3. Materials and Methods
3.1. General Experimental
Chemicals and reagents were purchased from commercial sources (Alfa Aesar, Sigma-Aldrich) and were used without further purification. In case of anhydrous solvents, these were prepared either by filtration through a column of activated alumina or by standing over activated 4Å molecular sieves and stored under argon atmosphere. Hexanes refer to a mixture of C-6 alkanes (b.p. 60–80 °C). Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous material, unless otherwise stated. Reactions were monitored by thin layer chromatography (TLC) carried out on aluminium sheets pre-coated with silica gel 60 F254 (Merck) or aluminium oxide 60 F254 (neutral, Merck). Visualisation was performed using shortwave UV light followed by dipping TLC plates in either basic solution of KMnO4, acidic solution of vanillin or acidic solution of ceric ammonium nitrate followed by heating with a heat gun. Flash column chromatography (FLC) was performed using Silica Gel 60 (particle size 0.040–0.063 mm). NMR spectra were recorded in CDCl3 on a Varian INOVA 300 (300 MHz for 1H, 75 MHz for 13C nuclei) or Varian VNMRS 600 (600 MHz for 1H, 151 MHz for 13C nuclei) NMR spectrometer and were correctly shifted using residual non-deuterated solvent or tetramethylsilane as an internal reference (CHCl3: δH = 7.26 ppm, δC = 77.16 ppm (central peak of a 1:1:1 triplet), TMS: δH = δC = 0.00 ppm). Chemical shifts (δ) are quoted in ppm. LC-MS analyses were performed on Agilent 1200 Series instrument equipped with a multimode MS detector using the MM ESI/APCI ionisation method (column Zorbax Eclipse XDB-18, 150 × 4.6 mm, particle size 5 μm, eluent water with 0.1% HCO2H/CH3CN, 70:30, flow 1.5 mL/min). GC analyses were performed on a gas chromatograph Agilent 7820A equipped with FID and a split–splitless injector (column DB-5 30 m × 0.25 mm × 0.25 µm, injection 0.1 µL, split 20:1, temperature gradient 40 °C (2 min) → 15 °C/min → 220 °C (15 min), carrier gas H2, flow 1.2 mL/min). Chiral GC analyses were performed on a gas chromatograph Agilent 7890A equipped with FID and a split–splitless injector (column Cyclosil-B 30 m × 0.32 mm × 0.25 µm, injection 0.2 µL, split 50:1, temperature gradient 40 °C (0 min) → 10 °C/min → 80 °C (0 min) → 25 °C/min → 220 °C (2 min), carrier gas H2, flow 2.0 mL/min). GC-MS analyses were performed on a gas chromatograph Agilent 7890A and coupled with Agilent 5975C inert MSD with Triple-Axis Detector (column DB-Wax 30 m × 0.25 mm × 0.15 µm, injection 1 µL, split 20:1, temperature gradient 40 °C (2 min) → 15 °C/min → 220 °C (15 min), carrier gas H2, flow 1.2 mL/min). High-resolution mass spectra (HR-MS) were recorded on a Thermo Scientific Orbitrap Velos mass spectrometer with a heated electrospray ionisation (HESI) source in positive and/or negative mode. FTIR spectra were obtained on a Nicolet 5700 spectrometer (Thermo Electron) equipped with a Smart Orbit (diamond crystal ATR) accessory using the reflectance technique (4000–400 cm–1). The sensory analysis was performed by authors in a clean and odourless environment at 22 °C. The prepared compounds were evaluated as 10% solutions in aqueous ethanol (95% w/w) by using testing strips.
