Chiral Chalcogen Bond Donors Based on the 4,4′-Bipyridine Scaffold


 ,
,  , ,
, , 

Abstract
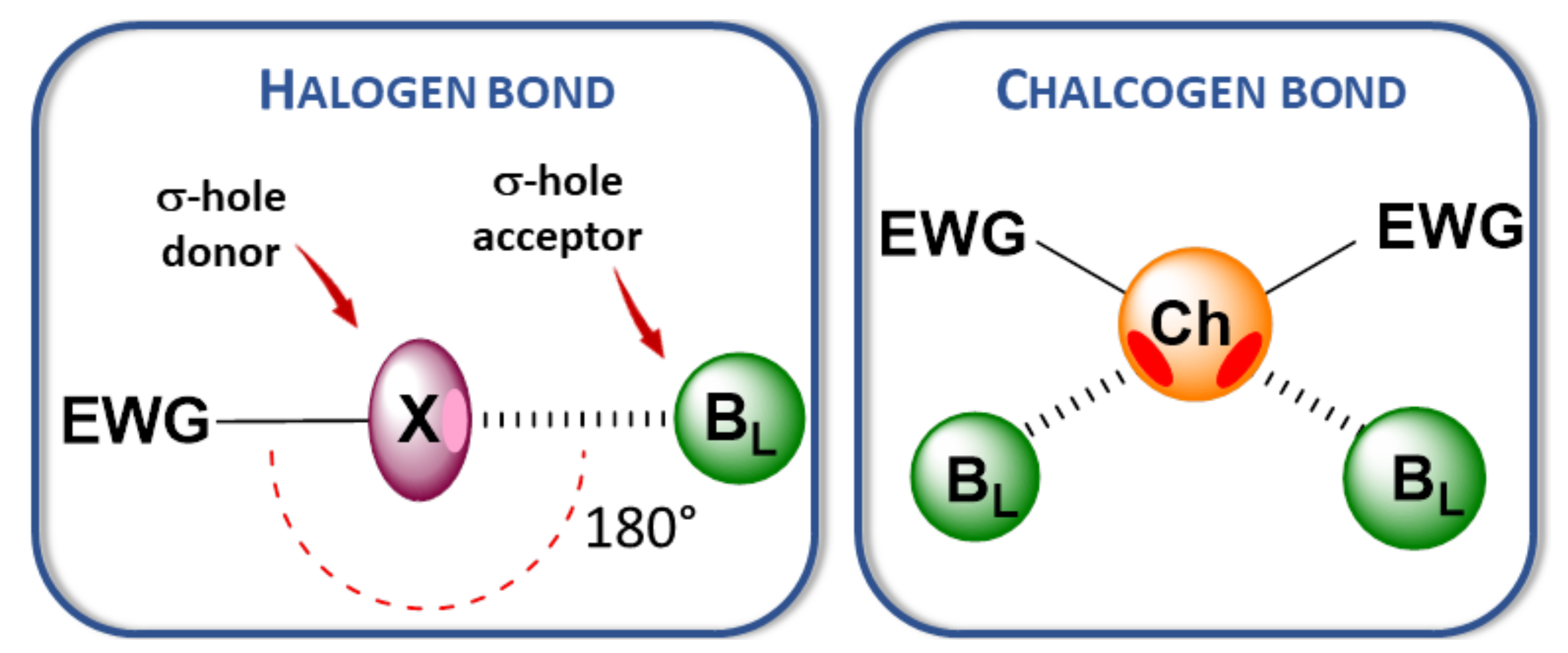
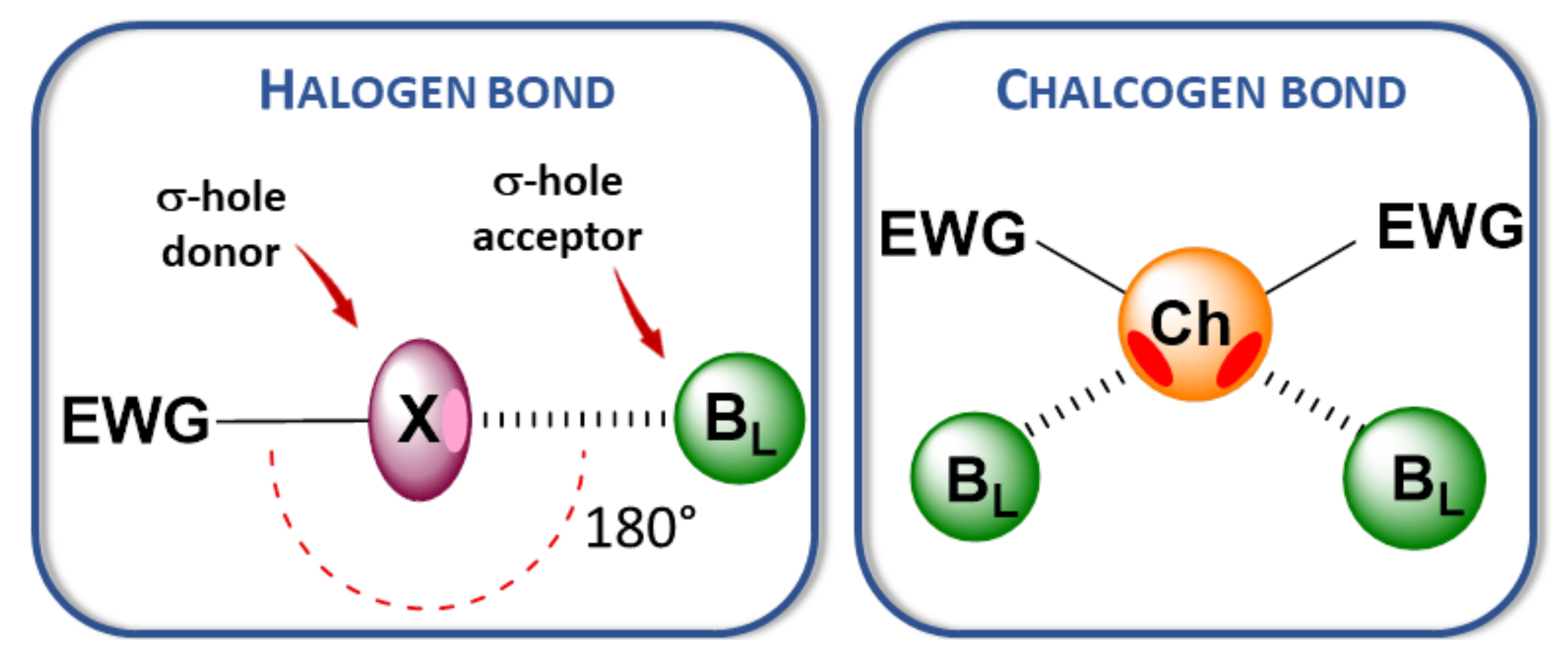
:1. Introduction
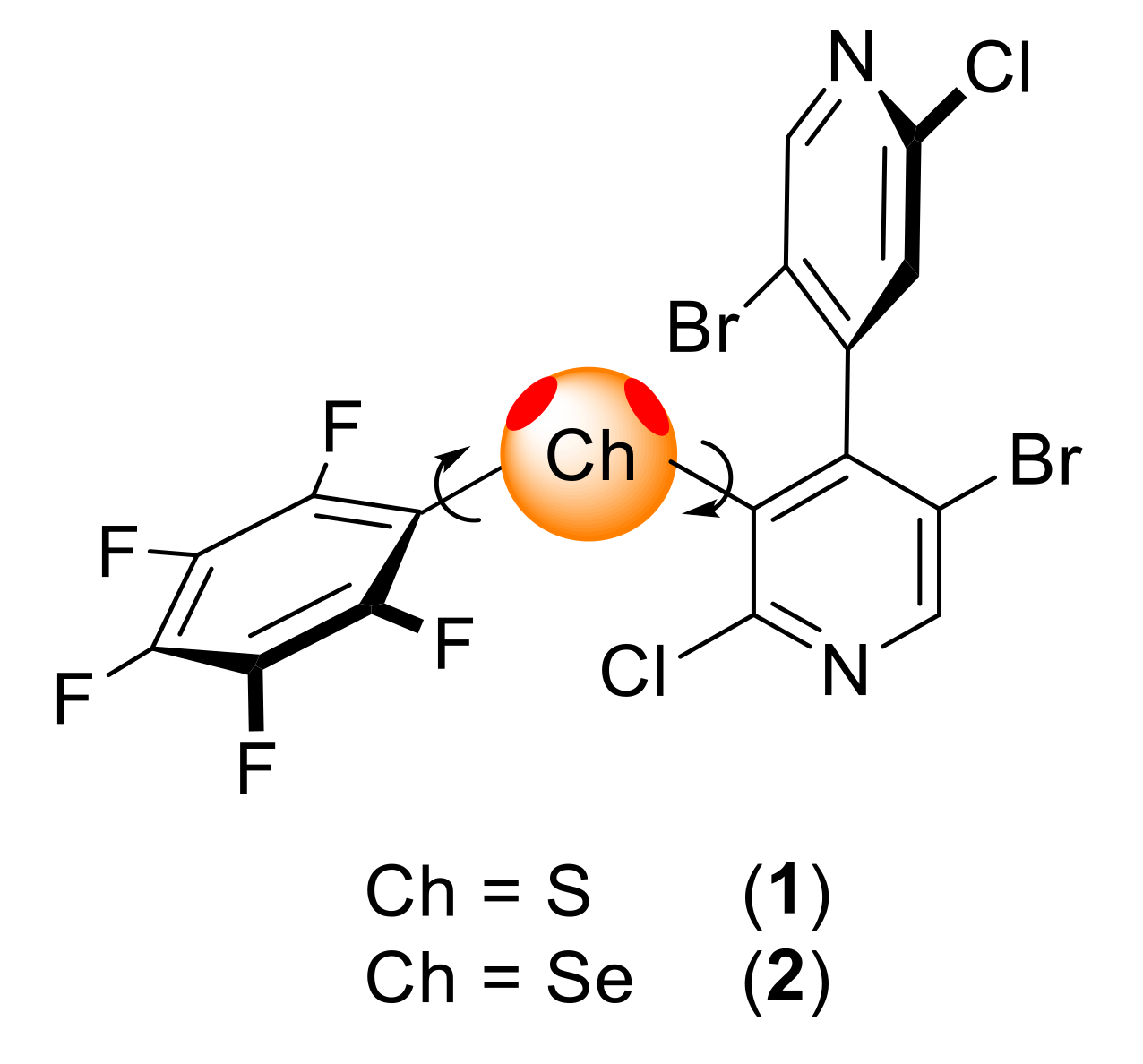
2. Results
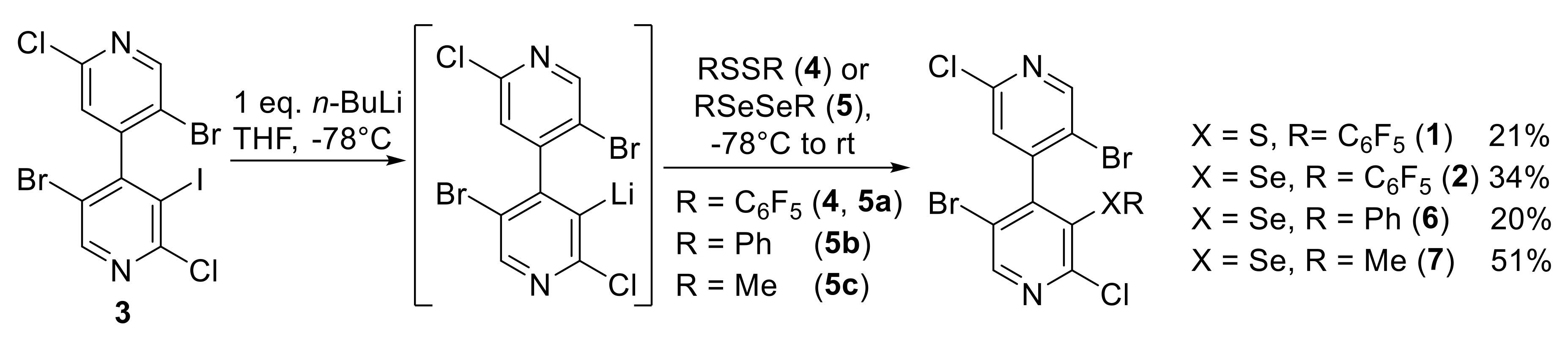
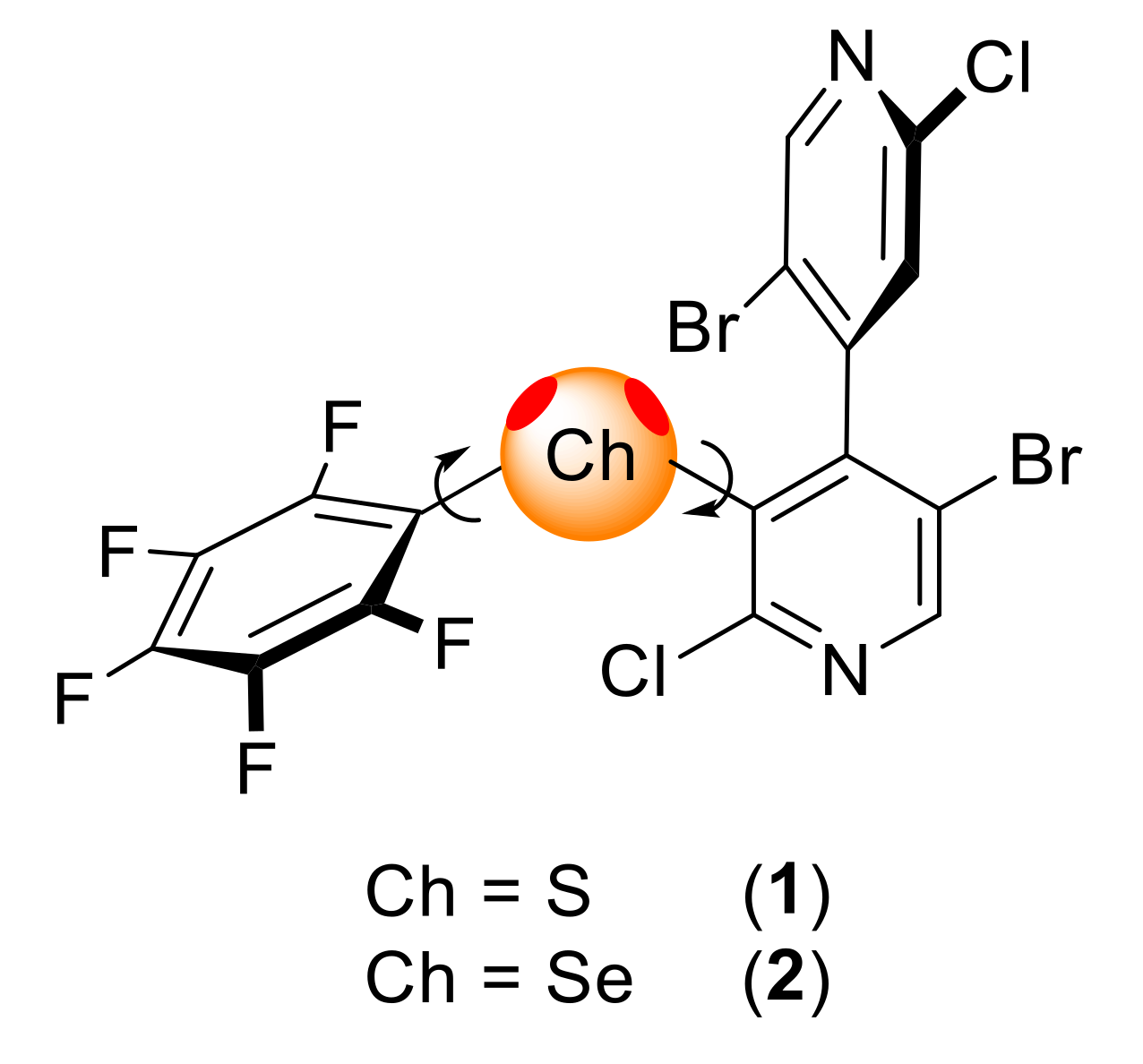
2.1. Synthesis
2.2. Analyses
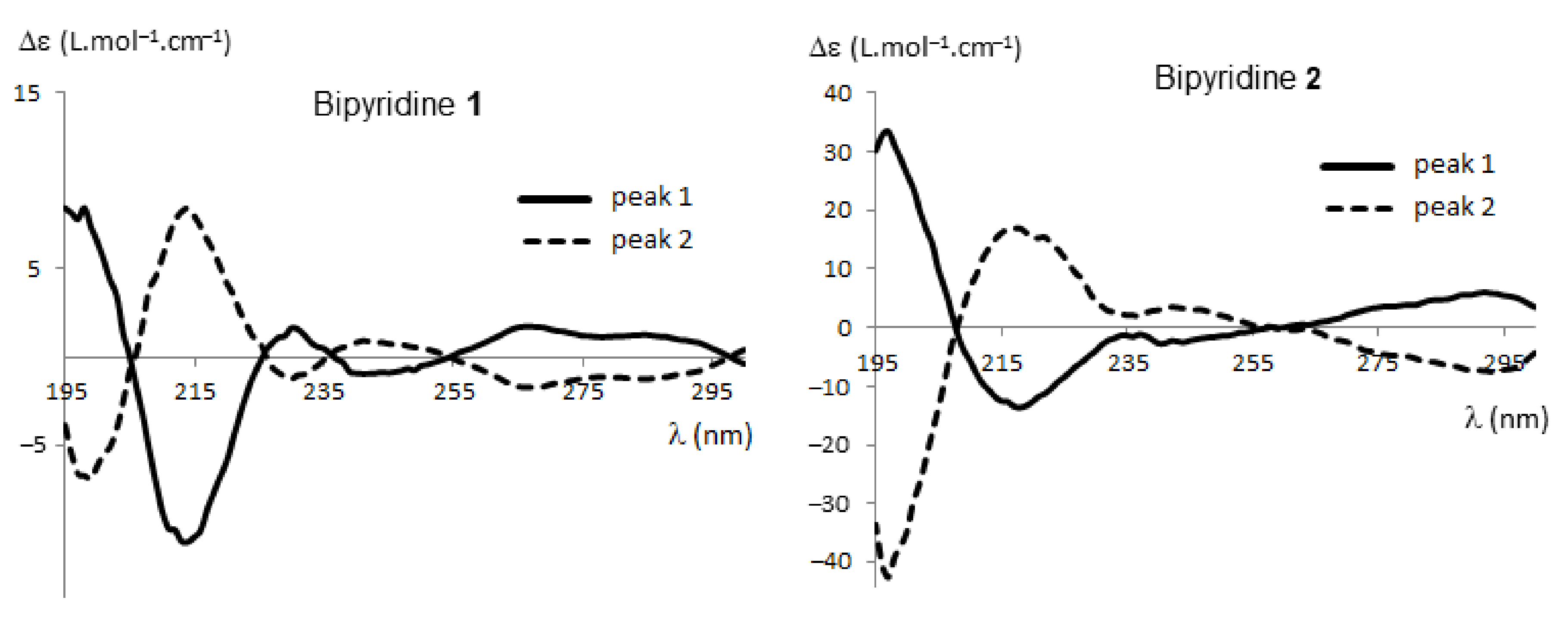
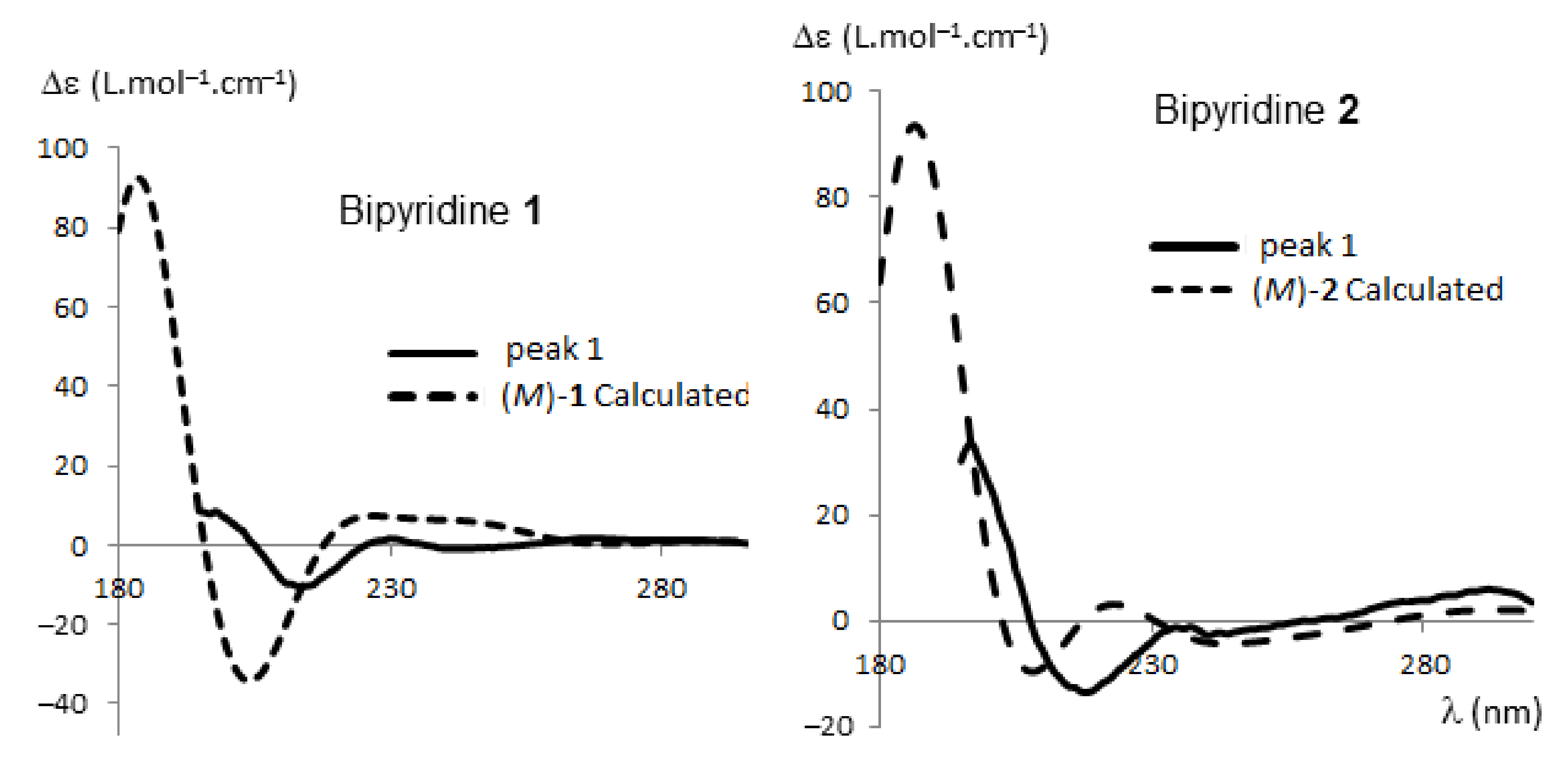
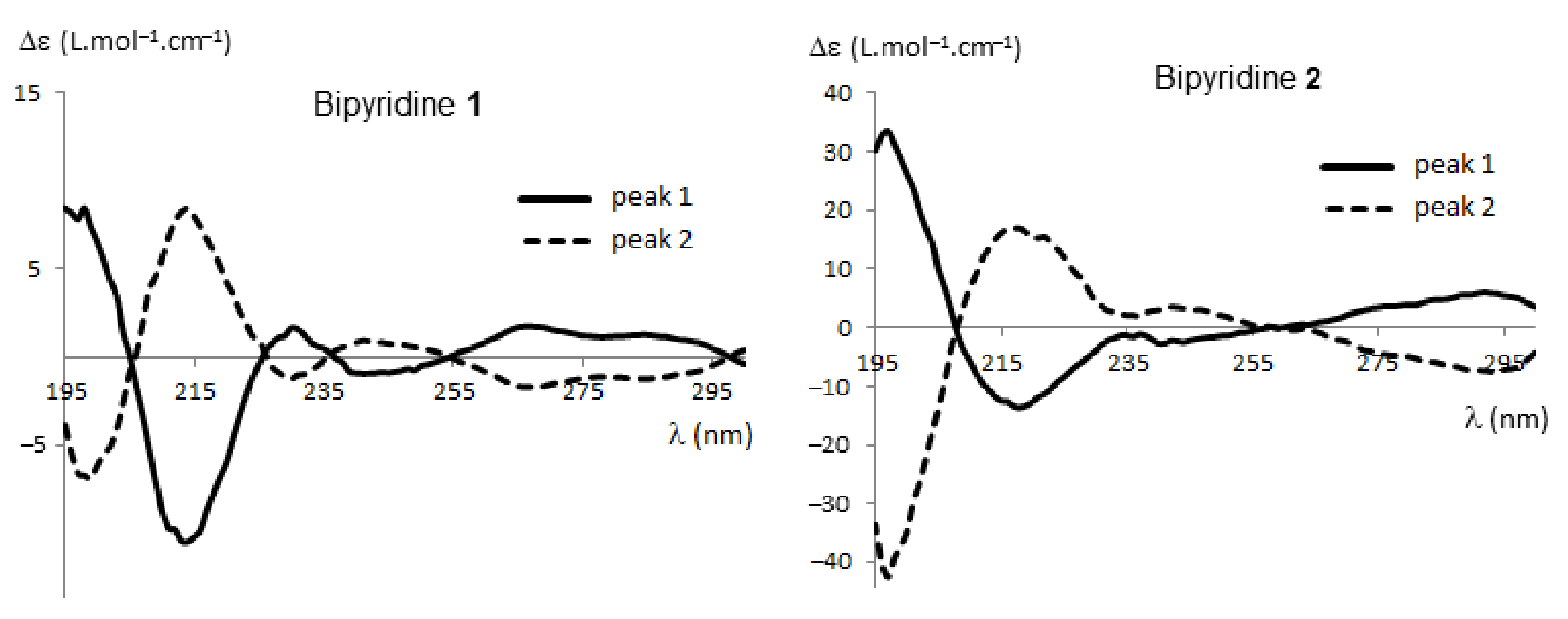
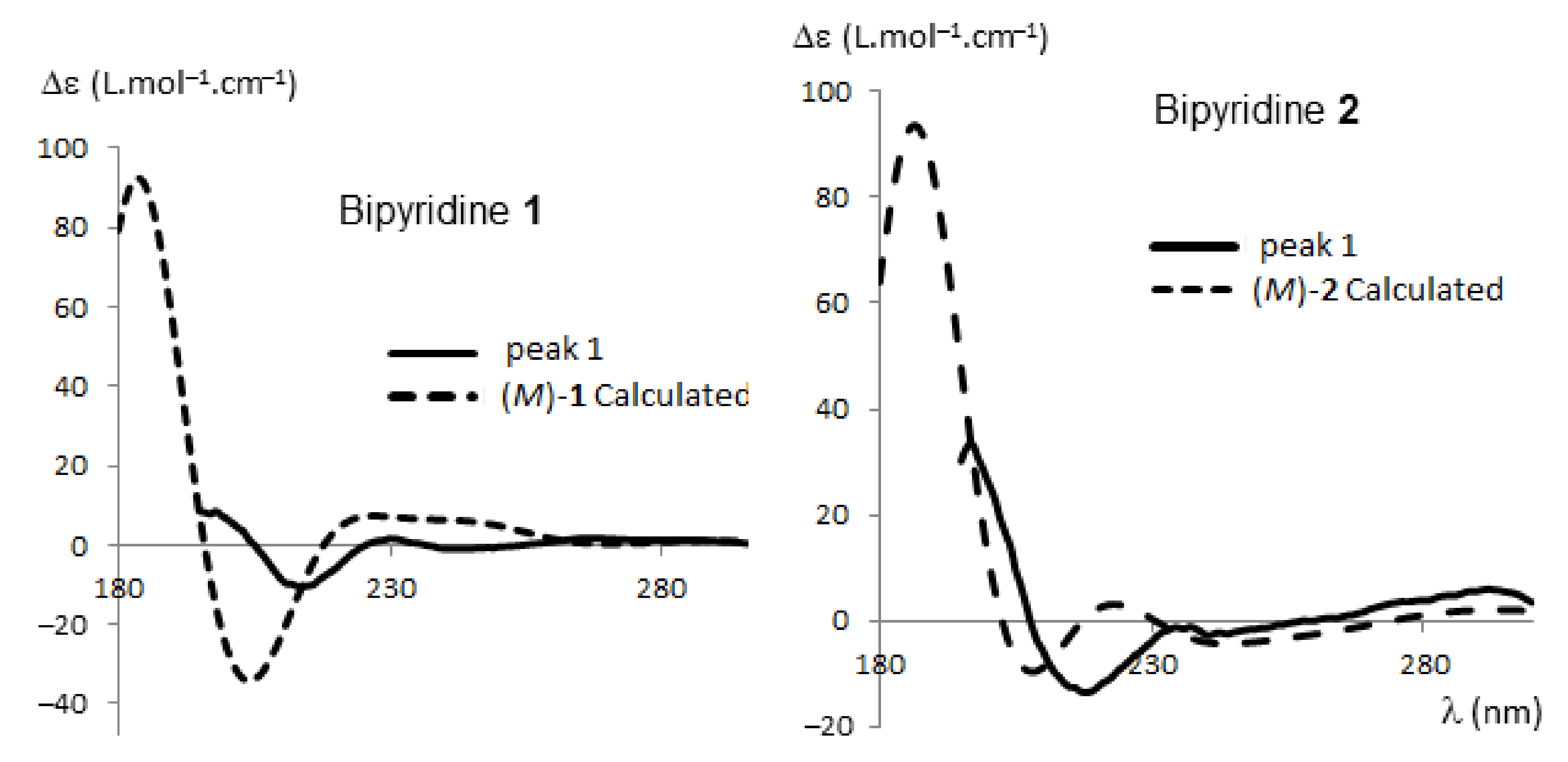
2.2.1. HPLC Enantioseparation and Electronic Circular Dichroism (ECD)
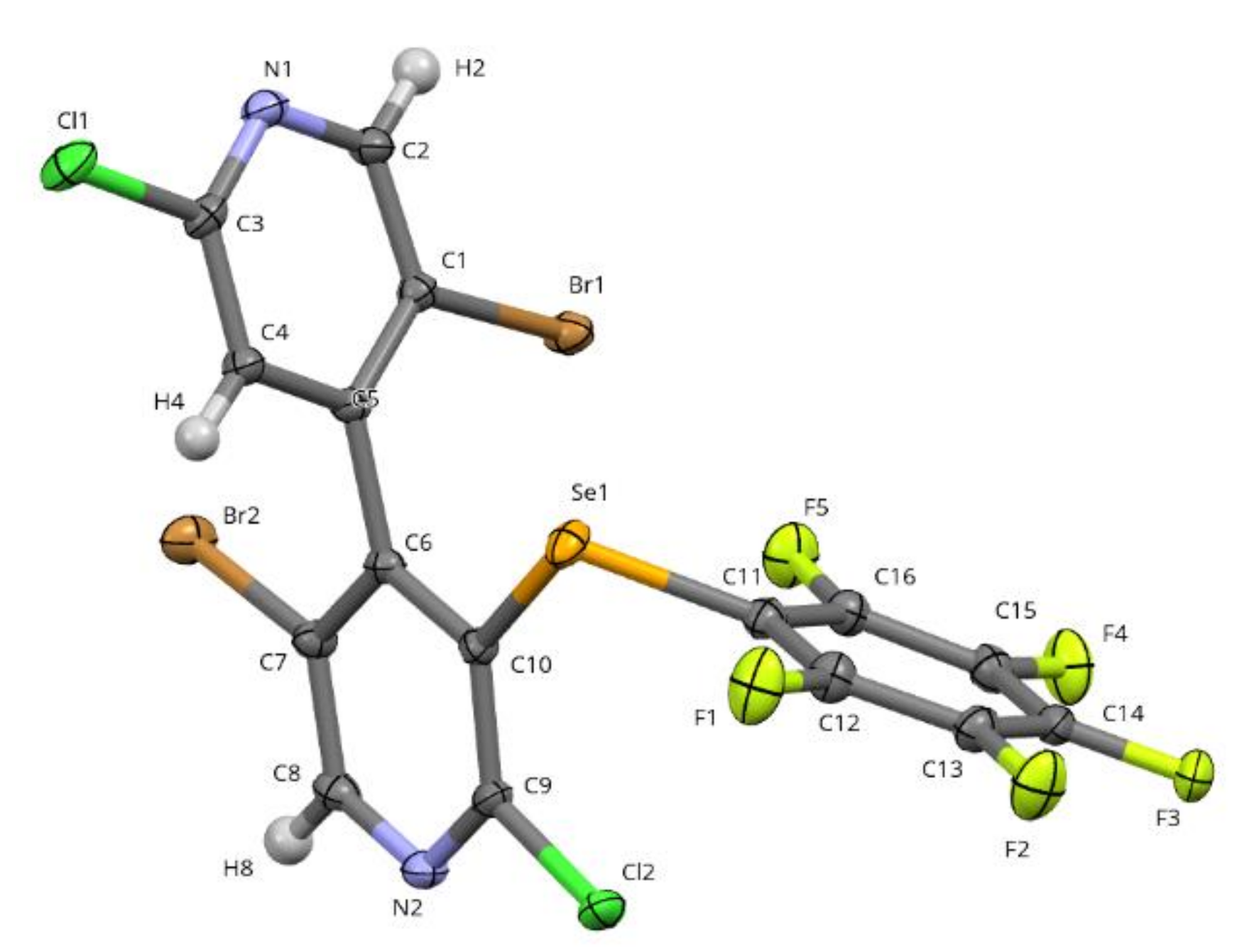
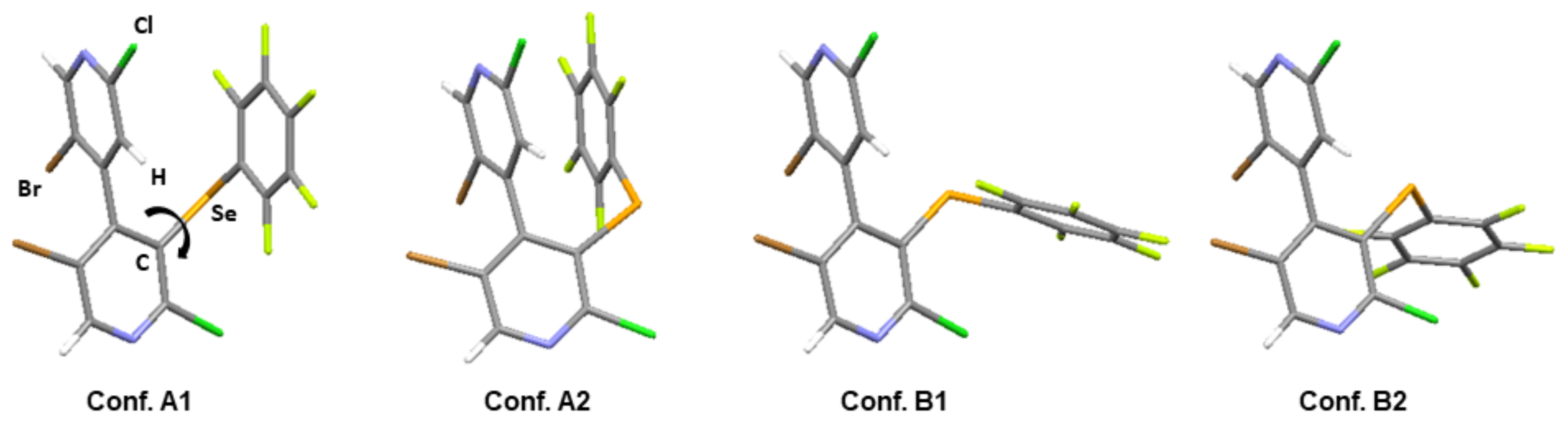
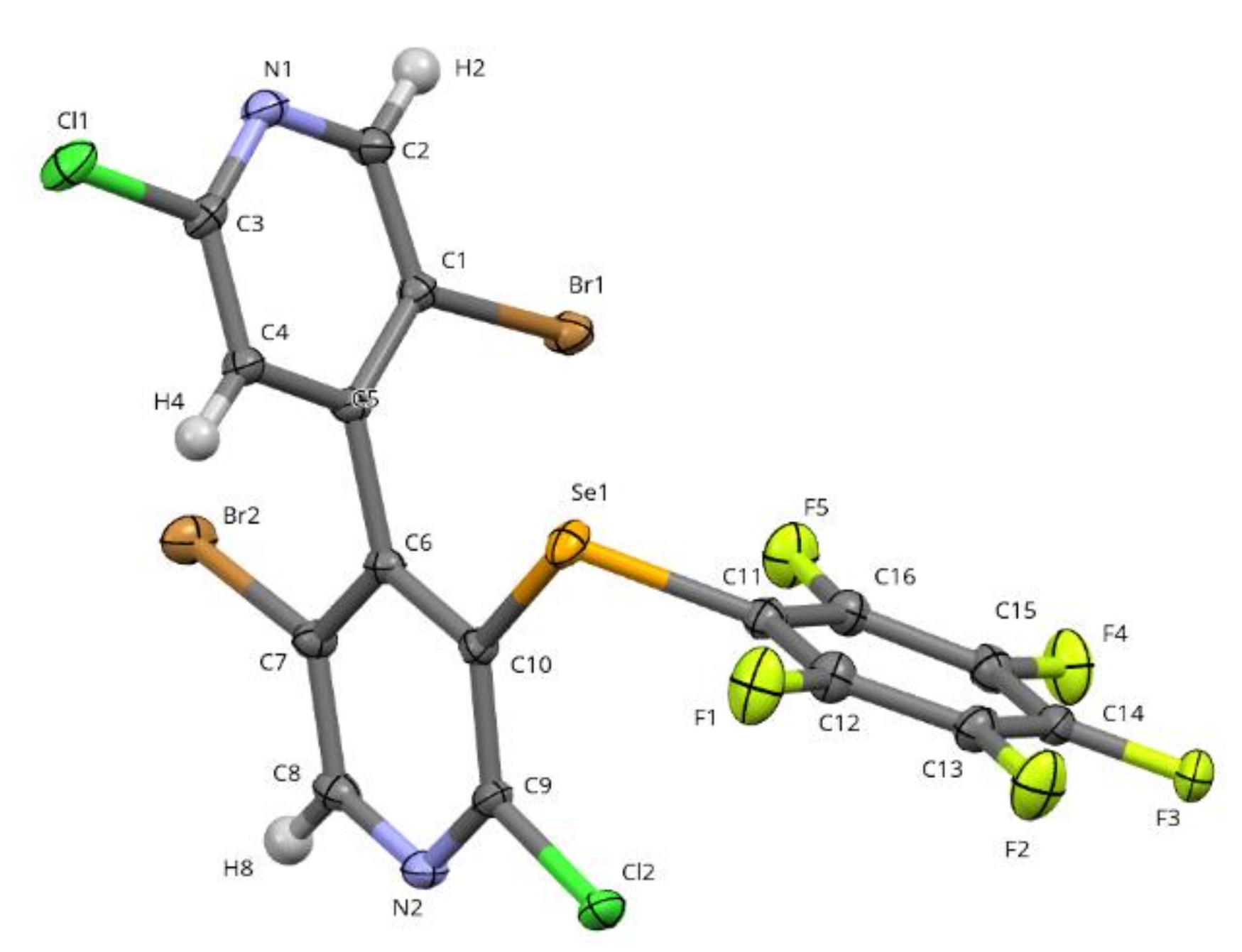
2.2.2. X-Ray Diffraction
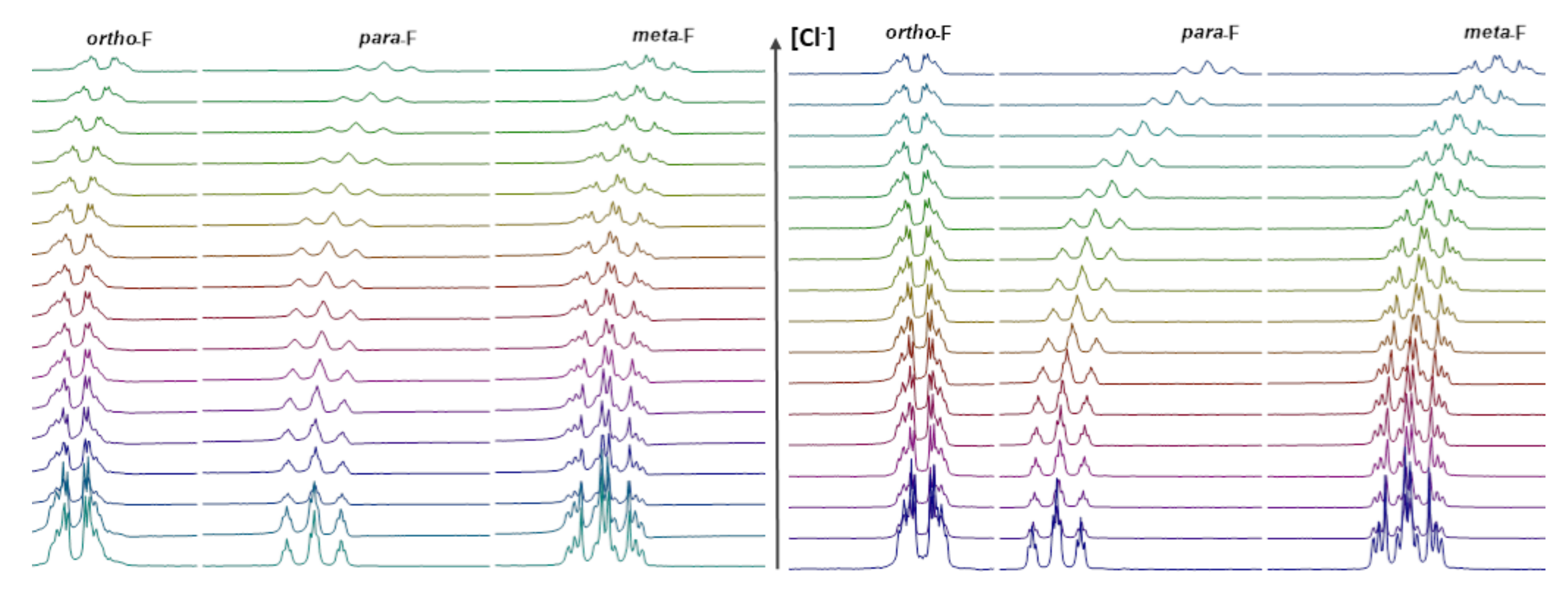
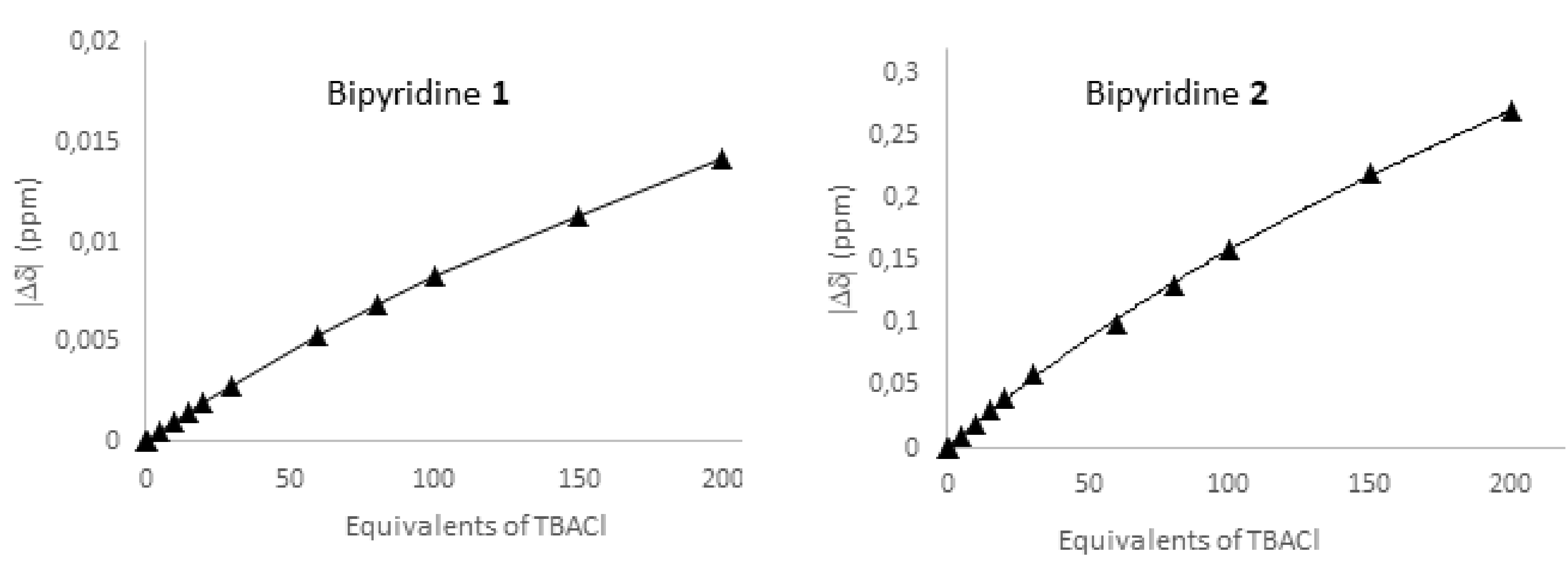
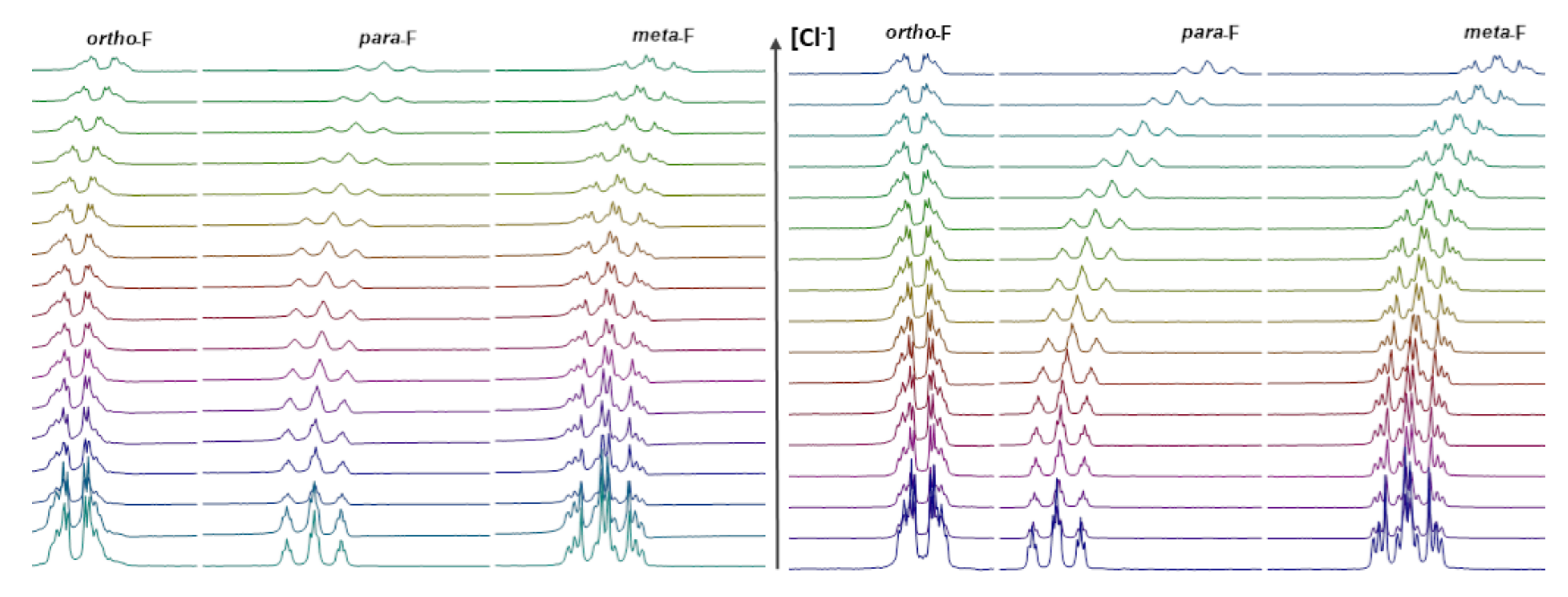
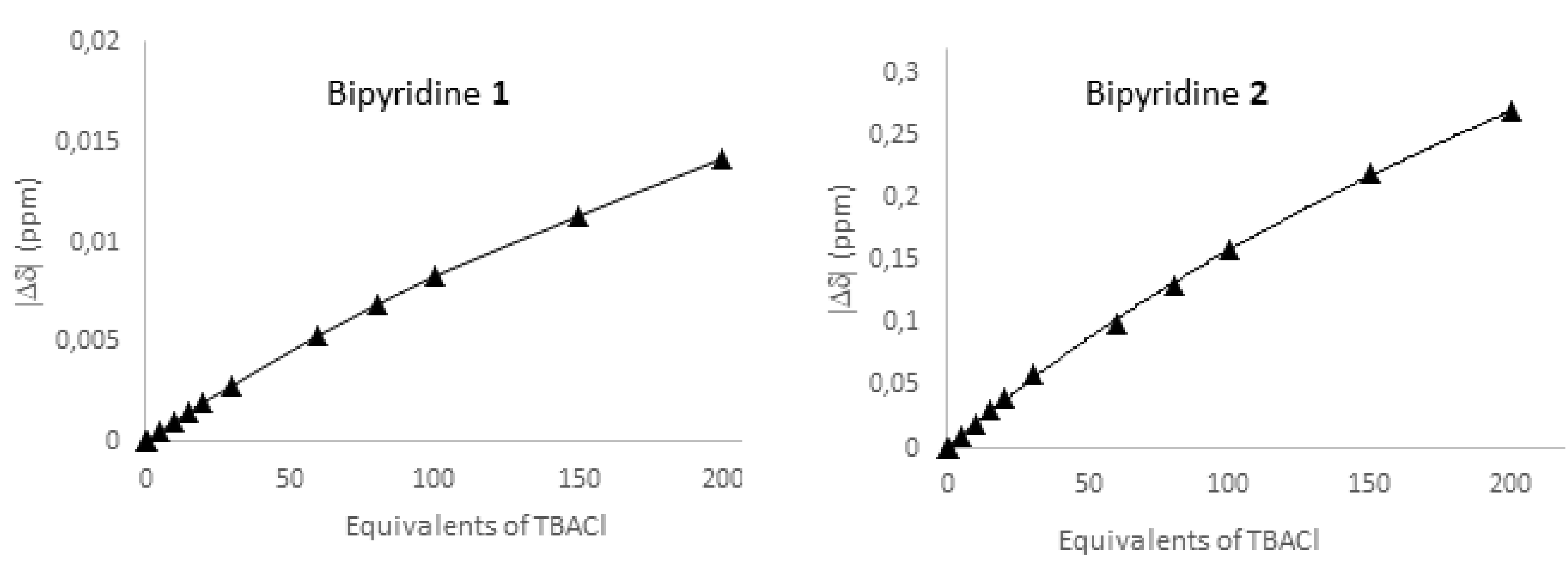
2.2.3. 19F NMR Spectroscopic Titration with Tetrabutylammonium Chloride (TBACl)
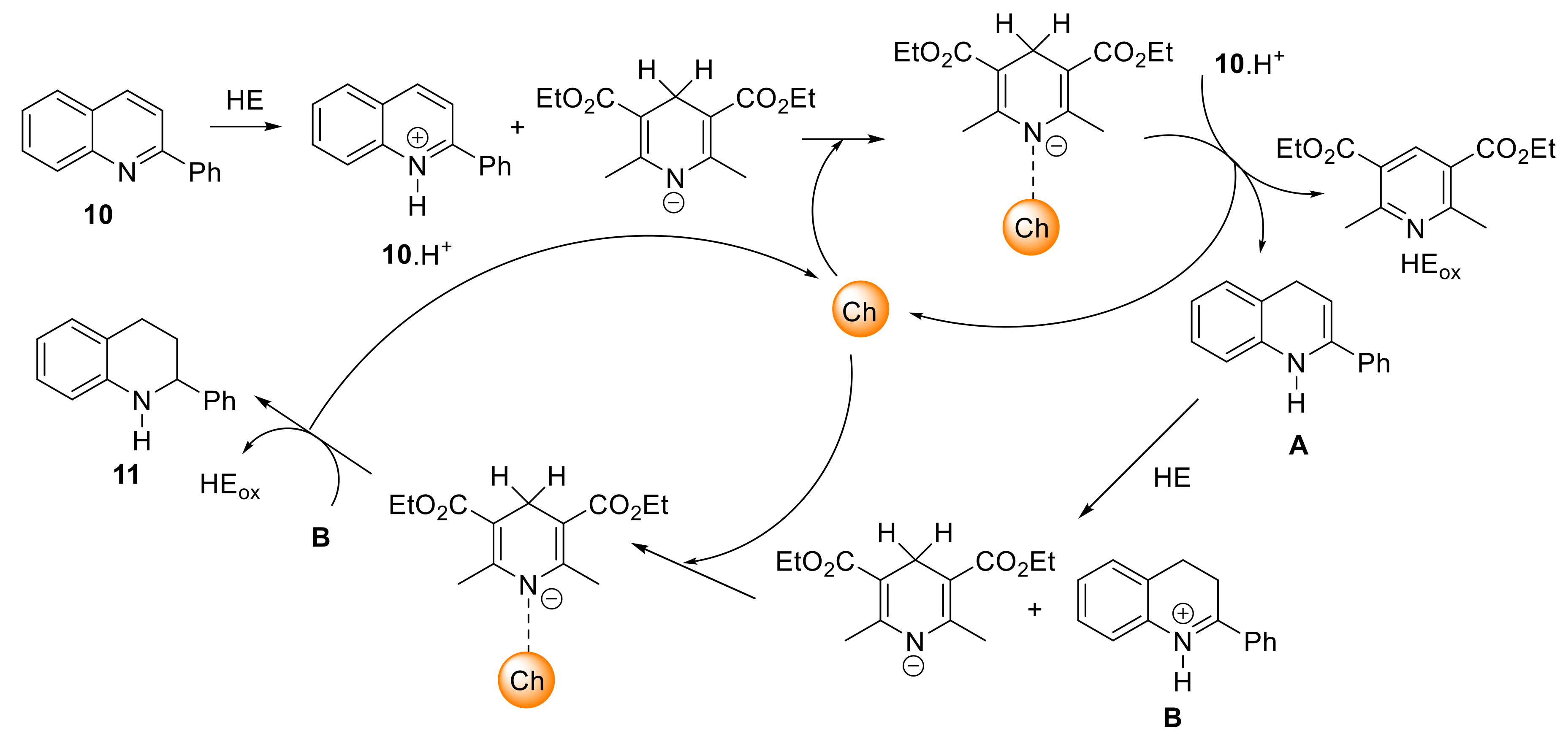
2.3. Catalysis
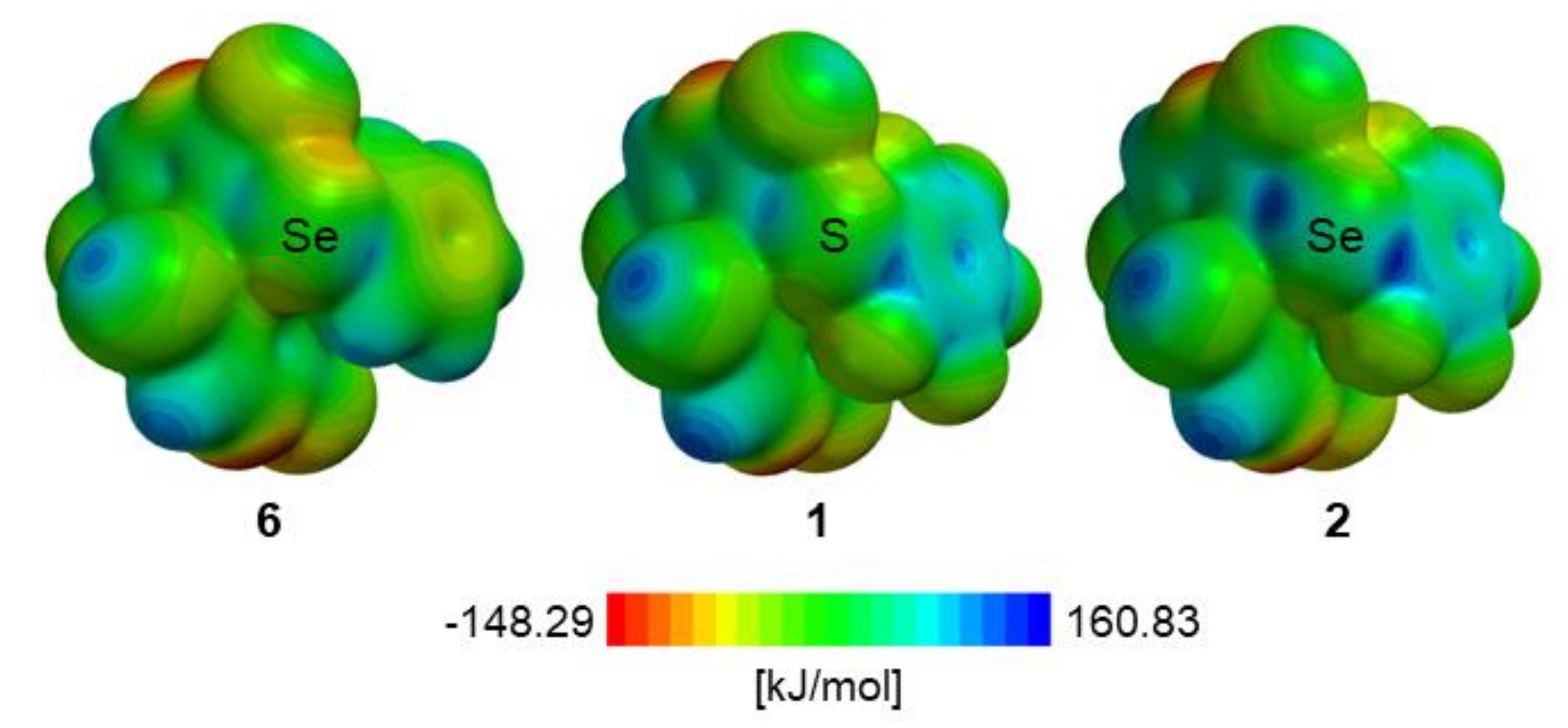
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. General Procedure for the Synthesis of 2, 6, and 7 from Bipyridine 3
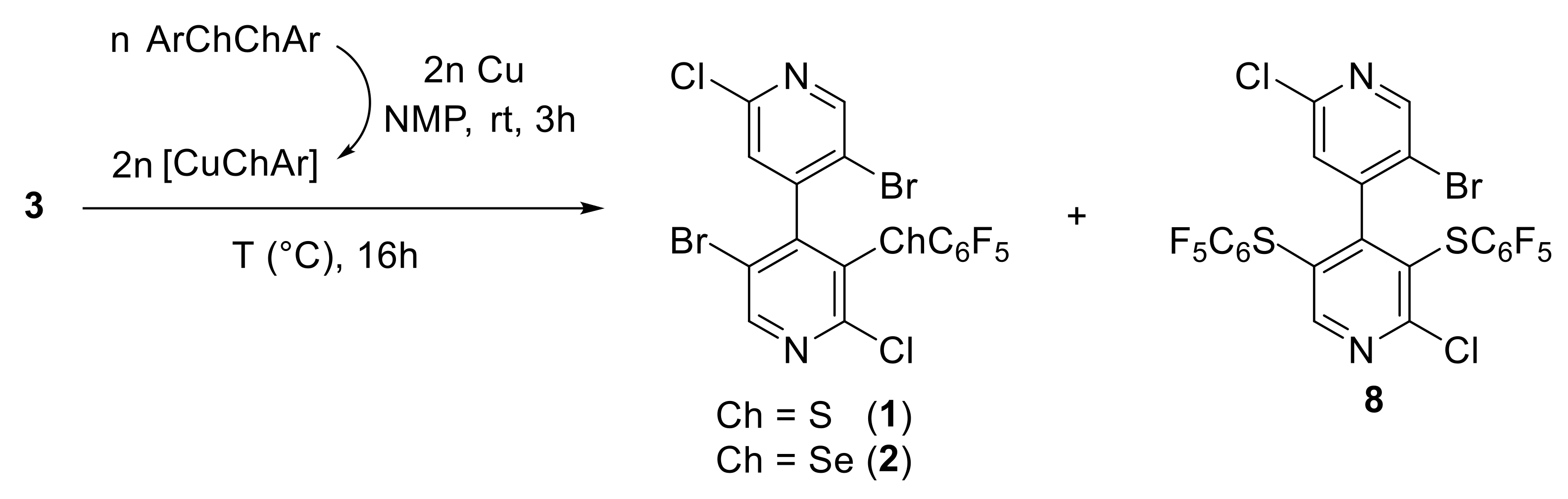
4.3. General Procedure for the Copper-Mediated Synthesis of 1, 2, and 8
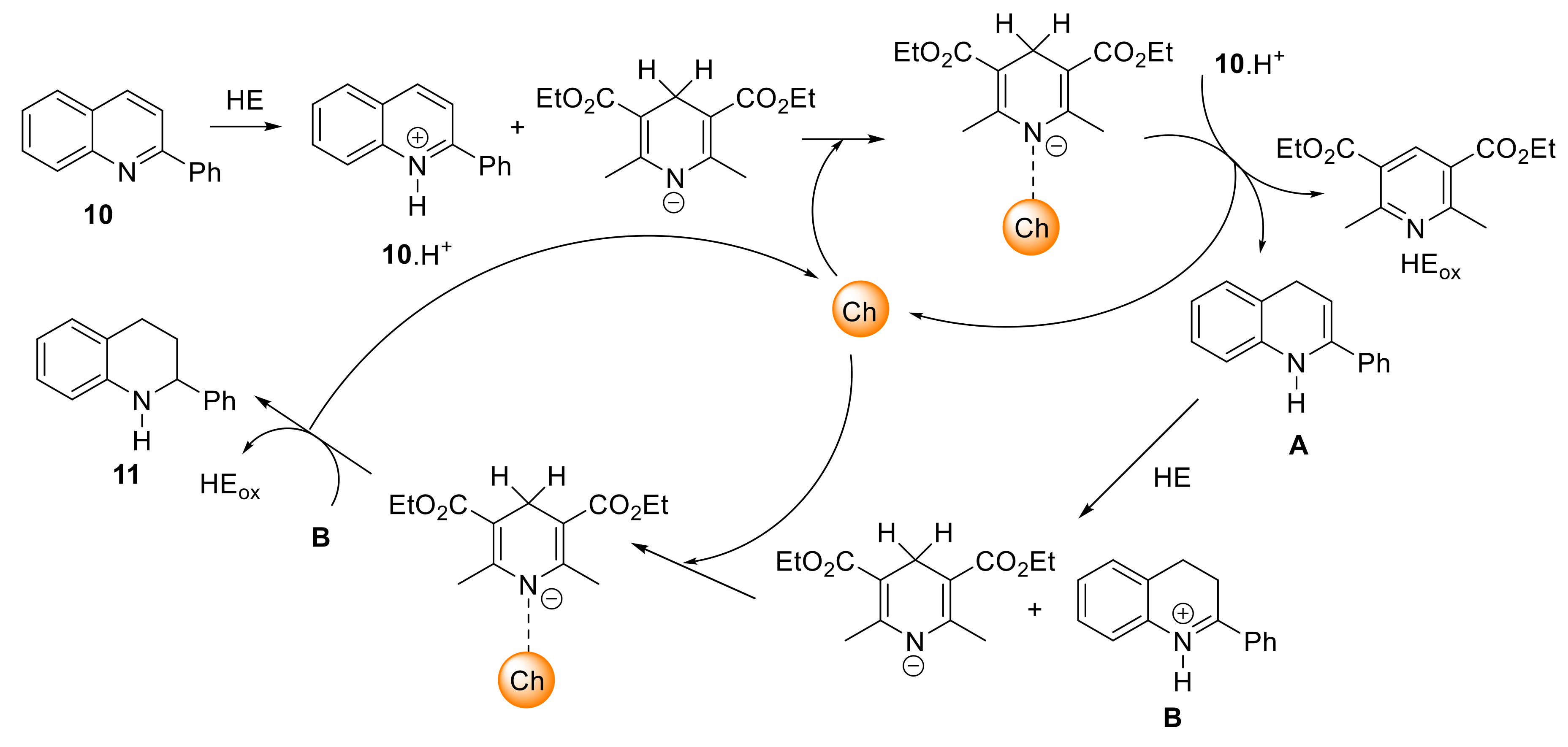
4.4. General Procedure for the Catalytic Reduction of 2-Phenylquinoline 10
4.5. Crystal Data for 2
