Regulation of Osteoclast Differentiation and Skeletal Maintenance by Histone Deacetylases


{kind=link}
{kind=link}
Abstract
1. Introduction
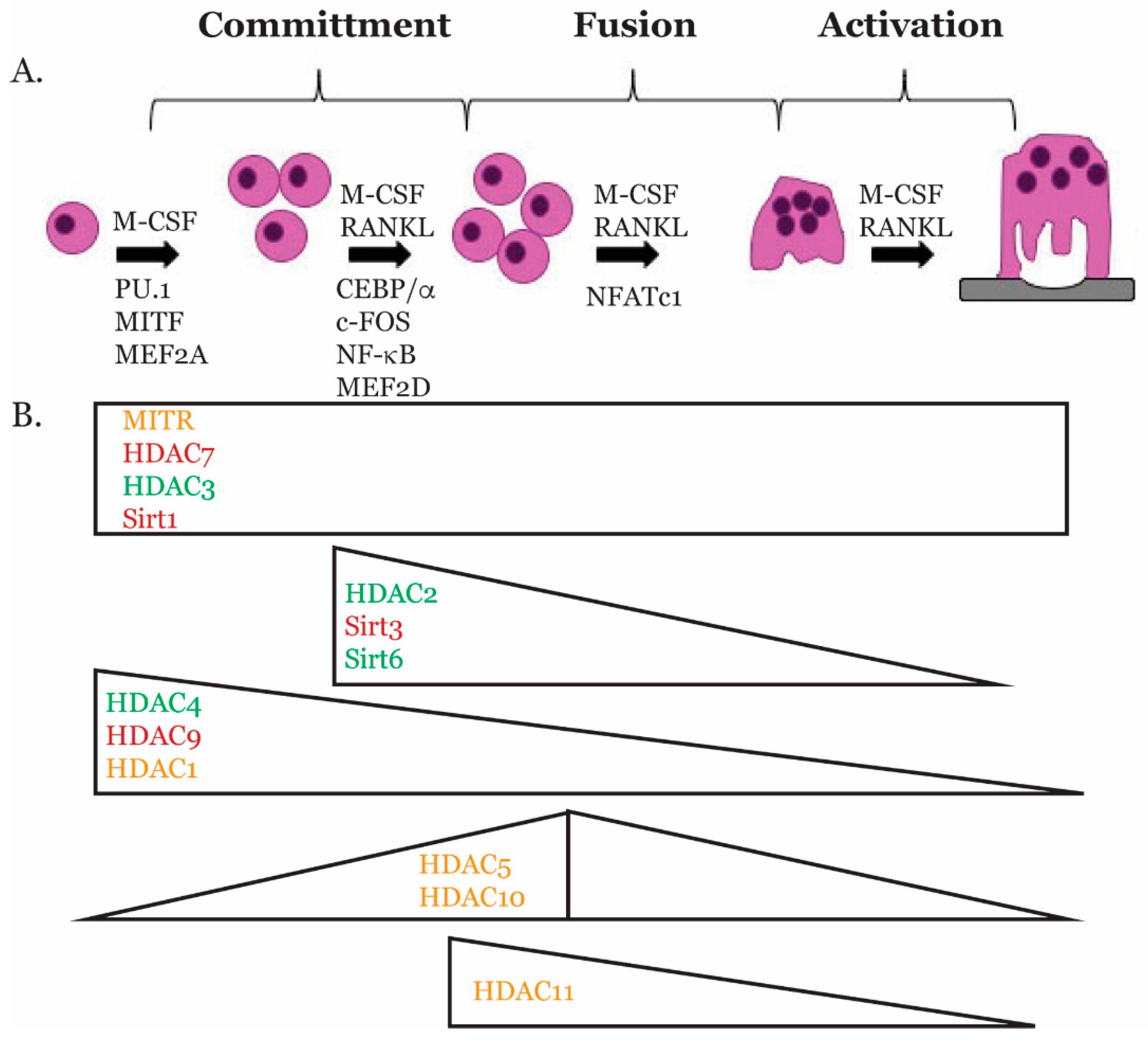
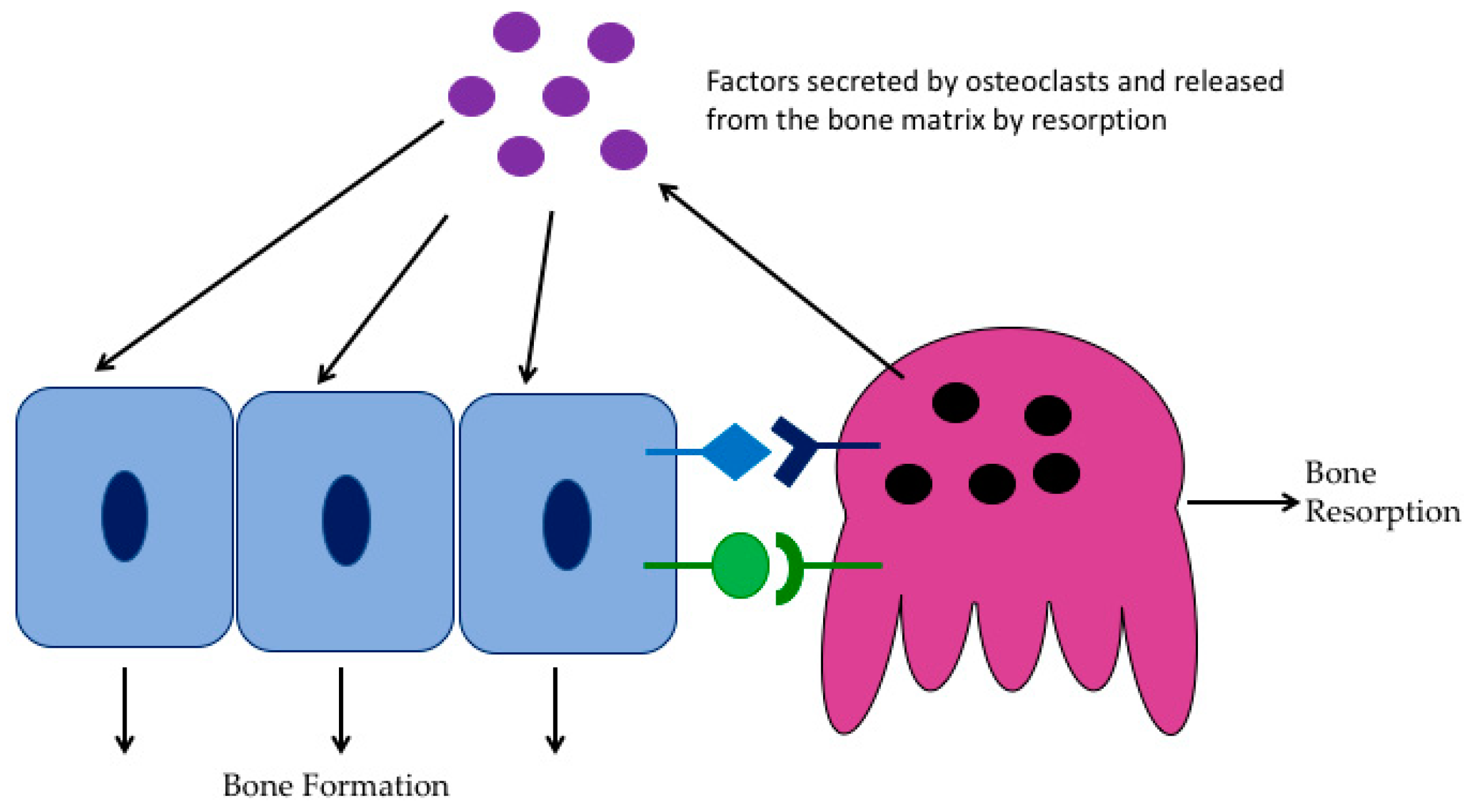
2. Osteoclast Biology
3. Transcriptional Regulators of Osteoclast Gene Expression
3.1. PU.1
3.2. MITF
3.3. CEBPα
3.4. MEF2
3.5. c-FOS
3.6. NF-κB
3.7. NFATc1
4. Histone Deacetylases
4.1. HDAC Classes
4.2. HDACs in Bone Development
4.3. HDACs and Skeletal Maintenance
4.4. HDACs and Skeletal Diseases
5. Role of HDACs in Osteoclasts
5.1. Class I HDACs
5.1.1. HDAC1
5.1.2. HDAC2
5.1.3. HDAC3
5.1.4. HDAC8
5.2. Class II HDACs
5.2.1. HDAC4
5.2.2. HDAC5
5.2.3. HDAC6
5.2.4. HDAC7
5.2.5. HDAC9
5.2.6. HDAC10
5.3. Class III HDACs
5.3.1. Sirtuin 1
5.3.2. Sirtuin 3
5.3.3. Sirtuin 6
5.4. Class IV HDACs
6. Effects of HDAC Inhibitors on the Skeleton
HDAC Inhibitors and Fracture Healing
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Westendorf, J.J.; Modder, U.I. Concise review: Insights from normal bone remodeling and stem cell-based therapies for bone repair. Stem Cells 2010, 28, 2124–2128. [Google Scholar] [PubMed]
- Crockett, J.C.; Rogers, M.J.; Coxon, F.P.; Hocking, L.J.; Helfrich, M.H. Bone remodelling at a glance. J. Cell Sci. 2011, 124, 991–998. [Google Scholar] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. 2011, 6, 121–145. [Google Scholar] [CrossRef]
- Bi, H.; Chen, X.; Gao, S.; Yu, X.; Xiao, J.; Zhang, B.; Liu, X.; Dai, M. Key triggers of osteoclast-related diseases and available strategies for targeted therapies: A review. Front. Med. (Lausanne) 2017, 4, 234. [Google Scholar] [CrossRef] [PubMed]
- Novack, D.V.; Mbalaviele, G. Osteoclasts-key players in skeletal health and disease. Microbiol. Spectr. 2016, 4. [Google Scholar] [PubMed]
- Mellis, D.J.; Itzstein, C.; Helfrich, M.H.; Crockett, J.C. The skeleton: A multi-functional complex organ: The role of key signalling pathways in osteoclast differentiation and in bone resorption. J. Endocrinol. 2011, 211, 131–143. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACS): Characterization of the classical hdac family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Wade, P.A. Transcriptional control at regulatory checkpoints by histone deacetylases: Molecular connections between cancer and chromatin. Hum. Mol. Genet. 2001, 10, 693–698. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. Hdac and hdac inhibitor: From cancer to cardiovascular diseases. Chonnam. Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef]
- Ikeda, K.; Takeshita, S. The role of osteoclast differentiation and function in skeletal homeostasis. J. Biochem. 2016, 159, 1–8. [Google Scholar] [CrossRef]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [PubMed]
- Novack, D.V.; Teitelbaum, S.L. The osteoclast: Friend or foe? Annu. Rev. Pathol. 2008, 3, 457–484. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Vaananen, H.K.; Laitala-Leinonen, T. Osteoclast lineage and function. Arch. Biochem. Biophys. 2008, 473, 132–138. [Google Scholar] [CrossRef]
- Blair, H.C.; Teitelbaum, S.L.; Ghiselli, R.; Gluck, S. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science 1989, 245, 855–857. [Google Scholar] [CrossRef]
- Scott, E.W.; Simon, M.C.; Anastasi, J.; Singh, H. Requirement of transcription factor pu.1 in the development of multiple hematopoietic lineages. Science 1994, 265, 1573–1577. [Google Scholar] [CrossRef]
- Tondravi, M.M.; McKercher, S.R.; Anderson, K.; Erdmann, J.M.; Quiroz, M.; Maki, R.; Teitelbaum, S.L. Osteopetrosis in mice lacking haematopoietic transcription factor pu.1. Nature 1997, 386, 81–84. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential role of p38 mitogen-activated protein kinase in cathepsin k gene expression during osteoclastogenesis through association of nfatc1 and pu.1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef]
- DeKoter, R.P.; Walsh, J.C.; Singh, H. Pu.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J. 1998, 17, 4456–4468. [Google Scholar] [CrossRef]
