
Engineering Peptide-Based Polyelectrolyte Complexes with Increased Hydrophobicity


Abstract

1. Introduction
2. Results and Discussion
2.1. Peptide Design
2.2. Characterization of Polypeptides
2.2.1. Matrix-Assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) Mass Spectroscopy
2.2.2. Proton Nuclear Magnetic Resonance (H-NMR) Spectroscopy
2.2.3. Circular Dichroism (CD) Spectroscopy
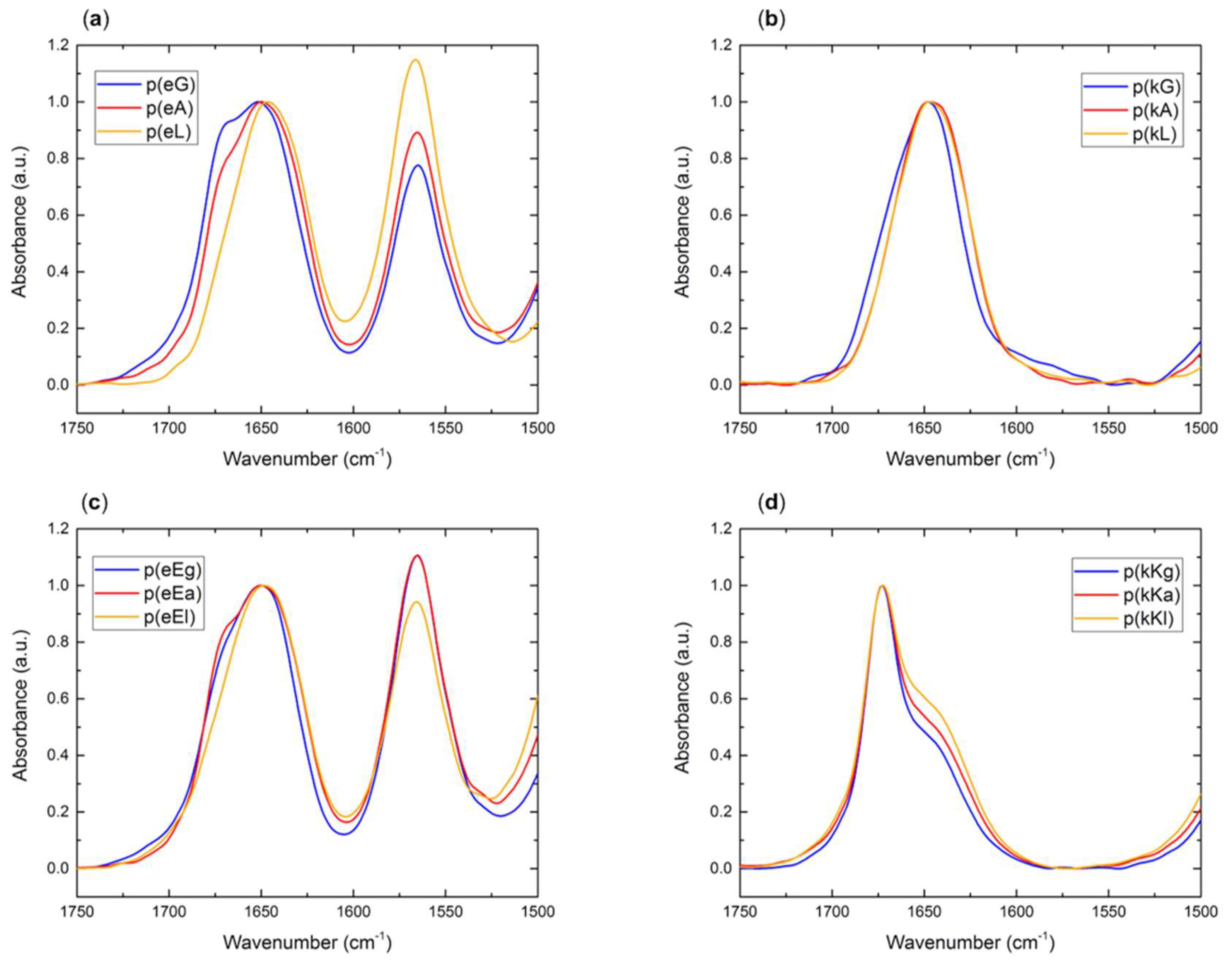
2.2.4. Fourier Transform Infrared (FTIR) Spectroscopy
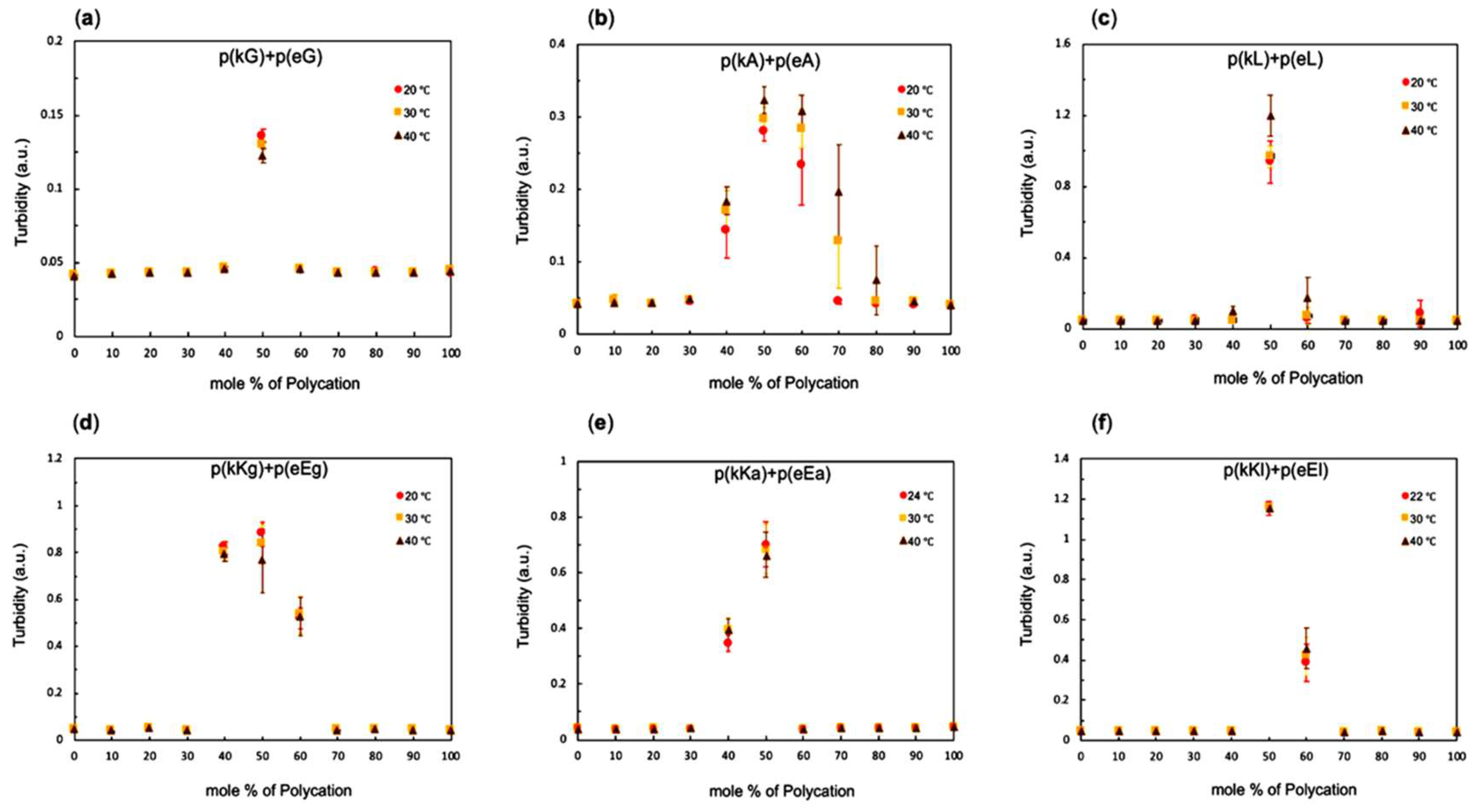
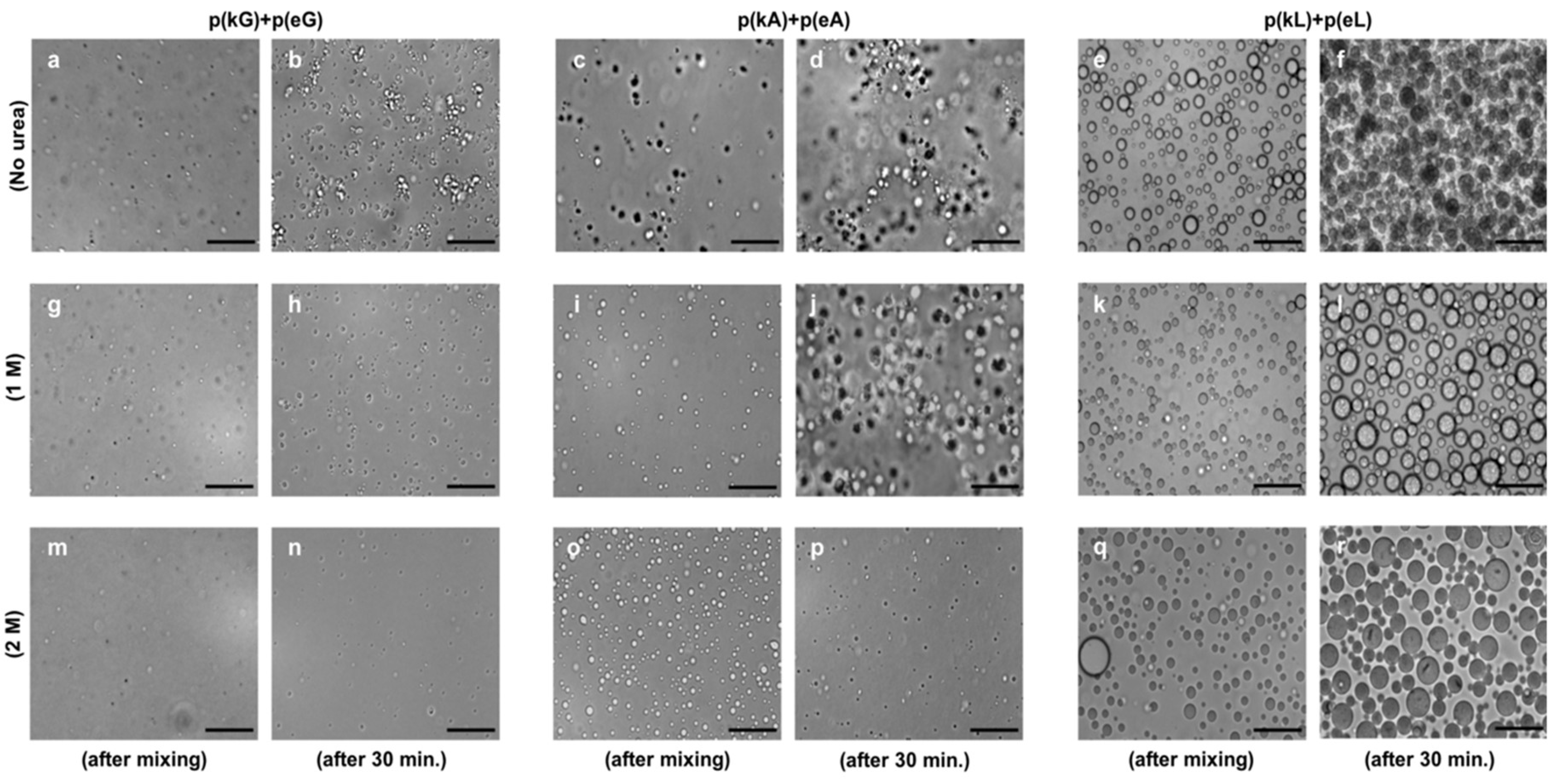
2.3. Stoichiometry and Temperature Effect on Complex Formation
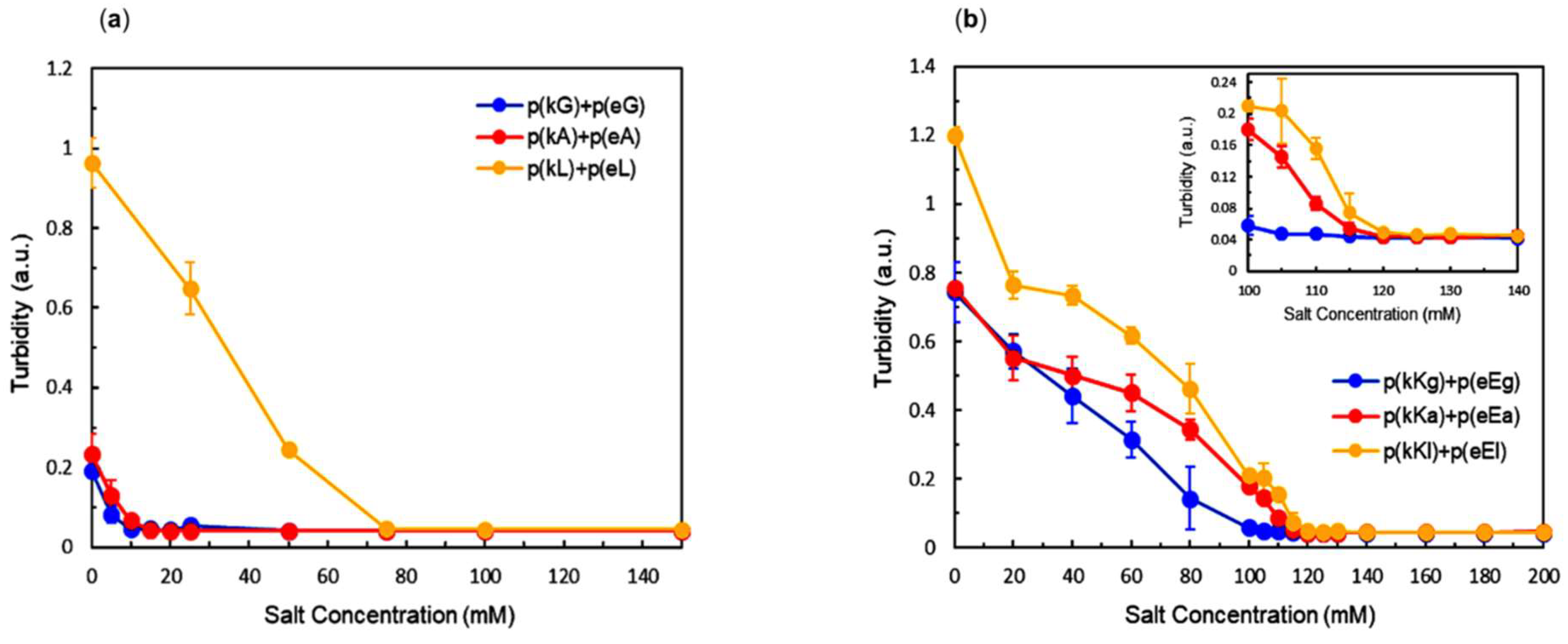
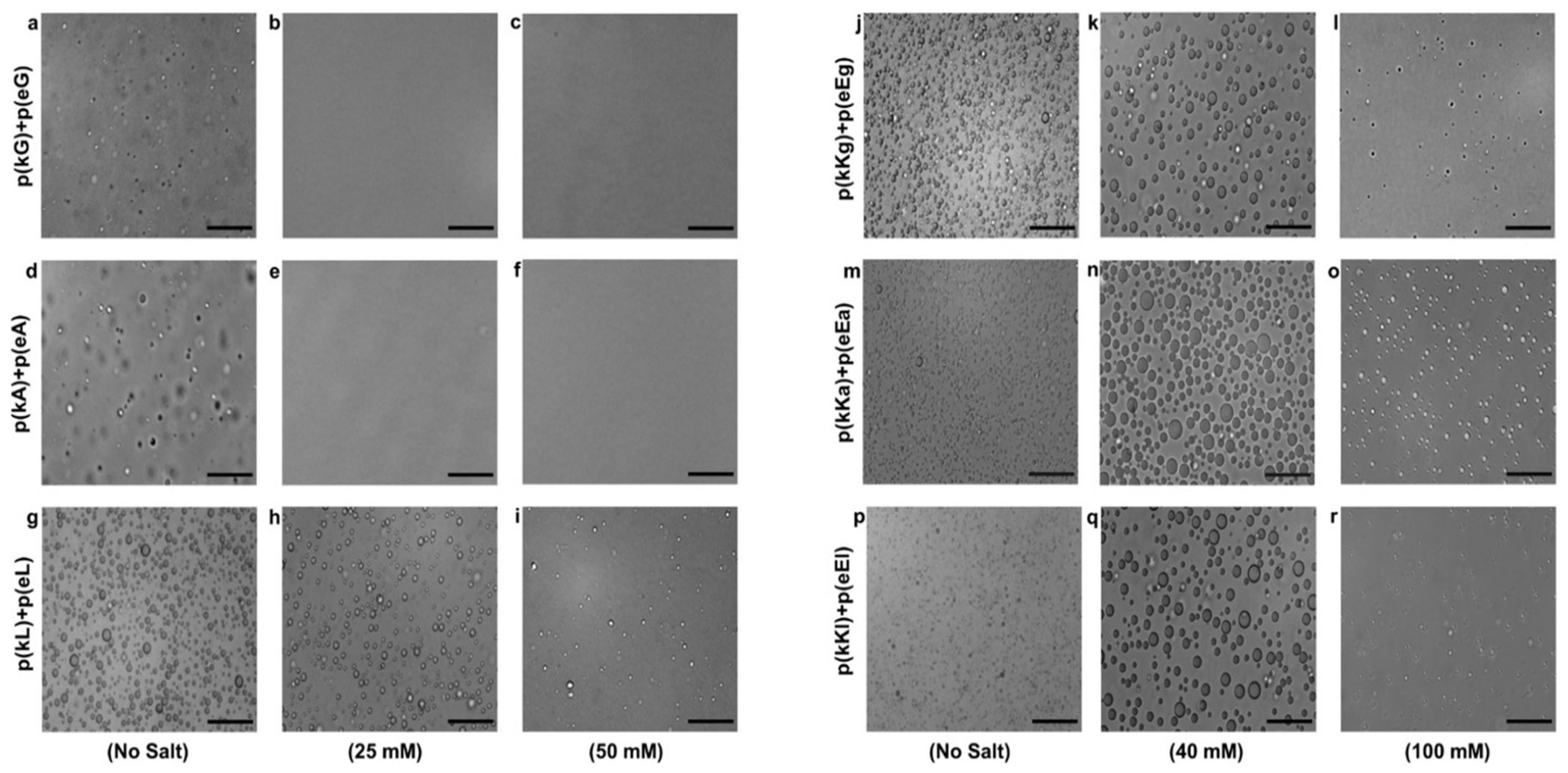
2.4. Effect of Ionic Strength and Charge Density on Complex Formation
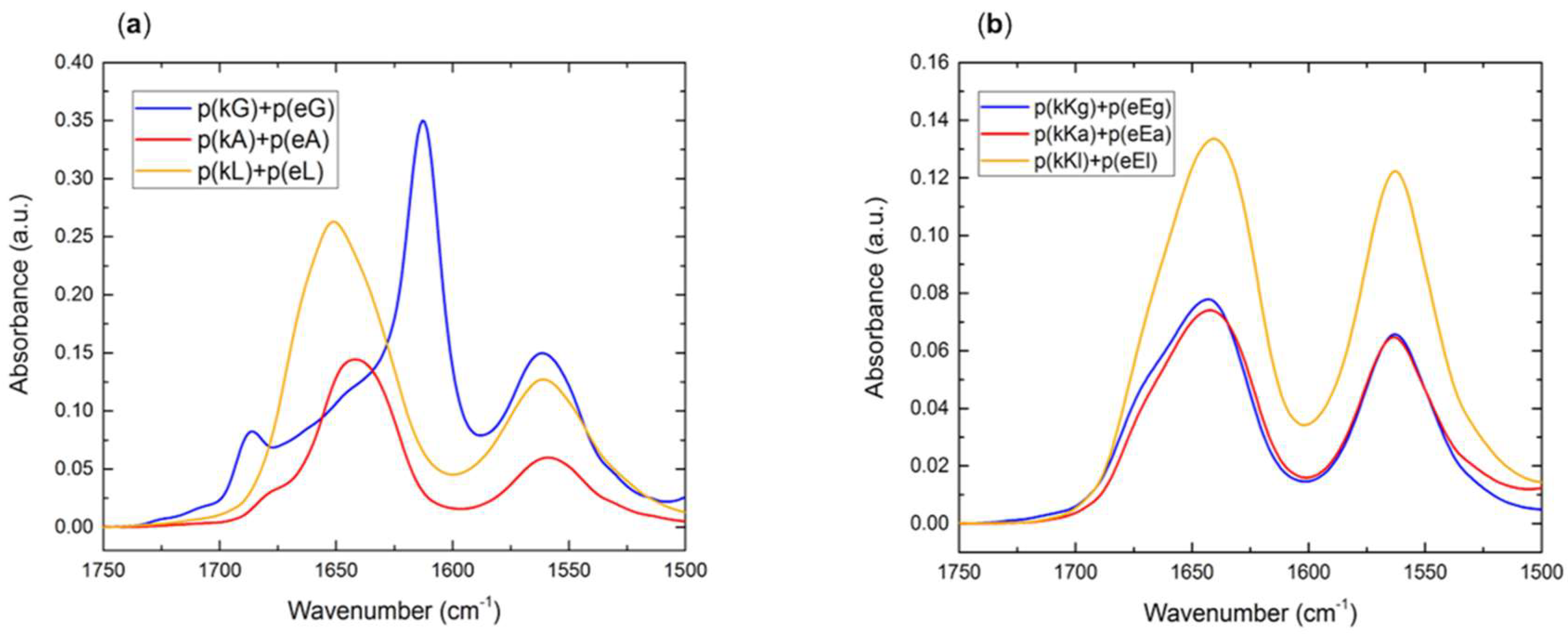
2.5. Secondary Structure of Polypeptide Complexes
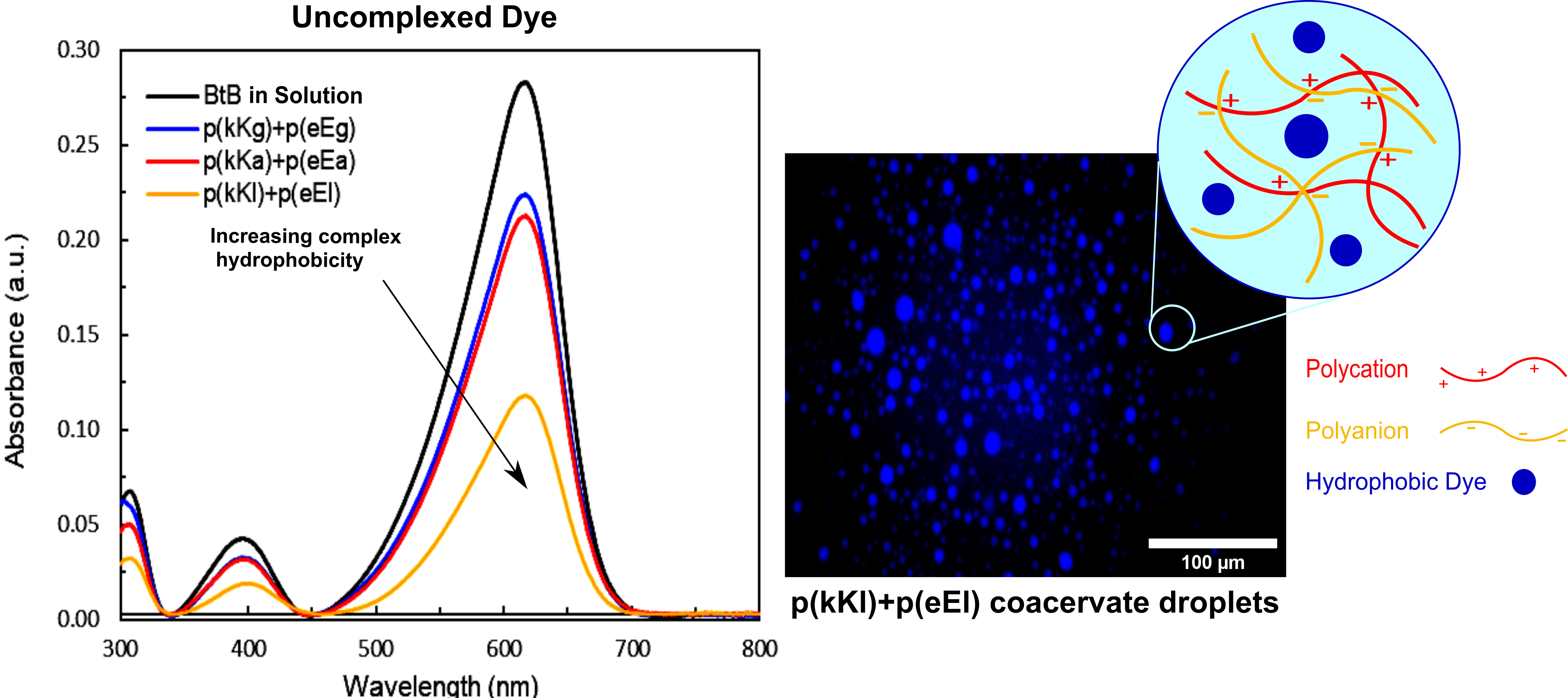
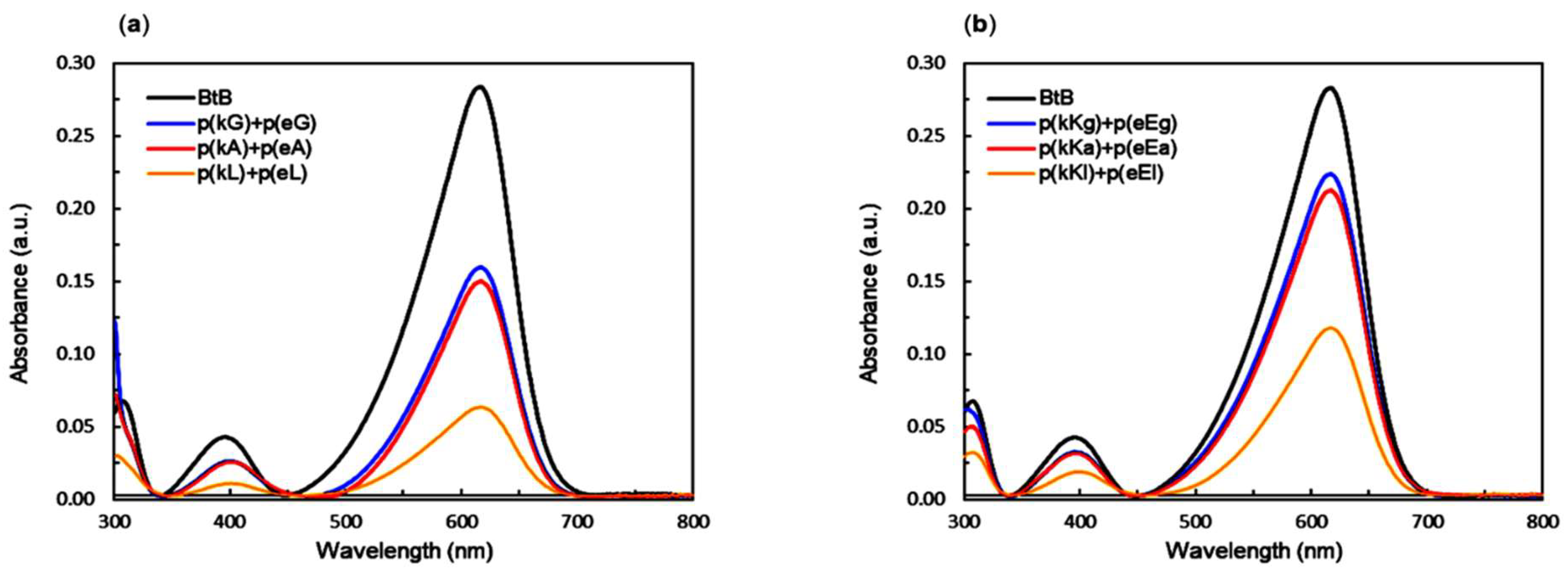
2.6. Encapsulation Behavior of Complexes
3. Materials and Methods
3.1. Materials
3.2. Peptide Synthesis
3.3. Preparation of Polypeptide and Complex Solutions
3.4. Characterization and Analysis of Polypeptides and Complex
3.4.1. Mass Spectroscopy
3.4.2. Nuclear Magnetic Resonance (NMR) Spectroscopy
3.4.3. Secondary Structure Characterization of Polypeptides and Complexes
3.4.4. Turbidity Measurements
3.4.5. UV-vis Spectroscopy
3.4.6. Optical Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Q.; Schlenoff, J.B. The Polyelectrolyte Complex/Coacervate Continuum. Macromolecules 2014, 47, 3108–3116. [Google Scholar] [CrossRef]
- Perry, S.L.; Leon, L.; Hoffmann, K.Q.; Kade, M.J.; Priftis, D.; Black, K.A.; Wong, D.; Klein, R.A.; Pierce, C.F.; Margossian, K.O.; et al. Chirality-selected phase behaviour in ionic polypeptide complexes. Nat. Commun. 2015, 6, 6052. [Google Scholar] [CrossRef] [PubMed]
- Pacalin, N.M.; Leon, L.; Tirrell, M. Directing the phase behavior of polyelectrolyte complexes using chiral patterned peptides. Eur. Phys. J. Spec. Top. 2016, 225, 1805–1815. [Google Scholar] [CrossRef]