3.2. Synthetic Procedures and Analytical Data
(
S)-Ethyl 4-methyl-3-oxohexanoate (
3) A mixture of potassium ethyl malonate (100.0 g, 0.587 mol, 1.5 equiv) was treated with magnesium chloride (56.0 g, 0.587 mol, 1.5 equiv) in tetrahydrofuran (360 mL), and the resulting grey slurry was stirred at 60 °C for 5 h. During that time, in a separate reaction vessel, a solution of (
S)-2-methylbutanoic acid
2 (88%
ee, 40.0 g, 0.343 mol) in THF (160 mL) was added to a solution of carbonyldiimidazole (66.0 g, 0.407 mol, 1.2 equiv) in THF (140 mL) and the resulting yellow solution was stirred at 28 °C for 4 h. Then, after 5 h reaction time, a THF solution containing a mixture of malonate and MgCl
2 was cooled to RT and a THF solution of crude acyl imidazole formed from acid
2 was added dropwise over 30 min. The resulting white suspension was stirred at 50 °C for 5 h and subsequently at RT overnight. The reaction mixture was then added to 1M aqueous HCl solution (1600 mL). The resulting pale-yellow solution was stirred at RT for 30 min, ethyl acetate (600 mL) was added, phases were separated, and aq ueous layer was extracted with EtOAc (600 mL). Combined organic extracts were sequentially washed with 1M aq ueous HCl solution (400 mL), water (500 mL), 2% aqueous Na
2CO
3 solution (700 mL), water (500 mL) and brine (500 mL), subsequently dried over anhydrous Na
2SO
4 and concentrated in vacuo to give pale-yellow oil (76.23 g). The crude product was purified by vacuum distillation (b.p. 64–67 °C/3.4 mbar) to yield (
S)-ketoester
3 (60.54 g, 90%) as a colourless liquid; δ
H (300 MHz, CDCl
3) 4.19 (q,
J = 7.1 Hz, 2H, O
CH2CH
3), 3.47 (s, 2H, H-2), 2.58 (m,
J = 6.8 Hz, 1H, H-4), 1.79–1.62 (m, 1H, H-5A), 1.42 (m, 1H, H-5B), 1.27 (t,
J = 7.1 Hz, 3H, OCH
2CH3), 1.10 (d,
J = 6.9 Hz, 3H, Me), 0.90 (t,
J = 7.4 Hz, 3H, H-6); δ
C (75 MHz, CDCl
3) 206.6 (C-3), 167.4 (C-1), 61.3 (O
CH2), 48.1 (C-4), 47.8 (C-2), 25.7 (C-5), 15.5 (Me), 14.2 (OCH
2CH3), 11.5 (C-6), NMR spectra of (
S)-
3 are in full accordance with literature data [
25,
26,
27] for racemic
3; in addition, signals of the enol form of
3 are clearly detectable in both proton and carbon spectra measured in deuterochloroform: δ
H (300 MHz, CDCl
3) 12.11 (d,
J = 0.7 Hz, 1H, OH), 4.97 (s, 1H, H-2), 4.18 (q,
J = 7.1 Hz, 2H, O
CH2CH
3), 2.15 (m,
J = 7.0 Hz, 1H, H-4), 1.79–1.62 (m, 1H, H-5A), 1.42 (m, 1H, H-5B), 1.29 (t,
J = 7.1 Hz, 3H, OCH
2CH3), 1.12 (d,
J = 6.9 Hz, 3H, Me), 0.89 (t,
J = 7.4 Hz, 3H, H-6); δ
C (75 MHz, CDCl
3) 182.7 (C-3), 173.0 (C-1), 88.1 (C-2), 60.0 (O
CH2), 41.2 (C-4), 27.1 (C-5), 17.7 (Me), 14.4 (OCH
2CH3), 11.7 (C-6); GC: t
R = 7.73 min (keto-form), t
R = 7.54 min (enol-form); GC-MS:
m/z (%) 172 (6, M
+), 157 (1), 144 (9), 127 (3), 115 (27), 98 (5), 85 (31), 69 (12), 57 (100), 43 (36).
