
Mono- and Di-Quaternized 4,4′-Bipyridine Derivatives as Key Building Blocks for Medium- and Environment-Responsive Compounds and Materials


Abstract
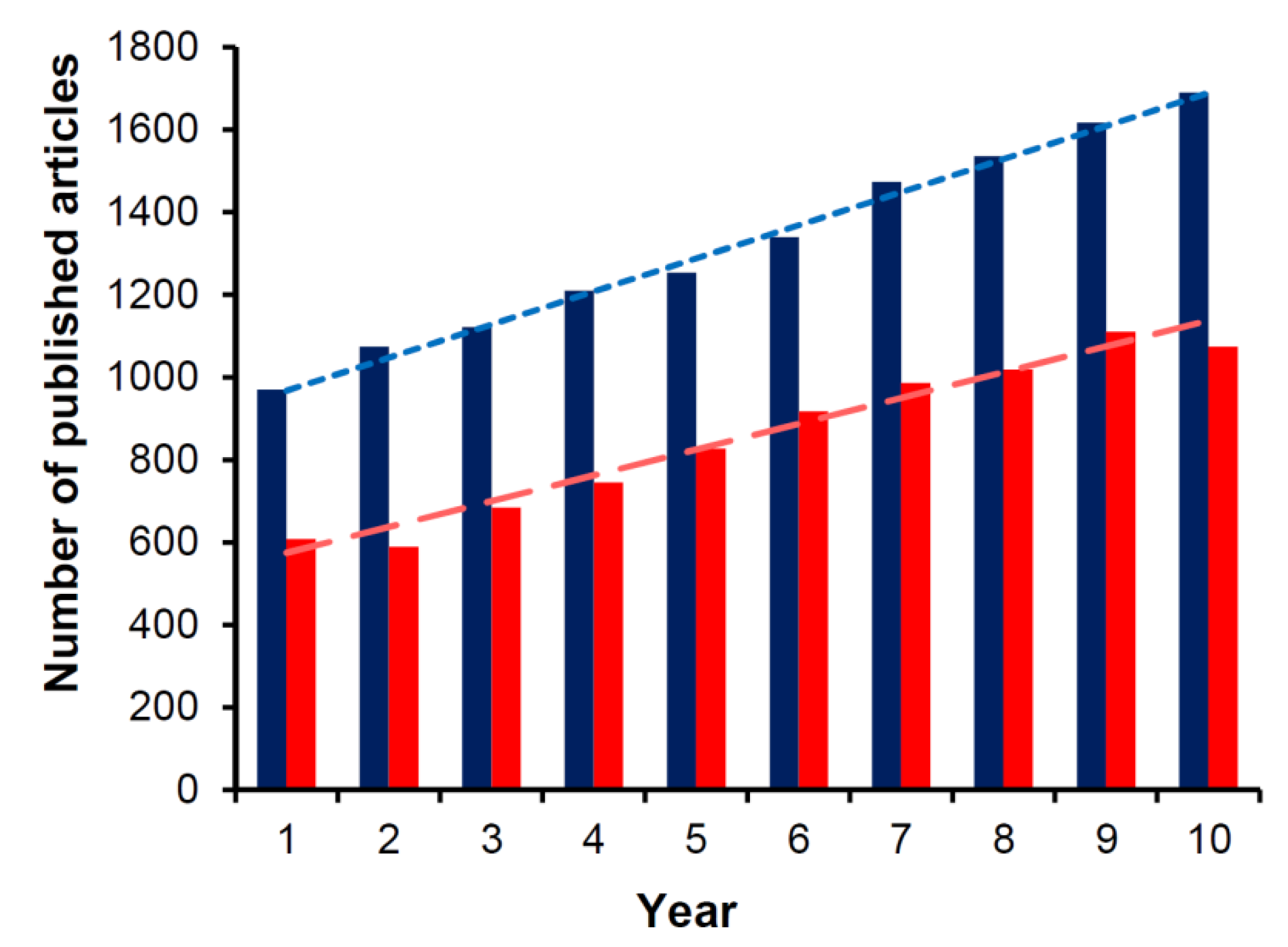
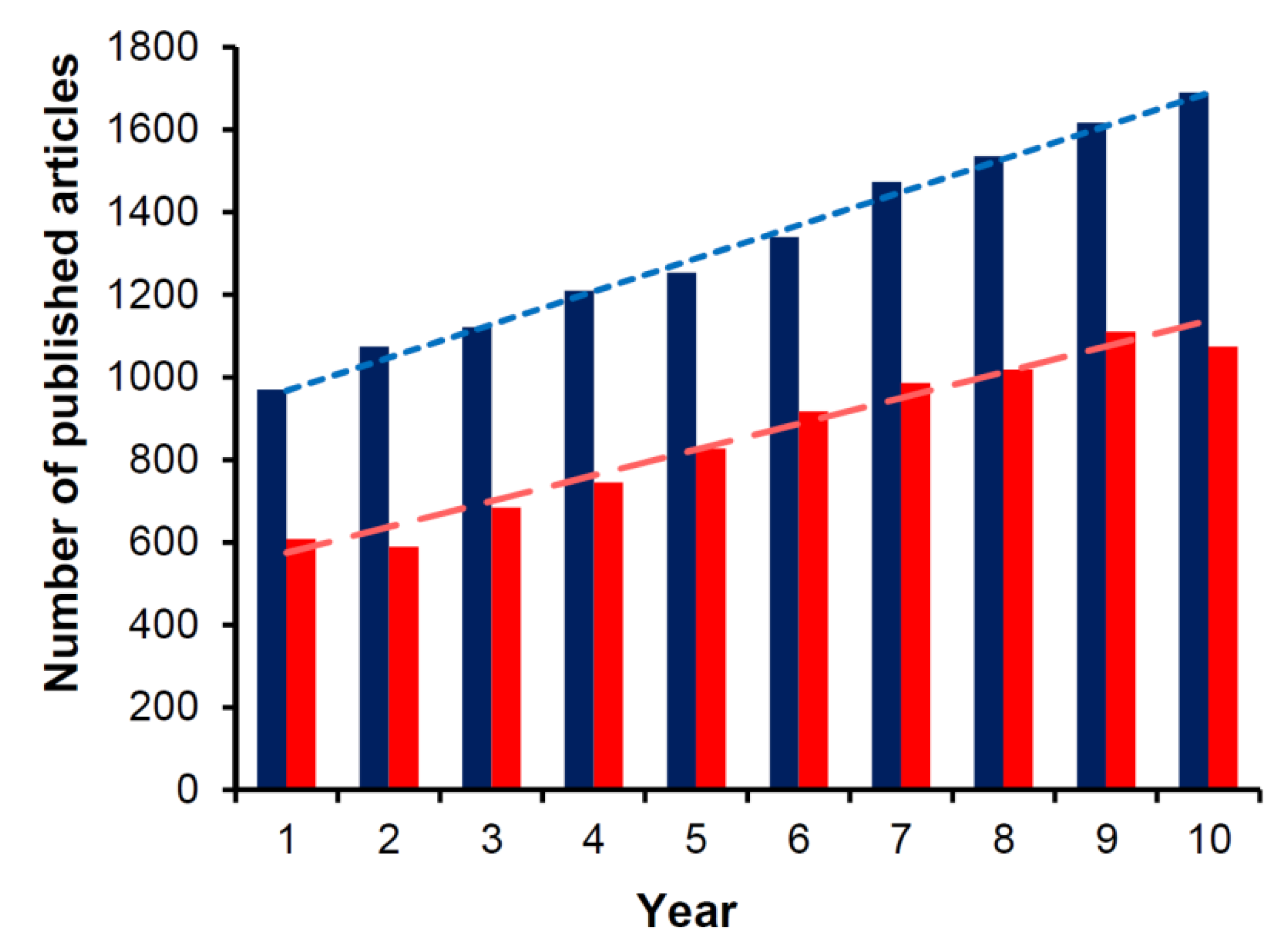
:1. Introduction
2. Structure and Properties of Viologens
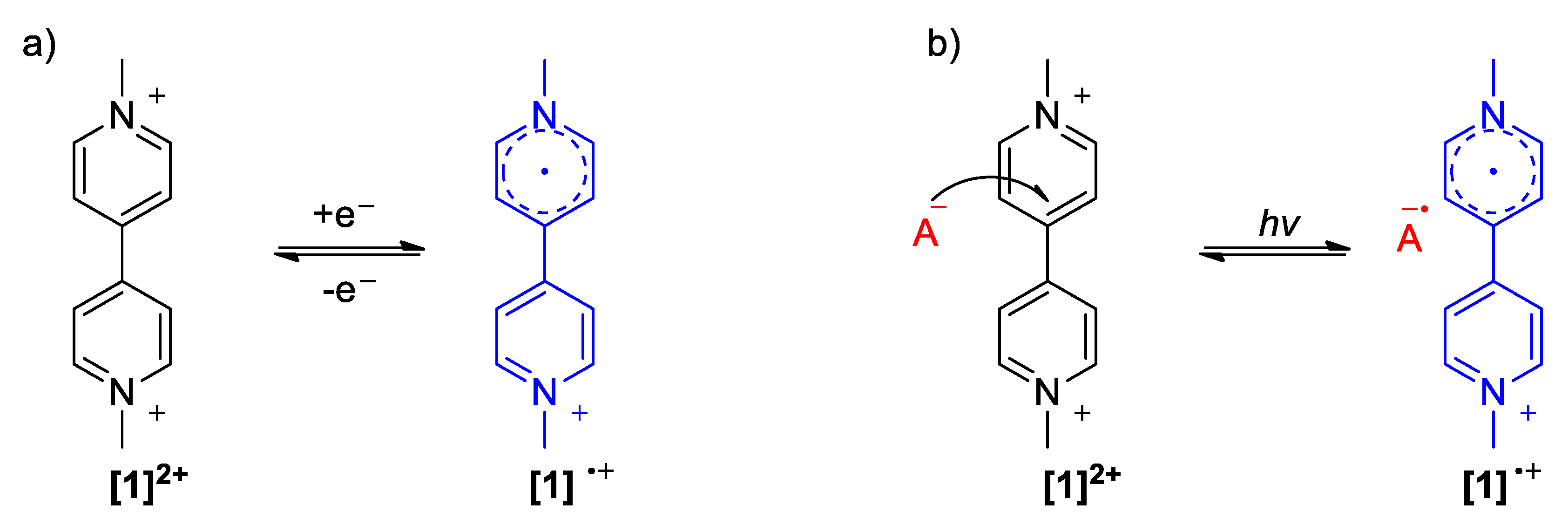
