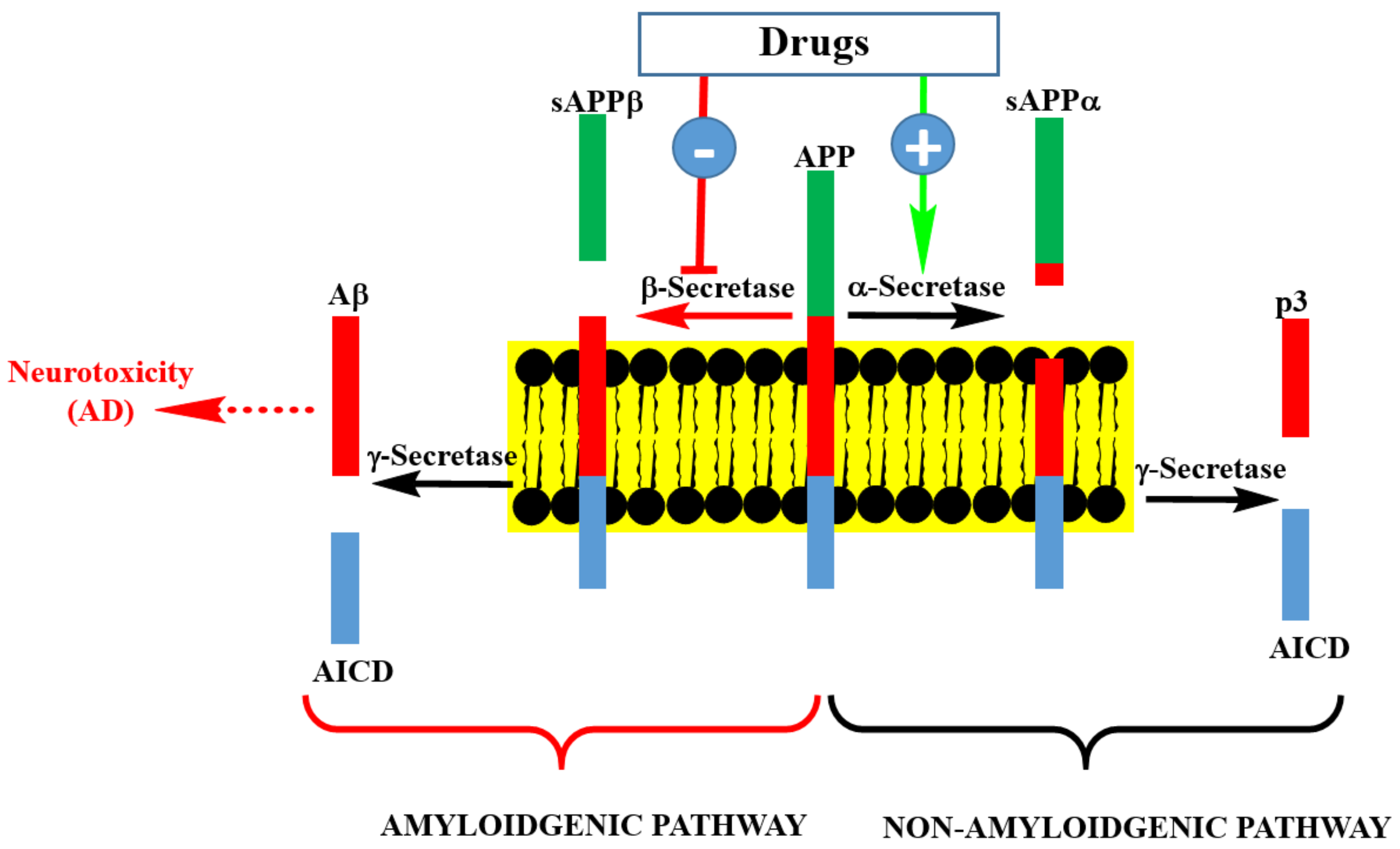
3. The Aβ Therapeutic Approach
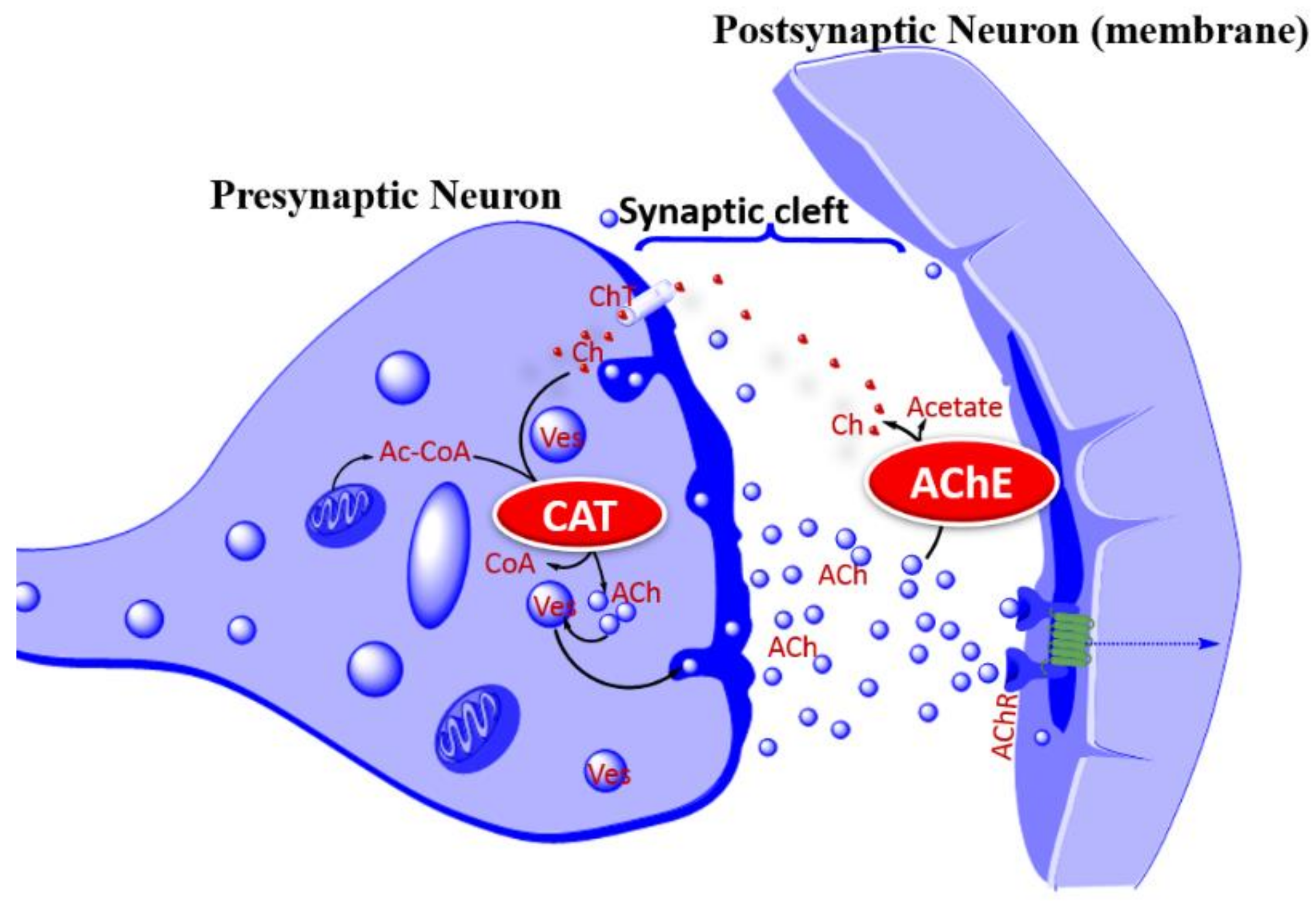
4. The Cholinergic Hypothesis
5. Other Neurotransmitters
6. Tau Hyperphosphorylation and Aggregation
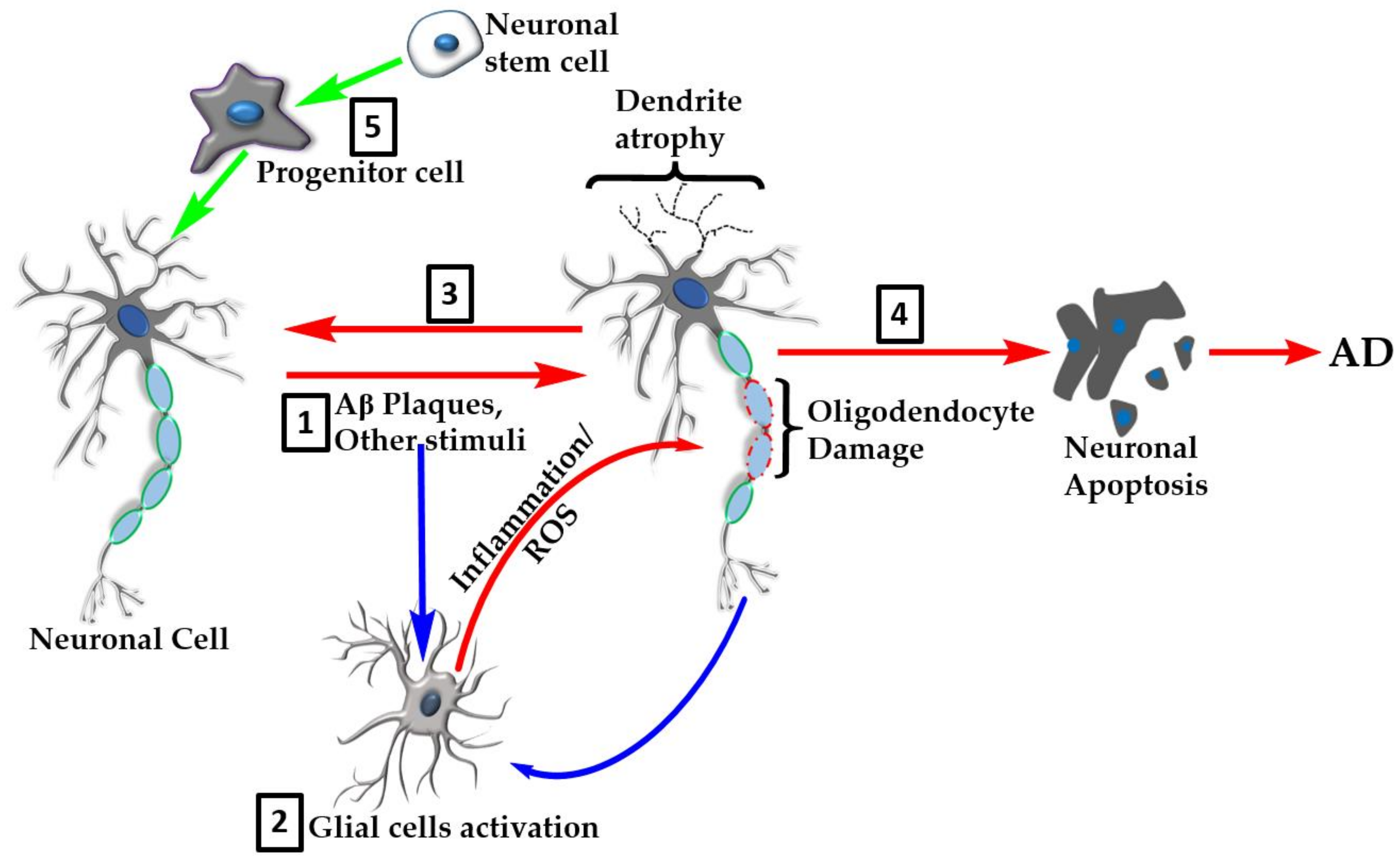
7. Neuroinflammation
8. Oxidative Stress
9. Methods of AD Efficacy Evaluations