- Vieregg, J.R.; Lueckheide, M.; Marciel, A.B.; Leon, L.; Bologna, A.J.; Rivera, J.R.; Tirrell, M.V. Oligonucleotide-peptide complexes: Phase control by hybridization. J. Am. Chem. Soc. 2018, 140, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Momani, B.; Winter, H.H.; Perry, S.L. Rheological characterization of liquid-to-solid transitions in bulk polyelectrolyte complexes. Soft Matter 2017, 13, 7332–7340. [Google Scholar] [CrossRef] [PubMed]
- Bungenberg de Jong, H.G.; Kruyt, H.R. Coacervation (partial miscibility in colloid systems). Proc. Acad. Sci. Amsterdam 1929, 32, 849–856. [Google Scholar]
- Qin, J.; Priftis, D.; Farina, R.; Perry, S.L.; Leon, L.; Whitmer, J.; Hoffmann, K.; Tirrell, M.; De Pablo, J.J. Interfacial tension of polyelectrolyte complex coacervate phases. ACS Macro Lett. 2014, 3, 565–568. [Google Scholar] [CrossRef]
- Spruijt, E.; Sprakel, J.; Cohen Stuart, M.A.; Van Der Gucht, J. Interfacial tension between a complex coacervate phase and its coexisting aqueous phase. Soft Matter 2009, 6, 172–178. [Google Scholar] [CrossRef]
- Schmitt, C.; Turgeon, S.L. Protein/polysaccharide complexes and coacervates in food systems. Adv. Colloid Interface Sci. 2011, 167, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Weinbreck, F.; de Vries, R.; Schrooyen, P.; de Kruif, C.G. Complex coacervation of whey proteins and gum arabic. Biomacromolecules 2003, 4, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Vecchies, F.; Sacco, P.; Decleva, E.; Menegazzi, R.; Porrelli, D.; Donati, I.; Turco, G.; Paoletti, S.; Marsich, E. Complex Coacervates between a Lactose-Modified Chitosan and Hyaluronic Acid as Radical-Scavenging Drug Carriers. Biomacromolecules 2018, 19, 3936–3944. [Google Scholar] [CrossRef] [PubMed]
- Black, K.A.; Priftis, D.; Perry, S.L.; Yip, J.; Byun, W.Y.; Tirrell, M. Protein encapsulation via polypeptide complex coacervation. ACS Macro Lett. 2014, 3, 1088–1091. [Google Scholar] [CrossRef]