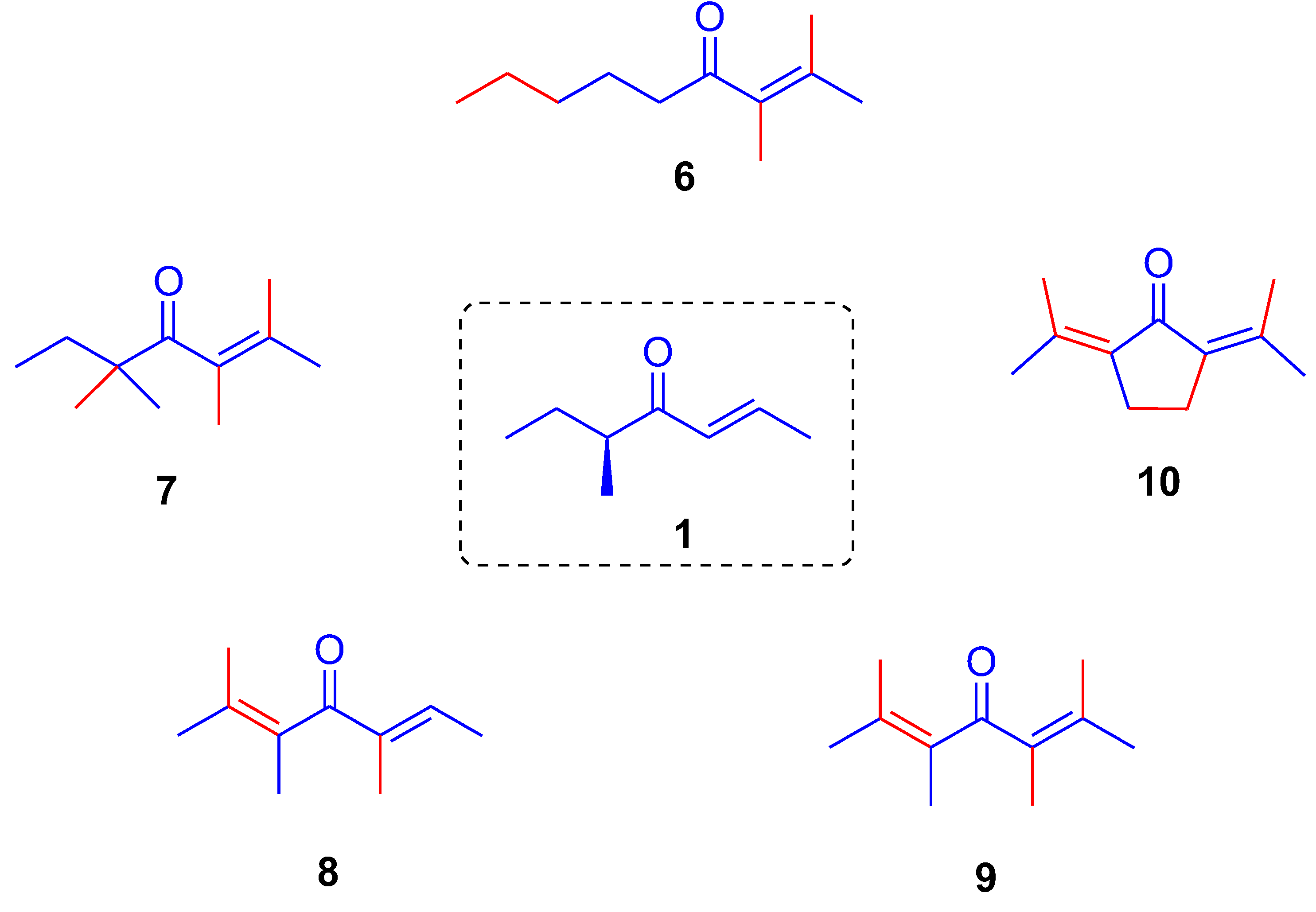
(S)-4-Methyl-3-oxohexanoic acid (4) To a solution of ketoester 3 (8.0 g, 46.5 mmol) in an aqueous sodium phosphate buffer (32 mL, pH~7) was added Novozym 435 (400 mg) and the suspension was stirred at RT for 22 h, while the pH was kept neutral by addition of aqueous NaOH. Solids were filtered off and the filtrate containing ketoacid 4 (100% GC yield, 88% ee) was directly used for the subsequent Knoevenagel condensation with acetaldehyde. The enantiomeric purity of crude 4 was determined by chiral GC via (S)-3-methylpent-2-on (tR = 3.53 min) formed in situ by thermal decarboxylation of 4 during analysis.
