Towards Covalent Photosensitizer-Polyoxometalate Dyads-Bipyridyl-Functionalized Polyoxometalates and Their Transition Metal Complexes

 ,
, 
Abstract
1. Introduction
2. Results and Discussion
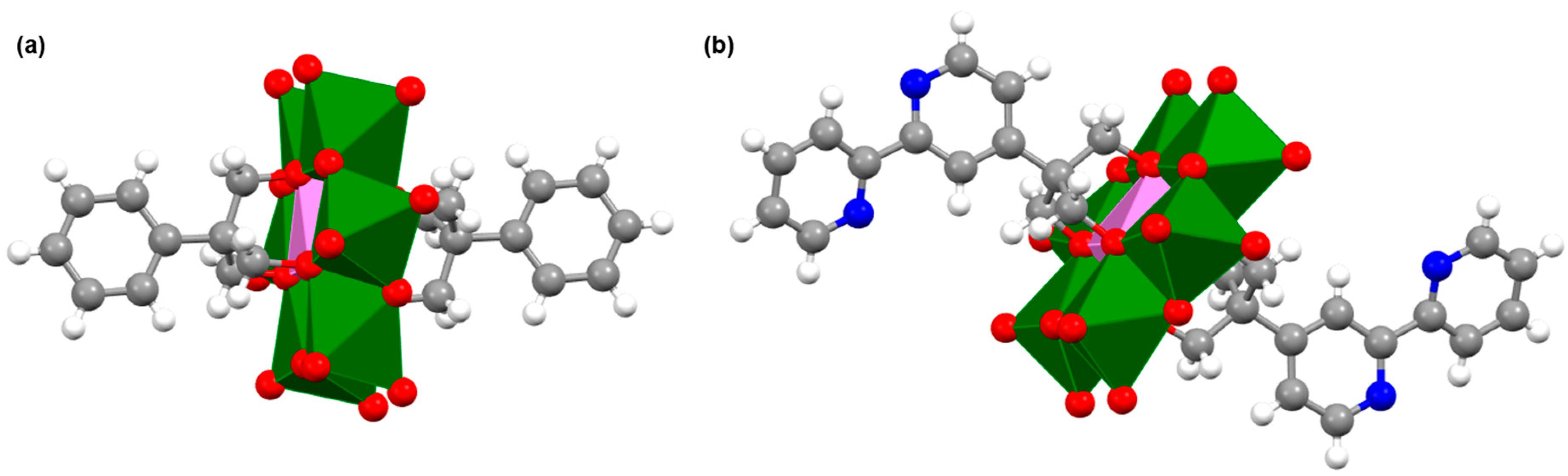
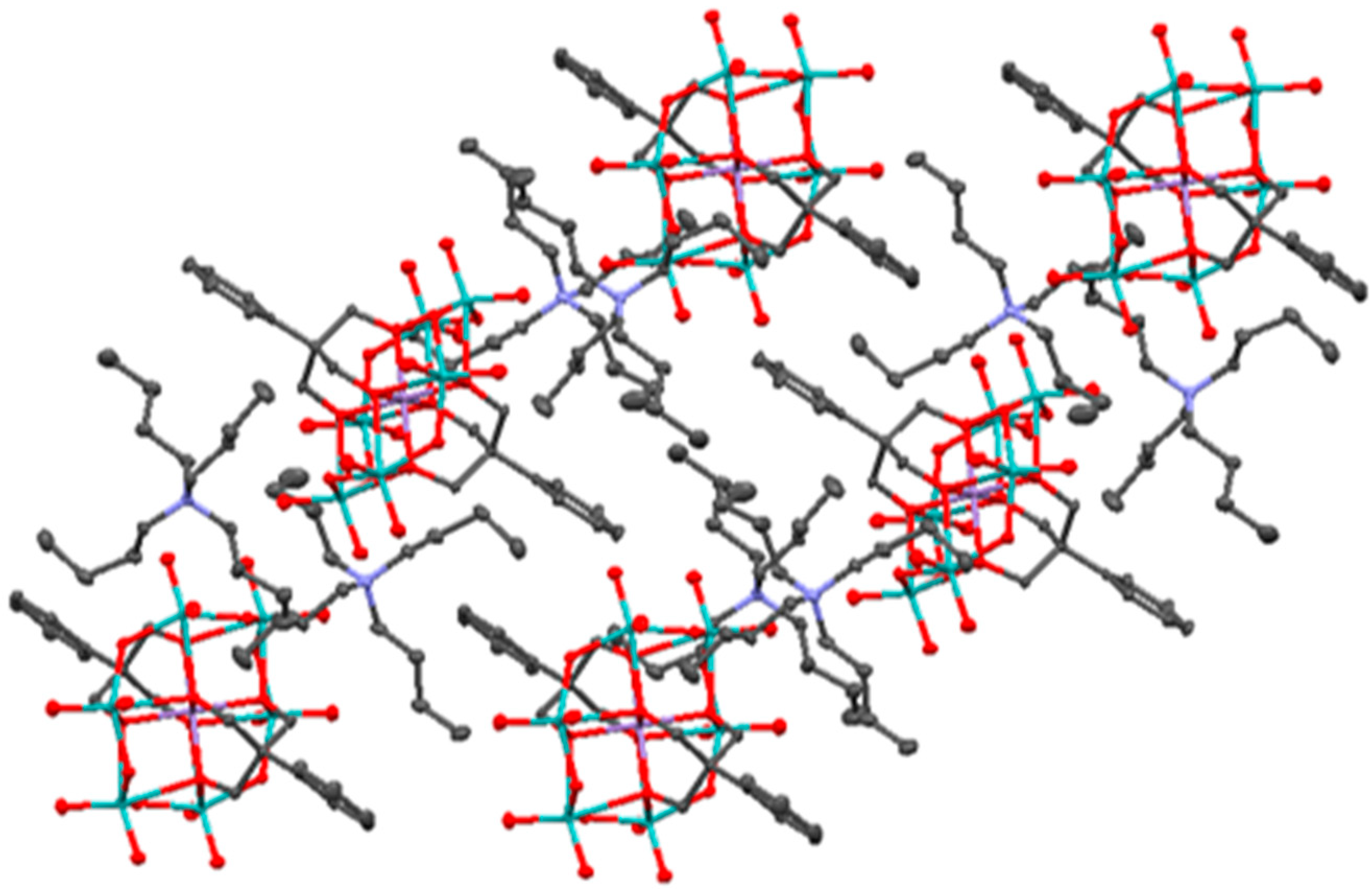
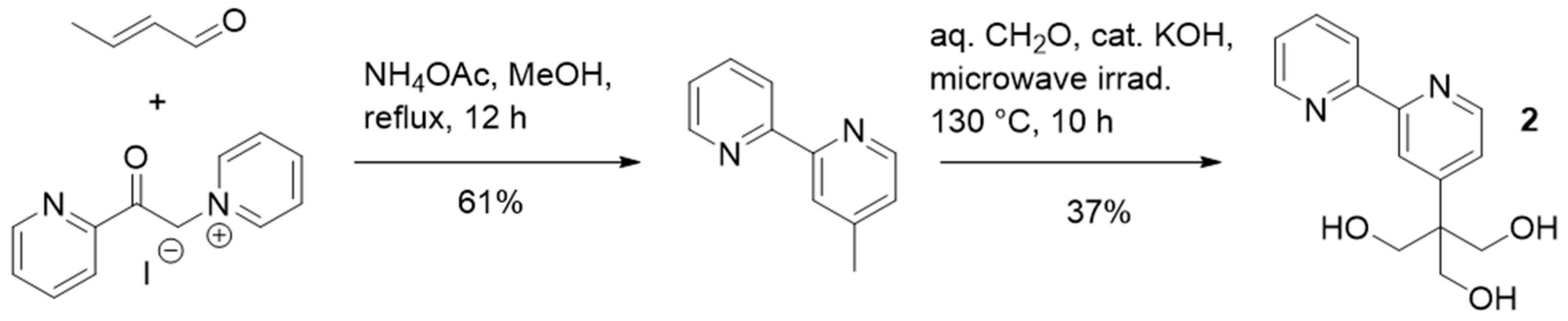
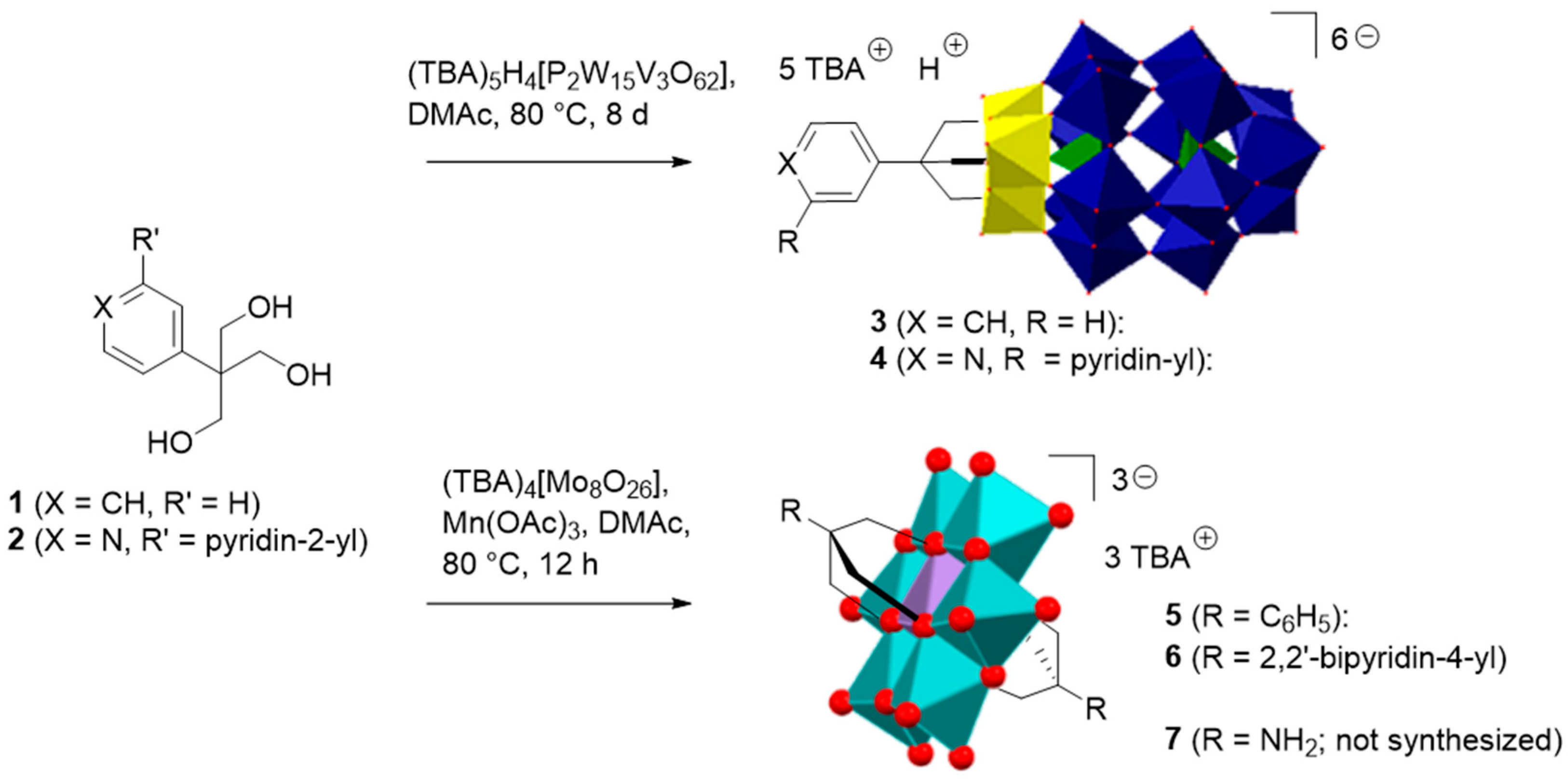
2.1. Synthesis and Characterization of the Organo-Functionalized POMs
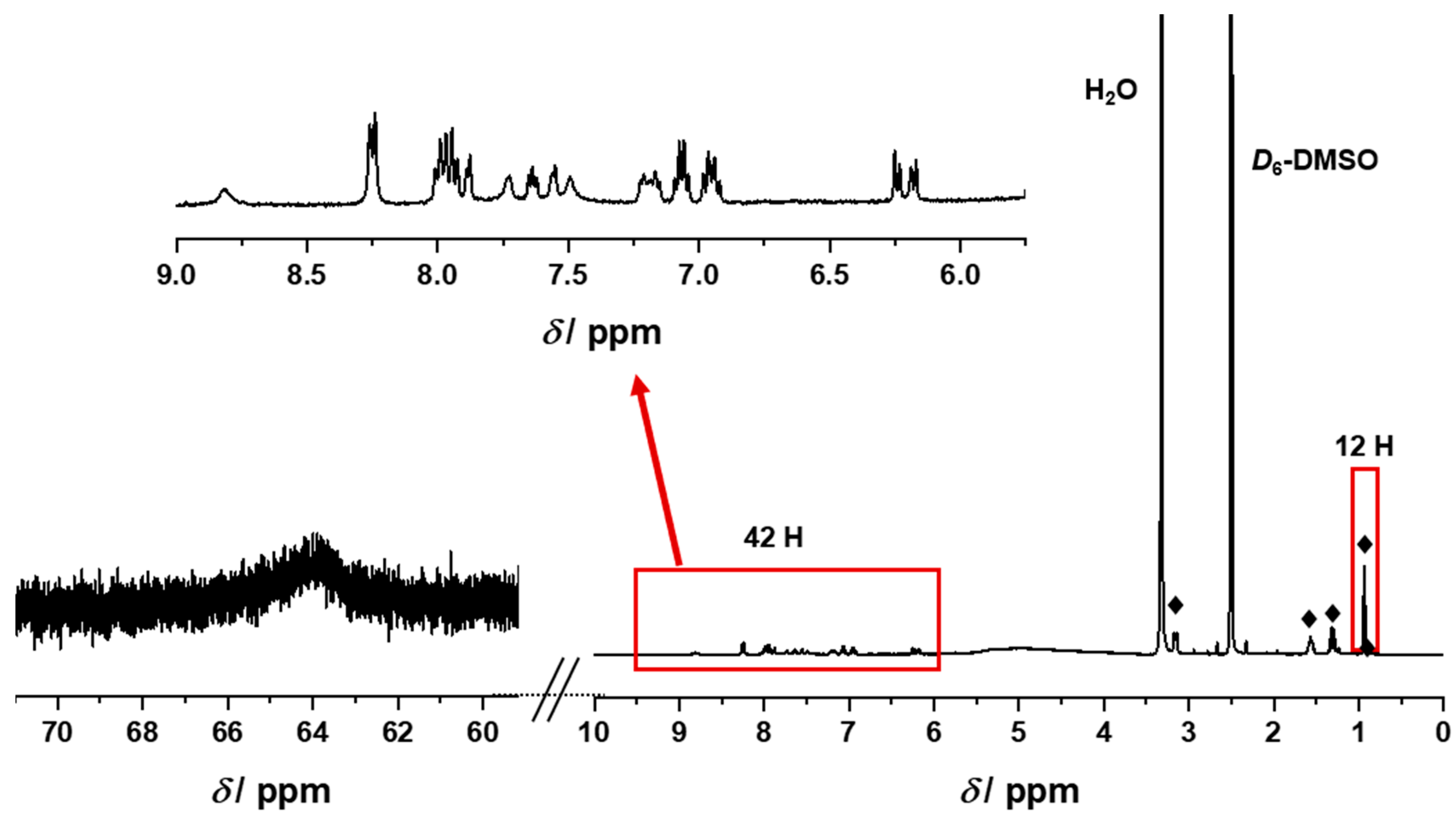
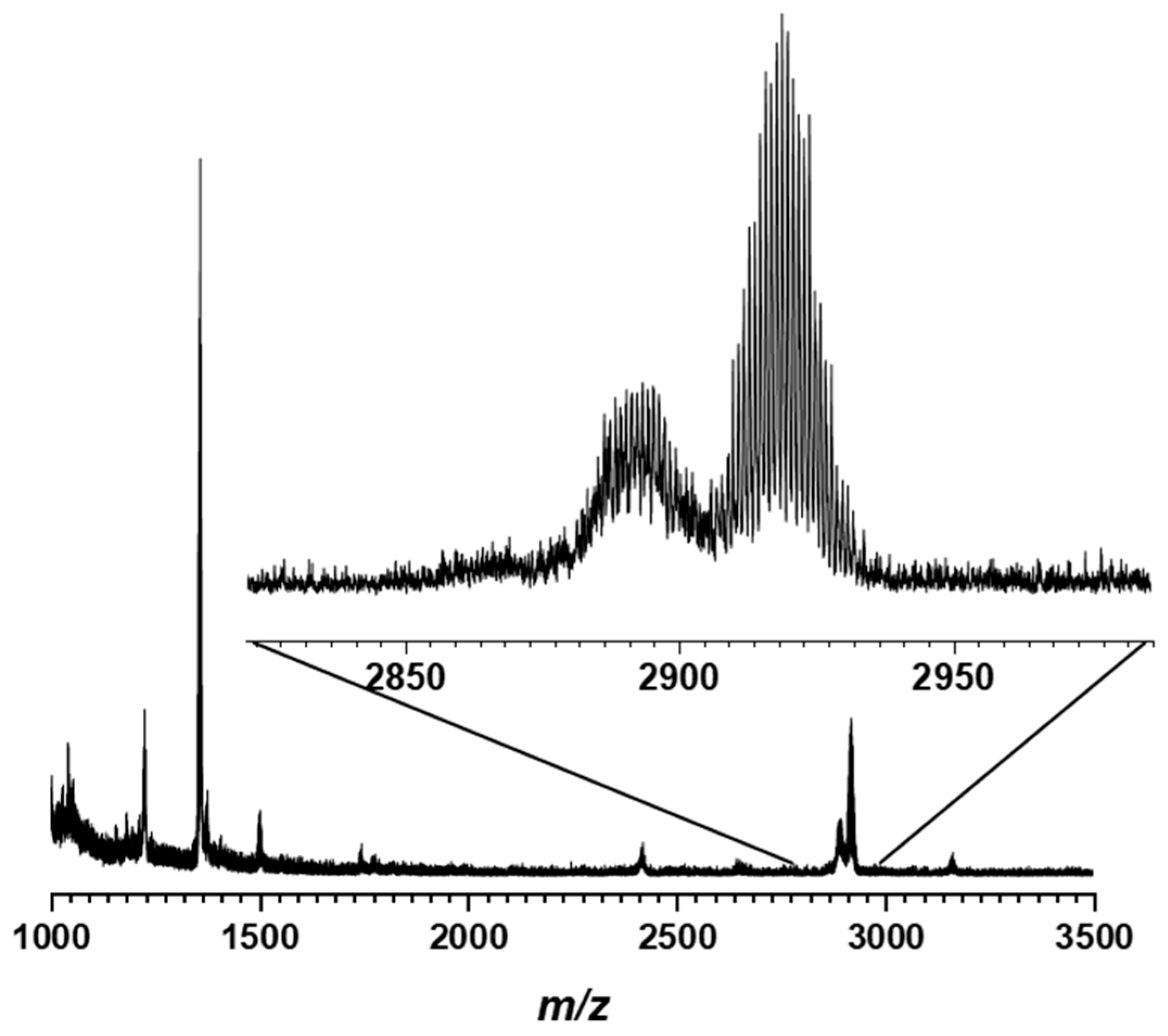
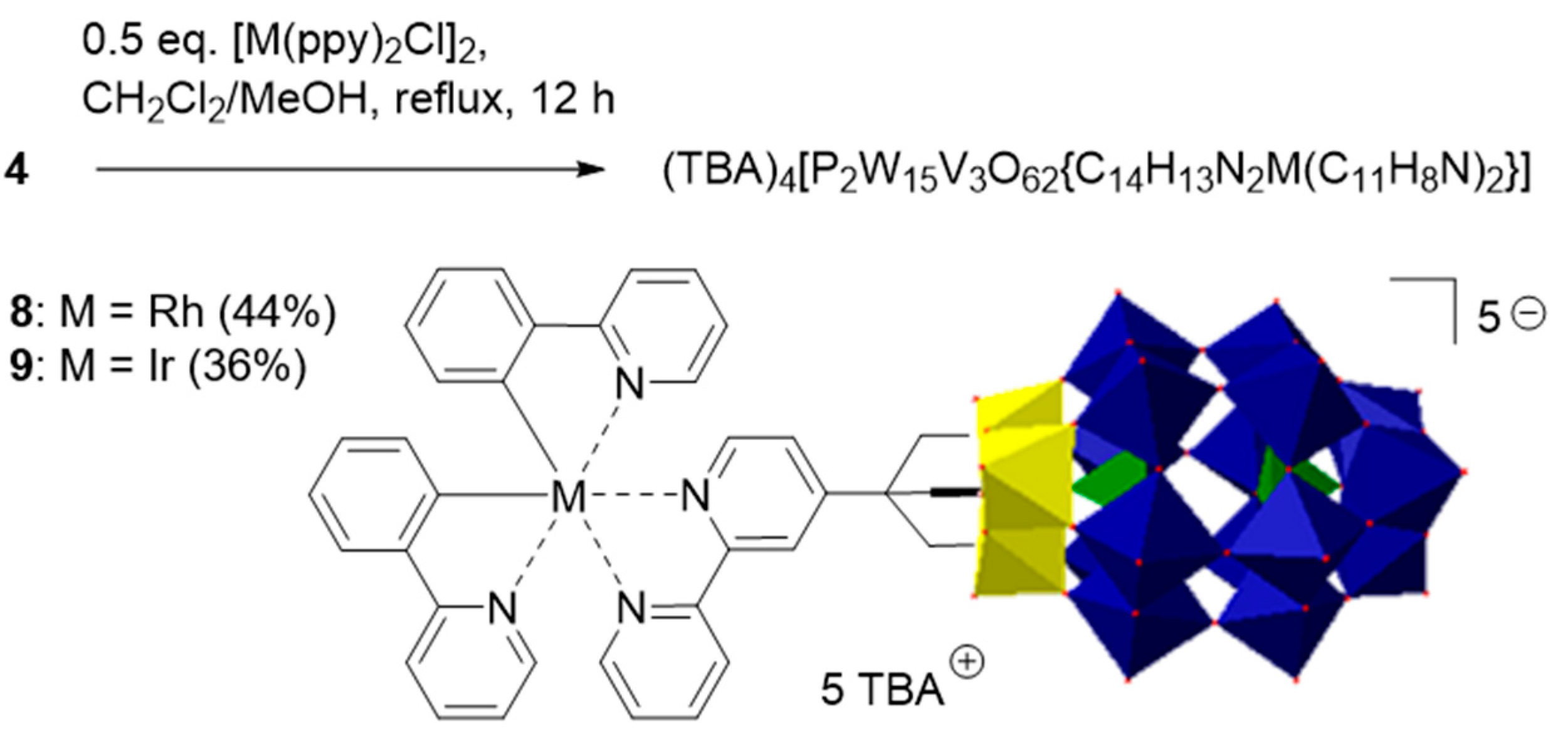
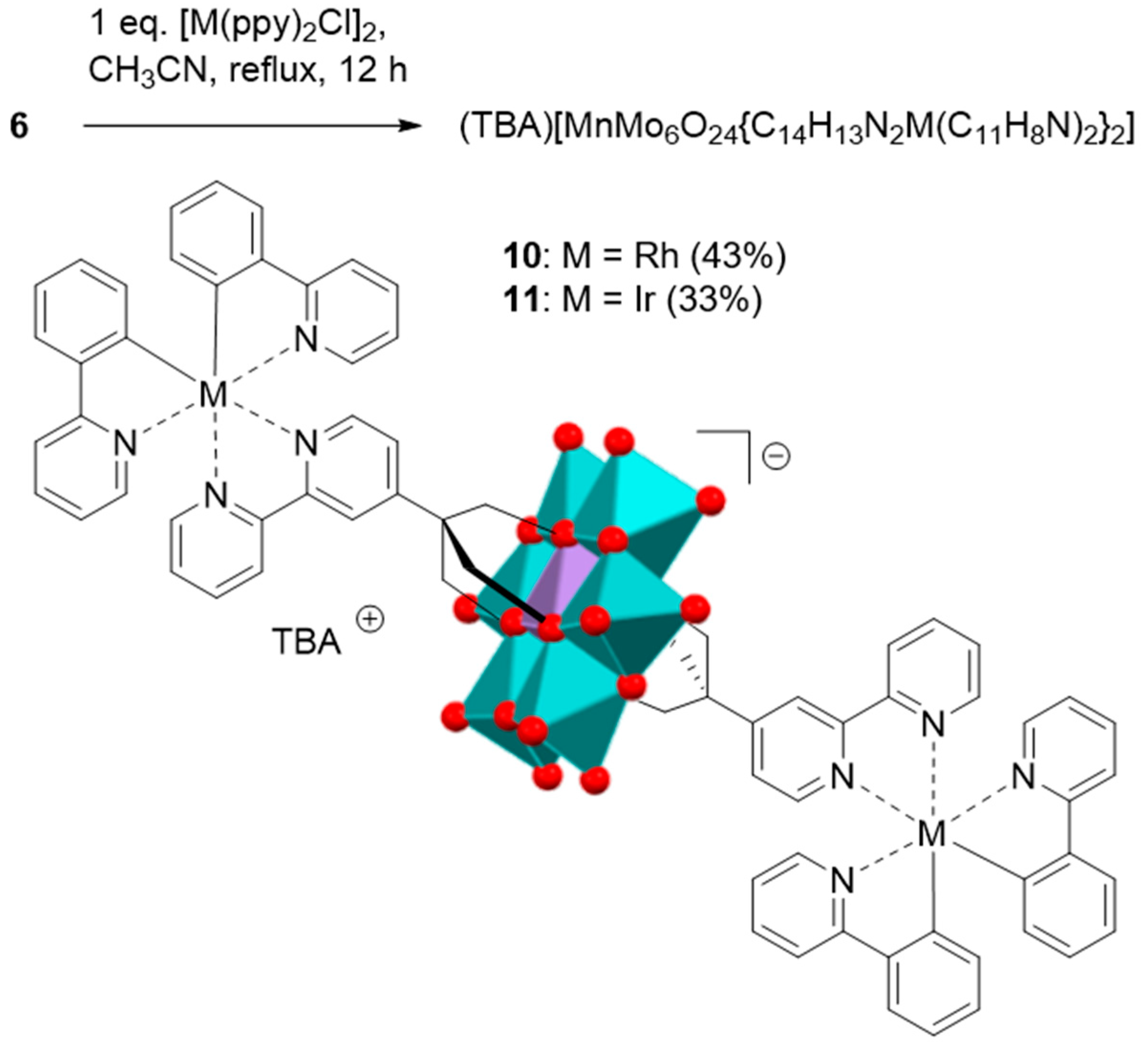
2.2. Synthesis and Characterization of the Photosensitizer-Polyoxometalate Dyads
2.3. Photophysical and Electrochemical Properties of the Dyads Based on the Wells–Dawson-Type POM
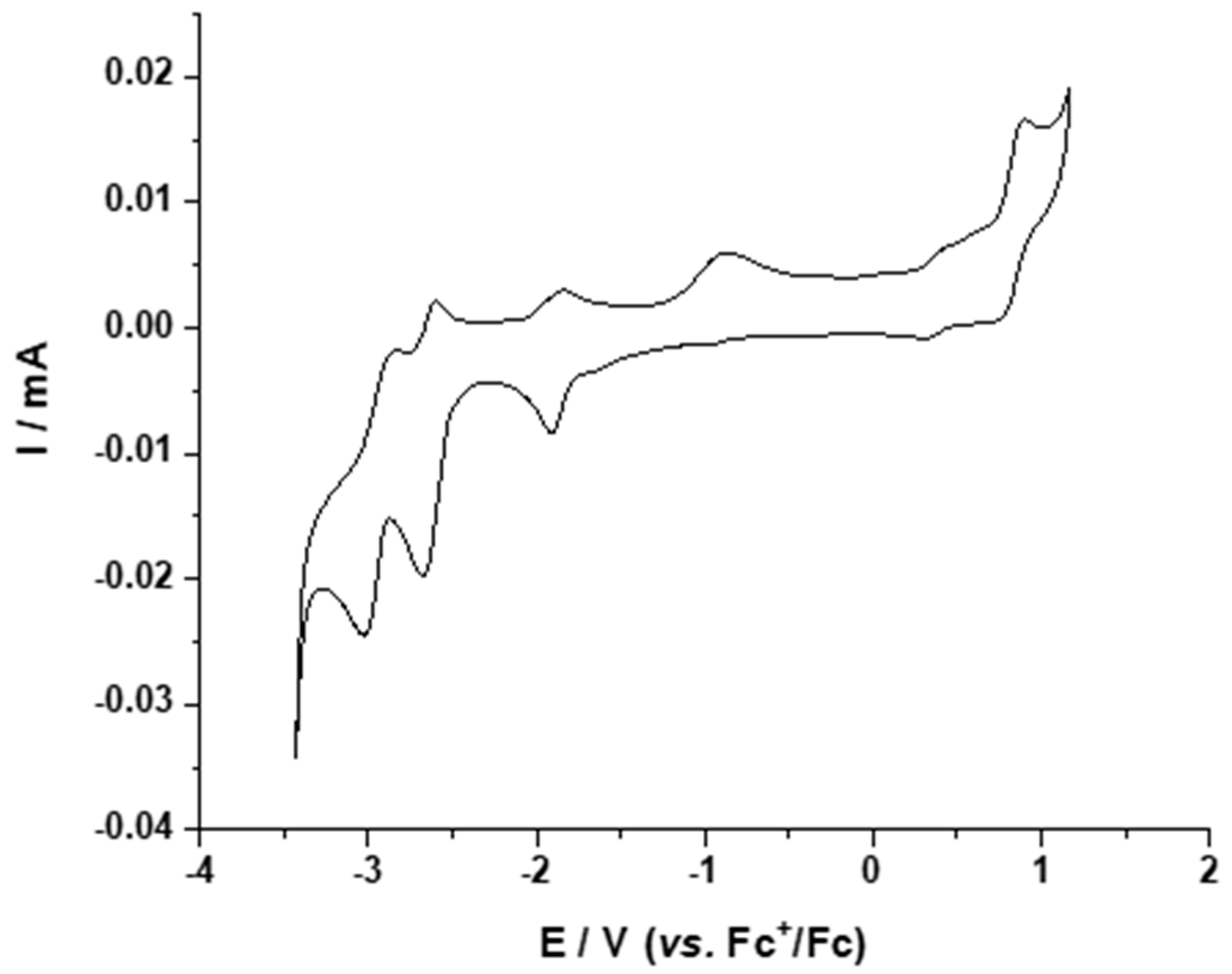
2.3.1. Electrochemical Properties
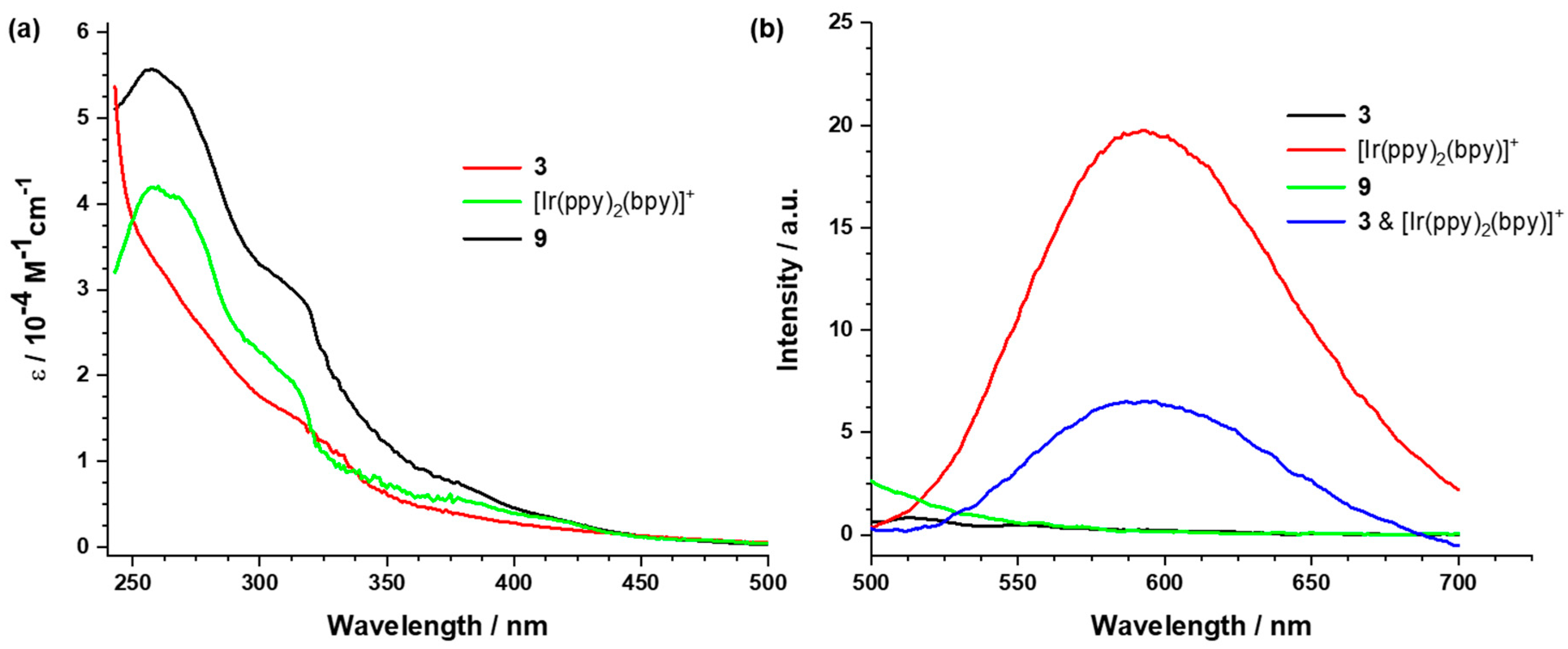
2.3.2. UV/vis Absorption and Emission Studies
2.4. Photophysical and Electrochemical Properties of the Dyads Based on the Anderson-Type POM
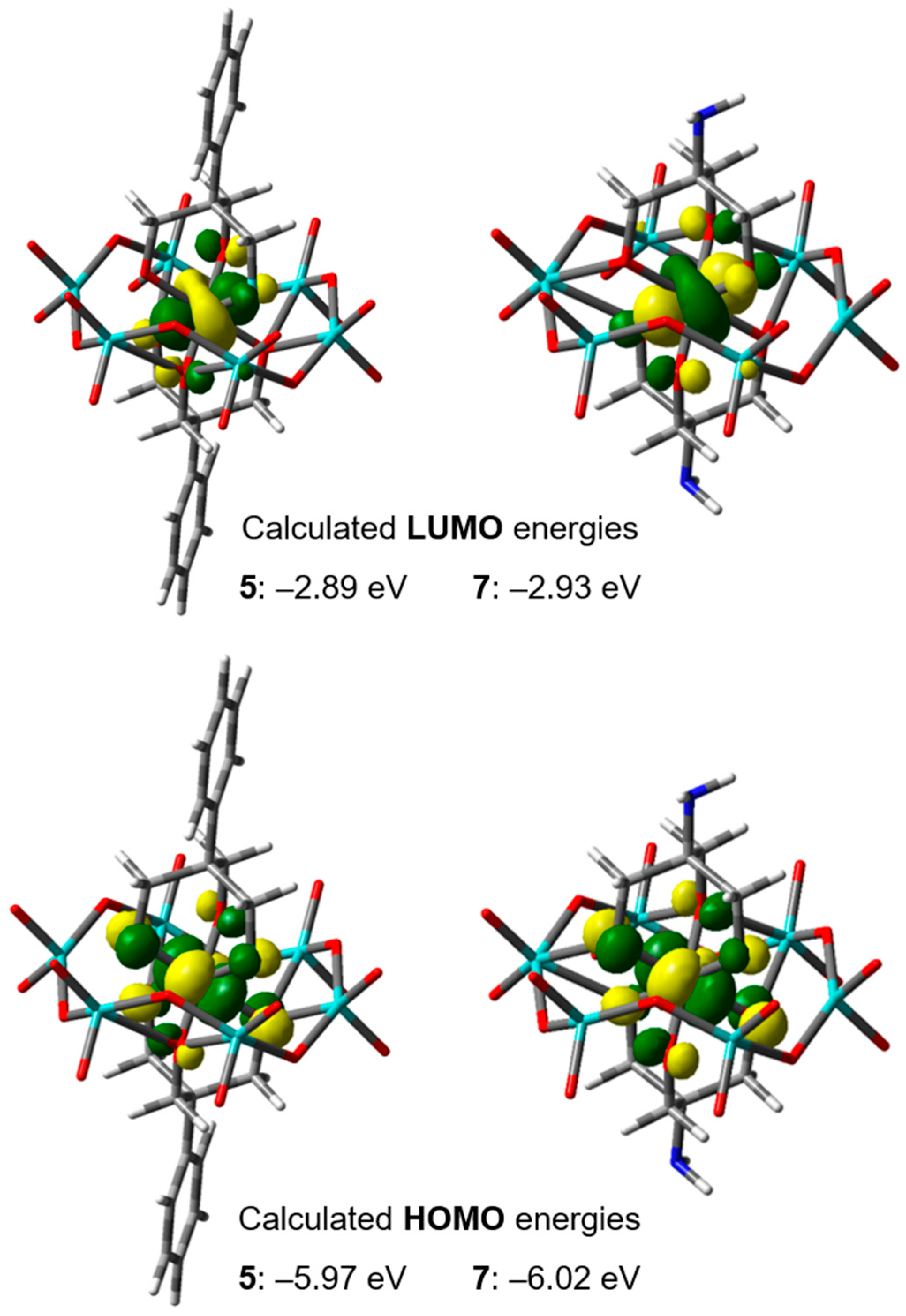
2.4.1. DFT Studies
2.4.2. Electrochemical Properties
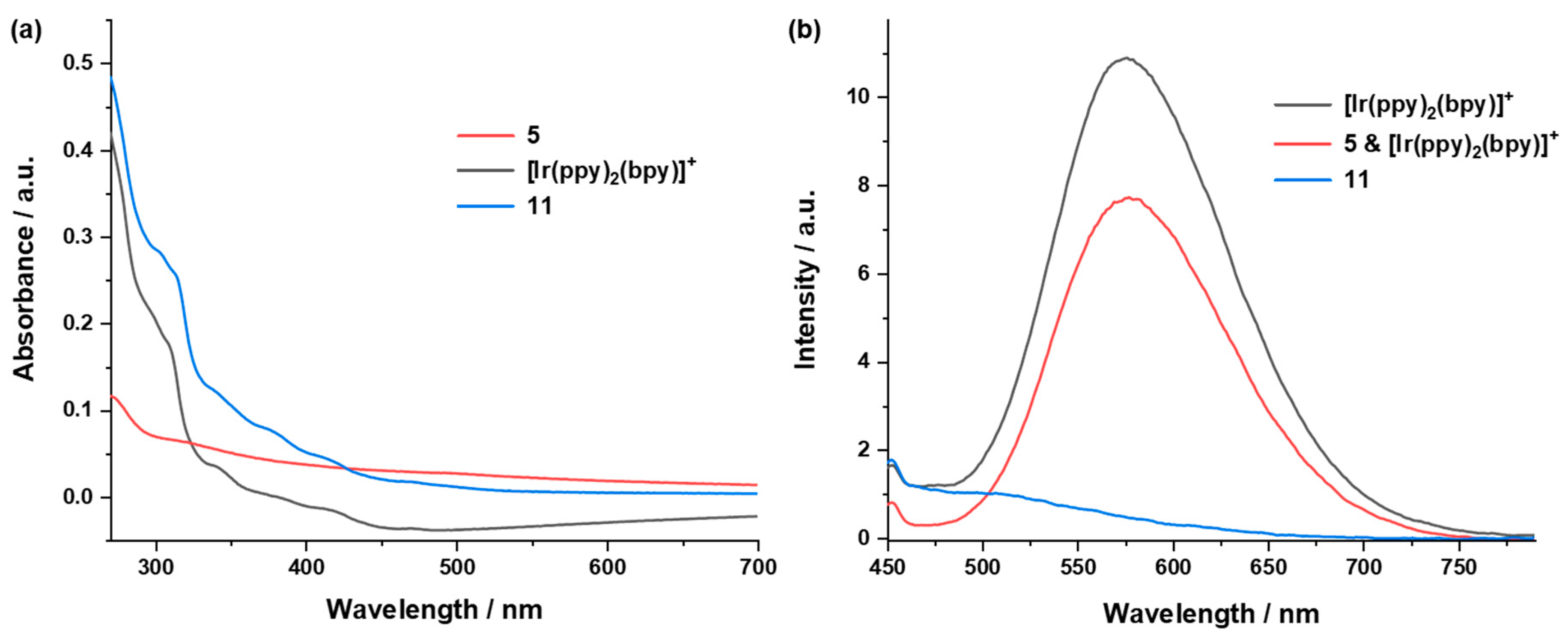
2.4.3. UV/vis Absorption and Emission Properties
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Qi, W.; Wu, L. Polyoxometalate/polymer hybrid materials: Fabrication and properties. Polym. Int. 2009, 58, 1217–1225. [Google Scholar] [CrossRef]
- Wang, S.-S.; Yang, G.-Y. Recent advances in polyoxometalate-catalyzed reactions. Chem. Rev. 2015, 115, 4893–4962. [Google Scholar] [CrossRef] [PubMed]
- Streb, C. New trends in polyoxometalate photoredox chemistry: From photosensitisation to water oxidation catalysis. Dalton Trans. 2012, 41, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.J.; Bond, A.M.; Forster, R.J.; Keyes, T.E. Hybrid polyoxometalate materials for photo(electro-)chemical applications. Coord. Chem. Rev. 2016, 306, 217–234. [Google Scholar] [CrossRef]
