
Site Selectivity in Pd-Catalyzed Reactions of α-Diazo-α-(methoxycarbonyl)acetamides: Effects of Catalysts and Substrate Substitution in the Synthesis of Oxindoles and β-Lactams

 ,
, 
Abstract
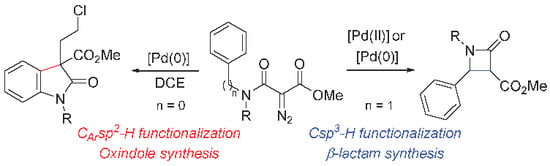
1. Introduction
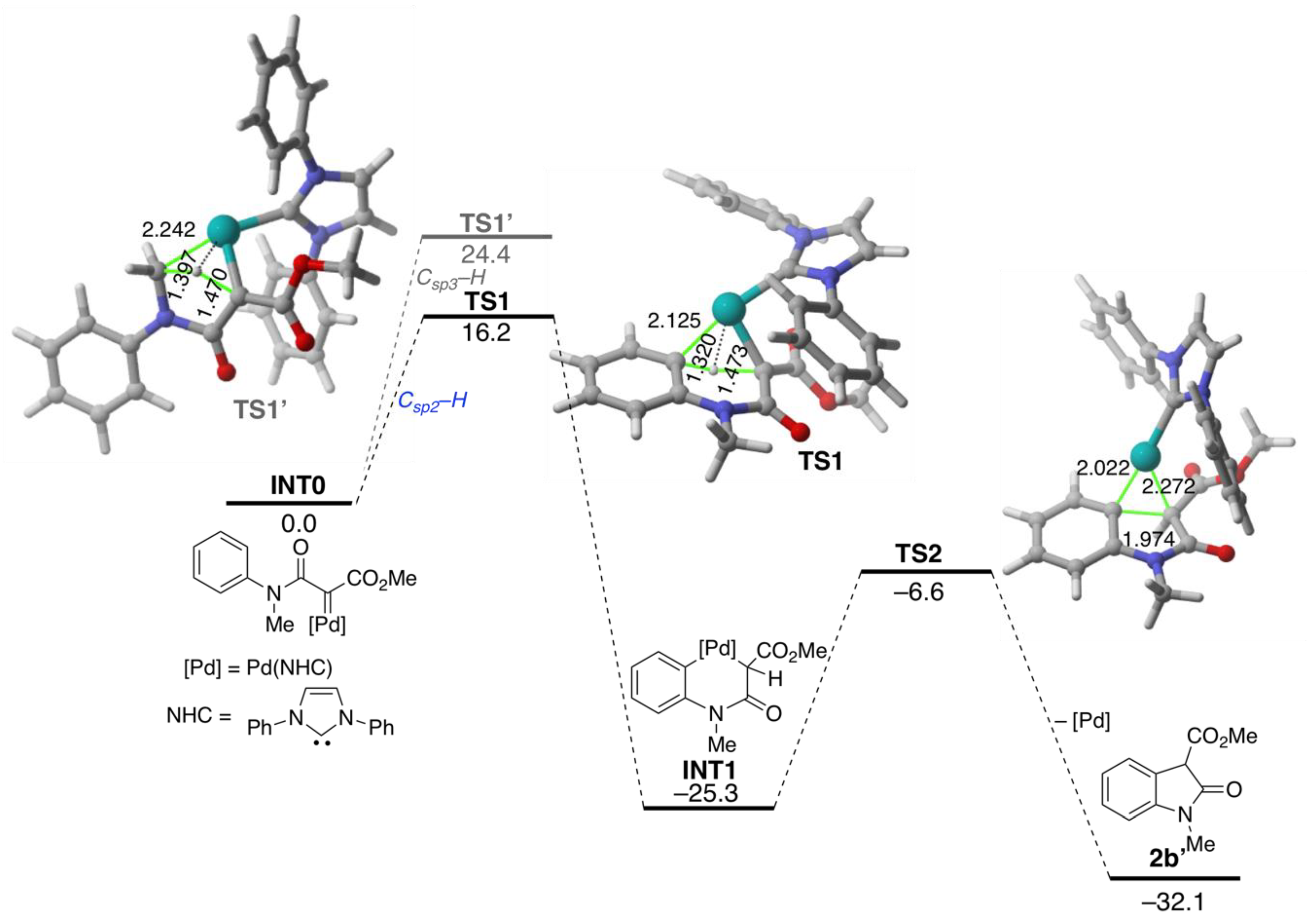
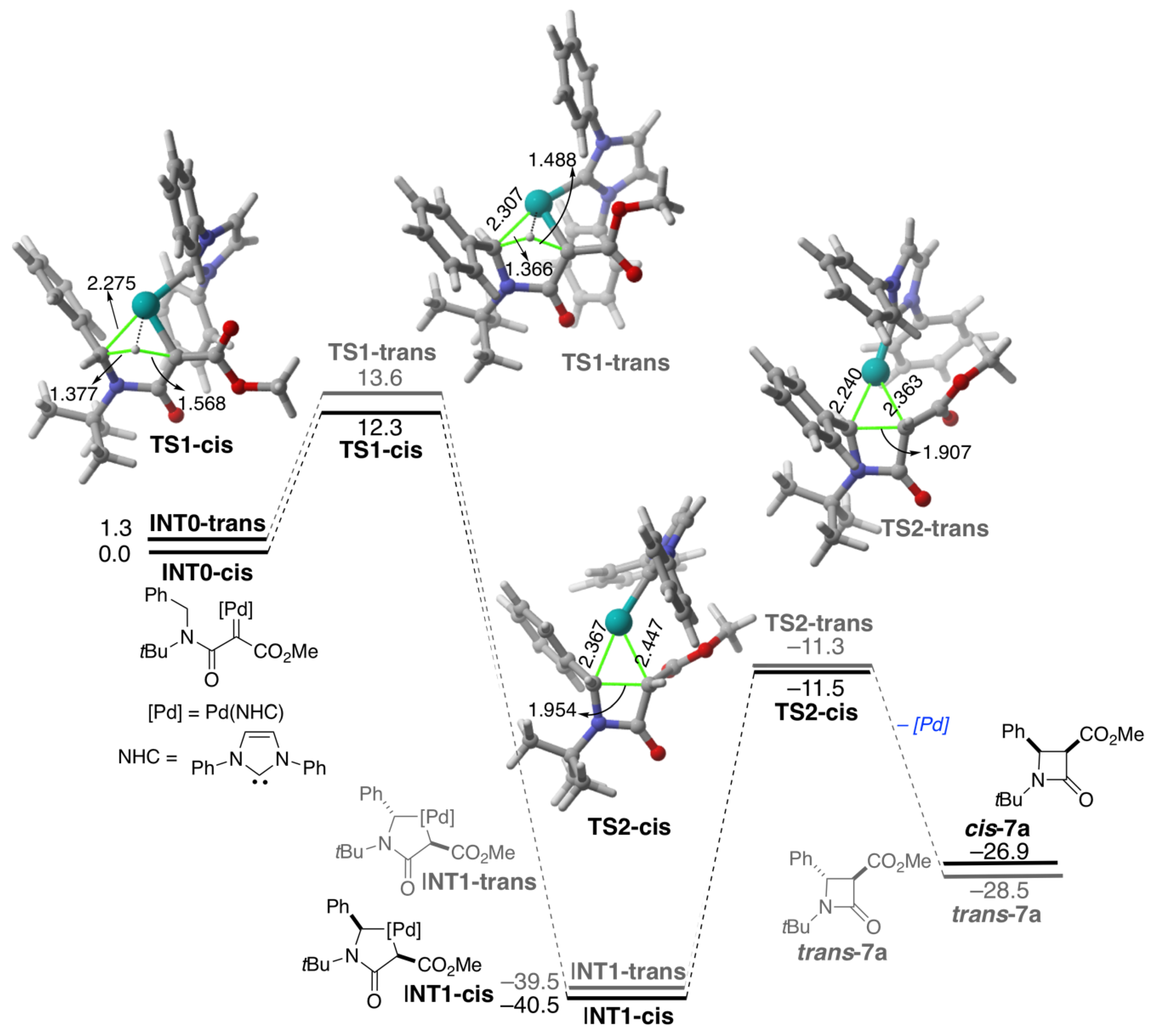
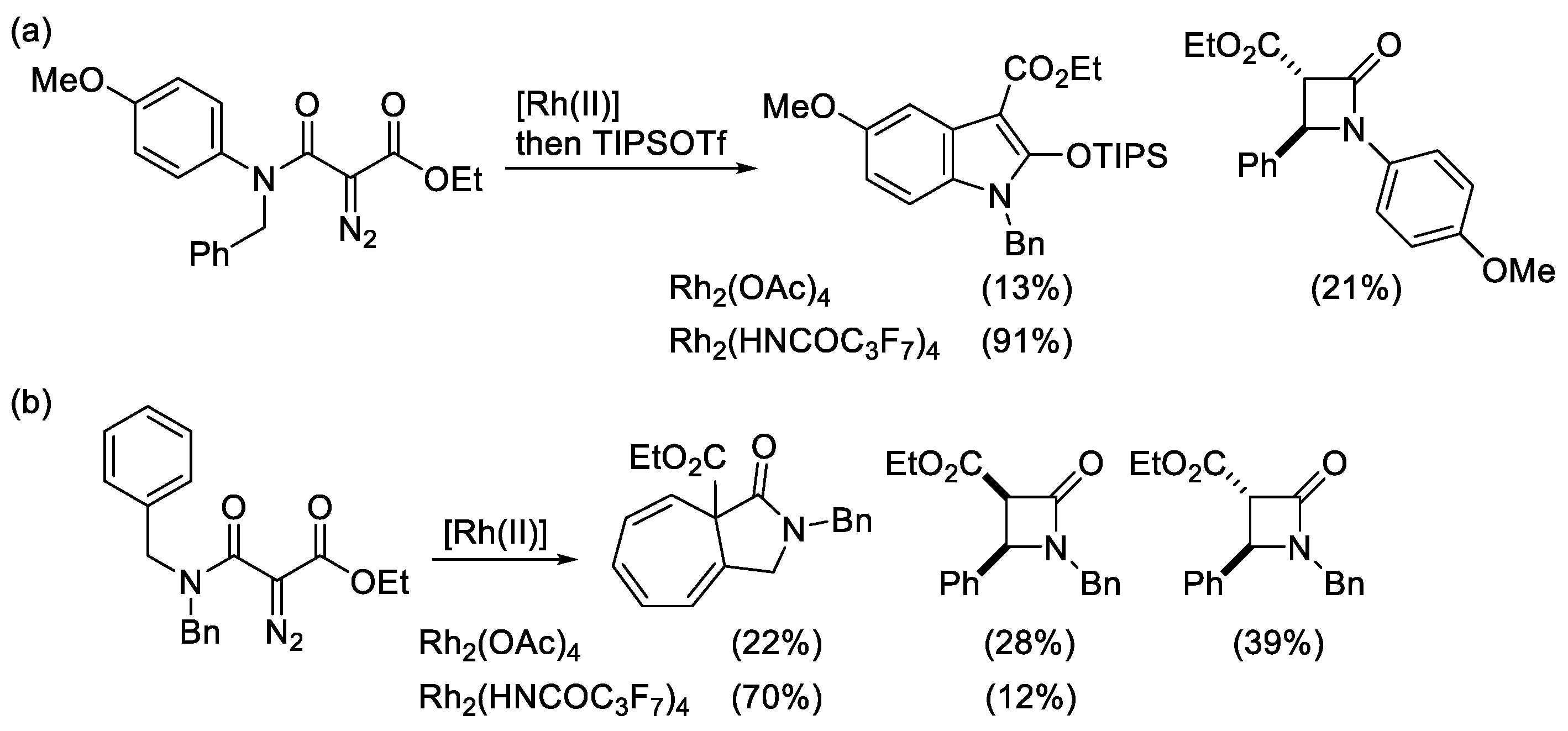