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen bonding: An overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P. Sigma-hole bonding: Molecules containing group VI atoms. J. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Tepper, R.; Schubert, U.S. Halogen bonding in solution: Anion recognition, templated self-assembly, and organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen bonding: A halogen-centered noncovalent interaction yet to be understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Kolar, M.H.; Hobza, P. Computer modeling of halogen bonds and other σ-hole interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [Green Version]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The chalcogen bond in crystalline solids: A world parallel to halogen bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.C.; Beer, P.D. Sigma-hole interactions in anion recognition. Chem 2018, 4, 731–783. [Google Scholar] [CrossRef]
- Sutar, R.; Huber, S.M. Catalysis of organic reactions through halogen bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Bamberger, J.; Ostler, F.; García Mancheño, O. Frontiers in halogen and chalcogen-bond donor organocatalysis. ChemCatChem 2019, 11. [Google Scholar] [CrossRef]
- Benz, S.; Lopez-Andarias, J.; Mareda, J.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds. Angew. Chem. Int. Ed. 2017, 56, 812–815. [Google Scholar] [CrossRef]
- Benz, S.; Mareda, J.; Besnard, C.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds: Neutral benzodiselenazole scaffolds with high-precision selenium donors of variable strength. Chem. Sci. 2017, 8, 8164–8169. [Google Scholar] [CrossRef] [Green Version]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with pnictogen, chalcogen, and halogen bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Benz, S.; Besnard, C.; Matile, S. Chalcogen-bonding catalysis: From neutral to cationic benzodiselenazole scaffolds. Helv. Chim. Acta 2018, 101, e1800075. [Google Scholar] [CrossRef]