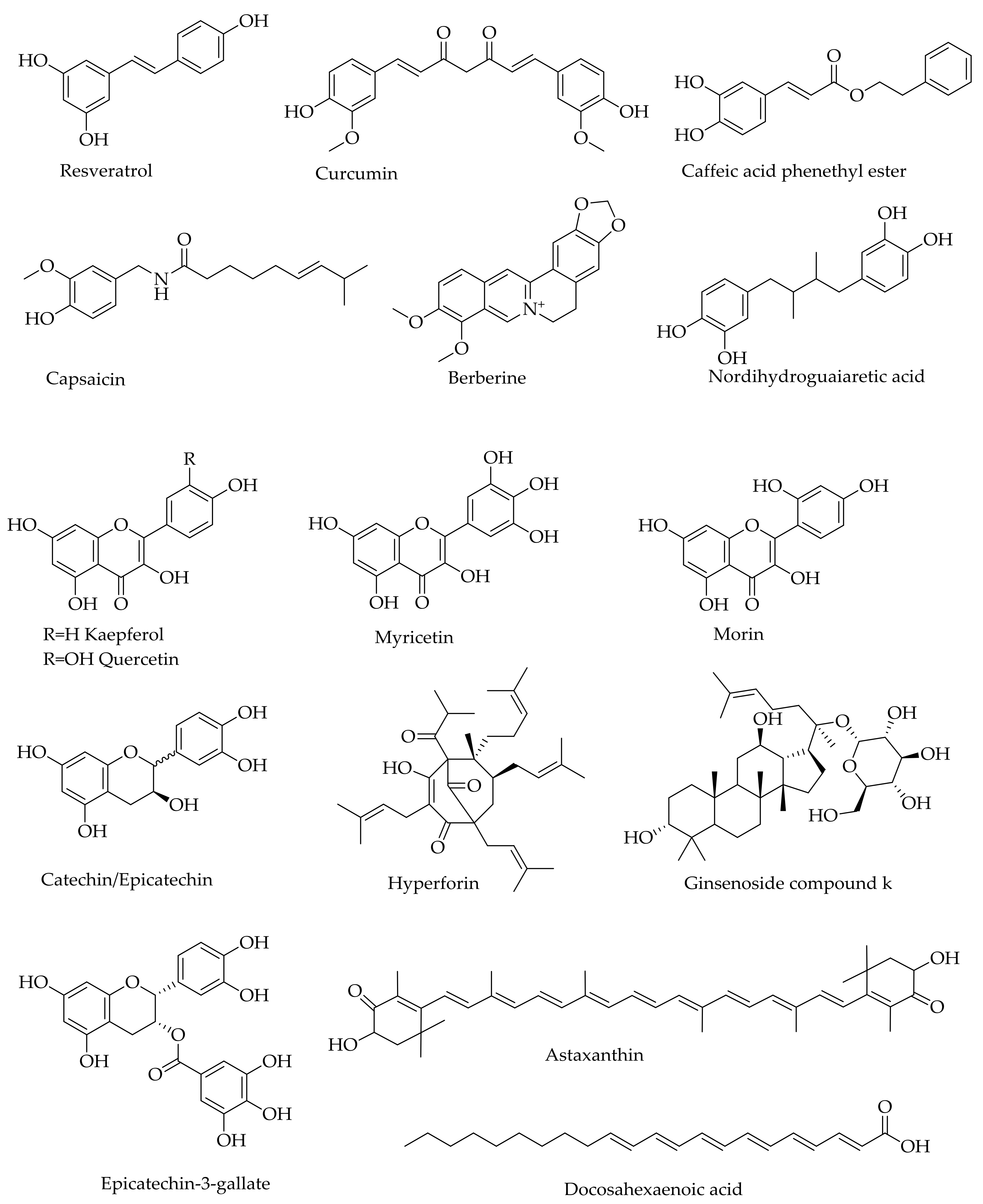
10. Multi-Target Approach—Lessons from Natural Products
11. Conclusions
Funding
Conflicts of Interest
References
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine Study Group. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.S.; Dagerman, K.S.; Higgins, J.P.; McShane, R. Lack of evidence for the efficacy of memantine in mild Alzheimer disease. Arch. Neurol. 2011, 68, 991–998. [Google Scholar] [CrossRef] [PubMed]
- ADA (Alzheimers Disease Association). Medications for Memory. Available online: https://www.alz.org/alzheimers-dementia/treatments/medications-for-memory (accessed on 28 March 2019).