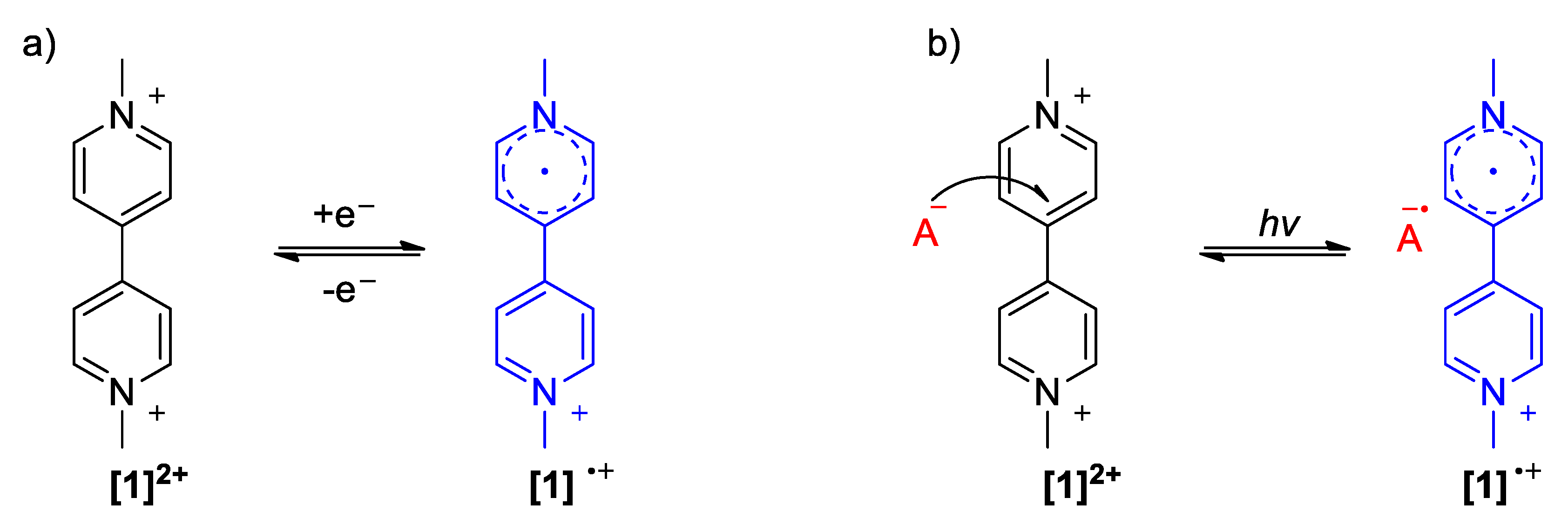
2.1. The Strong Electron-Withdrawing Character of Viologens
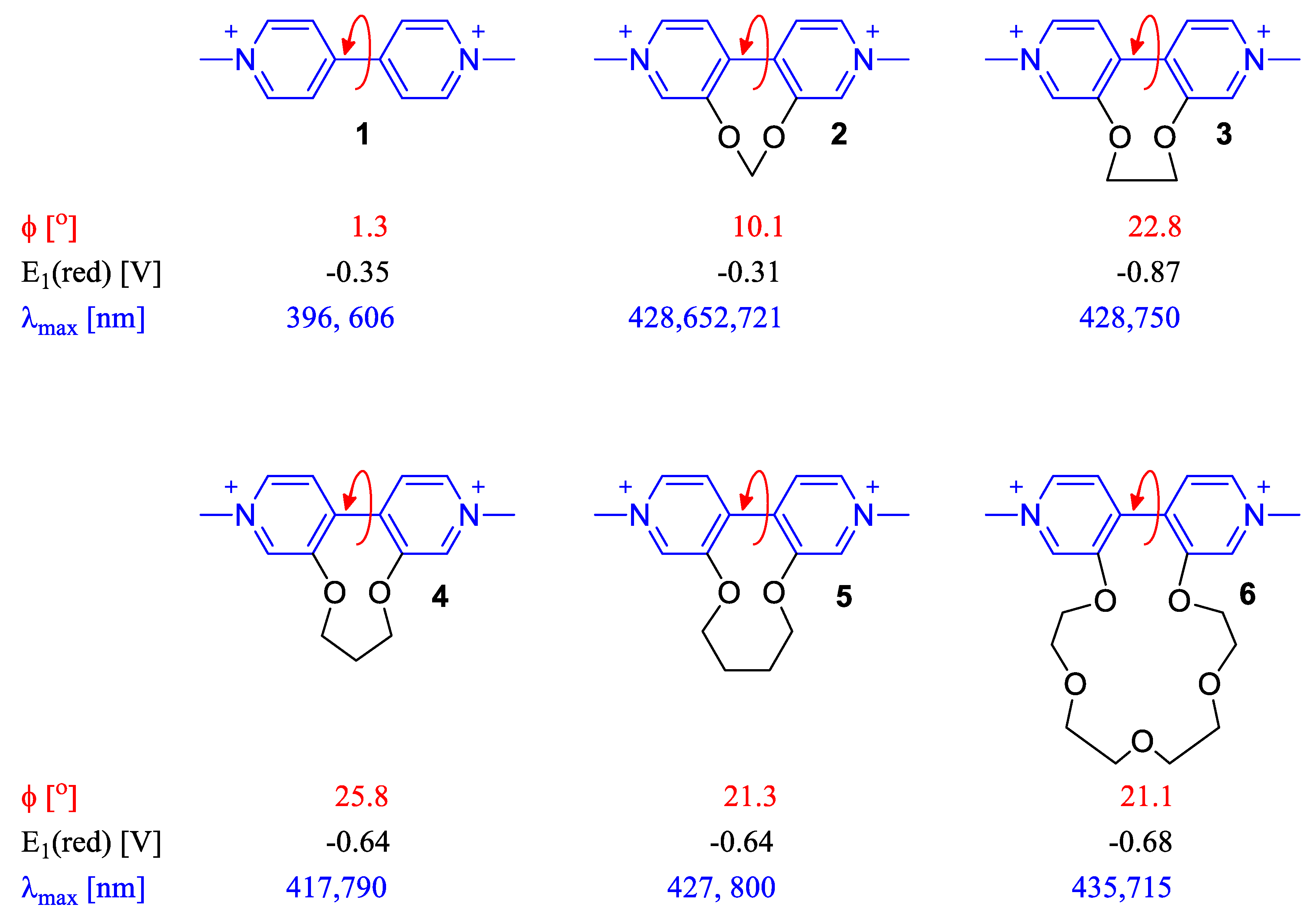
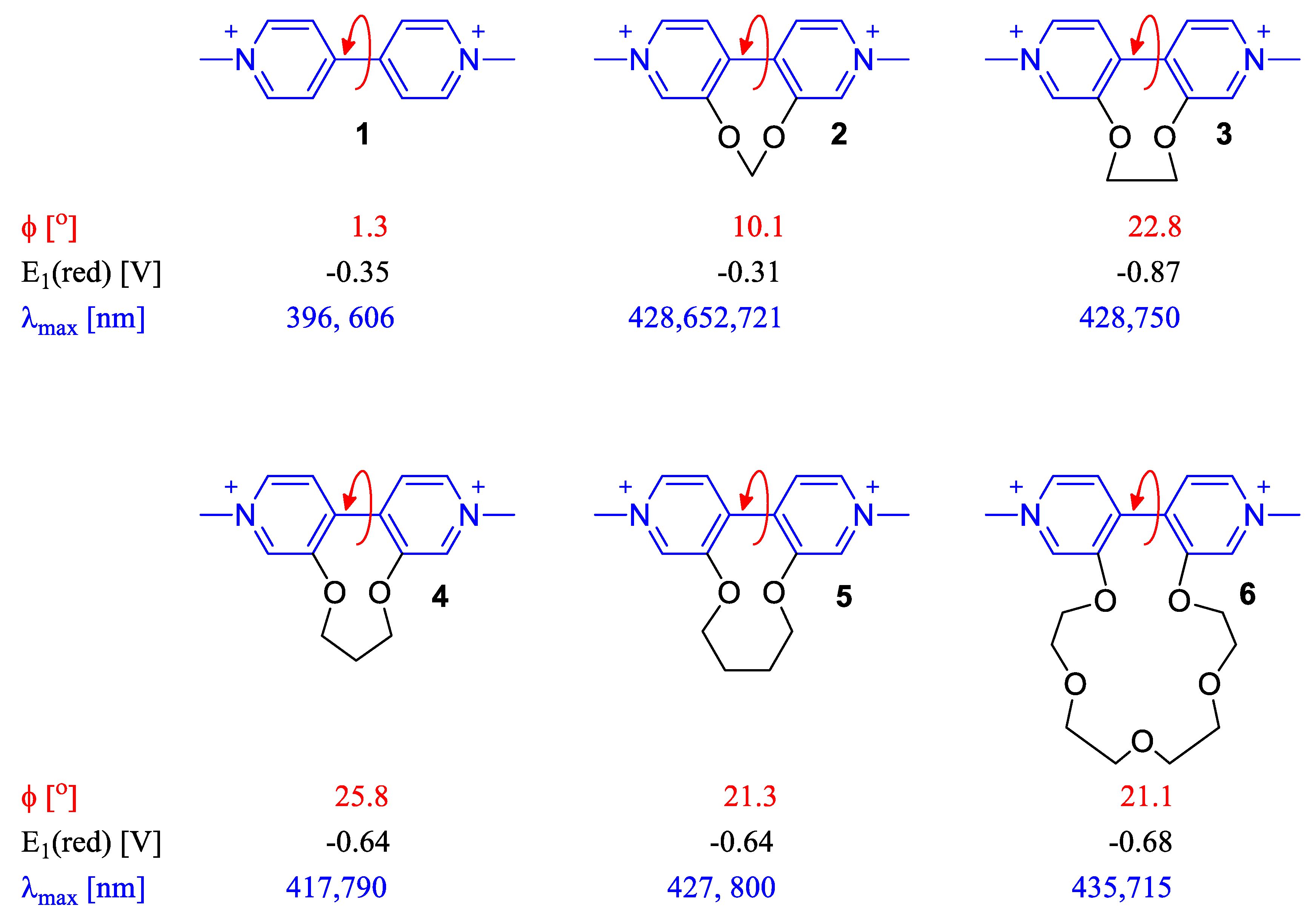