- Wonner, P.; Vogel, L.; Kniep, F.; Huber, S.M. Catalytic carbon–chlorine bond activation by selenium-based chalcogen bond donors. Chem. Eur. J. 2017, 23, 16972–16975. [Google Scholar] [CrossRef]
- Wonner, P.; Vogel, L.; Düser, M.; Gomes, L.; Kniep, F.; Mallick, B.; Werz, D.B.; Huber, S.M. Carbon–halogen bond activation by selenium-based chalcogen bonding. Angew. Chem. Int. Ed. 2017, 56, 12009–12012. [Google Scholar] [CrossRef]
- Wonner, P.; Dreger, A.; Engelage, E.; Huber, S.M. Chalcogen bonding catalysis in a nitro-michael reaction. Angew. Chem. Int. Ed. 2019, 58, 16923–16927. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhu, H.; Liu, S.; Zhao, Z.; Zhang, L.; Hao, J.; Wang, Y. Chalcogen-chalcogen bonding catalysis enables assembly of discrete molecules. J. Am. Chem. Soc. 2019, 141, 9175–9179. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.C.; Liew, J.Y.; Beer, P.D. Thermodynamics of anion binding by chalcogen bonding receptors. Chem. Eur. J. 2018, 24, 14560–14566. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Marques, I.; Félix, V.; Beer, P.D. Chiral halogen and chalcogen bonding receptors for discrimination of stereo- and geometric dicarboxylate isomers in aqueous media. Chem. Commun. 2018, 54, 10851–10854. [Google Scholar] [CrossRef]
- Benz, S.; Macchione, M.; Verolet, Q.; Mareda, J.; Sakai, N.; Matile, S. Anion transport with chalcogen bonds. J. Am. Chem. Soc. 2016, 138, 9093–9096. [Google Scholar] [CrossRef] [Green Version]