- Cameron, J.M.; Wales, D.J.; Newton, G.N. Shining a light on the photo-sensitisation of organic–inorganic hybrid polyoxometalates. Dalton Trans. 2018, 47, 5120–5136. [Google Scholar] [CrossRef] [PubMed]
- Yvon, C.; Surman, A.J.; Hutin, M.; Alex, J.; Smith, B.O.; Long, D.-L.; Cronin, L. Polyoxometalate clusters integrated into peptide chains and as inorganic amino acids: Solution- and solid-phase approaches. Angew. Chem. Int. Ed. 2014, 53, 3336–3341. [Google Scholar] [CrossRef]
- Stuckart, M.; Monakhov, K.Y. Polyoxometalates as components of supramolecular assemblies. Chem. Sci. 2019, 10, 4364–4376. [Google Scholar] [CrossRef]
- Matt, B.; Fize, J.; Moussa, J.; Amouri, H.; Pereira, A.; Artero, V.; Izzet, G.; Proust, A. Charge photo-accumulation and photocatalytic hydrogen evolution under visible light at an iridium(III)-photosensitized polyoxotungstate. Energy Environ. Sci. 2013, 6, 1504–1508. [Google Scholar] [CrossRef]
- Santoni, M.-P.; Hanan, G.S.; Hasenknopf, B. Covalent multi-component systems of polyoxometalates and metal complexes: Toward multi-functional organic–inorganic hybrids in molecular and material sciences. Coord. Chem. Rev. 2014, 281, 64–85. [Google Scholar] [CrossRef]
- Schönweiz, S.; Heiland, M.; Anjass, M.; Jacob, T.; Rau, S.; Streb, C. Experimental and theoretical investigation of the light-driven hydrogen evolution by polyoxometalate–photosensitizer dyads. Chem. Eur. J. 2017, 23, 15370–15376. [Google Scholar] [CrossRef]
- Herrmann, S.; Ritchie, C.; Streb, C. Polyoxometalate—Conductive polymer composites for energy conversion, energy storage and nanostructured sensors. Dalton Trans. 2015, 44, 7092–7104. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Huang, L.; Hu, J.; Streb, C.; Song, Y.-F. Polyoxometalate-functionalized nanocarbon materials for energy conversion, energy storage and sensor systems. Energy Environ. Sci. 2015, 8, 776–789. [Google Scholar] [CrossRef]