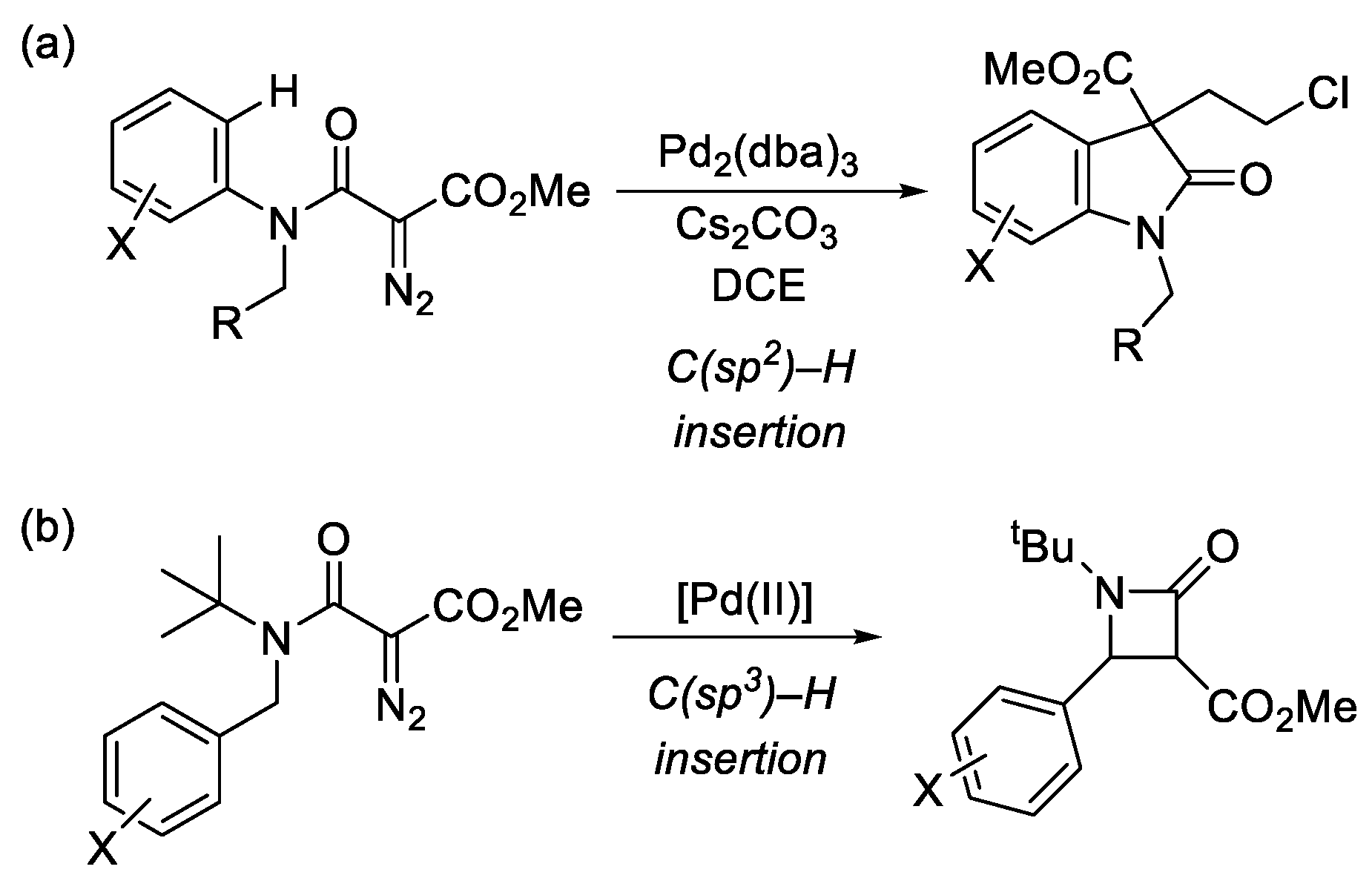
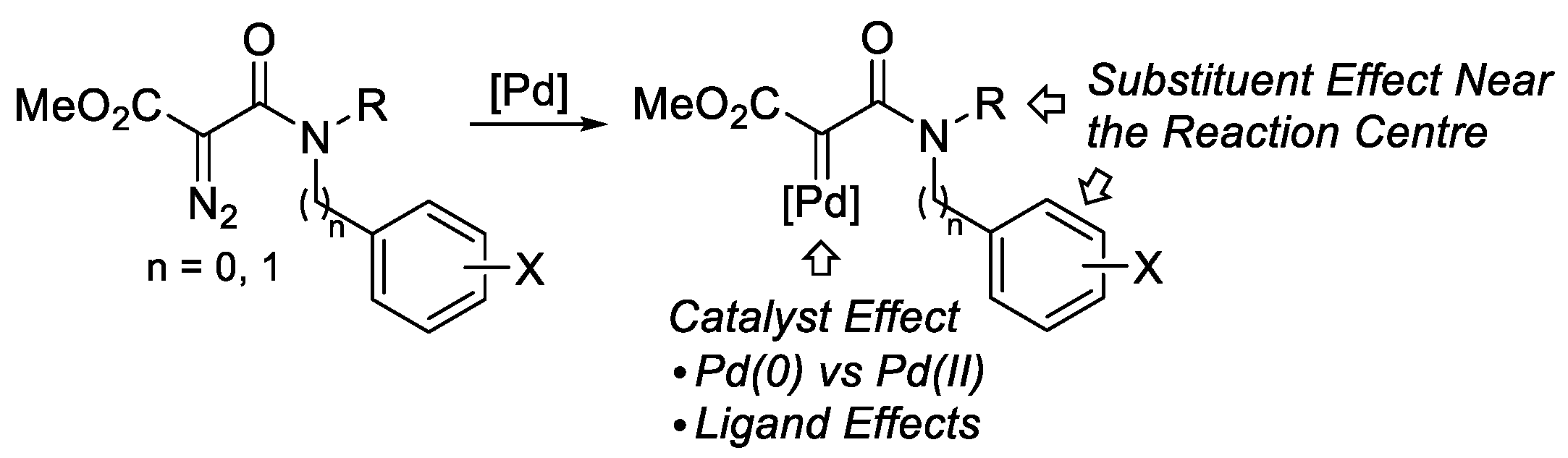
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of N-Benzyl-N-(2-pyridinyl)-α-(ethoxycarbonyl)-α-diazoacetamide (3).