- Johnson, N.R.; Ambe, T.; Wang, Y. Lysine-based polycation:heparin coacervate for controlled protein delivery. Acta Biomater. 2014, 10, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.R.; Wang, Y. Coacervate delivery systems for proteins and small molecule drugs. Expert Opin. Drug Deliv. 2014, 11, 1829–1832. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, T.; Carlisle, R.C.; Read, M.L.; Ogris, M.; Seymour, L.W. Peptide-mediated RNA delivery: A novel approach for enhanced transfection of primary and post-mitotic cells. Nucleic Acids Res. 2001, 29, 3882–3891. [Google Scholar] [CrossRef] [PubMed]
- Rhaese, S.; Von Briesen, H.; Rübsamen-Waigmann, H.; Kreuter, J.; Langer, K. Human serum albumin-polyethylenimine nanoparticles for gene delivery. J. Control. Release 2003, 92, 199–208. [Google Scholar] [CrossRef]
- Yasar, H.; Ho, D.K.; De Rossi, C.; Herrmann, J.; Gordon, S.; Loretz, B.; Lehr, C.M. Starch-chitosan polyplexes: A versatile carrier system for anti-infectives and gene delivery. Polymers 2018, 10, 252. [Google Scholar] [CrossRef]
- Stewart, R.J.; Wang, C.S.; Shao, H. Complex coacervates as a foundation for synthetic underwater adhesives. Adv. Colloid Interface Sci. 2011, 167, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lee, D.W.; Ahn, B.K.; Seo, S.; Kaufman, Y.; Israelachvili, J.N.; Waite, J.H. Underwater contact adhesion and microarchitecture in polyelectrolyte complexes actuated by solvent exchange. Nat. Mater. 2016, 15, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Choi, W.S.; Lee, H.-J. Nonstoichiometric Polyelectrolyte Complexes: Smart Nanoreactors for Alloy and Multimetallic Catalyst. Int. J. Mater. Mech. Manuf. 2014, 2, 1–4. [Google Scholar] [CrossRef]
- Anraku, Y.; Kishimura, A.; Kamiya, M.; Tanaka, S.; Nomoto, T.; Toh, K.; Matsumoto, Y.; Fukushima, S.; Sueyoshi, D.; Kano, M.R.; et al. Systemically Injectable Enzyme-Loaded Polyion Complex Vesicles as in Vivo Nanoreactors Functioning in Tumors. Angew. Chemie Int. Ed. 2016, 55, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Pak, C.W.; Kosno, M.; Holehouse, A.S.; Padrick, S.B.; Mittal, A.; Ali, R.; Yunus, A.A.; Liu, D.R.; Pappu, R.V.; Rosen, M.K. Sequence