- Macchione, M.; Tsemperouli, M.; Goujon, A.; Mallia, A.R.; Sakai, N.; Sugihara, K.; Matile, S. Mechanosensitive oligodithienothiophenes: transmembrane anion transport along chalcogen-bonding cascades. Helv. Chim. Acta 2018, 101, e1800014. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.M.; Tsemperouli, M.; Poblador-Bahamonde, A.I.; Benz, S.; Sakai, N.; Sugihara, K.; Matile, S. Anion transport with pnictogen bonds in direct comparison with chalcogen and halogen bonds. J. Am. Chem. Soc. 2019, 141, 810–814. [Google Scholar] [CrossRef]
- Knowles, R.R.; Jacobsen, E.N. Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. USA 2010, 107, 20678–20685. [Google Scholar] [CrossRef] [Green Version]
- By Liu, M.; Zhang, L.; Wang, T. Supramolecular chirality in self-assembled systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Cossu, S. Liquid chromatography enantioseparations of halogenated compounds on polysaccharide-based chiral stationary phases: Role of halogen substituents in molecular recognition. Chirality 2015, 27, 667–684. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Aubert, E.; Cossu, S. Insights into the impact of shape and electronic properties on the enantioseparation of polyhalogenated 4,4’-bipyridines on polysaccharide-type selectors. Evidence for stereoselective halogen bonding interactions. J. Chromatogr. A 2014, 1345, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Mamane, V.; Aubert, E.; Dessi, A.; Dallocchio, R.; Dore, A.; Pale, P.; Cossu, S. Insights into halogen bond driven enantioseparations. J. Chromatogr. A 2016, 1467, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Mamane, V.; Aubert, E.; Cossu, S. Recent trends and applications in liquid-phase chromatography enantioseparation of atropisomers. Electrophoresis 2017, 38, 1830–1850. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Mamane, V.; Dallocchio, R.; Dessi, A.; Villano, R.; Sanna, D.; Aubert, E.; Pale, P.; Cossu, S. Polysaccharide-based chiral stationary phases as halogen bond acceptors: A novel strategy for detection of stereoselective σ-hole bonds in solution. J. Sep. Sci. 2018, 41, 1247–1256. [Google Scholar] [CrossRef]
- Dallocchio, R.; Dessi, A.; Solinas, M.; Arras, A.; Cossu, S.; Aubert, E.; Mamane, V.; Peluso, P. Halogen bond in high-performance liquid chromatography enantioseparations: Description, features and modelling. J. Chromatogr. A 2018, 1563, 71–81. [Google Scholar] [CrossRef]
- Peluso, P.; Dessi, A.; Dallocchio, R.; Mamane, V.; Cossu, S. Recent studies of docking and molecular dynamics simulation for liquid-phase enantioseparations. Electrophoresis 2019, 40, 1881–1896. [Google Scholar] [CrossRef]
- Peluso, P.; Gatti, C.; Dessi, A.; Dallocchio, R.; Weiss, R.; Aubert, E.; Pale, P.; Cossu, S.; Mamane, V. Enantioseparation of fluorinated 3-arylthio-4,4’-bipyridines: insights into chalcogen and π-hole bonds in high-performance liquid chromatography. J. Chromatogr. A 2018, 1567, 119–129. [Google Scholar] [CrossRef]
- Mamane, V.; Aubert, E.; Peluso, P.; Cossu, S. Lithiation of prochiral 2,2’-dichloro-5,5’-dibromo-4,4’-bipyridine as a tool for the synthesis of chiral polyhalogenated 4,4’-bipyridines. J. Org. Chem. 2013, 78, 7683–7689. [Google Scholar] [CrossRef]
- Mamane, V.; Aubert, E.; Peluso, P.; Cossu, S. Synthesis, resolution, and absolute configuration of chiral 4,4′-bipyridines. J. Org. Chem. 2012, 77, 2579–2583. [Google Scholar] [CrossRef]
- Kondratenko, N.V.; Kolomeytsev, A.A.; Popov, V.I.; Yagupolskii, L.M. Synthesis and reactions of trifluoromethylthio(seleno)- and pentafluorophenylthio(seleno)-copper. Synthesis 1985, 667–669. [Google Scholar] [CrossRef]