(
S)-2-Hydroxy-5-methylhept-4-one (
5) To a solution of crude ketoacid
4 (8 g, 46.5 mmol) in an aqueous phosphate buffer (42.5 mL) was added tetrabutylammonium hydrogen sulphate (79 mg) and the pH was adjusted to approx. 8 by aqueous NaOH solution. Acetaldehyde (2.6 mL, 51.4 mmol, 1.1 equiv) was added and the resulting soln. was stirred at RT for 1.5 h and then at 40 °C for 21 h. The reaction mixture was extracted with diethyl ether (3 × 70 mL), separated organic layer was dried over anhydrous Na
2SO
4 and the solvent was evaporated in vacuo to yield pale-yellow liquid (6.865 g). The crude product was purified by vacuum distillation (b.p. 63-64 °C/3.6 mbar) to afford ketol
5 (3.577 g, 53%) as a colourless liquid; δ
H (600 MHz, CDCl
3) 4.23–4.17 (m, 1H, H-2), 3.01 (bs, 1H, exchange with D
2O, OH), 2.62 (ddd,
J = 17.8, 13.0, 2.8 Hz, 1H, H-3a), 2.51 (ddd,
J = 17.8, 13.5, 9.0 Hz, 1H, H-3b), 2.46–2.39 (m, 1H, H-5), 1.72–1.63 (m, 1H, H-6a), 1.43–1.35 (m, 1H, H-6b), 1.18 (dd,
J = 6.4, 0.7 Hz, 3H, H-1), 1.06 (dd,
J = 7.0, 1.3 Hz, 3H, Me), 0.87 (t,
J = 7.5 Hz, 3H, H-7), NMR spectrum is in accordance with the literature data [
28]; GC: t
R = 7.67 min; GC-MS:
m/z (%) 144 (2, M
+), 116 (4), 103 (18), 87 (85), 85 (26), 69 (23), 57 (75), 43 (100).
(
E,
S)-5-Methylhept-2-en-4-one (
1) To a mixture of ketol
5 (2.85 g, 19.8 mmol) in cyclohexane (49 mL) was added
p-toluenesulfonic acid monohydrate, (190 mg, 1.0 mmol, 0.05 equiv) and resulting mixture was stirred at 70 °C for 2.5 h. Subsequently, the mixture was washed with saturated aqueous NaHCO
3 solution (25 mL), aqueous phase was extracted with diethyl ether (3 × 60 mL) and organic phase was dried over anhydrous Na
2SO
4 and concentrated in vacuo to give a colourless liquid (2.60 g). The crude product was purified by bulb-to-bulb vacuum distillation (b.p. 75–76 °C/4 mbar) to afford a (
S)-enantioenriched enone
1 (2.03 g, 82% yield, 73%
ee) as a colourless liquid; (the (S)-configuration of
1 as the major enantiomer was determined by the comparison with the analytical sample of (S)-
1 prepared independently from (
S)-2-methylbutanol by previously reported stereoselective synthesis, cf. Ref. [
4]); δ
H (300 MHz, CDCl
3) 6.85 (dq,
J = 15.6, 6.9 Hz, 1H, H-2), 6.16 (dd,
J = 15.6, 1.7 Hz, 1H, H-3), 2.62 (m,
J = 6.8 Hz, 1H, H-5), 1.86 (dd,
J = 6.9, 1.7 Hz, 3H, H-1), 1.74–1.59 (m, 1H, H-6A), 1.46–1.25 (m, 1H, H-6B), 1.04 (d,
J = 6.9 Hz, 3H, Me), 0.84 (t,
J = 7.4 Hz, 3H, H-7); δ
C (75 MHz, CDCl
3) 203.9 (C-4), 142.3 (C-2), 130.7 (C-3), 45.4 (C-5), 26.2 (C-6), 18.3 (C-1), 16.2 (Me), 11.8 (C-7), NMR spectra of (
S)-
1 are in full accordance with the literature data [
2]; Chiral GC: t
R = 5.74 min for (
S)-
1 (t
R = 5.59 min for (
R)-
1); GC-MS:
m/z (%) 126 (1, M
+), 111 (11), 98 (12), 69 (100), 57 (4), 41 (21).