- Kwon, O.H.; Lee, C.K.; Lee, Y.I.; Paik, S.G.; Lee, H.J. The hematopoietic transcription factor pu.1 regulates rank gene expression in myeloid progenitors. Biochem. Biophys. Res. Commun. 2005, 335, 437–446. [Google Scholar] [CrossRef] [PubMed]
- So, H.; Rho, J.; Jeong, D.; Park, R.; Fisher, D.E.; Ostrowski, M.C.; Choi, Y.; Kim, N. Microphthalmia transcription factor and pu.1 synergistically induce the leukocyte receptor osteoclast-associated receptor gene expression. J. Biol. Chem. 2003, 278, 24209–24216. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Sato, K.; Asagiri, M.; Morita, I.; Soma, K.; Takayanagi, H. Contribution of nuclear factor of activated t cells c1 to the transcriptional control of immunoreceptor osteoclast-associated receptor but not triggering receptor expressed by myeloid cells-2 during osteoclastogenesis. J. Biol. Chem. 2005, 280, 32905–32913. [Google Scholar] [CrossRef]
- Hodgkinson, C.A.; Moore, K.J.; Nakayama, A.; Steingrimsson, E.; Copeland, N.G.; Jenkins, N.A.; Arnheiter, H. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 1993, 74, 395–404. [Google Scholar] [CrossRef]
- Hughes, M.J.; Lingrel, J.B.; Krakowsky, J.M.; Anderson, K.P. A helix-loop-helix transcription factor-like gene is located at the mi locus. J. Biol. Chem. 1993, 268, 20687–20690. [Google Scholar]
- Mansky, K.C.; Sankar, U.; Han, J.; Ostrowski, M.C. Microphthalmia transcription factor is a target of the p38 mapk pathway in response to receptor activator of nf-kappa b ligand signaling. J. Biol. Chem. 2002, 277, 11077–11083. [Google Scholar] [CrossRef] [PubMed]
- Marks, S.C., Jr.; Walker, D.G. The hematogenous origin of osteoclasts: Experimental evidence from osteopetrotic (microphthalmic) mice treated with spleen cells from beige mouse donors. Am. J. Anat. 1981, 161, 1–10. [Google Scholar] [CrossRef]
- Glowacki, J.; Cox, K.A.; Wilcon, S. Impaired osteoclast differentiation in subcutaneous implants of bone particles in osteopetrotic mutants. Bone Miner. 1989, 5, 271–278. [Google Scholar] [CrossRef]
- Ishii, J.; Kitazawa, R.; Mori, K.; McHugh, K.P.; Morii, E.; Kondo, T.; Kitazawa, S. Lipopolysaccharide suppresses rank gene expression in macrophages by down-regulating pu.1 and mitf. J. Cell Biochem. 2008, 105, 896–904. [Google Scholar] [CrossRef]
- Luchin, A.; Purdom, G.; Murphy, K.; Clark, M.Y.; Angel, N.; Cassady, A.I.; Hume, D.A.; Ostrowski, M.C. The microphthalmia transcription factor regulates expression of the tartrate-resistant acid phosphatase gene during terminal differentiation of osteoclasts. J. Bone Miner. Res. 2000, 15, 451–460. [Google Scholar] [CrossRef]
- Motyckova, G.; Weilbaecher, K.N.; Horstmann, M.; Rieman, D.J.; Fisher, D.Z.; Fisher, D.E. Linking osteopetrosis and pycnodysostosis: Regulation of cathepsin k expression by the microphthalmia transcription factor family. Proc. Natl. Acad. Sci. USA 2001, 98, 5798–5803. [Google Scholar] [CrossRef]
- Ramji, D.P.; Foka, P. Ccaat/enhancer-binding proteins: Structure, function and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef]
- Keeshan, K.; Santilli, G.; Corradini, F.; Perrotti, D.; Calabretta, B. Transcription activation function of c/ebpalpha is required for induction of granulocytic differentiation. Blood 2003, 102, 1267–1275. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, G.; Hao, L.; Wu, M.; Ci, H.; Li, Y.P. C/ebpalpha regulates osteoclast lineage commitment. Proc. Natl. Acad. Sci. USA 2013, 110, 7294–7299. [Google Scholar] [CrossRef]
- Jules, J.; Li, Y.P.; Chen, W. C/ebpalpha and pu.1 exhibit different responses to rank signaling for osteoclastogenesis. Bone 2018, 107, 104–114. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, G.; Tang, J.; Zhou, H.D.; Li, Y.P. C/ebpalpha controls osteoclast terminal differentiation, activation, function, and postnatal bone homeostasis through direct regulation of nfatc1. J. Pathol. 2018, 244, 271–282. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, G.; Jules, J.; Nguyen, D.; Li, Y.P. Monocyte-specific knockout of c/ebpalpha results in osteopetrosis phenotype, blocks bone loss in ovariectomized mice, and reveals an important function of c/ebpalpha in osteoclast differentiation and function. J. Bone Miner. Res. 2018, 33, 691–703. [Google Scholar] [CrossRef]