Determinants of Intracellular Phase Separation by Complex Coacervation of a Disordered Protein. Mol. Cell 2016, 63, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Marciel, A.B.; Chung, E.J.; Brettmann, B.K.; Leon, L. Bulk and nanoscale polypeptide based polyelectrolyte complexes. Adv. Colloid Interface Sci. 2017, 239, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Batys, P.; O’Neal, J.T.; Li, F.; Sammalkorpi, M.; Lutkenhaus, J.L. Molecular Origin of the Glass Transition in Polyelectrolyte Assemblies. ACS Cent. Sci. 2018, 4, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Fares, H.M.; Schlenoff, J.B. Ion-Pairing Strength in Polyelectrolyte Complexes. Macromolecules 2017, 50, 1066–1074. [Google Scholar] [CrossRef]
- Schaaf, P.; Schlenoff, J.B. Saloplastics: Processing compact polyelectrolyte complexes. Adv. Mater. 2015, 27, 2420–2432. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.W.; Lytle, T.K.; Radhakrishna, M.; Madinya, J.J.; Vélez, J.; Sing, C.E.; Perry, S.L. Sequence and entropy-based control of complex coacervates. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.Q.; Perry, S.L.; Leon, L.; Priftis, D.; Tirrell, M.; De Pablo, J.J. A molecular view of the role of chirality in charge-driven polypeptide complexation. Soft Matter 2015, 11, 1525–1538. [Google Scholar] [CrossRef] [PubMed]
- Priftis, D.; Leon, L.; Song, Z.; Perry, S.L.; Margossian, K.O.; Tropnikova, A.; Cheng, J.; Tirrell, M. Self-assembly of α-helical polypeptides driven by complex coacervation. Angew. Chem. Int. Ed. Engl. 2015, 54, 11128–11132. [Google Scholar] [CrossRef] [PubMed]
- Michaels, A.S. Polyelectrolyte Complexes. Ind. Eng. Chem. 1965, 57, 32–40. [Google Scholar] [CrossRef]
- Chollakup, R.; Beck, J.B.; Dirnberger, K.; Tirrell, M.; Eisenbach, C.D. Polyelectrolyte molecular weight and salt effects on the phase behavior and coacervation of aqueous solutions of poly(acrylic acid) sodium salt and poly(allylamine) hydrochloride. Macromolecules 2013, 46, 2376–2390. [Google Scholar] [CrossRef]