3.3. Synthesis of N-tert-Butyl-N-(4-pyridinylmethyl)-α-(methoxycarbonyl)-α-diazoacetamide (11).
3.4. Characterization Data for New Compounds of Scheme 4 and Tables 3 and 4
4. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Godula, K.; Sames, D. C-H Bond functionalization in complex organic synthesis. Science 2006, 312, 67–72. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C-H Bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Manning, J.R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef]
- Doyle, M.P.; Duffy, R.; Ratnikov, M.; Zhou, L. Catalytic carbene insertion into C-H bonds. Chem. Rev. 2010, 110, 704–724. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Recent development of direct asymmetric functionalization of inert C-H bonds. RSC Adv. 2014, 4, 6173–6214. [Google Scholar] [CrossRef]
- Ford, A.; Miel, H.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern organic synthesis with α-diazocarbonyl compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef]
- Lombard, F.J.; Coster, M.J. Rhodium(II)-catalysed intramolecular C-H insertion α- to oxygen: reactivity, selectivity and applications to natural product synthesis. Org. Biomol. Chem. 2015, 13, 6419–6431. [Google Scholar] [CrossRef]
- Hu, F.; Xia, Y.; Ma, C.; Zhang, Y.; Wang, J. C-H bond functionalization based on metal carbene migratory insertion. Chem. Commun. 2015, 51, 7986–7995. [Google Scholar] [CrossRef]
- Cai, Y.; Zhu, S.-F.; Wang, G.-P.; Zhou, Q.-L. Iron-catalyzed C-H functionalization of indoles. Adv. Synth. Catal. 2011, 353, 2939–2944. [Google Scholar] [CrossRef]
- Yao, T.; Hirano, K.; Satoh, T.; Miura, M. Nickel- and cobalt-catalyzed direct alkylation of azoles with N-tosylhydrazones bearing unactivated alkyl groups. Angew. Chem. Int. Ed. 2012, 51, 775–779. [Google Scholar] [CrossRef]
- Yu, Z.; Ma, B.; Chen, M.; Wu, H.-H.; Liu, L.; Zhang, J. Highly site-selective direct C-H bond functionalization of phenols with α-aryl-α-diazoacetates and diazooxindoles via gold catalysis. J. Am. Chem. Soc. 2014, 136, 6904–6907. [Google Scholar] [CrossRef]
- Liu, X.-G.; Zhang, S.-S.; Wu, J.-Q.; Li, Q.; Wang, H. Cp*Co(III)-catalyzed direct functionalization of aromatic C-H bonds with α-diazomalonates. Tetrahedron Lett. 2015, 56, 4093–4095. [Google Scholar] [CrossRef]
- Zhao, D.; Kim, J.H.; Stegemann, L.; Strassert, C.A.; Glorius, F. Cobalt(III)-catalyzed directed C-H coupling with diazo compounds: straightforward access towards extended π-systems. Angew. Chem. Int. Ed. 2015, 54, 4508–4511. [Google Scholar] [CrossRef]
- Fructos, M.R.; Díaz-Requejo, M.M.; Pérez, P.J. Gold and diazo reagents: a fruitful tool for developing molecular complexity. Chem. Commun. 2016, 52, 7326–7335. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, J. Gold-catalyzed transformations of α-diazocarbonyl compounds: selectivity and diversity. Chem. Soc. Rev. 2016, 45, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Conde, A.; Sabenya, G.; Rodríguez, M.; Postils, V.; Luis, J.M.; Díaz-Requejo, M.M.; Costas, M.; Pérez, P.J. Iron and manganese catalysts for the selective functionalization of arene C(sp2)-H bonds by carbene insertion. Angew. Chem. Int. Ed. 2016, 55, 6530–6534. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Parr, B.T. Rhodium carbenes. In Contemporary Carbene Chemistry; Wiley: Hoboken, NJ, USA, 2013; pp. 363–403. [Google Scholar]
- Yakura, T.; Nambu, H. Recent topics in application of selective Rh(II)-catalyzed C-H functionalization toward natural product synthesis. Tetrahedron Lett. 2018, 59, 188–202. [Google Scholar] [CrossRef]
- Díaz-Requejo, M.M.; Pérez, P.J. Coinage metal catalyzed C-H bond functionalization of hydrocarbons. Chem. Rev. 2008, 108, 3379–3394. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Wang, J. Recent developments in copper-catalyzed reactions of diazo compounds. Chem. Commun. 2012, 48, 10162–10173. [Google Scholar] [CrossRef]
- Choi, M.K.-W.; Yu, W.-Y.; Che, C.-M. Ruthenium-catalyzed stereoselective intramolecular carbenoid C-H insertion for β- and γ-lactam formations by decomposition of α-diazoacetamides. Org. Lett. 2005, 7, 1081–1084. [Google Scholar] [CrossRef]
- Grohmann, M.; Buck, S.; Schäffler, L.; Maas, G. Diruthenium(I,I) catalysts for the formation of β- and γ-lactams via carbenoid C-H insertion of α-diazoacetamides. Adv. Synth. Catal. 2006, 348, 2203–2211. [Google Scholar] [CrossRef]