- Jules, J.; Chen, W.; Feng, X.; Li, Y.P. Ccaat/enhancer-binding protein alpha (c/ebpalpha) is important for osteoclast differentiation and activity. J. Biol. Chem. 2016, 291, 16390–16403. [Google Scholar] [CrossRef]
- Pon, J.R.; Marra, M.A. Mef2 transcription factors: Developmental regulators and emerging cancer genes. Oncotarget 2016, 7, 2297–2312. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Olson, E.N. Mef2: A central regulator of diverse developmental programs. Development 2007, 134, 4131–4140. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Mef2: A calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 2002, 27, 40–47. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Ovitt, C.; Grigoriadis, A.E.; Mohle-Steinlein, U.; Ruther, U.; Wagner, E.F. Bone and haematopoietic defects in mice lacking c-fos. Nature 1992, 360, 741–745. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor nfatc1 (nfat2) integrate rankl signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Grigoriadis, A.E.; Wang, Z.Q.; Cecchini, M.G.; Hofstetter, W.; Felix, R.; Fleisch, H.A.; Wagner, E.F. C-fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 1994, 266, 443–448. [Google Scholar] [CrossRef]
- Soysa, N.S.; Alles, N. Nf-kappab functions in osteoclasts. Biochem. Biophys. Res. Commun. 2009, 378, 1–5. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xiu, Y.; Li, J.; Xing, L.; Yao, Z. Nf-kappab-mediated regulation of osteoclastogenesis. Endocrinol. Metab. 2015, 30, 35–44. [Google Scholar] [CrossRef]
- Novack, D.V. Role of nf-kappab in the skeleton. Cell Res. 2011, 21, 169–182. [Google Scholar] [CrossRef]
- Yamashita, T.; Yao, Z.; Li, F.; Zhang, Q.; Badell, I.R.; Schwarz, E.M.; Takeshita, S.; Wagner, E.F.; Noda, M.; Matsuo, K.; et al. Nf-kappab p50 and p52 regulate receptor activator of nf-kappab ligand (rankl) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-fos and nfatc1. J. Biol. Chem. 2007, 282, 18245–18253. [Google Scholar] [CrossRef]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the nfat family: Regulation and function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Galson, D.L.; Zhao, C.; Peng, L.; Laplace, C.; Wang, K.Z.; Bachler, M.A.; Amano, H.; Aburatani, H.; Ishikawa, H.; et al. Nuclear factor of activated t-cells (nfat) rescues osteoclastogenesis in precursors lacking c-fos. J. Biol. Chem. 2004, 279, 26475–26480. [Google Scholar] [CrossRef]
- Song, I.; Kim, J.H.; Kim, K.; Jin, H.M.; Youn, B.U.; Kim, N. Regulatory mechanism of nfatc1 in rankl-induced osteoclast activation. FEBS Lett. 2009, 583, 2435–2440. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.; Jin, H.M.; Song, I.; Youn, B.U.; Lee, S.H.; Choi, Y.; Kim, N. Negative feedback control of osteoclast formation through ubiquitin-mediated down-regulation of nfatc1. J. Biol. Chem. 2010, 285, 5224–5231. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.M.; Bronisz, A.; Hu, R.; Patel, K.; Mansky, K.C.; Sif, S.; Ostrowski, M.C. Mitf and pu.1 recruit p38 mapk and nfatc1 to target genes during osteoclast differentiation. J. Biol. Chem. 2007, 282, 15921–15929. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bertos, N.R.; Wang, A.H.; Yang, X.J. Class ii histone deacetylases: Structure, function, and regulation. Biochem. Cell Biol. 2001, 79, 243–252. [Google Scholar] [CrossRef]
- Martin, M.; Kettmann, R.; Dequiedt, F. Class iia histone deacetylases: Regulating the regulators. Oncogene 2007, 26, 5450–5467. [Google Scholar] [CrossRef]