- AS (Alzheimer’s Societry UK). Drug treatments for Alzheimer’s disease. Available online: https://www.alzheimers.org.uk/about-dementia/treatments/drugs/drug-treatments-alzheimers-disease (accessed on 28 March 2019).
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid precursor protein in Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2017, 7, pii–a024539. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Song, H.; Croteau, D.L.; Akbari, M.; Bohr, V.A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 2017, 161, 83–94. [Google Scholar] [CrossRef]
- Tai, L.M.; Bilousova, T.; Jungbauer, L.; Roeske, S.K.; Youmans, K.L.; Yu, C.; Poon, W.W.; Cornwell, L.B.; Miller, C.A.; Vinters, H.V.; et al. Levels of soluble apolipoprotein E/amyloid-β (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J. Biol. Chem. 2013, 288, 5914–5926. [Google Scholar] [CrossRef]
- WHO. Dementia. 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 28 March 2019).
- ADI (Alzheimeris Disease international). Dementia Statistics. 2019. Available online: https://www.alz.co.uk/research/statistics (accessed on 28 March 2019).
- Weintraub, S.; Wicklund, A.H.; Salmon, D.P. The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006171. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. Alzheimer’s disease research: The future of BACE inhibitors. Lancet 2018, 391, 2486. [Google Scholar] [CrossRef]
- Deshpande, A.; Kawai, H.; Metherate, R.; Glabe, C.G.; Busciglio, J. A role for synaptic zinc in activity-dependent Aβ oligomer formation and accumulation at excitatory synapses. J. Neurosci. 2009, 29, 4004–4015. [Google Scholar] [CrossRef] [PubMed]
- Uranga, R.M.; Giusto, N.M.; Salvador, G.A. Effect of transition metals in synaptic damage induced by amyloid beta peptide. Neuroscience 2010, 170, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Crouch, P.J.; Hung, L.W.; Adlard, P.A.; Cortes, M.; Lal, V.; Filiz, G.; Perez, K.A.; Nurjono, M.; Caragounis, A.; Du, T.; et al. Increasing Cu bioavailability inhibits Aβ oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 381–386. [Google Scholar] [CrossRef]
- Duce, J.A.; Bush, A.I. Biological metals and Alzheimer’s disease: Implications for therapeutics and diagnostics. Prog. Neurobiol. 2010, 92, 1–18. [Google Scholar] [CrossRef]
- Leskovjan, A.C.; Kretlow, A.; Lanzirotti, A.; Barrea, R.; Vogt, S.; Miller, L.M. Increased brain iron coincides with early plaque formation in a mouse model of Alzheimer’s disease. NeuroImage 2011, 55, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef] [PubMed]
- Hull, M.; Sadowsky, C.; Arai, H.; Le Prince Leterme, G.; Holstein, A.; Booth, K.; Peng, Y.; Yoshiyama, T.; Suzuki, H.; Ketter, N.; et al. Long-term extensions of randomized vaccination trials of ACC-001 and QS-21 in mild to moderate Alzheimer’s disease. Curr. Alzheimer Res. 2017, 14, 696–708. [Google Scholar] [CrossRef]
- Pasquier, F.; Sadowsky, C.; Holstein, A.; Leterme Gle, P.; Peng, Y.; Jackson, N.; Fox, N.C.; Ketter, N.; Liu, E.; Ryan, J.M.; et al. Two phase 2 multiple ascending-dose studies of Vanutide cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer’s disease. J. Alzheimers Dis. 2016, 51, 1131–1143. [Google Scholar] [CrossRef]
- Van Dyck, C.H.; Sadowsky, C.; Le Prince Leterme, G.; Booth, K.; Peng, Y.; Marek, K.; Ketter, N.; Liu, E.; Wyman, B.T.; Jackson, N.; et al. Vanutide Cridificar (ACC-001) and QS-21 Adjuvant in individuals with early Alzheimer’s disease: Amyloid imaging positron emission tomography and safety results from a phase 2 study. J. Prev. Alzheimers Dis. 2016, 3, 75–84. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef]
- Gold, P.E. Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol. Learn. Mem. 2003, 80, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.; Dean, R.; Beer, B.; Lippa, A. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Coyle, J.; Price, D.; deLong, M. Alzheimer’s disease: A disorder of cortical cholinergic innervations. Science 1983, 219, 1184–1189. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Cholinesterases: New roles in brain function and in Alzheimer’s disease. Neurochem. Res. 2003, 28, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Klinkenberg, I.; Blokland, A. The validity of scopolamine as a pharmacological model for cognitive impairment: A review of animal behavioral studies. Neurosci. Biobehav. Rev. 2010, 34, 1307–1350. [Google Scholar] [CrossRef]
- Gilles, C. Pharmacological models in Alzheimer’s disease research. Dialogues Clin. Neurosci. 2000, 2, 247–255. [Google Scholar] [PubMed]
- Pan, S.Y.; Guo, B.F.; Zhang, Y.; Yu, Q.; Yu, Z.L.; Dong, H.; Ye, Y.; Han, Y.F.; Ko, K.M. Tacrine treatment at high dose suppresses the recognition memory in juvenile and adult mice with attention to hepatotoxicity. Basic Clin. Pharmacol. Toxicol. 2011, 108, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Galisteo, M.; Rissel, M.; Sergent, O.; Chevanne, M.; Cillard, J.; Guillouzo, A.; Lagadic-Gossmann, D. Hepatotoxicity of tacrine: Occurrence of membrane fluidity alterations without involvement of lipid peroxidation. J. Pharmacol. Exp. Ther. 2000, 294, 160–167. [Google Scholar]