- Choi, M.K.-W.; Yu, W.-Y.; So, M.-H.; Zhou, C.-Y.; Deng, Q.-H.; Che, C.-M. A Non-cross-linked soluble polystyrene-supported ruthenium catalyst for carbenoid transfer reactions. Chem. Asian J. 2008, 3, 1256–1265. [Google Scholar] [CrossRef]
- Reddy, A.R.; Zhou, C.-Y.; Guo, Z.; Wei, J.; Che, C.-M. Ruthenium-porphyrin-catalyzed diastereoselective intramolecular alkyl carbene insertion into C-H bonds of alkyl diazomethanes generated in situ from N-tosylhydrazones. Angew. Chem. Int. Ed. 2014, 53, 14175–14180. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Chanthamath, S.; Liang, Y.; Shibatomi, K.; Iwasa, S. Regio- and enantioselective intramolecular amide carbene insertion into primary C-H bonds using Ru(II)-Pheoux catalyst. J. Org. Chem. 2019, 84, 2067–2618. [Google Scholar] [CrossRef] [PubMed]
- Solé, D.; Amenta, A.; Bennasar, M.-L.; Fernández, I. Grubbs catalysts in intramolecular carbene C(sp3)-H insertion reactions from α-diazoesters. Chem. Commun. 2019, 55, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Solé, D.; Amenta, A.; Bennasar, M.-L.; Fernández, I. Pd- and Ru-Catalyzed intramolecular carbene CAr-H functionalization of γ-amino-α-diazoesters for the synthesis of tetrahydroquinolines. Chem. Eur. J. 2019, 25, 10239–10245. [Google Scholar] [CrossRef] [PubMed]
- Taber, D.F.; Amedio, J.C.; Sherill, R.G. Palladium-mediated diazo insertion: Preparation of 3-alkyl-2-carbomethoxycyclopentenones. J. Org. Chem. 1986, 51, 3382–3384. [Google Scholar] [CrossRef]
- Matsumoto, M.; Watanabe, N.; Kobayashi, H. Metal-catalyzed intramolecular cyclization of 2-diazo-4-(4-indolyl)-3-oxobutanoic acid esters. Heterocycles 1987, 26, 1479–1482. [Google Scholar] [CrossRef]
- Rosenberg, M.L.; Aasheim, J.H.F.; Trebbin, M.; Uggerud, E.; Hansen, T. Synthesis of a 1,3,4,5-tetrahydrobenzindole β-ketoester. Tetrahedron Lett. 2009, 50, 6506–6508. [Google Scholar] [CrossRef]
- Goll, J.M.; Fillion, E. Tuning the reactivity of palladium carbenes derived from diphenylketene. Organometallics 2008, 27, 3622–3625. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J. Recent developments in Pd-catalyzed reactions of diazo compounds. Eur. J. Org. Chem. 2011, 1015–1026. [Google Scholar] [CrossRef]
- Barluenga, J.; Valdés, C. Tosylhydrazones: new uses for classic reagents in palladium-catalyzed cross-coupling and metal-free reactions. Angew. Chem. Int. Ed. 2011, 50, 7486–7500. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Zhang, H. N-Tosylhydrazones: versatile reagents for metal-catalyzed and metal-free cross-coupling reactions. Chem. Soc. Rev. 2012, 41, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhang, Y.; Wang, J. Diazo compounds and N-tosylhydrazones: novel cross-coupling partners in transition-metal-catalyzed reactions. Acc. Chem. Res. 2013, 46, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Reiser, O. Cyclopropanation and other reactions of palladium-carbene (and carbine) complexes. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; Wiley-Interscience: New York, NY, USA, 2002; Volume 1, pp. 1561–1577. [Google Scholar]
- Gois, P.M.P.; Afonso, C.A.M. Stereo- and regiocontrol in the formation of lactams by rhodium-carbenoid C-H insertion of α-diazoacetamides. Eur. J. Org. Chem. 2004, 3773–3788. [Google Scholar] [CrossRef]
- Ring, A.; Ford, A.; Maguire, A.R. Substrate and catalyst effects in C-H insertion reactions of α-diazoacetamides. Tetrahedron Lett. 2016, 57, 5399–5406. [Google Scholar] [CrossRef]
- Grohmann, M.; Maas, G. Ruthenium catalysts for carbenoid intramolecular C-H insertion of 2-diazoacetoacetamides and diazomalonic ester amides. Tetrahedron 2007, 63, 12172–12178. [Google Scholar] [CrossRef]
- Large, T.; Müller, T.; Kunkel, H.; Buck, S.; Maas, G. Ruthenium- and rhodium-catalyzed carbenoid reactions of diazoesters in hexaalkylguanidinium-based ionic liquids. Z. Naturforsch. B 2012, 67, 347–353. [Google Scholar] [CrossRef]
- Chan, W.-W.; Kwong, T.-L.; Yu, W.-Y. Ruthenium-catalyzed intramolecular cyclization of diazo-β-ketoanilides for the synthesis of 3-alkylideneoxindoles. Org. Biomol. Chem. 2012, 10, 3749–3755. [Google Scholar] [CrossRef]
- Liu, N.; Tian, Q.-P.; Yang, Q.; Yang, S.-D. Ruthenium-catalyzed intramolecular cyclization and fluorination to form 3-fluorooxindoles. Synlett 2016, 27, 2621–2625. [Google Scholar]
- Yamamoto, K.; Qureshi, Z.; Tsoung, J.; Pisella, G.; Lautens, M. Combining Ru-catalyzed C-H functionalization with Pd-catalyzed asymmetric allylic alkylation: synthesis of 3-allyl-3-aryl oxindoles derivatives from aryl α-diazoamides. Org. Lett. 2016, 18, 4954–4957. [Google Scholar] [CrossRef] [PubMed]