- Dai, Y.; Faller, D.V. Transcription regulation by class III histone deacetylases (hdacs)-sirtuins. Transl. Oncogenomics 2008, 3, 53–65. [Google Scholar]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of hdac11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gupta, M.; Trivedi, C.M.; Singh, M.K.; Li, L.; Epstein, J.A. Murine craniofacial development requires hdac3-mediated repression of msx gene expression. Dev. Biol 2013, 377, 333–344. [Google Scholar] [CrossRef]
- Haberland, M.; Mokalled, M.H.; Montgomery, R.L.; Olson, E.N. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009, 23, 1625–1630. [Google Scholar] [CrossRef]
- Bradley, E.W.; Carpio, L.R.; van Wijnen, A.J.; McGee-Lawrence, M.E.; Westendorf, J.J. Histone deacetylases in bone development and skeletal disorders. Physiol. Rev. 2015, 95, 1359–1381. [Google Scholar] [CrossRef]
- Lagger, G.; O’Carroll, D.; Rembold, M.; Khier, H.; Tischler, J.; Weitzer, G.; Schuettengruber, B.; Hauser, C.; Brunmeir, R.; Jenuwein, T.; et al. Essential function of histone deacetylase 1 in proliferation control and cdk inhibitor repression. EMBO J. 2002, 21, 2672–2681. [Google Scholar] [CrossRef]
- Chang, S.; Young, B.D.; Li, S.; Qi, X.; Richardson, J.A.; Olson, E.N. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006, 126, 321–334. [Google Scholar] [CrossRef]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in s phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef]
- Trivedi, C.M.; Luo, Y.; Yin, Z.; Zhang, M.; Zhu, W.; Wang, T.; Floss, T.; Goettlicher, M.; Noppinger, P.R.; Wurst, W.; et al. Hdac2 regulates the cardiac hypertrophic response by modulating gsk3 beta activity. Nat. Med. 2007, 13, 324–331. [Google Scholar] [CrossRef]
- Chang, S.; McKinsey, T.A.; Zhang, C.L.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell Biol. 2004, 24, 8467–8476. [Google Scholar] [CrossRef]
- Razidlo, D.F.; Whitney, T.J.; Casper, M.E.; McGee-Lawrence, M.E.; Stensgard, B.A.; Li, X.; Secreto, F.J.; Knutson, S.K.; Hiebert, S.W.; Westendorf, J.J. Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PLoS ONE 2010, 5, e11492. [Google Scholar] [CrossRef]
- Bradley, E.W.; McGee-Lawrence, M.E.; Westendorf, J.J. Hdac-mediated control of endochondral and intramembranous ossification. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 101–113. [Google Scholar]
- Jeon, E.J.; Lee, K.Y.; Choi, N.S.; Lee, M.H.; Kim, H.N.; Jin, Y.H.; Ryoo, H.M.; Choi, J.Y.; Yoshida, M.; Nishino, N.; et al. Bone morphogenetic protein-2 stimulates runx2 acetylation. J. Biol. Chem. 2006, 281, 16502–16511. [Google Scholar] [CrossRef]
- Westendorf, J.J.; Zaidi, S.K.; Cascino, J.E.; Kahler, R.; van Wijnen, A.J.; Lian, J.B.; Yoshida, M.; Stein, G.S.; Li, X. Runx2 (cbfa1, aml-3) interacts with histone deacetylase 6 and represses the p21(cip1/waf1) promoter. Mol. Cell Biol. 2002, 22, 7982–7992. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.M.; Kahler, R.A.; Li, X.; Westendorf, J.J. Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J. Biol. Chem. 2004, 279, 41998–42007. [Google Scholar] [CrossRef] [PubMed]
- Westendorf, J.J. Transcriptional co-repressors of runx2. J. Cell Biochem. 2006, 98, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Obri, A.; Makinistoglu, M.P.; Zhang, H.; Karsenty, G. Hdac4 integrates pth and sympathetic signaling in osteoblasts. J. Cell Biol. 2014, 205, 771–780. [Google Scholar] [CrossRef]
- Jensen, E.D.; Schroeder, T.M.; Bailey, J.; Gopalakrishnan, R.; Westendorf, J.J. Histone deacetylase 7 associates with runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. J. Bone Miner. Res. 2008, 23, 361–372. [Google Scholar] [CrossRef]
- Lee, H.W.; Suh, J.H.; Kim, A.Y.; Lee, Y.S.; Park, S.Y.; Kim, J.B. Histone deacetylase 1-mediated histone modification regulates osteoblast differentiation. Mol. Endocrinol. 2006, 20, 2432–2443. [Google Scholar] [CrossRef]
- Wein, M.N.; Spatz, J.; Nishimori, S.; Doench, J.; Root, D.; Babij, P.; Nagano, K.; Baron, R.; Brooks, D.; Bouxsein, M.; et al. Hdac5 controls mef2c-driven sclerostin expression in osteocytes. J. Bone Miner. Res. 2015, 30, 400–411. [Google Scholar] [CrossRef]