2.2. Dihedral Angle Importance in Optical and Electronic Applications
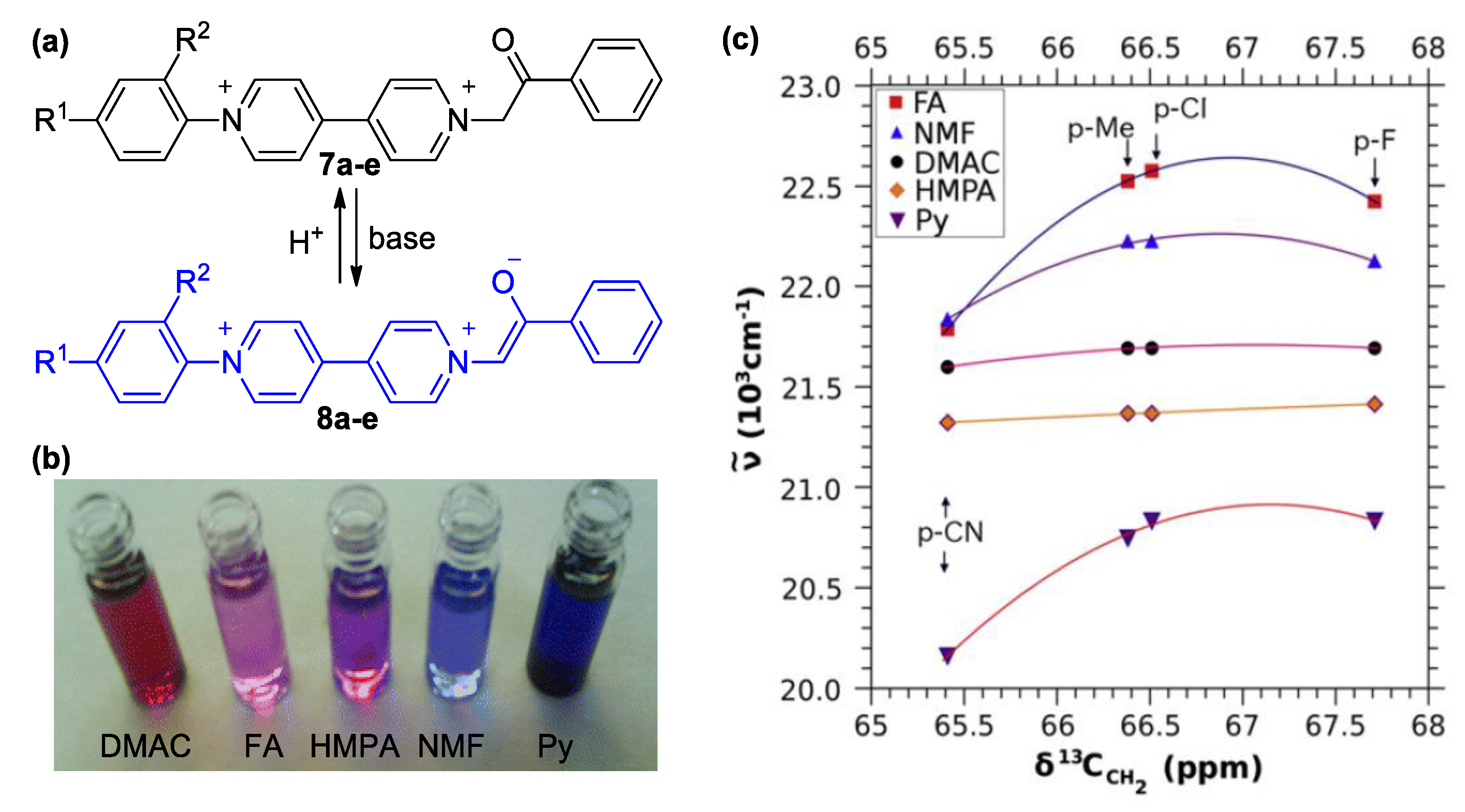
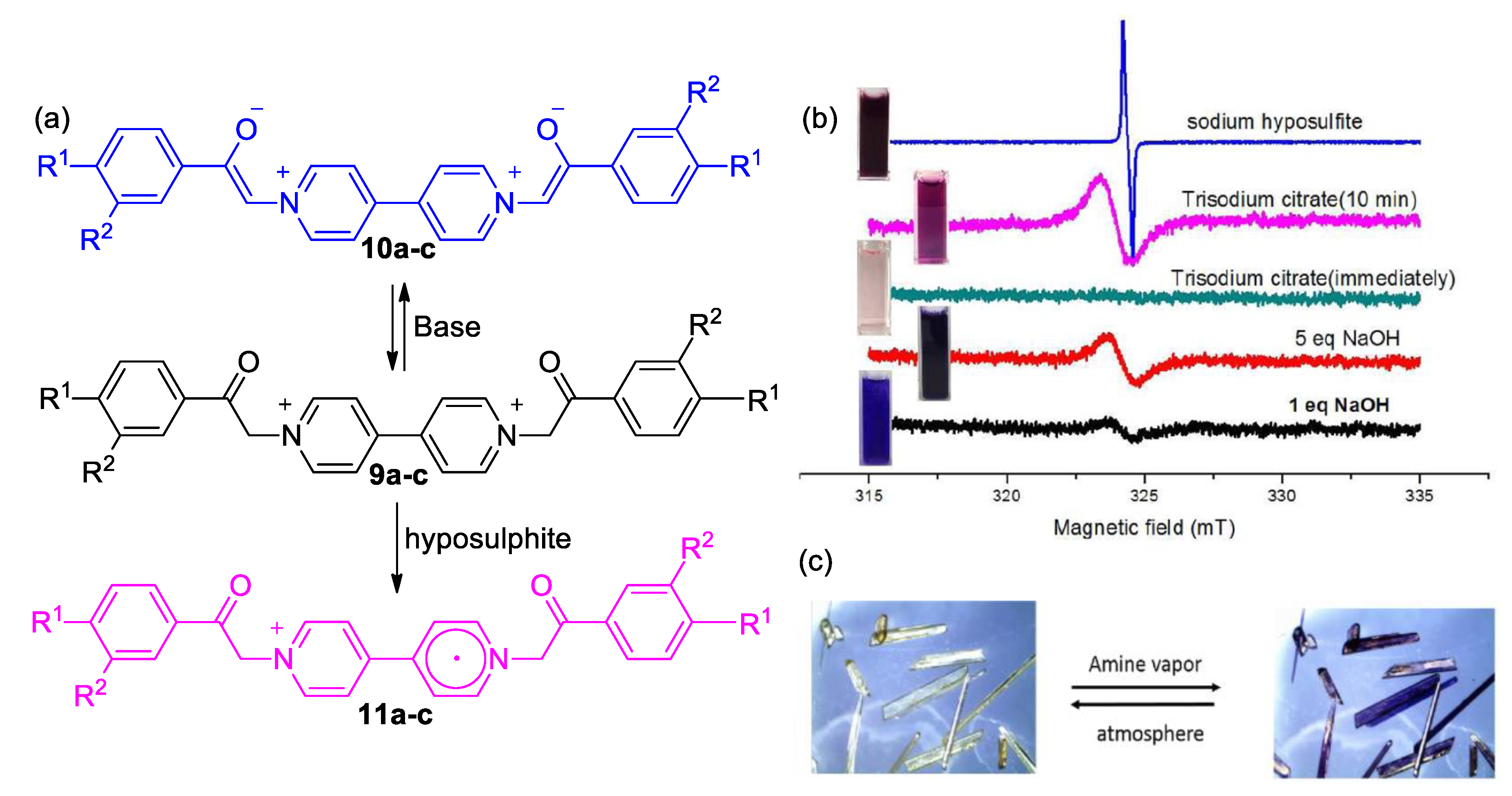