- Gracon, S.I.; Knapp, M.J.; Berghoff, W.G.; Pierce, M.; DeJong, R.; Lobbestael, S.J.; Symons, J.; Dombey, S.L.; Luscombe, F.A.; Kraemer, D. Safety of tacrine: Clinical trials, treatment IND, and postmarketing experience. Alzheimer Dis. Assoc. Disord. 1998, 12, 93–101. [Google Scholar] [CrossRef]
- Lanari, A.; Amenta, F.; Silvestrelli, G.; Tomassoni, D.; Parnetti, L. Neurotransmitter deficits in behavioural and psychological symptoms of Alzheimer’s disease. Mech. Ageing Dev. 2006, 127, 158–165. [Google Scholar] [CrossRef]
- Solas, M.; Puerta, E.; Ramirez, M.J. Treatment Options in Alzheimer´s Disease: The GABA Story. Curr. Pharm. Des. 2015, 21, 4960–4971. [Google Scholar] [CrossRef] [PubMed]
- Švob Štrac, D.; Pivac, N.; Mück-Šeler, D. The serotonergic system and cognitive function. Transl. Neurosci. 2016, 7, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef] [PubMed]
- Black, S.A.; Stys, P.K.; Zamponi, G.W.; Tsutsui, S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol. 2014, 2, 45. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef]
- Medina, M.; Garrido, J.J.; Wandosell, F.G. Modulation of GSK-3 as a therapeutic strategy on tau pathologies. Front. Mol. Neurosci. 2011, 4, 24. [Google Scholar] [CrossRef]
- Oliveira, J.; Costa, M.; de Almeida, M.S.C.; da Cruz, E.; Silva, O.A.B.; Henriques, A.G. Protein phosphorylation is a key mechanism in Alzheimer’s disease. J. Alzheimers Dis. 2017, 58, 953–978. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, ‘Uber eine eigenartige Erkankung der Hirnrinde’. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Roda, A.R.; Montoliu-Gaya, L.; Villegas, S. The role of apolipoprotein E isoforms in Alzheimer’s disease. J. Alzheimers Dis. 2019, 68, 459–471. [Google Scholar] [CrossRef]
- Boza-Serrano, A.; Yang, Y.; Paulus, A.; Deierborg, T. Innate immune alterations are elicited in microglial cells before plaque deposition in the Alzheimer’s disease mouse model 5xFAD. Sci. Rep. 2018, 8, 1550. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Durafourt, B.A.; Moore, C.S.; Zammit, D.A.; Johnson, T.A.; Zaguia, F.; Guiot, M.C.; Bar-Or, A.; Antel, J.P. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012, 60, 717–727. [Google Scholar] [CrossRef]
- Ueda, Y.; Gullipalli, D.; Song, W.-C. Modeling complement-driven diseases in transgenic mice: Values and limitations. Immunobiology 2016, 221, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- McQuade, A.; Blurton-Jones, M. Microglia in Alzheimer’s disease: Exploring how genetics and phenotype influence risk. J. Mol. Biol. 2019; 431, 1805–1817. [Google Scholar] [CrossRef]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci. 2006, 29, 506–510. [Google Scholar] [CrossRef]
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflamm. 2005, 2, 9. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Bousejra-ElGarah, F.; Bijani, C.; Coppel, Y.; Faller, P.; Hureau, C. Iron(II) binding to amyloid-β, the Alzheimer’s peptide. Inorg. Chem. 2011, 50, 9024–9030. [Google Scholar] [CrossRef]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef]
- Reybier, K.; Ayala, S.; Alies, B.; Rodrigues, J.V.; Bustos-Rodriguez, S.; La Penna, G.; Collin, F.; Gomes, C.M.; Hureau, C.; Faller, P. Free superoxide is an intermediate in the production of H2O2 by copper(I)-Aβ peptide and O2. Angew. Chem. Int. Ed. Engl. 2016, 55, 1085–1089. [Google Scholar] [CrossRef]
- Tahmasebinia, F.; Emadi, S. Effect of metal chelators on the aggregation of beta-amyloid peptides in the presence of copper and iron. Biometals 2017, 30, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Zawisza, I.; Rózga, M.; Bal, W. Affinity of copper and zinc ions to proteins and peptides related to neurodegenerative conditions (Aβ, APP, α-synuclein, PrP). Coord. Chem. Rev. 2012, 256, 2297–2307. [Google Scholar] [CrossRef]
- Migliorini, C.; Porciatti, E.; Luczkowski, M.; Valensin, D. Structural characterization of Cu2+, Ni2+ and Zn2+ binding sites of model peptides associated with neurodegenerative diseases. Coord. Chem. Rev. 2012, 256, 352–368. [Google Scholar] [CrossRef]
- Noel, S.; Bustos Rodriguez, S.; Sayen, S.; Guillon, E.; Faller, P.; Hureau, C. Use of a new water-soluble Zn sensor to determine Zn affinity for the amyloid-β peptide and relevant mutants. Metallomics 2014, 6, 1220–1222. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, P.; Martin-Aragon, S.; Benedi, J.; Susin, C.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from mild cognitive impairment. Free Radic. Res. 2008, 42, 162–170. [Google Scholar] [CrossRef]