- Baertschi, S.; Baur, N.; Lueders-Lefevre, V.; Voshol, J.; Keller, H. Class i and iia histone deacetylases have opposite effects on sclerostin gene regulation. J. Biol. Chem. 2014, 289, 24995–25009. [Google Scholar] [CrossRef] [PubMed]
- McGee-Lawrence, M.E.; Westendorf, J.J. Histone deacetylases in skeletal development and bone mass maintenance. Gene 2011, 474, 1–11. [Google Scholar] [CrossRef]
- Huynh, N.C.; Everts, V.; Ampornaramveth, R.S. Histone deacetylases and their roles in mineralized tissue regeneration. Bone Rep. 2017, 7, 33–40. [Google Scholar] [CrossRef]
- Harakalova, M.; van den Boogaard, M.J.; Sinke, R.; van Lieshout, S.; van Tuil, M.C.; Duran, K.; Renkens, I.; Terhal, P.A.; de Kovel, C.; Nijman, I.J.; et al. X-exome sequencing identifies a hdac8 variant in a large pedigree with x-linked intellectual disability, truncal obesity, gynaecomastia, hypogonadism and unusual face. J. Med. Genet. 2012, 49, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, F.J.; Ansari, M.; Braunholz, D.; Concepcion Gil-Rodriguez, M.; Decroos, C.; Wilde, J.J.; Fincher, C.T.; Kaur, M.; Bando, M.; Amor, D.J.; et al. Loss-of-function hdac8 mutations cause a phenotypic spectrum of cornelia de lange syndrome-like features, ocular hypertelorism, large fontanelle and x-linked inheritance. Hum. Mol. Genet. 2014, 23, 2888–2900. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. Hdac8 mutations in cornelia de lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Tassano, E.; Mirabelli-Badenier, M.; Veneselli, E.; Puliti, A.; Lerone, M.; Vaccari, C.M.; Morana, G.; Porta, S.; Gimelli, G.; Cuoco, C. Clinical and molecular characterization of a patient with interstitial 6q21q22.1 deletion. Mol. Cytogenet. 2015, 8, 31. [Google Scholar] [CrossRef]
- Villavicencio-Lorini, P.; Klopocki, E.; Trimborn, M.; Koll, R.; Mundlos, S.; Horn, D. Phenotypic variant of brachydactyly-mental retardation syndrome in a family with an inherited interstitial 2q37.3 microdeletion including hdac4. Eur. J. Hum. Genet. 2013, 21, 743–748. [Google Scholar] [CrossRef]
- Williams, S.R.; Aldred, M.A.; Der Kaloustian, V.M.; Halal, F.; Gowans, G.; McLeod, D.R.; Zondag, S.; Toriello, H.V.; Magenis, R.E.; Elsea, S.H. Haploinsufficiency of hdac4 causes brachydactyly mental retardation syndrome, with brachydactyly type e, developmental delays, and behavioral problems. Am. J. Hum. Genet. 2010, 87, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, H.; Liu, W.; Hu, R.; Huang, B.; Tan, Y.F.; Xu, K.; Sheng, Z.F.; Zhou, H.D.; Wu, X.P.; et al. A novel microrna targeting hdac5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J. Clin. Invest. 2009, 119, 3666–3677. [Google Scholar] [CrossRef]
- Rivadeneira, F.; Styrkarsdottir, U.; Estrada, K.; Halldorsson, B.V.; Hsu, Y.H.; Richards, J.B.; Zillikens, M.C.; Kavvoura, F.K.; Amin, N.; Aulchenko, Y.S.; et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat. Genet. 2009, 41, 1199–1206. [Google Scholar] [CrossRef]
- Simon, D.; Laloo, B.; Barillot, M.; Barnetche, T.; Blanchard, C.; Rooryck, C.; Marche, M.; Burgelin, I.; Coupry, I.; Chassaing, N.; et al. A mutation in the 3’-utr of the hdac6 gene abolishing the post-transcriptional regulation mediated by hsa-mir-433 is linked to a new form of dominant x-linked chondrodysplasia. Hum. Mol. Genet. 2010, 19, 2015–2027. [Google Scholar] [CrossRef]
- Hong, S.; Derfoul, A.; Pereira-Mouries, L.; Hall, D.J. A novel domain in histone deacetylase 1 and 2 mediates repression of cartilage-specific genes in human chondrocytes. FASEB J. 2009, 23, 3539–3552. [Google Scholar] [CrossRef]
- Higashiyama, R.; Miyaki, S.; Yamashita, S.; Yoshitaka, T.; Lindman, G.; Ito, Y.; Sasho, T.; Takahashi, K.; Lotz, M.; Asahara, H. Correlation between mmp-13 and hdac7 expression in human knee osteoarthritis. Mod. Rheumatol. 2010, 20, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Dvir-Ginzberg, M.; Gagarina, V.; Lee, E.J.; Hall, D.J. Regulation of cartilage-specific gene expression in human chondrocytes by sirt1 and nicotinamide phosphoribosyltransferase. J. Biol. Chem. 2008, 283, 36300–36310. [Google Scholar] [CrossRef]