- Esswein, A.J.; Nocera, D.G. Hydrogen production by molecular photocatalysis. Chem. Rev. 2007, 107, 4022–4047. [Google Scholar] [CrossRef] [PubMed]
- Matt, B.; Coudret, C.; Viala, C.; Jouvenot, D.; Loiseau, F.; Izzet, G.; Proust, A. Elaboration of covalently linked polyoxometalates with ruthenium and pyrene chromophores and characteriation of their photophysical properties. Inorg. Chem. 2011, 50, 7761–7768. [Google Scholar] [CrossRef]
- Matt, B.; Renaudineau, S.; Chamoreau, L.M.; Afonso, C.; Izzet, G.; Proust, A. Hybrid polyoxometalates: Keggin and Dawson silyl derivatives as versatile platforms. J. Org. Chem. 2011, 76, 3107–3112. [Google Scholar] [CrossRef]
- Matt, B.; Moussa, J.; Chamoreau, L.-M.; Afonso, C.; Proust, A.; Amouri, H.; Izzet, G. Elegant approach to the synthesis of a unique heteroleptic cyclometalated iridium(III)-polyoxometalate conjugate. Organometallics 2012, 31, 35–38. [Google Scholar] [CrossRef]
- Matt, B.; Xiang, X.; Kaledin, A.L.; Han, N.; Moussa, J.; Amouri, H.; Alves, S.; Hill, C.L.; Lian, T.; Musaev, D.G.; et al. Long lived charge separation in iridium(III)-photosensitized polyoxometalates: Synthesis, photophysical and computational studies of organometallic–redox tunable oxide assemblies. Chem. Sci. 2013, 4, 1737–1745. [Google Scholar] [CrossRef]
- Schönweiz, S.; Rommel, S.A.; Kübel, J.; Micheel, M.; Dietzek, B.; Rau, S.; Streb, C. Covalent photosensitizer–polyoxometalate-catalyst dyads for visible-light-driven hydrogen evolution. Chem. Eur. J. 2016, 22, 12002–12005. [Google Scholar] [CrossRef]
- Odobel, F.; Séverac, M.; Pellegrin, Y.; Blart, E.; Fosse, C.; Cannizzo, C.; Mayer, C.R.; Elliott, K.J.; Harriman, A. Coupled sensitizer–catalyst dyads: Electron-transfer reactions in a perylene–polyoxometalate conjugate. Chem. Eur. J. 2009, 15, 3130–3138. [Google Scholar] [CrossRef]