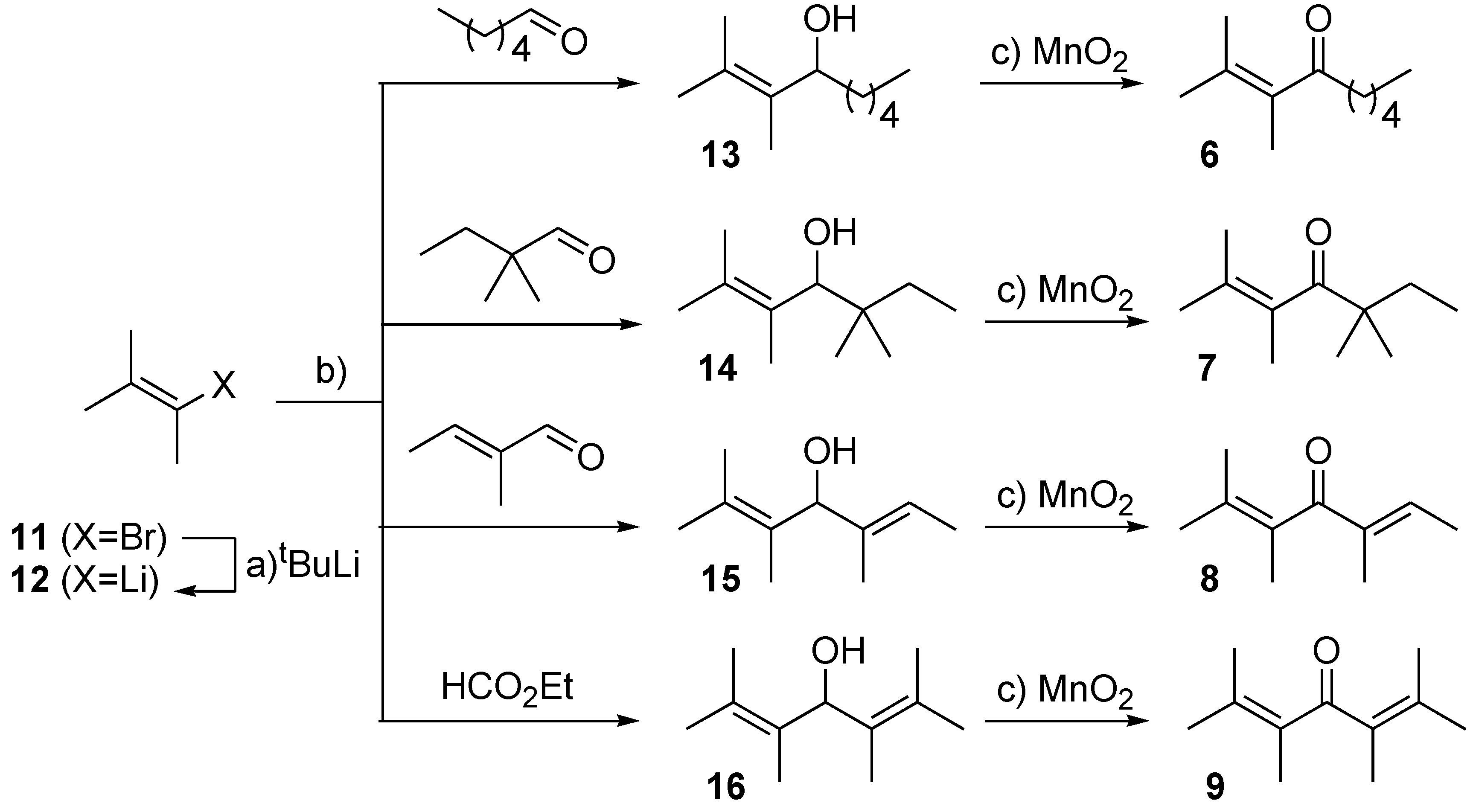
3.3. General experiment for the preparation of (di)allyl alcohols (13–16)
To a freshly prepared solution of 1,2-dimethylpropenyl lithium 12 in anhydrous THF was added the respective aldehyde or ester dropwise at low temperature over 5 min. under argon. The mixture was gradually warmed to RT and stirred overnight. The reaction was quenched with saturated aqueous NH4Cl solution, diluted with water and extracted with diethyl ether. Combined organic extracts were dried over MgSO4 and concentrated in vacuo (34 °C, 650 → 250 mbar). Crude product was purified by bulb-to-bulb vacuum distillation to furnish a corresponding allyl alcohol 13–16.