- Abed, E.; Couchourel, D.; Delalandre, A.; Duval, N.; Pelletier, J.P.; Martel-Pelletier, J.; Lajeunesse, D. Low sirtuin 1 levels in human osteoarthritis subchondral osteoblasts lead to abnormal sclerostin expression which decreases wnt/beta-catenin activity. Bone 2014, 59, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Gabay, O.; Zaal, K.J.; Sanchez, C.; Dvir-Ginzberg, M.; Gagarina, V.; Song, Y.; He, X.H.; McBurney, M.W. Sirt1-deficient mice exhibit an altered cartilage phenotype. Joint Bone Spine 2013, 80, 613–620. [Google Scholar] [CrossRef]
- Lee, H.A.; Song, M.J.; Seok, Y.M.; Kang, S.H.; Kim, S.Y.; Kim, I. Histone deacetylase 3 and 4 complex stimulates the transcriptional activity of the mineralocorticoid receptor. PLoS ONE 2015, 10, e0136801. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Schreiber, S.L. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA 2000, 97, 7835–7840. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Dequiedt, F.; Fillion, M.; Hendzel, M.J.; Voelter, W.; Verdin, E. Human hdac7 histone deacetylase activity is associated with hdac3 in vivo. J. Biol. Chem. 2001, 276, 35826–35835. [Google Scholar] [CrossRef]
- Jin, Z.; Wei, W.; Huynh, H.; Wan, Y. Hdac9 inhibits osteoclastogenesis via mutual suppression of ppargamma/rankl signaling. Mol. Endocrinol. 2015, 29, 730–738. [Google Scholar] [CrossRef]
- Kim, K.; Lee, J.; Kim, J.H.; Jin, H.M.; Zhou, B.; Lee, S.Y.; Kim, N. Protein inhibitor of activated stat 3 modulates osteoclastogenesis by down-regulation of nfatc1 and osteoclast-associated receptor. J. Immunol. 2007, 178, 5588–5594. [Google Scholar] [CrossRef]
- Hu, R.; Sharma, S.M.; Bronisz, A.; Srinivasan, R.; Sankar, U.; Ostrowski, M.C. Eos, mitf, and pu.1 recruit corepressors to osteoclast-specific genes in committed myeloid progenitors. Mol. Cell Biol. 2007, 27, 4018–4027. [Google Scholar] [CrossRef]
- Dou, C.; Li, N.; Ding, N.; Liu, C.; Yang, X.; Kang, F.; Cao, Z.; Quan, H.; Hou, T.; Xu, J.; et al. Hdac2 regulates foxo1 during rankl-induced osteoclastogenesis. Am. J. Physiol. Cell Physiol. 2016, 310, C780–C787. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.; Kaiser, B.; Romsa, A.; Schwarz, T.; Gopalakrishnan, R.; Jensen, E.D.; Mansky, K.C. Hdac3 and hdac7 have opposite effects on osteoclast differentiation. J. Biol. Chem. 2011, 286, 12056–12065. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Kukita, A.; Kukita, T.; Shobuike, T.; Nakamura, T.; Kohashi, O. Two histone deacetylase inhibitors, trichostatin a and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood 2003, 101, 3451–3459. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Ha, H.; Lee, J.H.; Jung, K.; Yang, D.; Woo, K.M.; Lee, Z.H. Trichostatin a inhibits osteoclastogenesis and bone resorption by suppressing the induction of c-fos by rankl. Eur. J. Pharmacol. 2009, 623, 22–29. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef]
- Chawla, S.; Vanhoutte, P.; Arnold, F.J.; Huang, C.L.; Bading, H. Neuronal activity-dependent nucleocytoplasmic shuttling of hdac4 and hdac5. J. Neurochem. 2003, 85, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Miska, E.A.; Karlsson, C.; Langley, E.; Nielsen, S.J.; Pines, J.; Kouzarides, T. Hdac4 deacetylase associates with and represses the mef2 transcription factor. EMBO J. 1999, 18, 5099–5107. [Google Scholar] [CrossRef]
- Blixt, N.C.; Faulkner, B.K.; Astleford, K.; Lelich, R.; Schering, J.; Spencer, E.; Gopalakrishnan, R.; Jensen, E.D.; Mansky, K.C. Class ii and iv hdacs function as inhibitors of osteoclast differentiation. PLoS ONE 2017, 12, e0185441. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.; Youn, B.U.; Jin, H.M.; Kim, J.Y.; Moon, J.B.; Ko, A.; Seo, S.B.; Lee, K.Y.; Kim, N. Rankl induces nfatc1 acetylation and stability via histone acetyltransferases during osteoclast differentiation. Biochem. J. 2011, 436, 253–262. [Google Scholar] [CrossRef]