- Haupt, A.; Lentz, D. Tuning the electron affinity and stacking properties of corannulene by introduction of fluorinated thioethers. Chem. Asian J. 2018, 13, 3022–3026. [Google Scholar] [CrossRef] [PubMed]
- Mamane, V.; Peluso, P.; Aubert, E.; Cossu, S.; Pale, P. Chiral hexahalogenated 4,4′-bipyridines. J. Org. Chem. 2016, 81, 4576–4587. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.Q.; Zhao, X.R.; Wang, H.; Jin, W.J. The Competition of σ-Hole·Cl− and π-Hole·Cl− bonds between C6F5X (X = F, Cl, Br, I) and the chloride anion and its potential application in separation science. J. Phys. Chem. B 2014, 118, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, S.-L. Transfer hydrogenation with Hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Antonchick, A.P.; Theissmann, T. A Highly enantioselective Brønsted acid catalyzed cascade reaction: Organocatalytic transfer hydrogenation of quinolines and their application in the synthesis of alkaloids. Angew. Chem. Int. Ed. 2006, 45, 3683–3686. [Google Scholar] [CrossRef]
- Bruckmann, A.; Pena, M.A.; Bolm, C. Organocatalysis through halogen-bond activation. Synlett 2008, 6, 900–902. [Google Scholar] [CrossRef]
- He, W.; Ge, Y.-C.; Tan, C.-H. Halogen-bonding-induced hydrogen transfer to C = N bond with Hantzsch ester. Org. Lett. 2014, 16, 3244–3247. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Uno, H.; Tokunaga, E.; Shibata, N. Fluorobissulfonylmethyl iodides: An efficient scaffold for halogen bonding catalysts with an sp3-hybridized carbon–iodine moiety. ACS Catal. 2018, 8, 6601–6605. [Google Scholar] [CrossRef]