- Santoni, M.-P.; Pal, A.K.; Hanan, G.S.; Tang, M.-C.; Furtos, A.; Hasenknopf, B. A light-harvesting polyoxometalate-polypyridine hybrid induces electron transfer as its Re(I) complex. Dalton Trans. 2014, 43, 6990–6993. [Google Scholar] [CrossRef]
- Luo, Y.; Wächtler, M.; Barthelmes, K.; Winter, A.; Schubert, U.S.; Dietzek, B. Direct detection of the photoinduced charge-separated state in a Ru(II) bis(terpyridine)–polyoxometalate molecular dyad. Chem. Commun. 2018, 54, 2970–2973. [Google Scholar] [CrossRef] [PubMed]
- Barthelmes, K.; Sittig, M.; Winter, A.; Schubert, U.S. Molecular dyads and triads based on phenothiazine and π-extended tetrathiafulvalene donors, bis(terpyridine)ruthenium(II) complexes, and polyoxometalates. Eur. J. Inorg. Chem. 2017, 2017, 3698–3706. [Google Scholar] [CrossRef]
- Proust, A.; Matt, B.; Villanneau, R.; Guillemot, G.; Gouzerh, P.; Izzet, G. Functionalization and post-functionalization: A step towards polyoxometalate-based materials. Chem. Soc. Rev. 2012, 41, 7605–7622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, Y.; Li, G.; Wei, Y. Recent advances in alkoxylation chemistry of polyoxometalates: From synthetic strategies, structural overviews to functional applications. Coord. Chem. Rev. 2019, 378, 395–414. [Google Scholar] [CrossRef]
- Santoni, M.-P.; Pal, A.K.; Hanan, G.S.; Proust, A.; Hasenknopf, B. Discrete covalent organic–inorganic hybrids: Terpyridine functionalized polyoxometalates obtained by a modular strategy and their metal complexation. Inorg. Chem. 2011, 50, 6737–6745. [Google Scholar] [CrossRef] [PubMed]
- Auvray, T.; Santoni, M.-P.; Hasenknopf, B.; Hanan, G.S. Covalent hybrids based on Re(I) tricarbonyl complexes and polypyridine-functionalized polyoxometalate: Synthesis, characterization and electronic properties. Dalton Trans. 2017, 46, 10029–10036. [Google Scholar] [CrossRef]