- Destaing, O.; Saltel, F.; Gilquin, B.; Chabadel, A.; Khochbin, S.; Ory, S.; Jurdic, P. A novel rho-mdia2-hdac6 pathway controls podosome patterning through microtubule acetylation in osteoclasts. J. Cell Sci. 2005, 118, 2901–2911. [Google Scholar] [CrossRef]
- Zilberman, Y.; Ballestrem, C.; Carramusa, L.; Mazitschek, R.; Khochbin, S.; Bershadsky, A. Regulation of microtubule dynamics by inhibition of the tubulin deacetylase hdac6. J. Cell Sci. 2009, 122, 3531–3541. [Google Scholar] [CrossRef]
- Jin, Z.; Wei, W.; Dechow, P.C.; Wan, Y. Hdac7 inhibits osteoclastogenesis by reversing rankl-triggered beta-catenin switch. Mol. Endocrinol. 2013, 27, 325–335. [Google Scholar] [CrossRef]
- Stemig, M.; Astelford, K.; Emery, A.; Cho, J.J.; Allen, B.; Huang, T.H.; Gopalakrishnan, R.; Mansky, K.C.; Jensen, E.D. Deletion of histone deacetylase 7 in osteoclasts decreases bone mass in mice by interactions with mitf. PLoS ONE 2015, 10, e0123843. [Google Scholar] [CrossRef]
- Kao, H.Y.; Lee, C.H.; Komarov, A.; Han, C.C.; Evans, R.M. Isolation and characterization of mammalian hdac10, a novel histone deacetylase. J. Biol. Chem. 2002, 277, 187–193. [Google Scholar] [CrossRef]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef]
- Kim, H.N.; Han, L.; Iyer, S.; de Cabo, R.; Zhao, H.; O’Brien, C.A.; Manolagas, S.C.; Almeida, M. Sirtuin1 suppresses osteoclastogenesis by deacetylating foxos. Mol. Endocrinol. 2015, 29, 1498–1509. [Google Scholar] [CrossRef]
- Huh, J.E.; Shin, J.H.; Jang, E.S.; Park, S.J.; Park, D.R.; Ko, R.; Seo, D.H.; Kim, H.S.; Lee, S.H.; Choi, Y.; et al. Sirtuin 3 (sirt3) maintains bone homeostasis by regulating ampk-pgc-1beta axis in mice. Sci. Rep. 2016, 6, 22511. [Google Scholar] [CrossRef]
- Lee, H.S.; Ka, S.O.; Lee, S.M.; Lee, S.I.; Park, J.W.; Park, B.H. Overexpression of sirtuin 6 suppresses inflammatory responses and bone destruction in mice with collagen-induced arthritis. Arthritis Rheum. 2013, 65, 1776–1785. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Cantley, M.D.; Zannettino, A.C.W.; Bartold, P.M.; Fairlie, D.P.; Haynes, D.R. Histone deacetylases (hdac) in physiological and pathological bone remodelling. Bone 2017, 95, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Sheth, R.D.; Wesolowski, C.A.; Jacob, J.C.; Penney, S.; Hobbs, G.R.; Riggs, J.E.; Bodensteiner, J.B. Effect of carbamazepine and valproate on bone mineral density. J. Pediatr. 1995, 127, 256–262. [Google Scholar] [CrossRef]
- Boluk, A.; Guzelipek, M.; Savli, H.; Temel, I.; Ozisik, H.I.; Kaygusuz, A. The effect of valproate on bone mineral density in adult epileptic patients. Pharmacol. Res. 2004, 50, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.O.; Jacobson, M.P.; Haneef, Z. Homocysteine and bone loss in epilepsy. Seizure 2007, 16, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, P.; Rejnmark, L.; Mosekilde, L. Fracture risk associated with use of antiepileptic drugs. Epilepsia 2004, 45, 1330–1337. [Google Scholar] [CrossRef]
- Sato, Y.; Kondo, I.; Ishida, S.; Motooka, H.; Takayama, K.; Tomita, Y.; Maeda, H.; Satoh, K. Decreased bone mass and increased bone turnover with valproate therapy in adults with epilepsy. Neurology 2001, 57, 445–449. [Google Scholar] [CrossRef]
- Oner, N.; Kaya, M.; Karasalihoglu, S.; Karaca, H.; Celtik, C.; Tutunculer, F. Bone mineral metabolism changes in epileptic children receiving valproic acid. J. Paediatr. Child. Health 2004, 40, 470–473. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, J.W.; Choi, K.G.; Chung, H.W.; Lee, H.W. A 6-month longitudinal study of bone mineral density with antiepileptic drug monotherapy. Epilepsy Behav. 2007, 10, 291–295. [Google Scholar] [CrossRef]
- Rieger-Wettengl, G.; Tutlewski, B.; Stabrey, A.; Rauch, F.; Herkenrath, P.; Schauseil-Zipf, U.; Schoenau, E. Analysis of the musculoskeletal system in children and adolescents receiving anticonvulsant monotherapy with valproic acid or carbamazepine. Pediatrics 2001, 108, E107. [Google Scholar] [CrossRef]