- Nayak, S.K.; Kumar, V.; Murray, J.S.; Politzer, P.; Terraneo, G.; Pilati, T.; Metrangolo, P.; Resnati, G. Fluorination promotes chalcogen bonding in crystalline solids. CrystEngComm 2017, 19, 4955–4959. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Ser, C.T.; Yang, H.; Wong, M.W. Iodoimidazolinium-catalyzed reduction of quinoline by Hantzsch ester: Halogen bond or Brønsted acid catalysis. J. Org. Chem. 2019, 84, 10338–10348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapötke, T.M.; Krumm, B.; Polborn, K. Synthesis, chemistry, and characterization of perfluoroaromatic selenium derivatives. Eur. J. Inorg. Chem. 1999, 8, 1359–1366. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1, 2, 6 and 7 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | ArChChAr | n (eq.) | T (°C) | NMR Ratio 3/1/8 or 3/2 | Yield (%) |
|---|---|---|---|---|---|
| 1 | (C6F5S)2 (4) | 1.3 | 80 | 33/67/0 | − |
| 2 | ‘’ | 1.3 | 100 | 12/77/11 | 74 (1) |
| 3 | ‘’ | 1.3 | 130 | 4/31/65 | 39 (8) |
| 4 | ‘’ | 0.5 | 100 | 78/20/2 | − |
| 5 | ‘’ | 0.8 | 100 | 53/43/4 | − |
| 6 | ‘’ | 1.0 | 100 | 39/57/4 | − |
| 7 | ‘’ | 2.0 | 100 | 0/75/25 | 67 (1) |
| 8 | ‘’ | 4.0 | 100 | 0/54/46 | |
| 9 | ‘’ | 0.5 | 130 | 49/42/9 | − |
| 10a | ‘’ | 1.3 | 100 | 17/63/20 | − |
| 11 | (C6F5Se)2 (5a) | 1.3 | 100 | 14/86 | 81 (2) |
| Bipyridine | Racemate [mg] | Absolute Configuration (e.e.b %) | Recovered Amounts [mg] (Recovery%) | ||
|---|---|---|---|---|---|
| 1st Eluted Peak | 2nd Eluted Peak | 1st Eluted Peak | 2nd Eluted Peak | ||
| 2 | 30.0 | >99 | 98.8 | 10.6 (70.7) | 13.0 (86.7) |
| 6 | 40.0 | >99 | 95.0 | 7.4 (37.0) | 18.0 (90.0) |
| 7 | 12.0 | >99 | 95.1 | 5.1 (85.0) | 5.9 (98.3) |
| 1 | 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| A1 | A2 | B1 | B2 | A1 | A2 | B1 | B2 | |