- Merlic, C.A.; Zechman, A.L. Selectivity in rhodium(II) catalyzed reactions of diazo compounds: effects of catalyst electrophilicity, diazo substitution, and substrate substitution. From chemoselectivity to enantioselectivity. Synthesis 2003, 1137–1156. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Morton, D. Guiding principles for site selective and stereoselective intermolecular C-H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev. 2011, 40, 1857–1869. [Google Scholar] [CrossRef]
- DeAngelis, A.; Panish, R.; Fox, J.M. Rh-catalyzed intermolecular reactions of α-alkyl-α-diazo carbonyl compounds with selectivity over β-hydride migration. Acc. Chem. Res. 2016, 49, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.S.; Elliott, M.C.; Moody, C.J.; Mowlem, T.J.; Marino, J.P.; Padwa, A. Ligand effects in the rhodium(II)-catalyzed reactions of α-diazoamides. Oxindole formation is promoted by the use of rhodium(II) perfluorocarboxamide catalysts. J. Org. Chem. 1994, 59, 2447–2455. [Google Scholar] [CrossRef]
- Miah, S.; Slawin, A.M.Z.; Moody, C.J.; Sheedan, S.M.; Marino, J.P., Jr.; Semones, M.A.; Padwa, A.; Richards, I.C. Ligand effects in the rhodium(II) catalyzed reactions of diazoamides and diazoimides. Tetrahedron 1996, 52, 2489–2514. [Google Scholar] [CrossRef]
- Solé, D.; Pérez-Janer, F.; Mancuso, R. Pd0-Catalyzed intramolecular α-arylation of sulfones: domino reactions in the synthesis of functionalized tetrahydroquinolines. Chem. Eur. J. 2015, 21, 4580–4584. [Google Scholar] [CrossRef]
- Solé, D.; Pérez-Janer, F.; Zulaica, E.; Guastavino, J.F.; Fernández, I. Pd-Catalyzed α-arylation of sulfones in a three-component synthesis of 3-[2-(phenyl/methylsulfonyl)ethyl]indoles. ACS Catal. 2016, 6, 1691–1700. [Google Scholar] [CrossRef]
- Solé, D.; Pérez-Janer, F.; García-Rodeja, Y.; Fernández, I. Exploring partners for the domino α-arylation/Michael addition reaction leading to tetrahydroquinolines. Eur. J. Org. Chem. 2017, 2017, 799–805. [Google Scholar] [CrossRef]
- Solé, D.; Fernández, I. Controlling the ambiphillic nature of σ-arylpalladium intermediates in intramolecular cyclization reactions. Acc. Chem. Res. 2014, 47, 168–179. [Google Scholar] [CrossRef]
- Solé, D.; Mariani, F.; Bennasar, M.-L.; Fernández, I. Palladium-catalyzed intramolecular carbene insertion into C(sp3)-H bonds. Angew. Chem. Int. Ed. 2016, 55, 6467–6470. [Google Scholar] [CrossRef] [PubMed]
- Solé, D.; Amenta, A.; Mariani, F.; Bennasar, M.-L.; Fernández, I. Transition metal-catalysed intramolecular carbenoid C-H insertion for pyrrolidine formation by decomposition of α-diazoesters. Adv. Synth. Catal. 2017, 359, 3654–3664. [Google Scholar] [CrossRef]
- Solé, D.; Pérez-Janer, F.; Fernández, I. Palladium-catalysed intramolecular carbenoid insertion of α-diazo-α-(methoxycarbonyl)acetanilides for oxindoles synthesis. Chem. Commun. 2017, 53, 3110–3113. [Google Scholar] [CrossRef] [PubMed]
- Solé, D.; Pérez-Janer, F.; Bennasar, M.-L.; Fernández, I. Palladium catalysis in the intramolecular carbene C-H insertion of α-diazo-α-(methoxycarbonyl)acetamides to forma β-lactams. Eur. J. Org. Chem. 2018, 4446–4455. [Google Scholar] [CrossRef]
- Gabriele, B.; Salerno, G.; Veltri, L.; Costa, M.; Massera, C. Stereoselective synthesis of (E)-3-(methoxycarbonyl)methylene-1,3-dihydroindol-2-ones by palladium-catalyzed oxidative carbonylation of 2-ethynylanilines. Eur. J. Org. Chem. 2001, 4607–4613. [Google Scholar] [CrossRef]
- Kischkewitz, M.; Daniliuc, C.-G.; Studer, A. 3-Alkyl-3-cyano-oxindoles from 2-cyano-2-diazo-N-phenylacetamides via cyclizing carbene insertion and subsequent radical oxidation. Org. Lett. 2016, 18, 1206–1209. [Google Scholar] [CrossRef]
- Ma, C.; Xing, D.; Hu, W. Catalyst-free halogenation of α-diazocarbonyl compounds with N-halosuccinimides: synthesis of 3-halooxindoles or vinyl halides. Org. Lett. 2016, 18, 3134–3137. [Google Scholar] [CrossRef]
- Mo, S.; Xu, C.; Xu, J. Expeditious and convenient synthesis of polycyclic difluoroboron complexes of 2-oxoindoline-3-carboxamides by tandem reaction. Adv. Synth. Catal. 2016, 358, 1767–1777. [Google Scholar] [CrossRef]
- Moody, C.J.; Miah, S.; Slawin, A.M.Z.; Mansfield, D.J.; Richards, I.C. Ligands effects in the metal catalyzed reactions of N-aryldiazoamides: ylide formation vs. insertion reactions. Tetrahedron 1998, 54, 9689–9700. [Google Scholar] [CrossRef]