- Priftis, D.; Tirrell, M. Phase behaviour and complex coacervation of aqueous polypeptide solutions. Soft Matter 2012, 8, 9396–9405. [Google Scholar] [CrossRef]
- Schmitt, C.; Sanchez, C.; Desobry-Banon, S.; Hardy, J. Structure and technofunctional properties of protein-polysaccharide complexes: A review. Crit. Rev. Food Sci. Nutr. 1998, 38, 689–753. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.L.; Dubin, P.L.; Kayitmazer, A.B.; Turksen, S. Polyelectrolyte-protein complexes. Curr. Opin. Colloid Interface Sci. 2005, 10, 52–78. [Google Scholar] [CrossRef]
- Perry, S.L.; Li, Y.; Priftis, D.; Leon, L.; Tirrell, M. The effect of salt on the complex coacervation of vinyl polyelectrolytes. Polymers 2014, 6, 1756–1772. [Google Scholar] [CrossRef]
- Galazka, V.B.; Smith, D.; Ledward, D.A.; Dickinson, E. Complexes of Bovine Serum Albumin with Sulphated Polysaccharides: Effects of pH, Ionic Strength and High Pressure Treatment. Food Chem. 1999, 64, 303–310. [Google Scholar] [CrossRef]
- Schlenoff, J.B.; Rmaile, A.H.; Bucur, C.B. Hydration contributions to association in polyelectrolyte multilayers and complexes: Visualizing hydrophobicity. J. Am. Chem. Soc. 2008, 130, 13589–13597. [Google Scholar] [CrossRef] [PubMed]
- Sadman, K.; Wang, Q.; Chen, Y.; Keshavarz, B.; Jiang, Z.; Shull, K.R. Influence of Hydrophobicity on Polyelectrolyte Complexation. Macromolecules 2017, 50, 9417–9426. [Google Scholar] [CrossRef]
- Mende, M.; Schwarz, S.; Zschoche, S.; Petzold, G.; Janke, A. Influence of the Hydrophobicity of Polyelectrolytes on Polyelectrolyte Complex Formation and Complex Particle Structure and Shape. Polymers 2011, 3, 1363–1376. [Google Scholar] [CrossRef]
- Monera, O.D.; Sereda, T.J.; Zhou, N.E.; Kay, C.M.; Hodges, R.S. Relationship of sidechain hydrophobicity and α-helical propensity on the stability of the single-stranded amphipathic α-helix. J. Pept. Sci. 1995, 1, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Baiz, C.R.; Reppert, M.; Tokmakoff, A. An introduction to protein 2D IR spectroscopy. Ultrafast Infrared Vib. Spectrosc. 2013, 361–403. [Google Scholar]