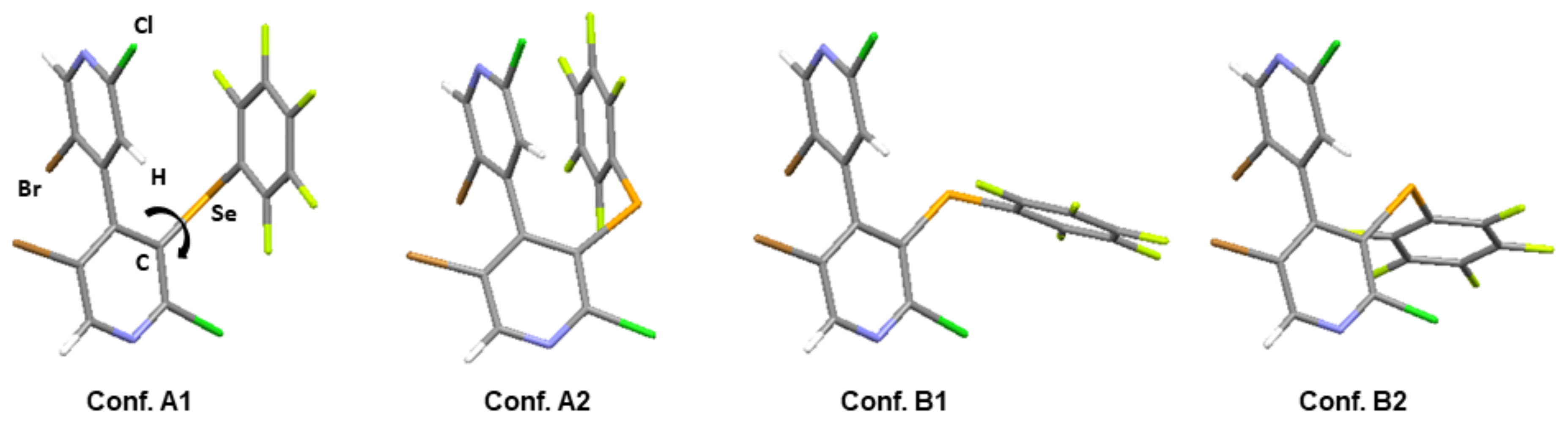
| ΔG (kJ/mol) | 0.66 | 0 | 0.74 | 8.09 | 0 | 3.17 | 0.96 | 7.53 |
| Pop. (%) | 30 | 39 | 29 | 2 | 50 | 14 | 34 | 2 |
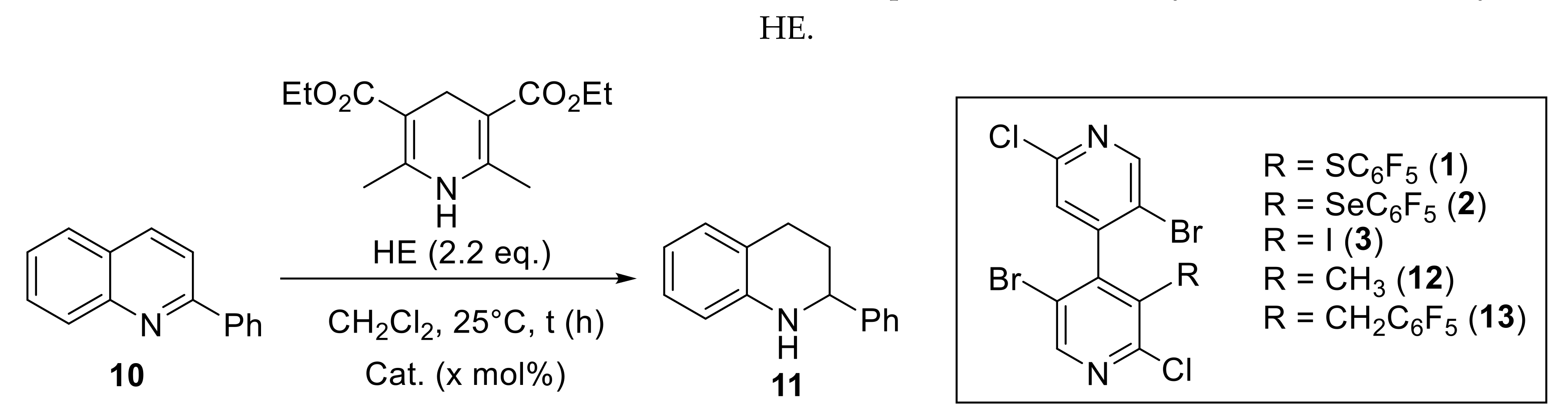
| Entry | Cat. | x (mol%) | Time (h) | Conv. (%)a | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1 | 20 | 48 | 100 | 87 |
| 2 | 2 | 20 | 48 | 100 | 94 |
| 3 | 1 | 5 | 72 | 81 | − |
| 4 | 2 | 5 | 72 | 100 | 88 |
| 5 | 2 | 5 | 18 | 54 | − |
| 6 | - | - | 72 | 0 | − |
| 7b | 2 | 5 | 18 | 38 | − |
| 8 | 12 | 20 | 72 | 62 | − |
| 9 | 13 | 20 | 72 | 78 | − |
| 10 | 3 | 20 | 48 | 68 | − |
| 11 | 3 | 20 | 72 | 100 | 86 |
| 12 | (P)-2 | 20 | 48 | 100 | 90 (0% ee) |
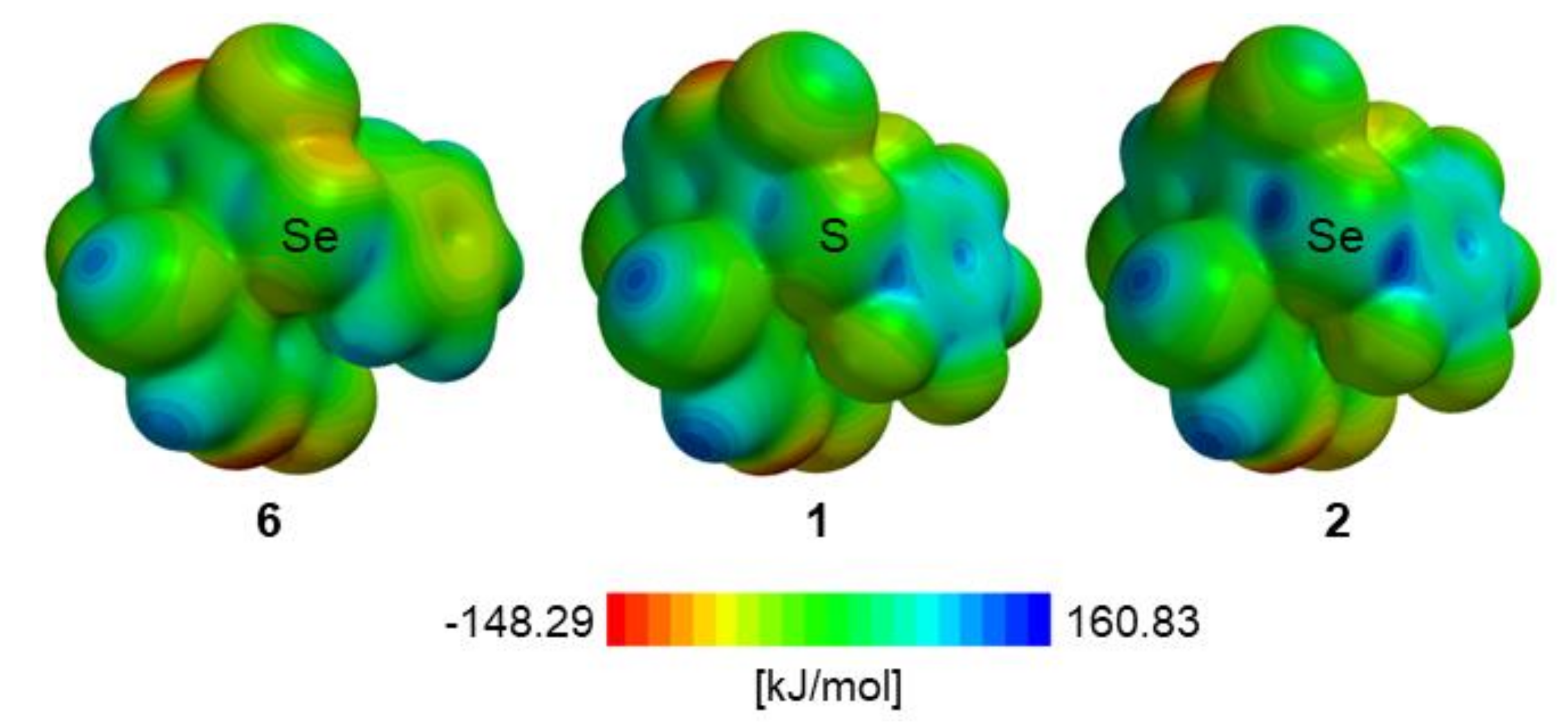
| σ-hole b | 6 | 1 | 2 |
| 1 | 88.0 | 113.2 | 136.7 |
| 2 | 73.6 | 98.6 | 127.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, R.; Aubert, E.; Peluso, P.; Cossu, S.; Pale, P.; Mamane, V. Chiral Chalcogen Bond Donors Based on the 4,4′-Bipyridine Scaffold. Molecules 2019, 24, 4484. https://doi.org/10.3390/molecules24244484
Weiss R, Aubert E, Peluso P, Cossu S, Pale P, Mamane V. Chiral Chalcogen Bond Donors Based on the 4,4′-Bipyridine Scaffold. Molecules. 2019; 24(24):4484. https://doi.org/10.3390/molecules24244484
Chicago/Turabian StyleWeiss, Robin, Emmanuel Aubert, Paola Peluso, Sergio Cossu, Patrick Pale, and Victor Mamane. 2019. "Chiral Chalcogen Bond Donors Based on the 4,4′-Bipyridine Scaffold" Molecules 24, no. 24: 4484. https://doi.org/10.3390/molecules24244484
APA StyleWeiss, R., Aubert, E., Peluso, P., Cossu, S., Pale, P., & Mamane, V. (2019). Chiral Chalcogen Bond Donors Based on the 4,4′-Bipyridine Scaffold. Molecules, 24(24), 4484. https://doi.org/10.3390/molecules24244484