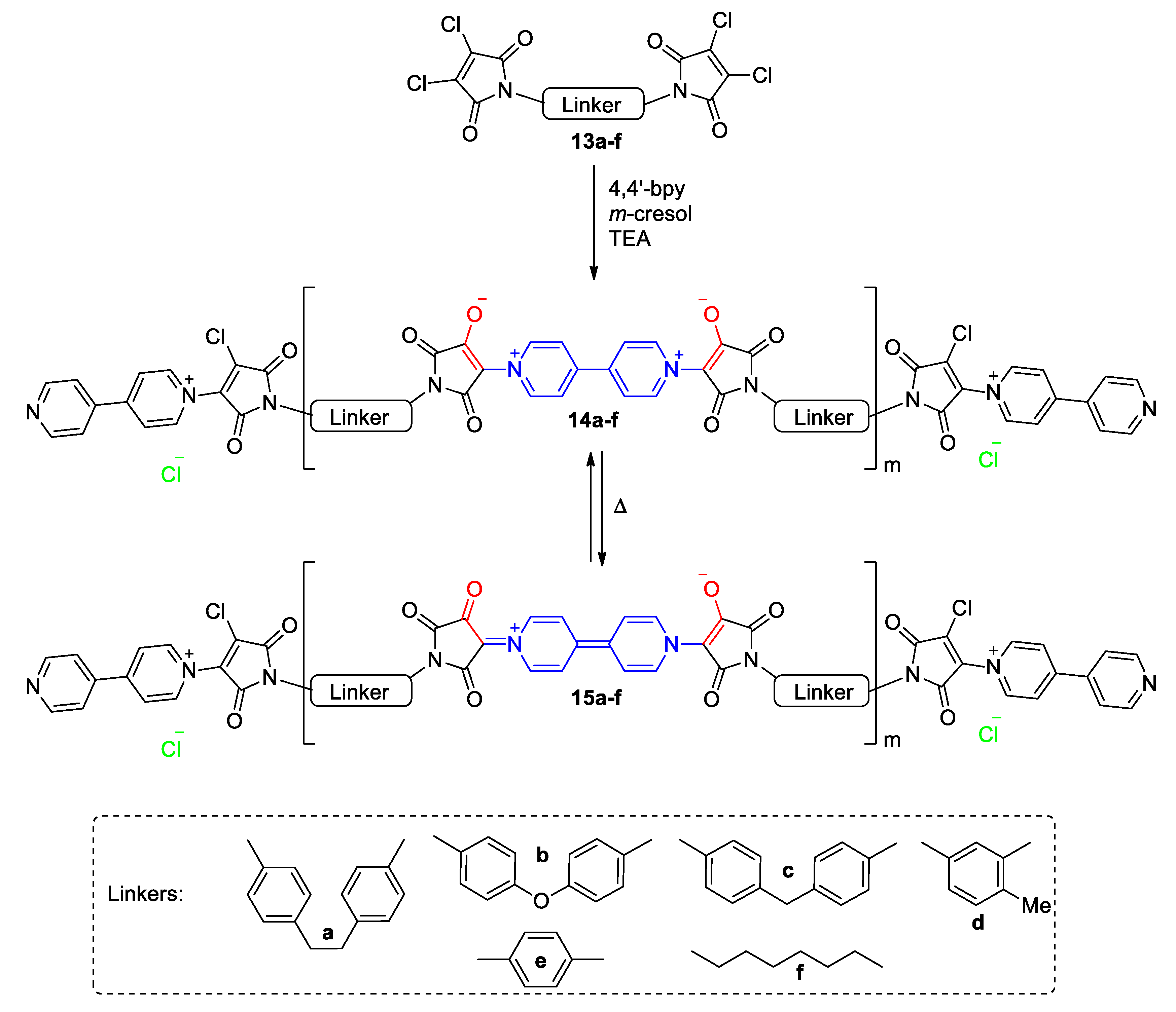
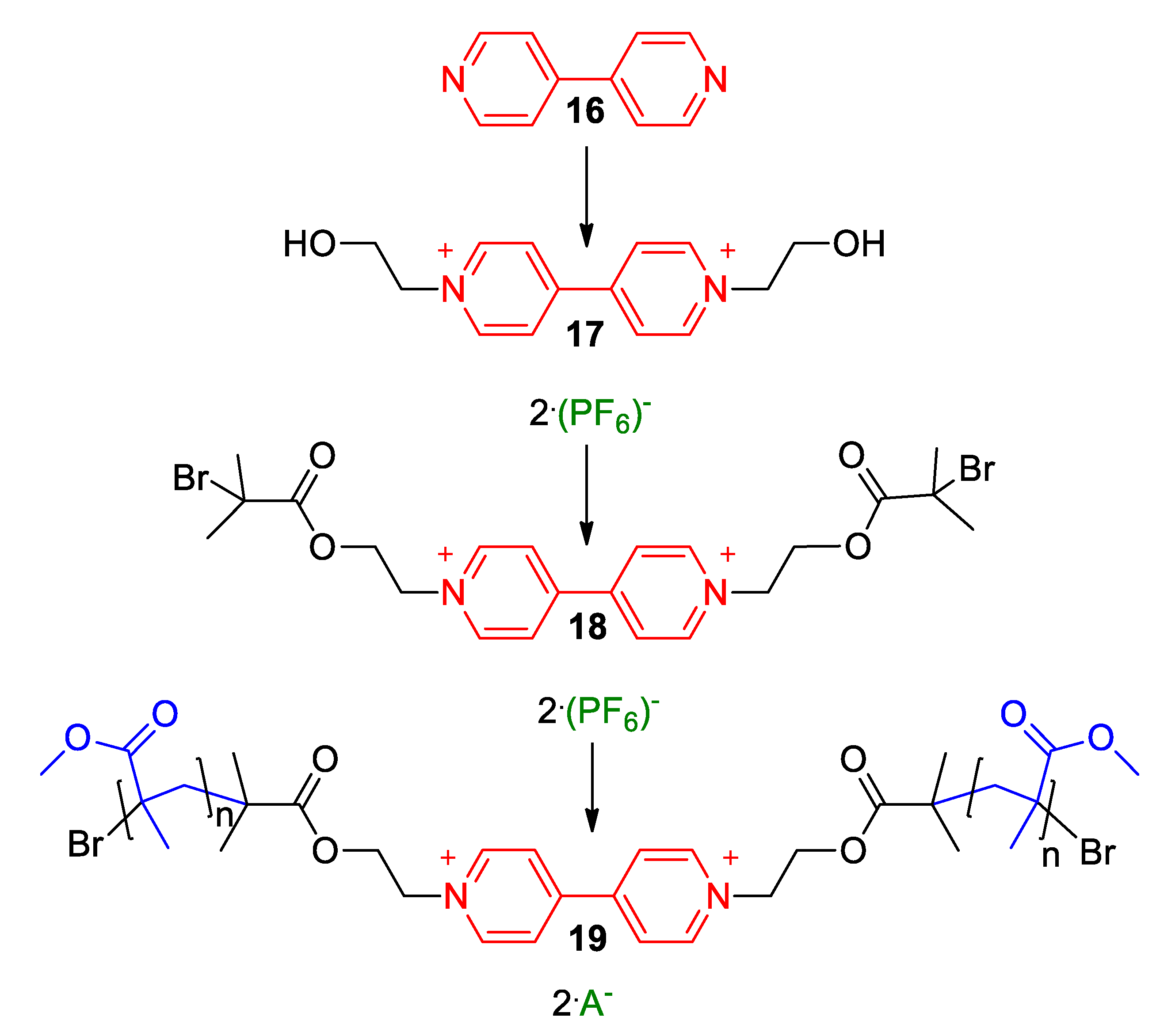
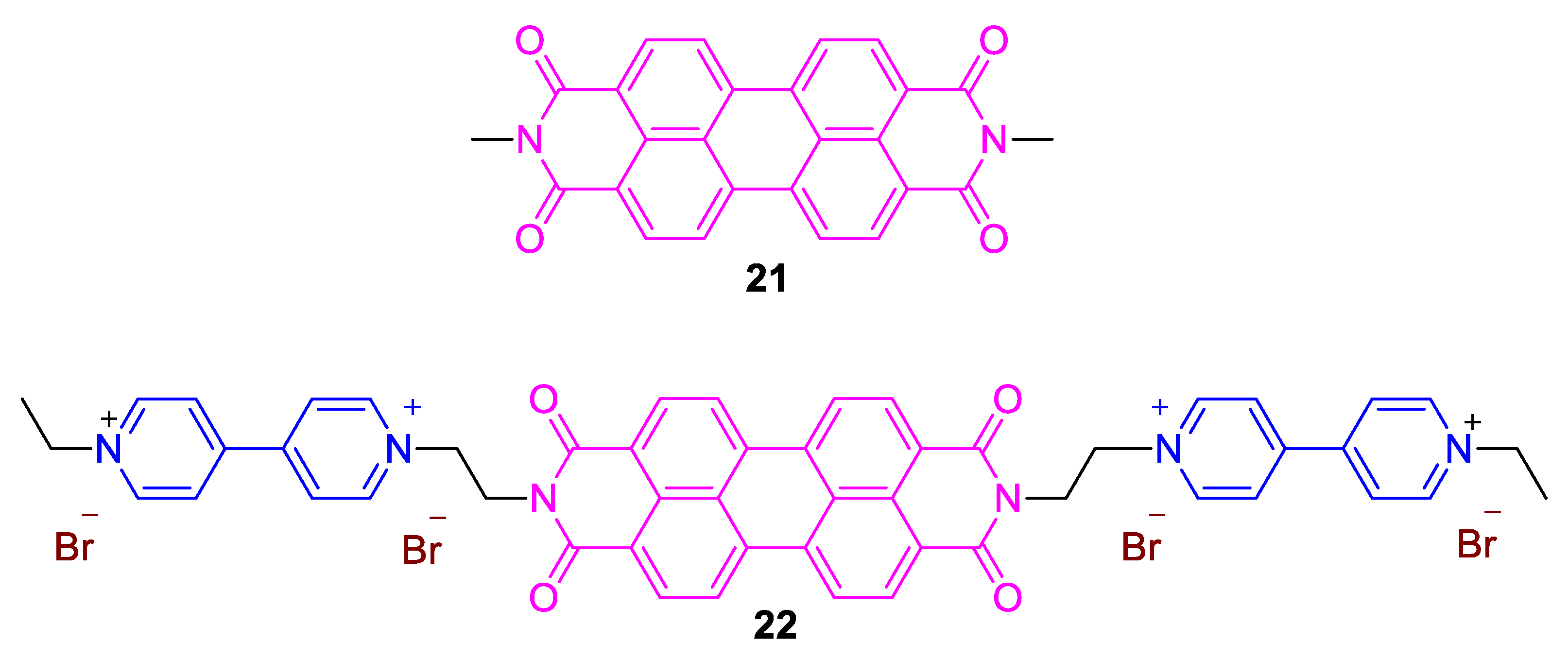
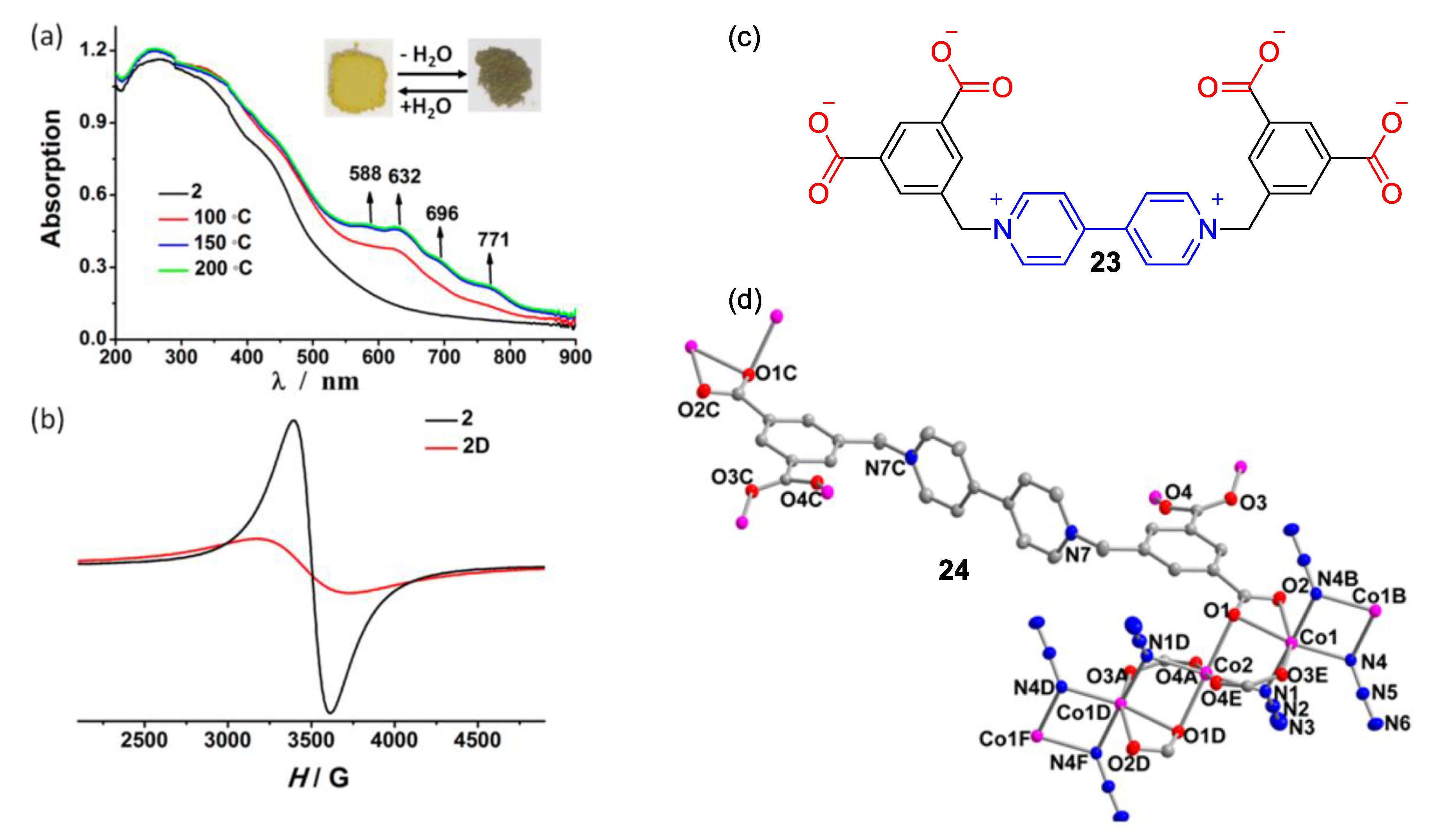