- Tsukahara, H.; Kimura, K.; Todoroki, Y.; Ohshima, Y.; Hiraoka, M.; Shigematsu, Y.; Tsukahara, Y.; Miura, M.; Mayumi, M. Bone mineral status in ambulatory pediatric patients on long-term anti-epileptic drug therapy. Pediatr. Int. 2002, 44, 247–253. [Google Scholar] [CrossRef]
- Van der Laan, J.W.; de Boer, T.; Bruinvels, J. Di-n-propylacetate and gaba degradation. Preferential inhibition of succinic semialdehyde dehydrogenase and indirect inhibition of gaba-transaminase. J. Neurochem. 1979, 32, 1769–1780. [Google Scholar] [CrossRef]
- Loscher, W. Valproate induced changes in gaba metabolism at the subcellular level. Biochem. Pharmacol. 1981, 30, 1364–1366. [Google Scholar] [CrossRef]
- Cantley, M.D.; Bartold, P.M.; Marino, V.; Fairlie, D.P.; Le, G.T.; Lucke, A.J.; Haynes, D.R. Histone deacetylase inhibitors and periodontal bone loss. J. Periodontal Res. 2011, 46, 697–703. [Google Scholar] [CrossRef]
- Chung, Y.L.; Lee, M.Y.; Wang, A.J.; Yao, L.F. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol. Ther. 2003, 8, 707–717. [Google Scholar] [CrossRef]
- Lin, H.S.; Hu, C.Y.; Chan, H.Y.; Liew, Y.Y.; Huang, H.P.; Lepescheux, L.; Bastianelli, E.; Baron, R.; Rawadi, G.; Clement-Lacroix, P. Anti-rheumatic activities of histone deacetylase (hdac) inhibitors in vivo in collagen-induced arthritis in rodents. Br. J. Pharmacol. 2007, 150, 862–872. [Google Scholar] [CrossRef]
- Lohman, R.J.; Iyer, A.; Fairlie, T.J.; Cotterell, A.; Gupta, P.; Reid, R.C.; Vesey, D.A.; Sweet, M.J.; Fairlie, D.P. Differential anti-inflammatory activity of hdac inhibitors in human macrophages and rat arthritis. J. Pharmacol. Exp. Ther. 2016, 356, 387–396. [Google Scholar] [CrossRef]
- Baumann, P.; Junghanns, C.; Mandl-Weber, S.; Strobl, S.; Oduncu, F.; Schmidmaier, R. The pan-histone deacetylase inhibitor cr2408 disrupts cell cycle progression, diminishes proliferation and causes apoptosis in multiple myeloma cells. Br. J. Haematol. 2012, 156, 633–642. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective hdac6 inhibitor, acy-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Deleu, S.; Lemaire, M.; Arts, J.; Menu, E.; Van Valckenborgh, E.; Vande Broek, I.; De Raeve, H.; Coulton, L.; Van Camp, B.; Croucher, P.; et al. Bortezomib alone or in combination with the histone deacetylase inhibitor jnj-26481585: Effect on myeloma bone disease in the 5t2mm murine model of myeloma. Cancer Res. 2009, 69, 5307–5311. [Google Scholar] [CrossRef]
- Stuhmer, T.; Arts, J.; Chatterjee, M.; Borawski, J.; Wolff, A.; King, P.; Einsele, H.; Leo, E.; Bargou, R.C. Preclinical anti-myeloma activity of the novel hdac-inhibitor jnj-26481585. Br. J. Haematol. 2010, 149, 529–536. [Google Scholar] [CrossRef]
- Marsell, R.; Einhorn, T.A. The biology of fracture healing. Injury 2011, 42, 551–555. [Google Scholar] [CrossRef]
- Lee, S.U.; Kwak, H.B.; Pi, S.H.; You, H.K.; Byeon, S.R.; Ying, Y.; Luesch, H.; Hong, J.; Kim, S.H. In vitro and in vivo osteogenic activity of largazole. ACS Med. Chem. Lett. 2011, 2, 248–251. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faulkner, B.; Astleford, K.; Mansky, K.C. Regulation of Osteoclast Differentiation and Skeletal Maintenance by Histone Deacetylases. Molecules 2019, 24, 1355. https://doi.org/10.3390/molecules24071355
Faulkner B, Astleford K, Mansky KC. Regulation of Osteoclast Differentiation and Skeletal Maintenance by Histone Deacetylases. Molecules. 2019; 24(7):1355. https://doi.org/10.3390/molecules24071355
Chicago/Turabian StyleFaulkner, Bora, Kristina Astleford, and Kim C. Mansky. 2019. "Regulation of Osteoclast Differentiation and Skeletal Maintenance by Histone Deacetylases" Molecules 24, no. 7: 1355. https://doi.org/10.3390/molecules24071355
APA StyleFaulkner, B., Astleford, K., & Mansky, K. C. (2019). Regulation of Osteoclast Differentiation and Skeletal Maintenance by Histone Deacetylases. Molecules, 24(7), 1355. https://doi.org/10.3390/molecules24071355