- Heil, M.; Hoffmeister, L.; Webber, M.; Ilg, K.; Goergens, U.; Turberg, A. Preparation of mesoionic imidazopyridines for use as insecticides. PCT Int. Appl. WO 2018192872 A1 20181025.
- Bonardi, A.; Costa, M.; Gabriele, B.; Salerno, G.; Chiusoli, G.P. Versatile synthesis of beta-lactams, gamma-lactams or oxazolidines by palladium-catalysed oxidative carbonylation of 1-substituted prop-2-ynylamines. Tetrahedron Lett. 1995, 36, 7495–7498. [Google Scholar] [CrossRef]
- Hosseyni, S.; Jarrahpour, A. Recent advances in β-lactam synthesis. Org. Biomol. Chem. 2018, 16, 6840–6852. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, J.; Dar’in, D.; Novikov, M.S.; Kantin, G.; Bakulina, O.; Krasavin, M. α-Acyl-α-diazoacetates in transition-metal-free β-lactam synthesis. J. Org. Chem. 2019, 84, 12101. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1998, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab-initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Pascual-Ahuir, J.L.; Silla, E.; Tuñón, I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comp. Chem. 1994, 15, 1127–1138. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst (mol%) | Time | Products (Yield (%)) 2 |
|---|---|---|---|
| 1 | Pd2(dba)3 (10) | 96 h 3 | 2a (66) |
| 2 | (Pd(allyl)Cl)2 (5) | 24 h | 1a/2a (1:2.2) 4 |
| 3 | (Pd(allyl)Cl)2 (5) | 60 h | 1a/2a (1:5.5) 4 |
| 4 | ((IMes)Pd(NQ))2 (4) | 24 h | 2a (65) |

| Entry | Substance 1 | Catalyst 1,2 | Product (Yield (%)) 3 | ||
|---|---|---|---|---|---|
| 1 |  | 1b (R:Me) | Pd2(dba)3 |  | 2b (71) |
| 2 | 1b (R:Me) | ((IMes)Pd(NQ))2 | 2b (79) | ||
| 3 | 1c (R:Et) | Pd2(dba)3 | 2c (54) | ||
| 4 | 1c (R:Et) | ((IMes)Pd(NQ))2 | 2c (78) | ||
| 5 | 1d (R:i-Pr) | Pd2(dba)3 | 2d (61) | ||
| 6 | 1d (R:i-Pr) | ((IMes)Pd(NQ))2 | 2d (64) | ||
| 7 | 1e (R:Ph) | Pd2(dba)3 4 | 2e (55) 5 | ||
| 8 | 1e (R:Ph) | ((IMes)Pd(NQ))2 | 2e (35) | ||
| 9 |  | 1f | Pd2(dba)3 |  | 2f (23) 6 |
| 10 | 1f | ((IMes)Pd(NQ))2 | 2f (0) 7 | ||
| 11 |  | 1g (R1:OMe) | Pd2(dba)3 |  | 2g (36) |
| 12 | 1g (R1:OMe) | ((IMes)Pd(NQ))2 | 2g (76) | ||
| 13 | 1h (R1F) | Pd2(dba)3 | 2h (64) | ||
| 14 | 1h (R1:F) | ((IMes)Pd(NQ))2 | 2h (39) 8 | ||
| 15 | 1i (R1:CO2Me) | Pd2(dba)3 4 | 2i (22) | ||
| 16 | 1i (R1:CO2Me) | ((IMes)Pd(NQ))2 | 2i (0) 9 | ||
| 17 |  | 1j | Pd2(dba)3 4 |  | 2j (27) |
| 18 | 1j | ((IMes)Pd(NQ))2 | 2j (23) 10 |

| Entry | 5 (X) | Catalyst (mol%) | 6/7/8 2,3 | cis-7/trans-7 2,3 | Products (Yield (%)) 4 |
|---|---|---|---|---|---|
| 1 | 5a (H) | Pd2(dba)3 (10) | 26/74/0 | 46/28 | 6a (20) cis-7a (9), trans-7a (42) |
| 2 | 5a (H) | ((IMes)Pd(NQ))2 (2.5) | 35/65/0 | 40/25 | 6a (28) cis-7a (35), trans-7a (23) |
| 3 | 5a (H) | (Pd(allyl)Cl)2 (5) | 0/100/0 | 47/53 | cis-7a (25), trans-7a (65) |
| 4 | 5a (H) | (SIPr)Pd(allyl)Cl (15) | 0/100/0 | 29/71 | cis-7a (17), trans-7a (59) |
| 5 | 5b (4-MeO) | Pd2(dba)3 (10) | 50/50/0 | 21/29 | 6b (24) cis-7b (9), trans-7b (20) |
| 6 | 5b (4-MeO) | ((IMes)Pd(NQ))2 (2.5) | 34/66/0 | 46/20 | 6b (22) cis-7b (21), trans-7b (24) |
| 7 | 5b (4-MeO) | (Pd(allyl)Cl)2 (5) | 14/86/0 | 29/57 | 6b (8) cis-7b (18), trans-7b (39) |
| 8 | 5b (4-MeO) | (SIPr)Pd(allyl)Cl (15) | 16/84/0 | 42/42 | 6b (10) cis-7b (8), trans-7b (52) |
| 9 | 5c (4-MeS) | Pd2(dba)3 (10) | 18/82/0 | 36/46 | 6c (6) cis-7c (4), trans-7c (30) |
| 10 | 5c (4-MeS) | ((IMes)Pd(NQ))2 (2.5) | 11/89/0 | 68/21 | 6c (7) cis-7c (30), trans-7c (50) |
| 11 | 5c (4-MeS) | (Pd(allyl)Cl)2 (5) | 5/95/0 | 42/53 | cis-7c (10), trans-7c (48) |
| 12 | 5c (4-MeS) | (SIPr)Pd(allyl)Cl (15) | 12/88/0 | 48/40 | 6c (7) cis-7c (4), trans-7c (60) |
| 13 | 5d (4-Cl) | Pd2(dba)3 (10) | 15/85/0 | 8/77 | 6d (9) cis-7d (5), trans-7d (50) |
| 14 | 5d (4-Cl) | ((IMes)Pd(NQ))2 (2.5) | 5/95/0 | 69/26 | 6d (5) cis-7d (53), trans-7d (19) |
| 15 | 5d (4-Cl) | (Pd(allyl)Cl)2 (5) | 0/100/0 | 52/48 | cis-7d (42), trans-7d (40) |
| 16 | 5d (4-Cl) | (SIPr)Pd(allyl)Cl (15) | 0/100/0 | 62/38 | cis-7d (27), trans-7d (42) |
| 17 | 5e (4-CN) | Pd2(dba)3 (10) | 3/97/0 | 67/30 | cis-7e (22), trans-7e (38) |
| 18 | 5e (4-CN) | ((IMes)Pd(NQ))2 (2.5) | 5/75/20 | 25/50 | cis-7e (15), trans-7e (33) 8e (15) |
| 19 | 5e (4-CN) | (Pd(allyl)Cl)2 (5) | 0/100/0 | 71/29 | cis-7e (34), trans-7e (36) |
| 20 | 5e (4-CN) | (SIPr)Pd(allyl)Cl (15) | 0/100/0 | 38/62 | cis-7e (27), trans-7e (51) |
| 21 | 5f (4-NMe2) | Pd2(dba)3 (10) | CM | --- | --- |
| 22 | 5f (4-NMe2) | ((IMes)Pd(NQ))2 (2.5) | CM | --- | cis-7f (9), trans-7f (10) 10 (15) 5 |
| 23 | 5f (4-NMe2) | (Pd(allyl)Cl)2 (5) | CM | --- | trans-7f (14), 10 (36) |