- Fändrich, M.; Dobson, C.M. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 2002, 21, 5682–5690. [Google Scholar] [CrossRef] [PubMed]
- Janek, K.; Behlke, J.; Zipper, J.; Fabian, H.; Georgalis, Y.; Beyermann, M.; Bienert, M.; Krause, E. Water-soluble beta-sheet models which self-assemble into fibrillar structures [In Process Citation]. Biochemistry 1999, 38, 8246–8252. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Zékri, E.; Rousseau, B.; Paternostre, M.; Cintrat, J.-C.; Fay, N. Elimination and exchange of trifluoroacetate counter-ion from cationic peptides: A critical evaluation of different approaches. J. Pept. Sci. 2008, 14, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.L.; Blocher, W.C. Complex coacervate-based materials for biomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 9, e1442. [Google Scholar]
- Chollakup, R.; Smitthipong, W.; Eisenbach, C.D.; Tirrell, M. Phase behavior and coacervation of aqueous poly(acrylic acid)-poly(allylamine) solutions. Macromolecules 2010, 43, 2518–2528. [Google Scholar] [CrossRef]
- Priftis, D.; Xia, X.; Margossian, K.O.; Perry, S.L.; Leon, L.; Qin, J.; De Pablo, J.J.; Tirrell, M. Ternary, tunable polyelectrolyte complex fluids driven by complex coacervation. Macromolecules 2014, 47, 3076–3085. [Google Scholar] [CrossRef]
- Lankalapalli, S.; Kolapalli, V.R.M. Polyelectrolyte Complexes: A Review of their Applicability in Drug Delivery Technology. Indian J. Pharm. Sci. 2009, 71, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, E.; Westphal, A.H.; Borst, J.W.; Cohen Stuart, M.A.; van der Gucht, J. Binodal Compositions of Polyelectrolyte Complexes. Macromolecules 2010, 43, 6476–6484. [Google Scholar] [CrossRef]
- O’Brien, E.P.; Dima, R.I.; Brooks, B.; Thirumalai, D. Interactions between hydrophobic and ionic solutes in aqueous guanidinium chloride and urea solutions: Lessons for protein denaturation mechanism. J. Am. Chem. Soc. 2007, 129, 7346–7353. [Google Scholar] [CrossRef] [PubMed]