- Li, X.-X.; Wang, Y.-X.; Wang, R.-H.; Cui, C.-Y.; Tian, C.-B.; Yang, G.-Y. Designed assembly of heterometallic cluster organic frameworks based on Anderson-type polyoxometalate clusters. Angew. Chem. Int. Ed. 2016, 55, 6462–6466. [Google Scholar] [CrossRef]
- Yazigi, F.-J.; Wilson, C.; Long, D.-L.; Forgan, R.S. Synthetic considerations in the self-assembly of coordination polymers of pyridine-functionalized hybrid Mn-Anderson polyoxometalates. Cryst. Growth Des. 2017, 17, 4739–4748. [Google Scholar] [CrossRef]
- Nachtigall, O.; Hagenbach, A.; Wiecko, J.; Lentz, D.; Abram, U.; Spandl, J. Functional polyoxometalates from solvothermal reactions of VOSO4 with tripodal alkoxides—A study on the reactivity of different “tris” derivatives. Dalton Trans. 2017, 46, 509–516. [Google Scholar] [CrossRef]
- Dermer, O.C.; Solomon, P.W. Extensions of the Tollens condensation. J. Am. Chem. Soc. 1954, 76, 1697–1699. [Google Scholar] [CrossRef][Green Version]
- Hellmann, K.W.; Friedrich, S.; Gade, L.H.; Li, W.-S.; McPartlin, M. Tripodal triamidostannates as building blocks in the generation of Sn-M-bonded heterobimetallics (M = Fe, Ru). Chem. Ber. 1995, 128, 29–34. [Google Scholar] [CrossRef]
- Constable, E.C.; Figgemeier, E.; Housecroft, C.E.; Olsson, J.; Zimmermann, Y.C. Electrochemical probing of ground state electronic interactions in polynuclear complexes of a new heteroditopic ligand. Dalton Trans. 2004, 2004, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Menozzi, D.; Biavardi, E.; Massera, C.; Schmidtchen, F.-P.; Cornia, A.; Dalcanale, E. Thermodynamics of host–guest interactions between methylpyridinium salts and phosphonate cavitands. Supramol. Chem. 2010, 22, 768–775. [Google Scholar] [CrossRef]