- Liu, H.; Wang, H.; Shenvi, S.; Hagen, T.M.; Liu, R.M. Glutathione metabolism during aging and in Alzheimer disease. Ann. N. Y. Acad. Sci. 2014, 1019, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Ryan, T.M.; Bush, A.I.; Masters, C.L.; Duce, J.A. The role of metallobiology and amyloid-beta peptides in Alzheimer’s disease. J. Neurochem. 2012, 120 (Suppl. 1), 149–166. [Google Scholar] [CrossRef] [PubMed]
- Rottkamp, C.A.; Raina, A.K.; Zhu, X.; Gaier, E.; Bush, A.I.; Atwood, C.S.; Chevion, M.; Perry, G.; Smith, M.A. Redox-active iron mediates amyloid-beta toxicity. Free Radic. Biol. Med. 2001, 30, 447–450. [Google Scholar] [CrossRef]
- Schubert, D.; Chevion, M. The role of iron in beta amyloid toxicity. Biochem. Biophys. Res. Commun. 1995, 216, 702–707. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Preclinical models of Alzheimer’s disease: Relevance and translational validity. Curr. Protoc. Pharmacol. 2019, 84, e57. [Google Scholar] [CrossRef]
- Franco Bocanegra, D.K.; Nicoll, J.A.R.; Boche, D. Innate immunity in Alzheimer’s disease: The relevance of animal models? J. Neural. Transm. 2018, 125, 827–846. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Bodea, L.G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A.; Aggarwal, A. Therapeutic potentials of herbal drugs for Alzheimer’s disease—An overview. Indian J. Exp. Biol. 2017, 55, 63–73. [Google Scholar]
- Elufioye, T.O.; Berida, T.I.; Habtemariam, S. Plants-derived neuroprotective agents: Cutting the cycle of cell death through multiple mechanisms. eCAM 2017, 2017, 3574012. [Google Scholar] [CrossRef]
- Habtemariam, S. Protective effects of caffeic acid and the Alzheimer’s brain: An update. Mini Rev. Med. Chem. 2017, 17, 667–674. [Google Scholar] [CrossRef]
- Braidy, N.; Behzad, S.; Habtemariam, S.; Ahmed, T.; Daglia, M.; Nabavi, S.M.; Sobarzo-Sanchez, E.; Nabavi, S.F. Neuroprotective effects of citrus fruit-derived flavonoids, nobiletin and tangeretin in Alzheimer’s and Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2016, 16, 387–397. [Google Scholar] [CrossRef]
- Habtemariam, S. Rutin as a natural therapy for Alzheimer’s disease: Insights into its mechanisms of action. Curr. Med. Chem. 2016, 23, 860–873. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Khan, H.; D’onofrio, G.; Šamec, D.; Shirooie, S.; Dehpour, A.R.; Castilla, S.A.; Habtemariam, S.; Sobarzo-Sanchez, E. Apigenin as neuroprotective agent: Of mice and men. Pharmacol. Res. 2018, 128, 359–365. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Sureda, A.; Manayi, A.; Nabavi, S.M. Neuroprotective effects of fisetin in Alzheimer’s and Parkinson’s Diseases: From chemistry to medicine. Curr. Top. Med. Chem. 2016, 16, 1910–1915. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Orhan, I.E.; Daglia, M.; Manayi, A.; Gortzi, O.; Nabavi, S.M. Neuroprotective effects of chrysin: From chemistry to medicine. Neurochem. Int. 2015, 90, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Roselli, M.; Cavalluzzi, M.M.; Bruno, C.; Lovece, A.; Carocci, A.; Franchini, C.; Habtemariam, S.; Lentini, G. Synthesis and evaluation of berberine derivatives and analogues as potential anti-acetylcholinesterase and antioxidant agents. Phytochem. Lett. 2016, 18, 150–156. [Google Scholar] [CrossRef]
- Habtemariam, S. The therapeutic potential of Berberis darwinii stem-bark: Quantification of berberine and in vitro evidence for Alzheimer’s disease therapy. Nat. Prod. Commun. 2011, 6, 1089–1090. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Iridoids and other monoterpenes in the Alzheimer’s brain: Recent development and future prospects. Molecules 2018, 23, 117. [Google Scholar] [CrossRef]
- Habtemariam, S. The therapeutic potential of rosemary (Rosmarinus officinalis) diterpenes for Alzheimer’s disease. eCAM 2016, 2016, 2680409. [Google Scholar] [CrossRef]
- Ishii, K.; Klunk, W.E.; Arawaka, S.; Debnath, M.L.; Furiya, Y.; Sahara, N.; Shoji, S.; Tamaoka, A.; Pettegrew, J.W.; Mori, H. Chrysamine G and its derivative reduce amyloid β-induced neurotoxicity in mice. Neurosci. Lett. 2002, 333, 5–8. [Google Scholar] [CrossRef]
- Adessi, C.; Frossard, M.; Boissard, C.; Fraga, S.; Bieler, S.; Ruckle, T.; Vilbois, F.; Robinson, S.M.; Mutter, M.; Banks, W.A.; et al. Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13905–13911. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.J.; Tappe, R.; Meredith, S.C. Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Aβ1-40 fibrillogenesis. J. Pept. Res. 2002, 60, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Näslund, J.; Lindqvist, F.; Johansson, J.; Karlstörm, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef]