2,3-Dimethylnon-2-en-4-ol (13) 12 (1.35 mmol, 1.05 equiv), THF (2 mL), −50 °C, hexanal (0.16 mL, 1.28 mmol), RT, overnight, NH4Cl (5 mL), H2O (5 mL), Et2O (3 × 10 mL), vacuum distillation (130 °C, 50 mbar), alcohol 13 (165 mg, 84%) as colourless oil; Rf (hexanes/AcOEt 4:1) 0.43; νmax (ATR) 3342 (OH), 2955, 2927, 2859, 1457, 1375, 1010 cm−1; δH (300 MHz, CDCl3) 4.63 (t, J = 6.96 Hz, 1H, H-4), 1.71, 1.67, 1.61 (s, 2 × m, 3 × 3H, H-1, 2 × Me), 1.40 (m, 8H, H-5, H-6, H-7, H-8), 0.89 (m, 3H, H-9); δC (75 MHz, CDCl3) 129.6, 127.4 (C-2, C-3), 71.2 (C-4), 35.1 (C-5), 31.9 (C-7), 25.6 (C-6), 22.7 (C-8), 21.1, 19.8 (C-1, Me); 14.1 (C-9), 11.6 (Me); m/z (ESI) 153 (100, M-OH+), 154 (14%); HR-MS (HESI): M+, found 170.1665. C11H22O requires 170.1665.
2,3,5,5-Tetramethylhept-2-en-4-ol (14) 12 (2.62 mmol, 1.05 equiv), THF (2 mL), −30 °C, 2,2-dimethylbutanal (0.31 mL, 2.5 mmol), RT, overnight, NH4Cl (10 mL), H2O (10 mL), Et2O (3 × 15 mL), vacuum distillation (130 °C, 80 mbar), alcohol 14 (285 mg, 67%) as yellowish oil; Rf (hexanes/AcOEt 4:1) 0.59; νmax (ATR) 3398 (OH), 2962, 2917, 2879, 1462, 1373, 1002 cm−1; δH (300 MHz, CDCl3) 4.42 (d, J = 3.75 Hz, 1H, H-4), 1.69, 1.66 (2 × m, 3H, 6H, H-1, 2 × Me), 1.35 (d, J = 3.75 Hz, OH), 1.31 (m, 2H, H-6), 0.86 (m, 3H, H-7), 0.88, 0.80 (2 × s,2 × 3H, 2 × Me); δC (75 MHz, CDCl3) 128.4, 129.2 (C-2, C-3), 76.9 (C-4), 39.7 (C-5), 31.9 (C-6), 23.5, 22.6 (2 × Me), 21.4, 21.3 (C-1, Me); 14.6 (Me), 8.5 (C-7); m/z (ESI) 153 (100, M-OH+), 154 (12%); HR-MS (HESI): M+, found 170.1665. C11H22O requires 170.1665.
(E)-2,3,5-Trimethylhept-2,5-dien-4-ol (15) 12 (1.35 mmol, 1.05 equiv), THF (2 mL), −45 °C, (E)-2-methylbutanal (0.13 mL, 1.28 mmol), RT, overnight, NH4Cl (5 mL), H2O (5 mL), Et2O (3 × 10 mL), vacuum distillation (155 °C, 27 mbar), alcohol 15 (165 mg, 84%) as yellowish oil; Rf (hexanes/AcOEt 8:1, 2x) 0.45; νmax (ATR) 3363 (OH), 2916, 2861, 1444, 1375, 1047, 998 cm−1; δH (300 MHz, CDCl3) 5.58 (m, 1H, H-6), 5.00 (s, 1H, H-4), 1.77 (m, 3H, Me), 1.69 (s, 3H, H-1), 1.64 (m, 3H, H-7), 1.50, 1.47 (2 × m, 2 × 3H, 2 × Me); δC (75 MHz, CDCl3) 136.3 (C-5), 128.7, 120.0 (C-2, C-3); 117.2 (C-6), 74.3 (C-4), 21.2 (C-1), 20.1, 13.3, (2 × Me), 13.0 (C-7), 11.8 (Me); m/z (ESI) 137 (100, M-OH+), 138 (11%); HR-MS (HESI): M+, found 154.1351. C10H18O requires 154.1352.