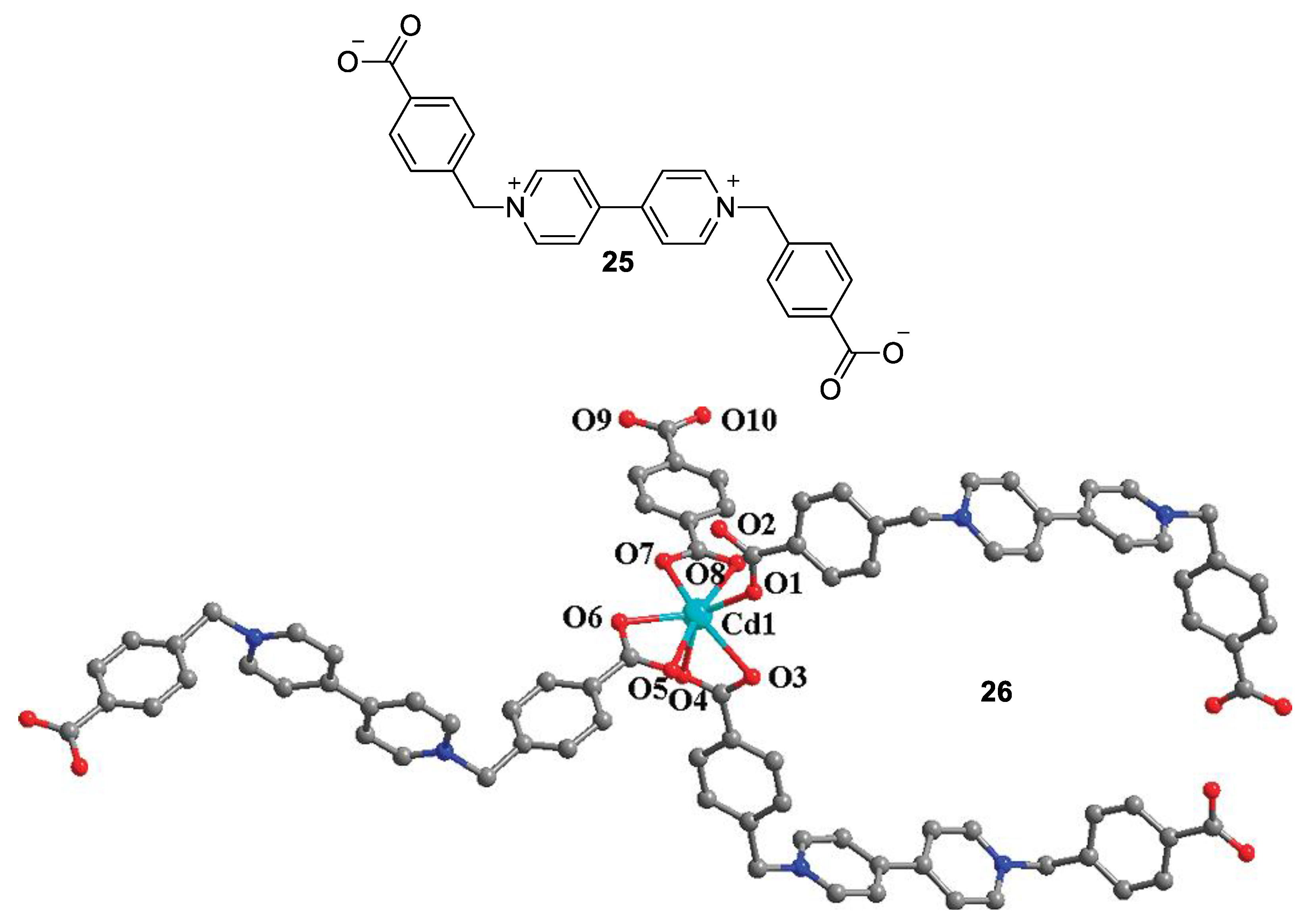
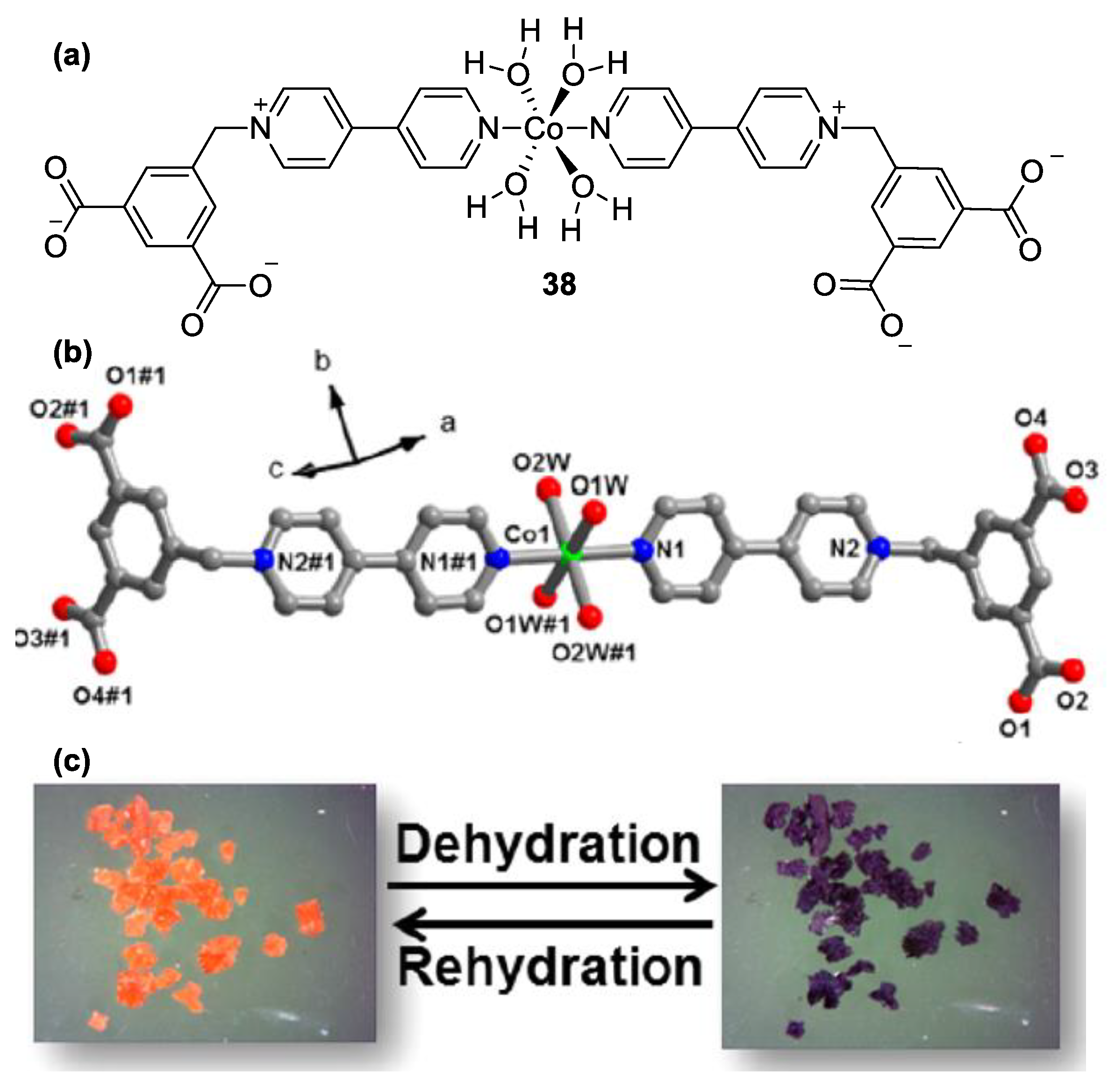
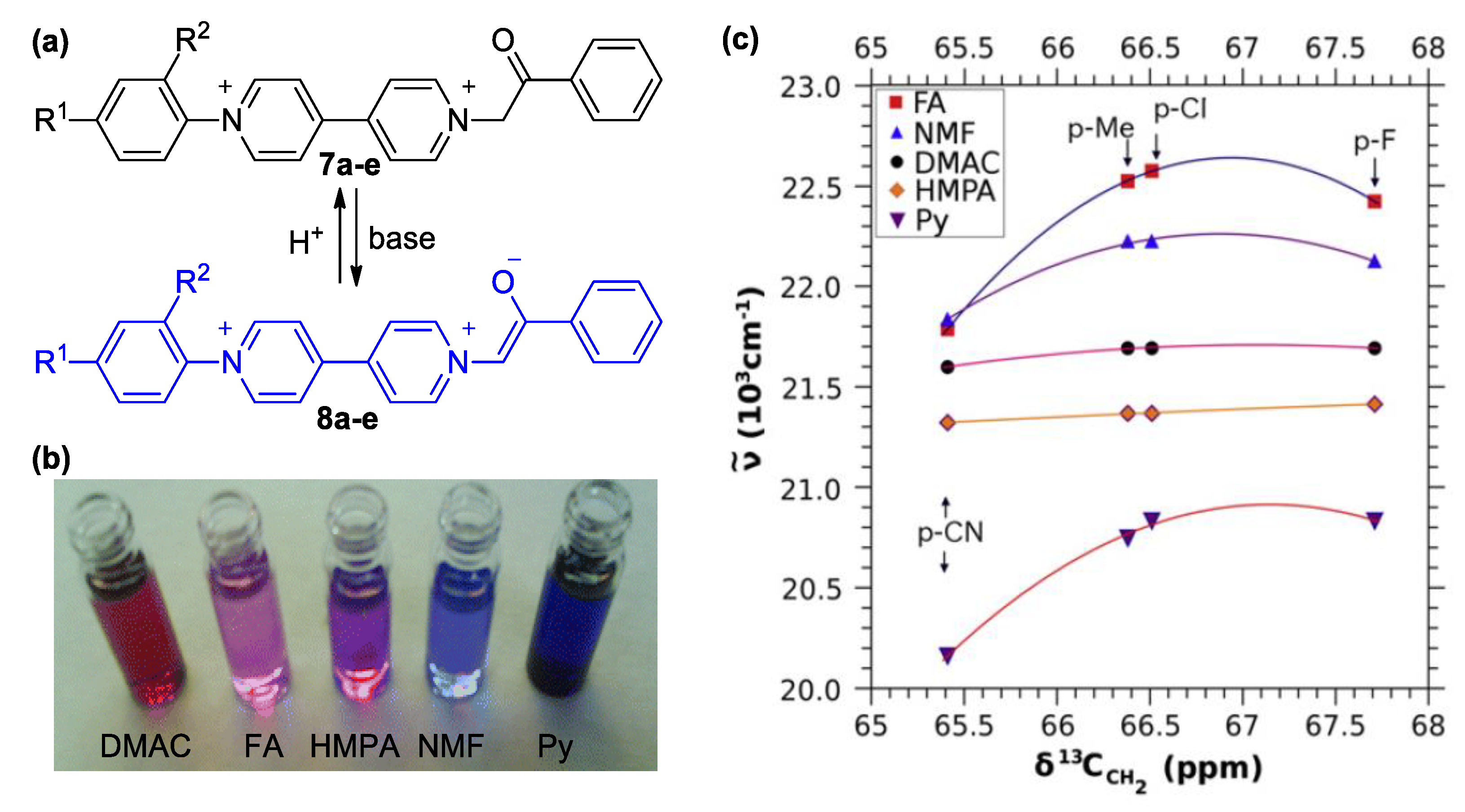
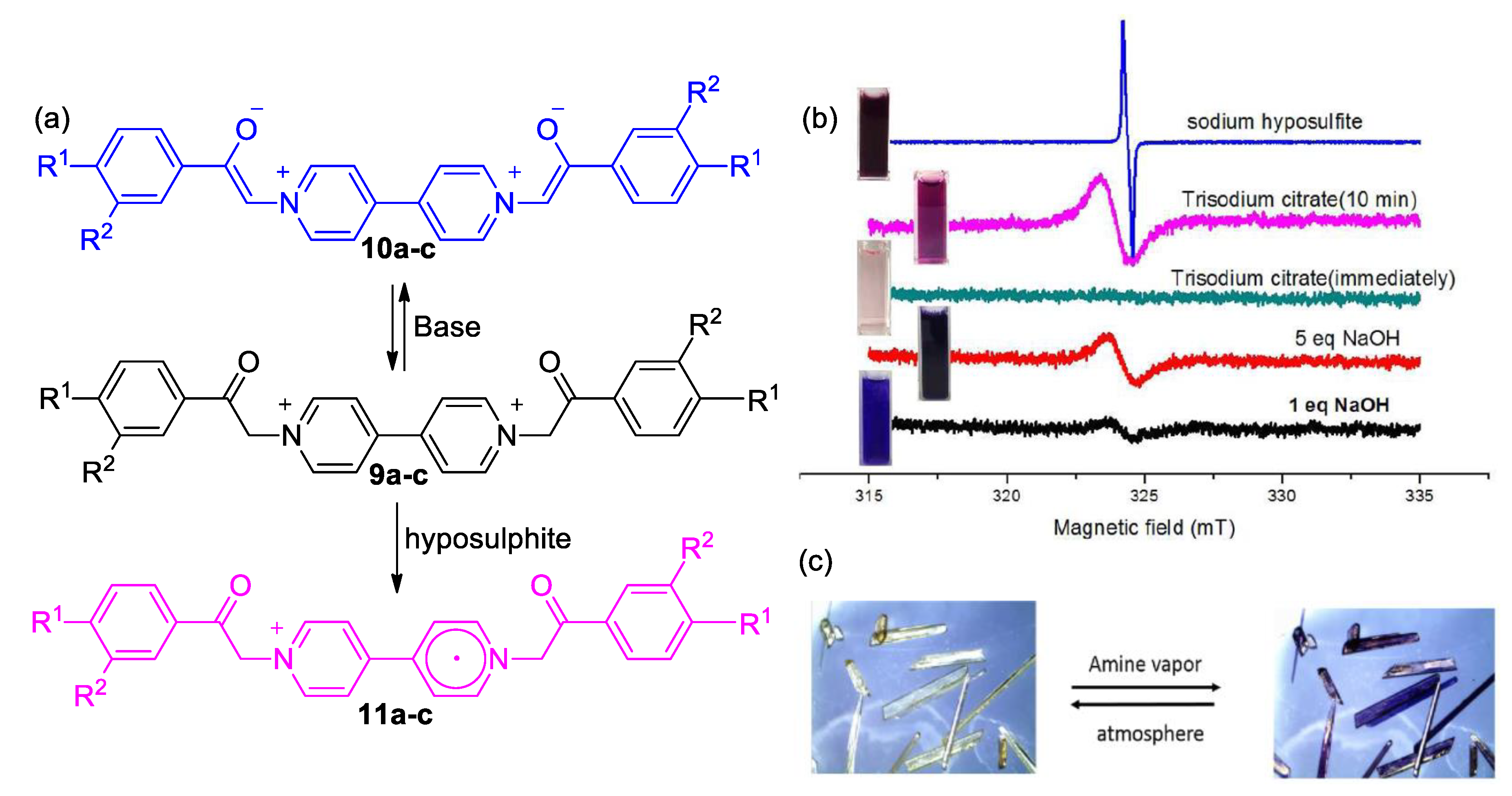