- Finke, R.G.; Rapko, B.; Saxton, R.J.; Domaille, P.J. Trisubstituted heteropolytungstates as soluble metal oxide analogs. Part III. Synthesis, characterization, phosphorus-31, silicon-29, vanadium-51, and 1- and 2-D tungsten-183 NMR, deprotonation, and proton mobility studies of organic solvent solute forms of HxSiW9V3O40x−7 and HxP2W15V3O62x−9. J. Am. Chem. Soc. 1986, 108, 2947–2960. [Google Scholar] [CrossRef]
- Ikegami, S.; Yagasaki, A. Efficient syntheses of [(n-C4H9)4N]4[α-Mo8O26] and [(n-C4H9)4N]2[Mo2O7]. Materials 2009, 2, 869. [Google Scholar] [CrossRef]
- Pradeep, C.P.; Long, D.-L.; Newton, G.N.; Song, Y.-F.; Cronin, L. Supramolecular metal oxides: Programmed hierarchical assembly of a protein-sized 21 kDa [(C16H36N)19{H2NC(CH2O)3P2V3W15O59}4]5− polyoxometalate assembly. Angew. Chem. Int. Ed. 2008, 47, 4388–4391. [Google Scholar] [CrossRef]
- Hasenknopf, B.; Delmont, R.; Herson, P.; Gouzerh, P. Anderson-type heteropolymolybdates containing tris(alkoxo) ligands: Synthesis and structural characterization. Eur. J. Inorg. Chem. 2002, 2002, 1081–1087. [Google Scholar] [CrossRef]
- Fedotov, M.; Maksimovskaya, R. NMR structural aspects of the chemistry of V, Mo, W polyoxometalates. J. Struct. Chem. 2006, 47, 952–978. [Google Scholar] [CrossRef]
- Blazevic, A.; Rompel, A. The Anderson–Evans polyoxometalate: From inorganic building blocks via hybrid organic–inorganic structures to tomorrows “Bio-POM”. Coord. Chem. Rev. 2016, 307, 42–64. [Google Scholar] [CrossRef]
- Song, Y.-F.; Long, D.-L.; Cronin, L. Hybrid polyoxometalate clusters with appended aromatic platforms. CrystEngComm 2010, 12, 109–115. [Google Scholar] [CrossRef]
- Azcarate, I.; Ahmed, I.; Farha, R.; Goldmann, M.; Wang, X.; Xu, H.; Hasenknopf, B.; Lacôte, E.; Ruhlmann, L. Synthesis and characterization of conjugated Dawson-type polyoxometalate–porphyrin copolymers. Dalton Trans. 2013, 42, 12688–12698. [Google Scholar] [CrossRef] [PubMed]