2,3,5,6-Tetramethylhept-2,5-dien-4-ol (16) 12 (2.62 mmol, 2.05 equiv), THF (2 mL), −30 °C, ethyl formate (0.12 mL, 1.47 mmol), RT, overnight, NH4Cl (10 mL), H2O (10 mL), Et2O (3 × 15 mL), vacuum distillation (120 °C, 80 mbar), alcohol 16 (175 mg, 71%) as yellowish oil; Rf (hexanes/AcOEt 8:1, 2x) 0.43; νmax (ATR) 3339(OH), 2915, 2862, 1446, 1372, 998 cm−1; δH (300 MHz, CDCl3) 5.42 (s, 1H, H-4), 1.70, 1.67, 1.65 (2 × m, s, 3 × 6H, H-1, H-7, 4 × Me); δC (75 MHz, CDCl3) 129.8, 127.0 (C-2, C-3, C-5, C-6), 71.1 (C-4), 21.2, 20.0 (C-1, C-7, 2 × Me); 13.7 (2 × Me); m/z (ESI) 151 (100, M-OH+), 152 (14%); HR-MS (HESI): M+, found 168.1508. C11H20O requires 168.1509.
3.4. General experiment for the preparation of (di)enones (6–9)
To a solution of allyl alcohol in pentane was added activated MnO2 (heated at 140 °C/10 Torr for 30 min) at RT under Ar. The suspension was stirred at RT for the indicated time, diluted with diethyl ether, filtered through Celite pad and solids were repeatedly washed with Et2O. Filtrate was concentrated in vacuo (34 °C, 550 mbar) to furnish a corresponding enone pure by NMR. For analytical purposes, an aliquot was purified by either FLC on silica gel or bulb-to-bulb vacuum distillation.
2,3-Dimethylnon-2-en-4-one (6) Alcohol 13 (200 mg, 1.18 mmol), pentane (4 mL), MnO2 (2.05 g, 23.60 mmol, 20 equiv), RT, 4 d, Et2O (10 mL), Celite (2 × 1 cm), Et2O (4 × 10 mL), vacuum distillation (120 °C, 80 mbar), enone 6 (140 mg, 71%) as a colourless oil; Rf (hexanes/AcOEt 10:1) 0.50; νmax (ATR) 2956, 2928, 2860, 1685 (C=O), 1456, 1375, 1043, 1013 cm−1; δH (300 MHz, CDCl3) 2.50 (dd, J = 7.3 Hz, 2H, H-5), 1.81, 1.73 (m, 6H, s, 3H, H-1, 2 × Me), 1.30 (m, 6H, H-6, H-7, H-8), 0.89 (t, J = 6.8 Hz, 3H, H-9); δC (75 MHz, CDCl3) 208.9 (C=O), 136.0, 131.8, (C-2, C-3), 41.9 (C-5), 31.7 (C-6), 23.9 (C-7), 22.7 (C-8), 22.4, 21.3, 15.6, 14.1 (4 × Me); m/z (ESI) 169 (100, M + H+), 170 (11%); HR-MS (HESI): M+, found 168.1508. C11H20O requires 168.1508.