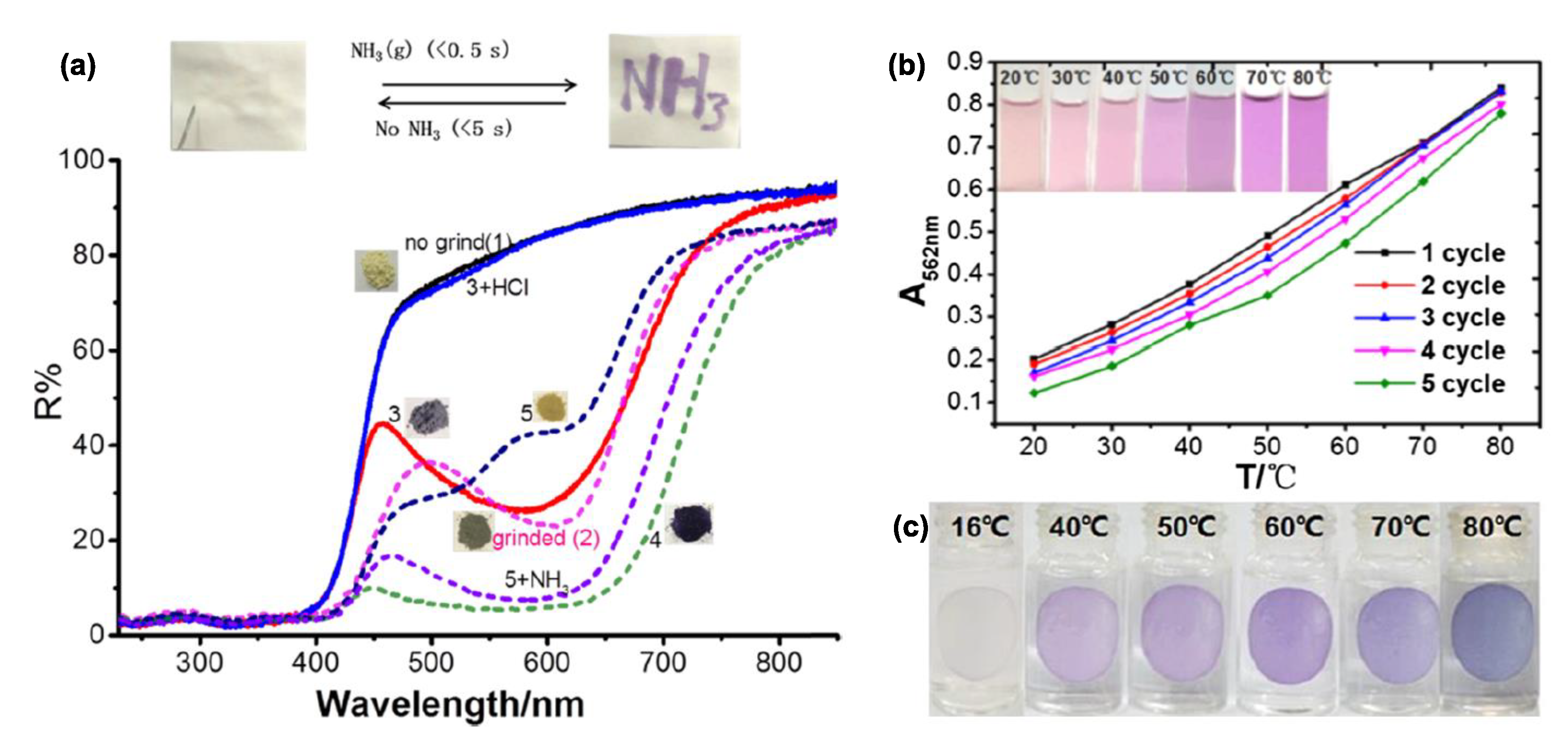
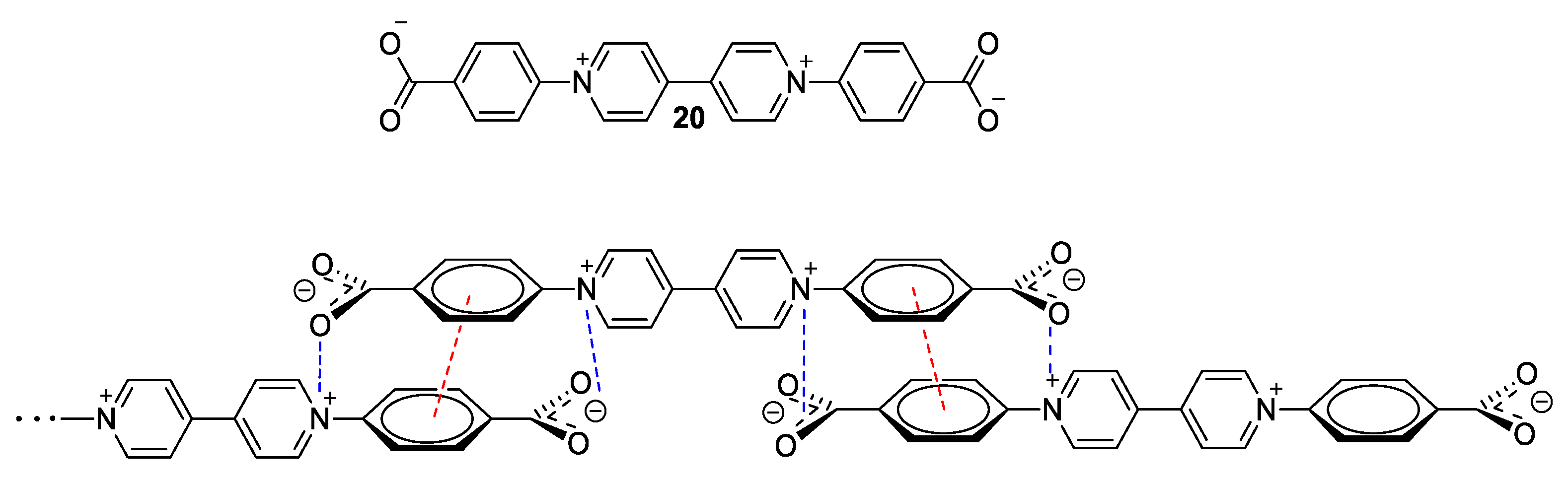
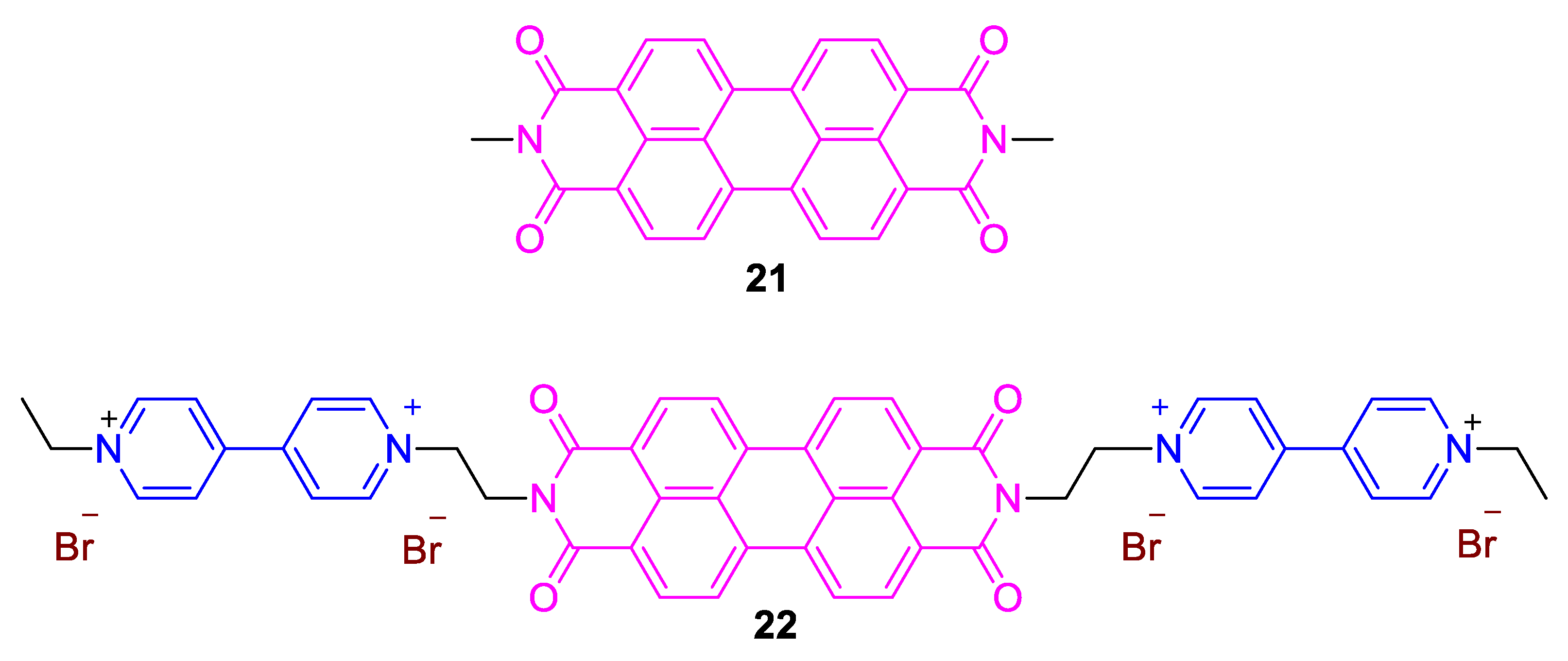
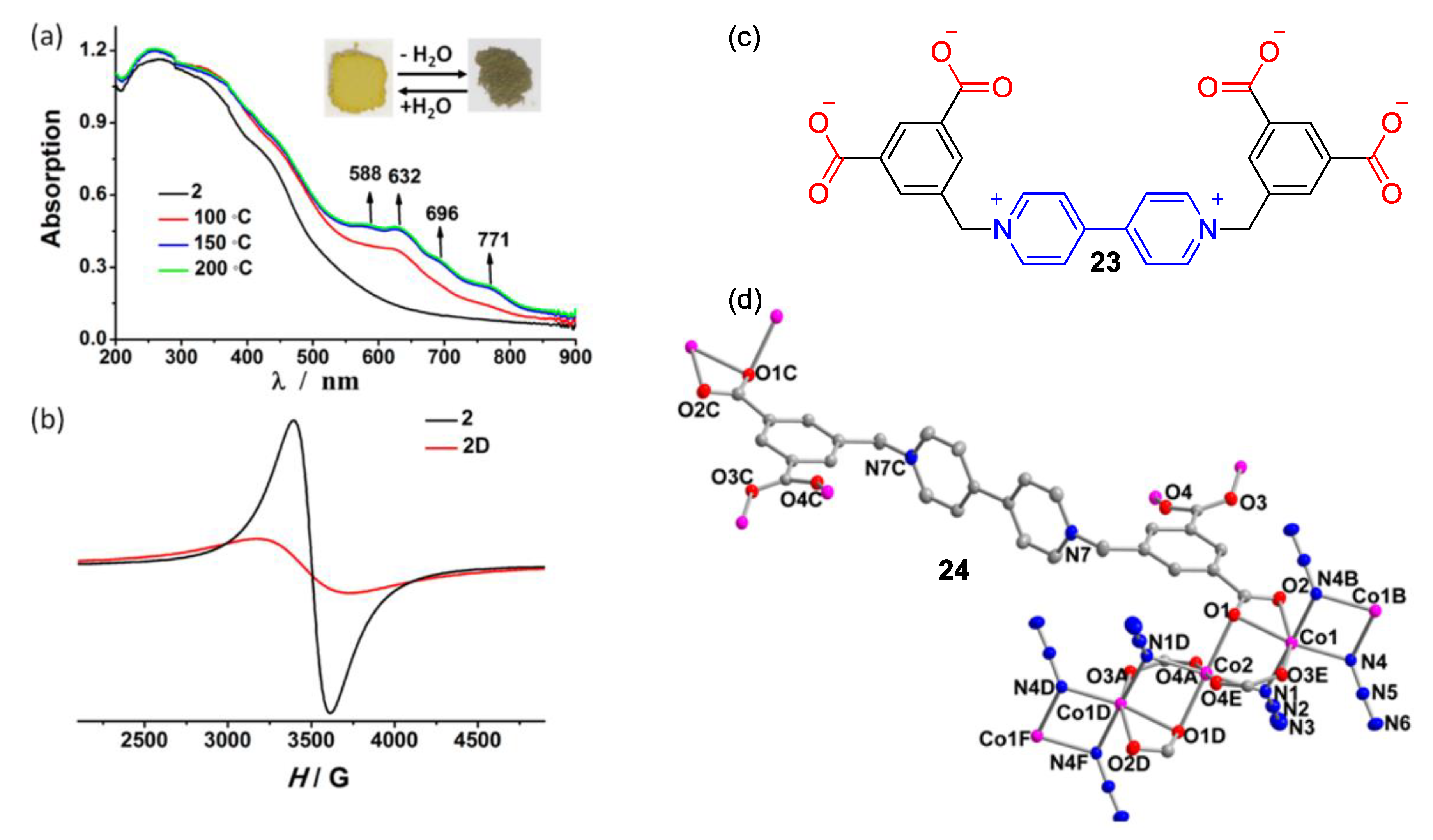