- Auton, M.; Holthauzen, L.M.F.; Bolen, D.W. Anatomy of energetic changes accompanying urea-induced protein denaturation. Proc. Natl. Acad. Sci. 2007, 104, 15317–15322. [Google Scholar] [CrossRef] [PubMed]
- Politi, M.J.; Chaimovich, H.; Liu, C.; Triboni, E.R.; Briotto Filho, D.; Cuccovia, I.M. Effect of urea on ion pair formation. The hydrophilic effect of urea. Colloids Surfaces A Physicochem. Eng. Asp. 2017, 520, 173–177. [Google Scholar] [CrossRef]
- Dzwolak, W.; Ravindra, R.; Nicolini, C.; Jansen, R.; Winter, R. The Diastereomeric Assembly of Polylysine is the Low-Volume Pathway for Preferential Formation of β-Sheet Aggregates. J. Am. Chem. Soc. 2004, 126, 3762–3768. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Hasegawa, T. Determination of equilibrium structures of bromothymol blue revealed by using quantum chemistry with an aid of multivariate analysis of electronic absorption spectra. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 185, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Eghtesadi, S.A.; Dawadi, M.B.; Wang, C.; Huang, S.; Seymore, A.E.; Vogt, B.D.; Modarelli, D.A.; Liu, T.; Zacharia, N.S. Partitioning of Small Molecules in Hydrogen-Bonding Complex Coacervates of Poly(acrylic acid) and Poly(ethylene glycol) or Pluronic Block Copolymer. Macromolecules 2017, 50, 3818–3830. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Wang, H.Y.; Zhang, X.Z.; Zhuo, R.X. Synthesis of thermo- and pH-sensitive polyion complex micelles for fluorescent imaging. Chem. A Eur. J. 2012, 18, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Lueckheide, M.; Vieregg, J.R.; Bologna, A.J.; Leon, L.; Tirrell, M.V. Structure-Property Relationships of Oligonucleotide Polyelectrolyte Complex Micelles. Nano Lett. 2018, 18, 7111–7117. [Google Scholar] [CrossRef] [PubMed]
- Voets, I.K.; de Keizer, A.; Cohen Stuart, M.A. Complex coacervate core micelles. Adv. Colloid Interface Sci. 2009, 147–148, 300–318. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-H.; Leon, L.; Chung, E.J.; Huang, R.