- Bokarev, S.I.; Bokareva, O.S.; Kühn, O. Electronic excitation spectrum of the photosensitizer [Ir(ppy)2(bpy)]+. J. Chem. Phys. 2012, 136, 214305. [Google Scholar] [CrossRef] [PubMed]
- Deaton, J.C.; Castellano, F.N. Archetypal iridium(III) compounds for optoelectronic and photonic applications. In Iridium(III) in Optoelectronic and Photonics Applications; Zysman-Coleman, E., Ed.; John Wiley & Sons: Chichester, UK, 2017; pp. 1–69. [Google Scholar]
- Ramlot, D.; Rebarz, M.; Volker, L.; Ovaere, M.; Beljonne, D.; Dehaen, W.; Van Meervelt, L.; Moucheron, C.; Kirsch-De Mesmaeker, A. An experimental and theoretical approach to the photophysical properties of some Rh and Ir complexes incorporating the dipyrromethene ligand. Eur. J. Inorg. Chem. 2013, 2013, 2031–2040. [Google Scholar] [CrossRef]
- Holder, E.; Marin, V.; Alexeev, A.; Schubert, U.S. Greenish-yellow-, yellow-, and orange-light-emitting iridium(III) polypyridyl complexes with poly(ε-caprolactone)–bipyridine macroligands. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 2765–2776. [Google Scholar] [CrossRef]
- Hooft, R.W.W. COLLECT Data Collection Software; Nonius B.V.: Delft, The Netherlands, 1998. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology; Carter, C.W., Sweet, R.M., Eds.; Academic Press: Cambridge, MA, USA, 1997; Volume 276, pp. 307–326. [Google Scholar]
- SADABS 2.10; Bruker-AXS Inc.: Madison, WI, USA, 2003.
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–11 are available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Rh-to-P Ratio 1 | Ir-to-P Ratio 1 | Rh-to-Mo Ratio 1 | Ir-to-Mo Ratio 1 |
|---|---|---|---|---|
| 8 | 1:(2.3 ± 0.2) | – | – | – |
| 9 | – | 1:(2.1 ± 0.2) | – | – |
| 10 | – | – | 1:(2.9 ± 0.2) | – |
| 11 | – | – | – | 1:(3.1 + 0.2) |
| Compound | Oxidation Potentials (V vs. Fc+/Fc) | Reduction Potentials (V vs. Fc+/Fc) |