- Tjernberg, L.O.; Lilliehöök, C.; Callaway, D.J.E.; Näslund, J.; Hahne, S.; Terenius, T.J.L.; Nordstedt, C. Controlling amyloid β-peptide fibril formation with protease-stable ligands. J. Biol. Chem. 1997, 272, 12601–12605. [Google Scholar] [CrossRef]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castaño, E.M.; Frangione, B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat. Med. 1998, 4, 826–882. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti-amiloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef]
- Ono, K.; Naiki, H.K.H.; Yamada, M. Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer’s β-amyloid fibrils in vitro. Biochim. Biophys. Acta 2004, 1690, 193–202. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Yoshiike, Y.; Takashima, A.; Yamada, M.; Naiki, H. Nordihydroguaigaretic acid potently breaks down pre-formed Alzheimer’s β-amyloid fibrils in vitro. J. Neurochem. 2002, 81, 434–440. [Google Scholar] [CrossRef]
- Kurt, B.Z.; Gazioglu, I.; Dag, A.; Salmas, R.E.; Kayık, G.; Durdagi, S.; Sonmez, F. Synthesis, anticholinesterase activity and molecular modelling study of novel carbamate-substituted thymol/carvacrol derivatives. Bioorg. Med. Chem. 2017, 25, 1352–1363. [Google Scholar] [CrossRef]
- Stavrakov, G.; Philipova, I.; Zheleva-Dimitrova, D.; Valkova, I.; Salamanova, E.; Konstantinov, S.; Doytchinova, I. Docking-based design and synthesis of galantamine-camphane hybrids as inhibitors of acetylcholinesterase. Chem. Biol. Drug Des. 2017, 90, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Sureda, A.; Habtemariam, S.; Nabavi, S.M. Ginsenoside Rd and ischemic stroke; a short review of literatures. J. Ginseng Res. 2015, 39, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.M.; Habtemariam, S.; Daglia, M.; Braidy, N.; Loizzo, M.R.; Tundis, R.; Nabavi, S.F. Neuroprotective effects of ginkgolide B against ischemic stroke: A review of current literature. Curr. Top. Med. Chem. 2015, 15, 2222–2232. [Google Scholar] [CrossRef] [PubMed]
- Bahn, G.; Jo, DG. Therapeutic approaches to Alzheimer’s disease through modulation of NRF2. Neuromol. Med. 2019, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, L.; Weng, Q.; Xiao, L.; Li, Q. Curcumin analogues attenuate Aβ25-35-induced oxidative stress in PC12 cells via Keap1/Nrf2/HO-1 signaling pathways. Chem. Biol. Interact. 2019, 305, 171–179. [Google Scholar] [CrossRef]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective effect of caffeic acid phenethyl ester in a mouse model of Alzheimer’s disease involves Nrf2/HO-1 pathway. Aging Dis. 2018, 9, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, N. Berberine: Pathways to protect neurons. Phytother. Res. 2018, 32, 1501–1510. [Google Scholar] [CrossRef]
- Hui, Y.; Chengyong, T.; Cheng, L.; Haixia, H.; Yuanda, Z.; Weihua, Y. Resveratrol Attenuates the Cytotoxicity Induced by Amyloid-β1-42 in PC12 Cells by Upregulating Heme Oxygenase-1 via the PI3K/Akt/Nrf2 Pathway. Neurochem. Res. 2018, 43, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Lin, J.; Zhang, H.; Liu, Y.; Kan, M.; Xiu, Z.; Chen, X.; Lan, X.; Li, X.; Shi, X.; et al. Ginsenoside compound K regulates amyloid β via the Nrf2/Keap1 signaling pathway in mice with scopolamine hydrobromide-induced memory impairments. J. Mol. Neurosci. 2019, 67, 62–71. [Google Scholar] [CrossRef]
- Zaplatic, E.; Bule, M.; Shah, S.Z.A.; Uddin, M.S.; Niaz, K. Molecular mechanisms underlying protective role of quercetin in attenuating Alzheimer’s disease. Life Sci. 2019, 224, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Jhang, K.A.; Park, J.S.; Kim, H.S.; Chong, Y.H. Resveratrol Ameliorates Tau Hyperphosphorylation at Ser396 Site and Oxidative Damage in Rat Hippocampal Slices Exposed to Vanadate: Implication of ERK1/2 and GSK-3β Signaling Cascades. J. Agric. Food. Chem. 2017, 65, 9626–9634. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, X.; Wang, C.; Teng, Z.; Li, Y. Curcumin decreases hyperphosphorylation of tau by down-regulating caveolin-1/GSK-3β in N2a/APP695swe cells and APP/PS1 double transgenic Alzheimer’s disease mice. Am. J. Chin. Med. 2017, 45, 1667–1682. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Cheng, P.; Yu, K.; Han, Y.; Song, M.; Li, Y. Hyperforin attenuates aluminum-induced Aβ production and tau phosphorylation via regulating Akt/GSK-3β signaling pathway in PC12 cells. Biomed. Pharmacother. 2017, 96, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, J.; Ma, D.; Yuan, G.; Lu, Y.; Yang, Y. Capsaicin reduces Alzheimer-associated tau changes in the hippocampus of type 2 diabetes rats. PLoS ONE 2017, 12, e0172477. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. The brain-derived neurotrophic factor in neuronal plasticity and neuroregeneration: New pharmacological concepts for old and new drugs. Neural Regen. Res. 2018, 13, 983–984. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. PNAS 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- Wurzelmann, M.; Romeika, J.; Sun, D. Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen Res. 2017, 12, 7–12. [Google Scholar] [CrossRef]