2,3,5,5-Tetramethylhept-2-en-4-one (7) Alcohol 14 (230 mg, 1.35 mmol), pentane (4 mL), MnO2 (2.35 g, 27.0 mmol, 20 equiv), RT, 72 h, Et2O (10 mL), Celite (2 × 1 cm), Et2O (4 × 10 mL), enone 7 (175 mg, 76%) as a colourless oil; Rf (hexanes/AcOEt 10:1) 0.53; νmax (ATR) 2967, 2932, 2880, 1682 (C=O), 1462, 1376, 1002, 973 cm−1; δH (300 MHz, CDCl3) 1.72, 1.63, 1.56 (3 × m, 3 × 3H, 3 × Me), 1.55 (q, 2H, J = 7.4 Hz, H-6), 1.1 (s, 6H, 2 × Me), 0.82 (t, J = 7.4 Hz, 3H, H-7); δC (75 MHz, CDCl3) 218.1 (C=O), 131.9, 128.9 (C-2, C-3), 47.7 (C-5), 32.8 (C-6), 24.5 (2 × Me), 22.4, 19.4, 16.2 (C-1, 2 × Me), 8.9 (C-7); m/z (ESI) 169 (100, M + H+), 170 (12%); HR-MS (HESI): M+, found 168.1506. C11H20O requires 168.1509.
(E)-2,3,5-Trimethylhept-2,5-dien-4-one (8) Alcohol 15 (113 mg, 0.73 mmol), pentane (3 mL), MnO2 (1.28 g, 14.68 mmol, 20 equiv), RT, 48 h, Et2O (5 mL), Celite (2 × 1 cm), Et2O (3 × 10 mL), enone 8 (89 mg, 74%) as a colourless oil; Rf (pentane/Et2O 10:1) 0.64; νmax (ATR) 2979, 2918, 2860, 1639 (C=O), 1444, 1376, 1285, 1045 cm−1; δH (300 MHz, CDCl3) 6.64 (m, 1H, H-6), 1.86 (m, 3H, H-7), 1.80, 1.75, 1.72, 1.54 (3 × m, s, 4 × 3H, H-1, 3 × Me); δC (75 MHz, CDCl3) 204.3 (C=O), 141.6 (C-6), 137.7, 130.6, 130.0 (C-2, C-3, C-5), 22.2, 19.8, 17.0, 15.1, 10.5 (C-1, C-7, 3 × Me); m/z (ESI) 153 (100, M + H+), 154 (10%); HR-MS (HESI): M+, found 152.1194. C10H16O requires 152.1196.
(E)-2,3,5,6-Tetramethylhept-2,5-dien-4-one (9) Alcohol 16 (113 mg, 0.67 mmol), pentane (2 mL), MnO2 (1.17 g, 13.45 mmol, 20 equiv), RT, 4 d, Et2O (5 mL), Celite (2 × 1 cm), Et2O (3 × 10 mL), enone 9 (85 mg, 76%) as a colourless oil; Rf (pentane/Et2O 10:1) 0.64; νmax (ATR) 2915, 2863, 1629 (C=O), 1445, 1373, 1294, 1003 cm−1; δH (300 MHz, CDCl3) 1.82, 1.79, 1.76 (3 × m, 3 × 6H, H-1, H-7, 4 × Me); δC (75 MHz, CDCl3) 204.7 (C=O), 138.1, 132.1 (C-2, C-3, C-5, C-6), 22.0, 21.7 (C-1, C-7, 2 × Me), 15.6 (2 × Me); m/z (ESI) 167 (100, M + H+), 168 (11%); HR-MS (HESI): M+, found 166.1952. C11H18O requires 166.1952.
2,5-Diisopropylidene-cyclopentanone (10) Prepared according to the reported procedure [9c] as a colourless oil; Rf (hexanes/AcOEt 10:1) 0.51; νmax (ATR) 2956, 2905, 2849 (C-H), 1682, 1613 (C=O), 1435, 1363, 1267, 1195, 978, 796 cm−1; δH (300 MHz, CDCl3) 2.51 (s, 4H, H-3, H-4), 2.27 (s, 6H, 2 × Me), 1.81 (s, 6 H, 2 × Me); δC (75 MHz, CDCl3) 196.6 (C=O), 146.2, 134.4 (4 × Cq), 25.4 (2 × CH2), 24.4, 20.4 (4 × Me); m/z (ESI) 165 (100, M + H+), 166 (13%); HR-MS (HESI): M+, found 164.1195. C11H16O requires 164.1196.





{kind=link}
{kind=link}
{kind=link}
{kind=link}