|---|---|---|
| 3 | – | −0.32, −0.62, −1.37, −1.83 2 |
| 8 | – | −1.35, −1.95, −2.68 2 |
| 9 | – | −1.23, −1.60, −1.91, −2.02, −2.81 2 |
| 5 | 0.24 3 | −1.34 3 |
| 6 | 0.39 3 | −1.28, −2.71 3 |
| 10 | 0.42 3 | −1.09, −1.94, −2.56, −2.88 3 |
| 11 | 0.86, 0.42 3 | −1.23, −1.85, −2.55, −2.91 3 |
| Compound | Absorption (wavelength/nm) |
|---|---|
| [Rh(ppy)2(bpy)]+ | 258, 297, 3672 |
| [Ir(ppy)2(bpy)]+ | 307, 341, 380, 4192 |
| 8 | 258, 303, 370 |
| 9 | 258, 310, 380 |
| 10 | 260, 302, 369 |
| 11 | 310, 338, 378, 417 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winter, A.; Endres, P.; Schröter, E.; Jäger, M.; Görls, H.; Neumann, C.; Turchanin, A.; Schubert, U.S. Towards Covalent Photosensitizer-Polyoxometalate Dyads-Bipyridyl-Functionalized Polyoxometalates and Their Transition Metal Complexes. Molecules 2019, 24, 4446. https://doi.org/10.3390/molecules24244446
Winter A, Endres P, Schröter E, Jäger M, Görls H, Neumann C, Turchanin A, Schubert US. Towards Covalent Photosensitizer-Polyoxometalate Dyads-Bipyridyl-Functionalized Polyoxometalates and Their Transition Metal Complexes. Molecules. 2019; 24(24):4446. https://doi.org/10.3390/molecules24244446
Chicago/Turabian StyleWinter, Andreas, Patrick Endres, Erik Schröter, Michael Jäger, Helmar Görls, Christof Neumann, Andrey Turchanin, and Ulrich S. Schubert. 2019. "Towards Covalent Photosensitizer-Polyoxometalate Dyads-Bipyridyl-Functionalized Polyoxometalates and Their Transition Metal Complexes" Molecules 24, no. 24: 4446. https://doi.org/10.3390/molecules24244446
APA StyleWinter, A., Endres, P., Schröter, E., Jäger, M., Görls, H., Neumann, C., Turchanin, A., & Schubert, U. S. (2019). Towards Covalent Photosensitizer-Polyoxometalate Dyads-Bipyridyl-Functionalized Polyoxometalates and Their Transition Metal Complexes. Molecules, 24(24), 4446. https://doi.org/10.3390/molecules24244446