- Stonesifer, C.; Corey, S.; Ghanekar, S.; Diamandis, Z.; Acosta, S.A.; Borlongan, C.V. Stem cell therapy for abrogating stroke-induced neuroinflammation and relevant secondary cell death mechanisms. Prog. Neurobiol. 2017, 94–131. [Google Scholar] [CrossRef]
- Li, Q.; Che, H.X.; Wang, C.C.; Zhang, L.Y.; Ding, L.; Xue, C.H.; Zhang, T.T.; Wang, Y.M. Cerebrosides from Sea Cucumber Improved Aβ1-42-Induced Cognitive Deficiency in a Rat Model of Alzheimer’s Disease. Mol. Nutr. Food Res. 2019, 63, e1800707. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Z.; Liu, J.W.; Wang, X.; Jeong, I.H.; Ahn, Y.J.; Zhang, C.J. Anti-BACE1 and antimicrobial activities of steroidal compounds isolated from marine Urechis unicinctus. Mar. Drugs 2018, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Syad, A.N.; Devi, K.P. Assessment of anti-amyloidogenic activity of marine red alga G. acerosa against Alzheimer’s beta-amyloid peptide 25–35. Neurol. Res. 2015, 37, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Moodie, L.W.K.; Sepčić, K.; Turk, T.; Frange, Ž.R.; Svenson, J. Natural cholinesterase inhibitors from marine organisms. Nat. Prod. Rep. 2019. [Google Scholar] [CrossRef]
- Che, H.; Li, Q.; Zhang, T.; Wang, D.; Yang, L.; Xu, J.; Yanagita, T.; Xue, C.; Chang, Y.; Wang, Y. Effects of astaxanthin and docosahexaenoic-acid-acylated astaxanthin on Alzheimer’s disease in APP/PS1 double-transgenic mice. J. Agric. Food Chem. 2018, 66, 4948–4957. [Google Scholar] [CrossRef]
- Wang, L.; Fan, H.; He, J.; Wang, L.; Tian, Z.; Wang, C. Protective effects of omega-3 fatty acids against Alzheimer’s disease in rat brain endothelial cells. Brain Behav. 2018, 8, e01037. [Google Scholar] [CrossRef]
- Bazan, N.G. Docosanoids and elovanoids from omega-3 fatty acids are pro-homeostatic modulators of inflammatory responses, cell damage and neuroprotection. Mol. Asp. Med. 2018, 64, 18–33. [Google Scholar] [CrossRef]
- Ajith, TA. A recent update on the effects of omega-3 fatty acids in Alzheimer’s disease. Curr. Clin. Pharmacol. 2018, 13, 252–260. [Google Scholar] [CrossRef]
- Nolan, J.M.; Mulcahy, R.; Power, R.; Moran, R.; Howard, A.N. Nutritional intervention to prevent Alzheimer’s disease: Potential benefits of xanthophyll carotenoids and omega-3 fatty acids combined. J. Alzheimers Dis. 2018, 64, 367–378. [Google Scholar] [CrossRef]
- Sharman, M.J.; Gyengesi, E.; Liang, H.; Chatterjee, P.; Karl, T.; Li, Q.X.; Wenk, M.R.; Halliwell, B.; Martins, R.N.; Münch, G. Assessment of diets containing curcumin, epigallocatechin-3-gallate, docosahexaenoic acid and α-lipoic acid on amyloid load and inflammation in a male transgenic mouse model of Alzheimer’s disease: Are combinations more effective? Neurobiol. Dis. 2019, 124, 505–519. [Google Scholar] [CrossRef]
- Rahman, S.O.; Panda, B.P.; Parvez, S.; Kaundal, M.; Hussain, S.; Akhtar, M.; Najmi, A.K. Neuroprotective role of astaxanthin in hippocampal insulin resistance induced by Aβ peptides in animal model of Alzheimer’s disease. Biomed. Pharmacother. 2019, 110, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Grimmig, B.; Daly, L.; Subbarayan, M.; Hudson, C.; Williamson, R.; Nash, K.; Bickford, P.C. Astaxanthin is neuroprotective in an aged mouse model of Parkinson’s disease. Oncotarget 2017, 9, 10388–10401. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, R. Bioavailable curcumin formulations: A review of pharmacokinetic studies in healthy volunteers. J. Integr. Med. 2018, 16, 367–374. [Google Scholar] [CrossRef]
- Pandareesh, M.D.; Mythri, R.B.; Srinivas Bharath, M.M. Bioavailability of dietary polyphenols: Factors contributing to their clinical application in CNS diseases. Neurochem. Int. 2015, 89, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.M.; Catanzaro, M.; Rosini, M.; Racchi, M.; Lanni, C. Curcumin in Alzheimer’s disease: Can we think to new strategies and perspectives for this molecule? Pharmacol. Res. 2017, 124, 146–155. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habtemariam, S. Natural Products in Alzheimer’s Disease Therapy: Would Old Therapeutic Approaches Fix the Broken Promise of Modern Medicines? Molecules 2019, 24, 1519. https://doi.org/10.3390/molecules24081519
Habtemariam S. Natural Products in Alzheimer’s Disease Therapy: Would Old Therapeutic Approaches Fix the Broken Promise of Modern Medicines? Molecules. 2019; 24(8):1519. https://doi.org/10.3390/molecules24081519
Chicago/Turabian StyleHabtemariam, Solomon. 2019. "Natural Products in Alzheimer’s Disease Therapy: Would Old Therapeutic Approaches Fix the Broken Promise of Modern Medicines?" Molecules 24, no. 8: 1519. https://doi.org/10.3390/molecules24081519
APA StyleHabtemariam, S. (2019). Natural Products in Alzheimer’s Disease Therapy: Would Old Therapeutic Approaches Fix the Broken Promise of Modern Medicines? Molecules, 24(8), 1519. https://doi.org/10.3390/molecules24081519