-T.; Sontag, T.J.; Reardon, C.a.; Getz, G.S.; Tirrell, M.; Fang, Y. Inhibition of atherosclerosis-promoting microRNAs via targeted polyelectrolyte complex micelles. J. Mater. Chem. B 2014, 2, 8142–8153. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the hydrophobically patterned peptides and polyelectrolyte complexes discussed in this paper are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glycine | Alanine | Leucine |
|---|---|---|
 |  |  |
| Peptide Patterns | Polycations | Polyanions |
|---|---|---|
| First generation | (kX)15 | (eX)15 |
| Second generation | (kKxKkX)5 | (eExEeX)5 |
| Peptide | m/z | Theoretical Mass |
|---|---|---|
| (kG)15 | 2810.71 | 2795.40 |
| (eG)15 | 2825.66 | 2809.51 |
| (kA)15 | 3021.94 | 3005.79 |
| (eA)15 | 3064.13 | 3019.91 |
| (kL)15 | 3665.76 | 3636.98 |
| (eL)15 | 3698.42 | 3651.1 |
| (kKg)10 | 3149.27 | 3150.99 |
| (eEg)10 | 3174.31 | 3169.82 |
| (kKa)10 | 3296.25 | 3291.26 |
| (eEa)10 | 3327.89 | 3310.09 |
| (kKl)10 | 3710.85 | 3712.05 |
| (eEl)10 | 3752.90 | 3730.88 |
| Supernatant Solution | Dye Concentration (µM) | Sequestration in the Complex Phase (%) |
|---|---|---|
| Bromothymol blue | 10 | - |
| p(kG)+p(eG) | 5.59 | 44.1 |
| p(kA)+p(eA) | 5.25 | 47.5 |
| p(kL)+p(eL) | 2.15 | 78.4 |
| p(kKg)+p(eEg) | 7.89 | 21.1 |
| p(kKa)+p(eEa) | 7.49 | 25.1 |
| p(kKl)+p(eEl) | 4.12 | 58.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabandeh, S.; Leon, L. Engineering Peptide-Based Polyelectrolyte Complexes with Increased Hydrophobicity. Molecules 2019, 24, 868. https://doi.org/10.3390/molecules24050868
Tabandeh S, Leon L. Engineering Peptide-Based Polyelectrolyte Complexes with Increased Hydrophobicity. Molecules. 2019; 24(5):868. https://doi.org/10.3390/molecules24050868
Chicago/Turabian StyleTabandeh, Sara, and Lorraine Leon. 2019. "Engineering Peptide-Based Polyelectrolyte Complexes with Increased Hydrophobicity" Molecules 24, no. 5: 868. https://doi.org/10.3390/molecules24050868
APA StyleTabandeh, S., & Leon, L. (2019). Engineering Peptide-Based Polyelectrolyte Complexes with Increased Hydrophobicity. Molecules, 24(5), 868. https://doi.org/10.3390/molecules24050868