| 24 | 5f (4-NMe2) | (SIPr)Pd(allyl)Cl (15) | CM | --- | trans-7f (10), 10 (45) |
| 25 | 5g (3-Cl) | Pd2(dba)3 (10) | 8/92/0 | 54/38 | 6g6 (7) cis-7g (30), trans-7g (43) |
| 26 | 5g (3-Cl) | ((IMes)Pd(NQ))2 (2.5) | 6/94/0 | 73/21 | 6g6 (5) cis-7g (50), trans-7g (20) |
| 27 | 5g (3-Cl) | (Pd(allyl)Cl)2 (5) | 0/100/0 | 64/36 | cis-7g (44), trans-7g (36) |
| 28 | 5g (3-Cl) | (SIPr)Pd(allyl)Cl (15) | 0/100/0 | 50/50 | cis-7g (30), trans-7g (31) |
| 29 | 5h (3-CN) | Pd2(dba)3 (10) | 0/100/0 | 33/67 | cis-7h (25), trans-7h (48) |
| 30 | 5h (3-CN) | ((IMes)Pd(NQ))2 (2.5) | 4/74/22 | 30/44 | cis-7h (15), trans-7h (37) 8h (6) |
| 31 | 5h (3-CN) | (Pd(allyl)Cl)2 (5) | 0/100/0 | 47/53 | cis-7h (22), trans-7h (43) |
| 32 | 5h (3-CN) | (SIPr)Pd(allyl)Cl (15) | 0/100/0 | 29/71 | cis-7h (15), trans-7h (55) |
| 33 | 5i (2-MeO) | Pd2(dba)3 (10) | 14/86/0 | 43/43 | 6i (8) cis-7i (12), trans-7i (19) |
| 34 | 5i (2-MeO) | ((IMes)Pd(NQ))2 (2.5) | 20/80/0 | 57/23 | 6i (10) cis-7i (45), trans-7i (24) |
| 35 | 5i (2-MeO) | (Pd(allyl)Cl)2 (5) | 5/95/0 | 42/53 | cis-7i (23), trans-7i (31) |
| 36 | 5i (2-MeO) | (SIPr)Pd(allyl)Cl (15) | 6/94/0 | 39/55 | cis-7i (10), trans-7i (63) |
| 37 | 5j (2-F) | Pd2(dba)3 (10) | 8/92/0 | 8/84 | cis-7j (5), trans-7j (66) |
| 38 | 5j (2-F) | ((IMes)Pd(NQ))2 (2.5) | 5/83/12 | 24/59 | cis-7j (10), trans-7j (62) 8j (11) |
| 39 | 5j (2-F) | (Pd(allyl)Cl)2 (5) | 0/100/0 | 69/31 | cis-7j (26), trans-7j (56) |
| 40 | 5j (2-F) | (SIPr)Pd(allyl)Cl (15) | 0/100/0 | 0/100 | trans-7j (66) |
| 41 | 5k (2-Br) | Pd2(dba)3 (10) | 7/93/0 | 70/23 | 6k (6) cis-7k (46), trans-7k (15) |
| 42 | 5k (2-Br) | ((IMes)Pd(NQ))2 (2.5) | 0/85/15 | 66/19 | cis-7k (43), trans-7k (20) 8k (12) |
| 43 | 5k (2-Br) | (Pd(allyl)Cl)2 (5) | 14/86/0 | 52/34 | 6k (9) cis-7k (34), trans-7k (31) |
| 44 | 5k (2-Br) | (SIPr)Pd(allyl)Cl (15) | 8/92/0 | 21/71 | 6k (4) cis-7k (8), trans-7k (42) |
| 45 | 5l (2-I) | Pd2(dba)3 (10) | 0/100/0 | 9/91 | cis-7l (5), trans-7l (45) |
| 46 | 5l (2-I) | ((IMes)Pd(NQ))2 (2.5) | 0/78/22 | 66/12 | cis-7l (64), trans-7l (12) 8l (15) |
| 47 | 5l (2-I) | (Pd(allyl)Cl)2 (5) | 8/92/0 | 50/42 | 6l (9) cis-7l (49), trans-7l (38) |
| 48 | 5l (2-I) | (SIPr)Pd(allyl)Cl (15) | 0/100/0 | 77/23 | cis-7l (53), trans-7l (15) |
| 49 | 5m (3-MeO, 4-MeO) | Pd2(dba)3 (10) | 50/50/0 | 21/29 | 6m (25) cis-7m (11), trans-7m (20) |
| 50 | 5m (3-MeO, 4-MeO) | ((IMes)Pd(NQ))2 (2.5) | 32/68/0 | 44/24 | 6m (13) cis-7m (23), trans-7m (30) |
| 51 | 5m (3-MeO, 4-MeO) | (Pd(allyl)Cl)2 (5) | 15/85/0 | 8/77 | 6m (6) trans-7m (39) |
| 52 | 5m (3-MeO, 4-MeO) | (SIPr)Pd(allyl)Cl (15) | 24/76/0 | 43/33 | 6m (9) cis-7m (6), trans-7m (38) |

| Entry | Catalyst (mol%) | Solvent | Temp. | Time | Products (Yield (%)) 2 |
|---|---|---|---|---|---|
| 1 | ((IMes)Pd(NQ))2 (2.5) | DCE | reflux | 24 h | --- |
| 2 | ((IMes)Pd(NQ))2 (2.5) | CH2Cl2 | reflux | 24 h | 11 |
| 3 | (Pd(allyl)Cl)2 (5) | DCE | reflux | 24 h | cis-12/trans-12 (1:10, 30%) |
| 4 | (SIPr)Pd(allyl)Cl (15) | DCE | reflux | 48 h | cis-12/trans-12 (1:2.2, 54%) |
| 5 | (Rh(OAc)2)2 (3) | CH2Cl2 | r.t. | 24 h | 11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solé, D.; Pérez-Janer, F.; Amenta, A.; Bennasar, M.-L.; Fernández, I. Site Selectivity in Pd-Catalyzed Reactions of α-Diazo-α-(methoxycarbonyl)acetamides: Effects of Catalysts and Substrate Substitution in the Synthesis of Oxindoles and β-Lactams. Molecules 2019, 24, 3551. https://doi.org/10.3390/molecules24193551
Solé D, Pérez-Janer F, Amenta A, Bennasar M-L, Fernández I. Site Selectivity in Pd-Catalyzed Reactions of α-Diazo-α-(methoxycarbonyl)acetamides: Effects of Catalysts and Substrate Substitution in the Synthesis of Oxindoles and β-Lactams. Molecules. 2019; 24(19):3551. https://doi.org/10.3390/molecules24193551
Chicago/Turabian StyleSolé, Daniel, Ferran Pérez-Janer, Arianna Amenta, M.-Lluïsa Bennasar, and Israel Fernández. 2019. "Site Selectivity in Pd-Catalyzed Reactions of α-Diazo-α-(methoxycarbonyl)acetamides: Effects of Catalysts and Substrate Substitution in the Synthesis of Oxindoles and β-Lactams" Molecules 24, no. 19: 3551. https://doi.org/10.3390/molecules24193551
APA StyleSolé, D., Pérez-Janer, F., Amenta, A., Bennasar, M.-L., & Fernández, I. (2019). Site Selectivity in Pd-Catalyzed Reactions of α-Diazo-α-(methoxycarbonyl)acetamides: Effects of Catalysts and Substrate Substitution in the Synthesis of Oxindoles and β-Lactams. Molecules, 24(19), 3551. https://doi.org/10.3390/molecules24193551