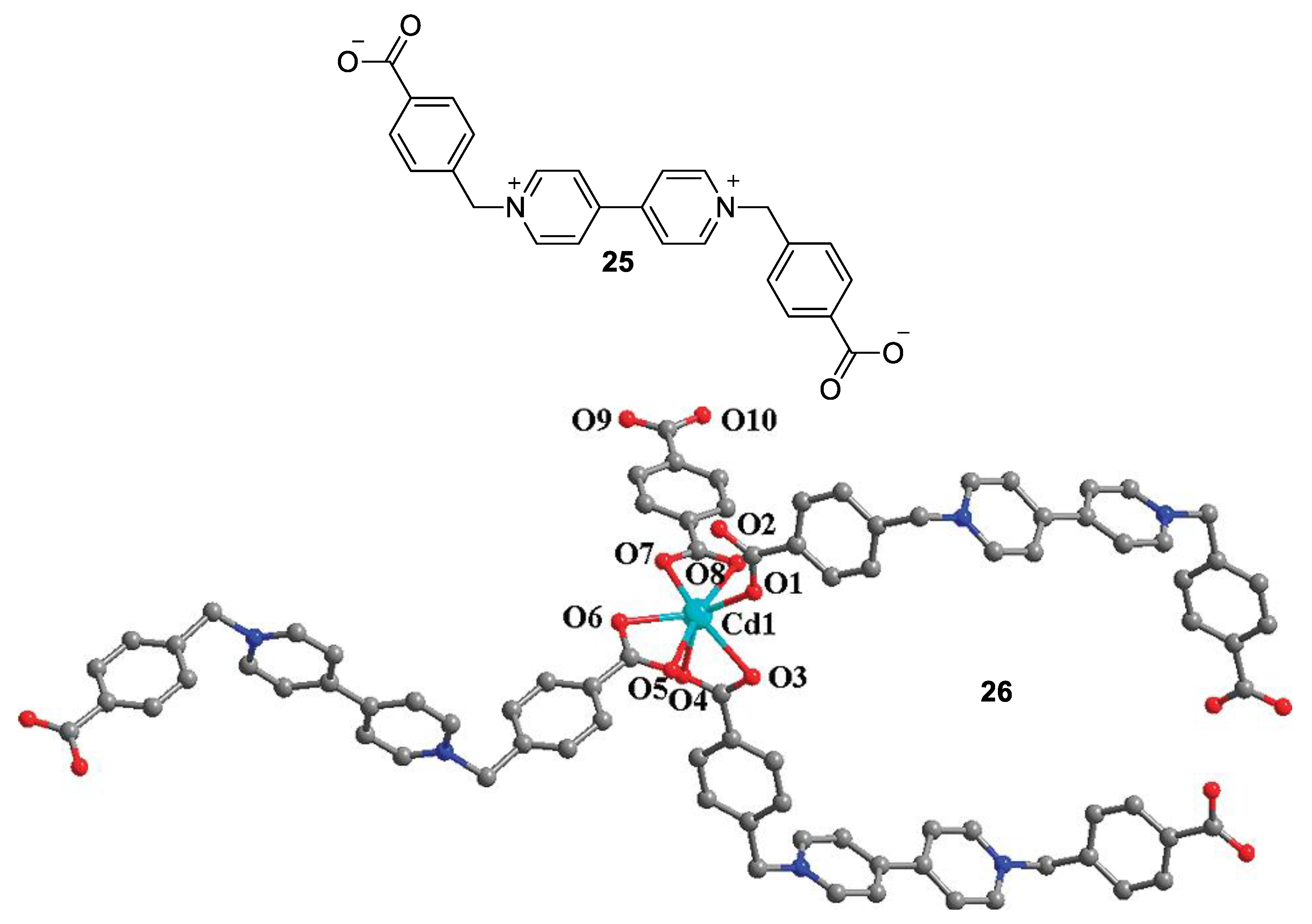
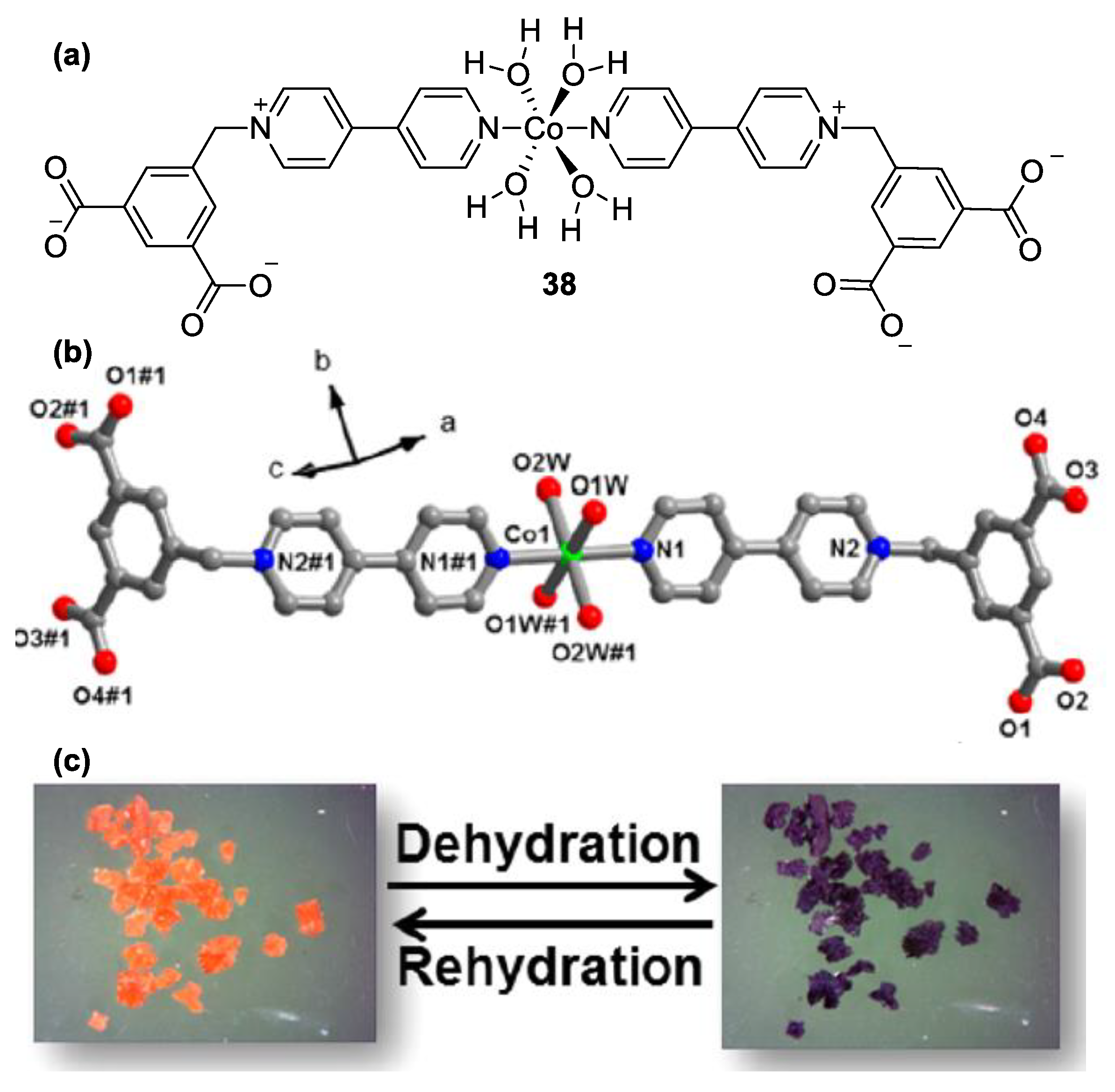
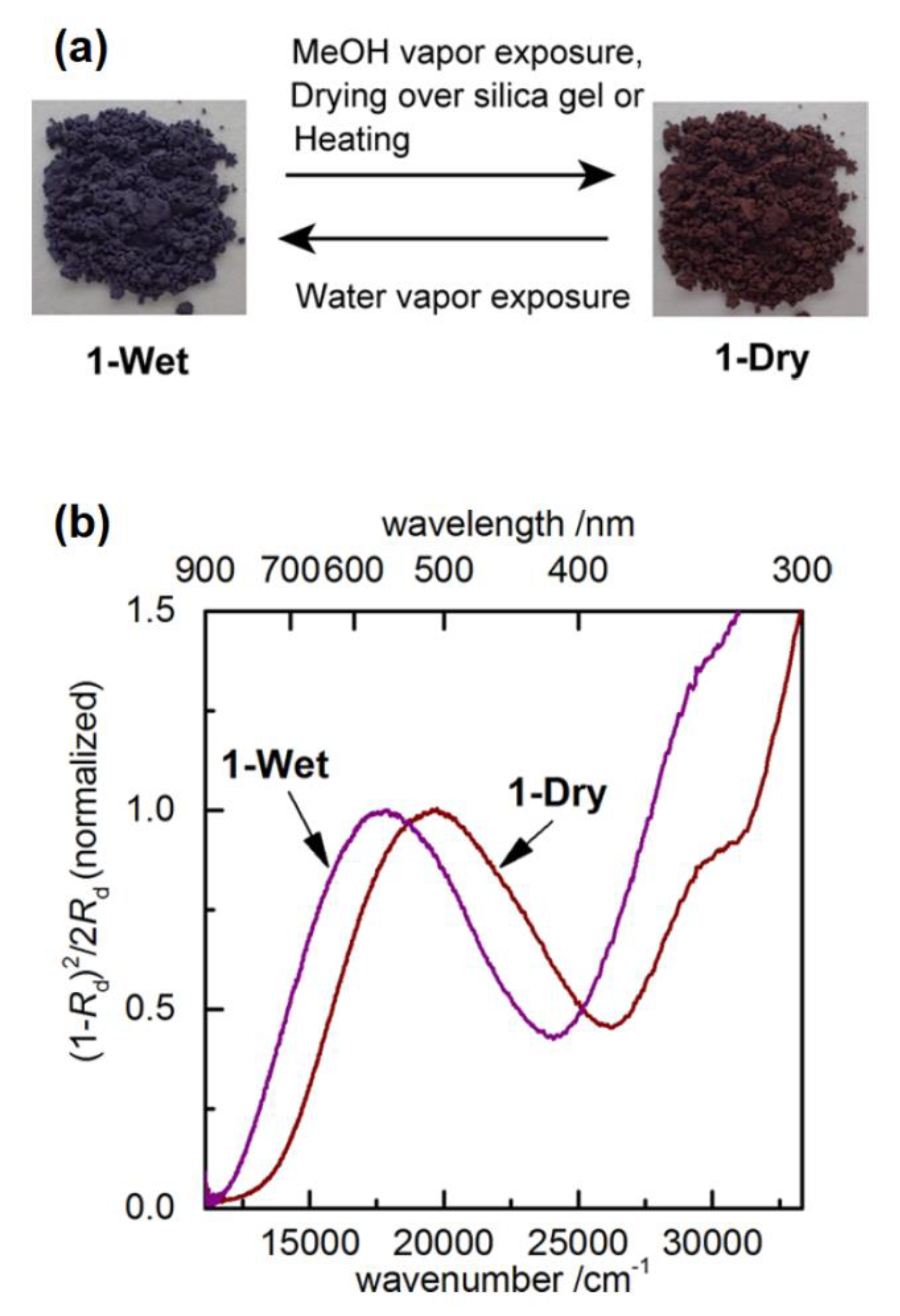
3. Direct Medium- and Environment-Responsive Viologens
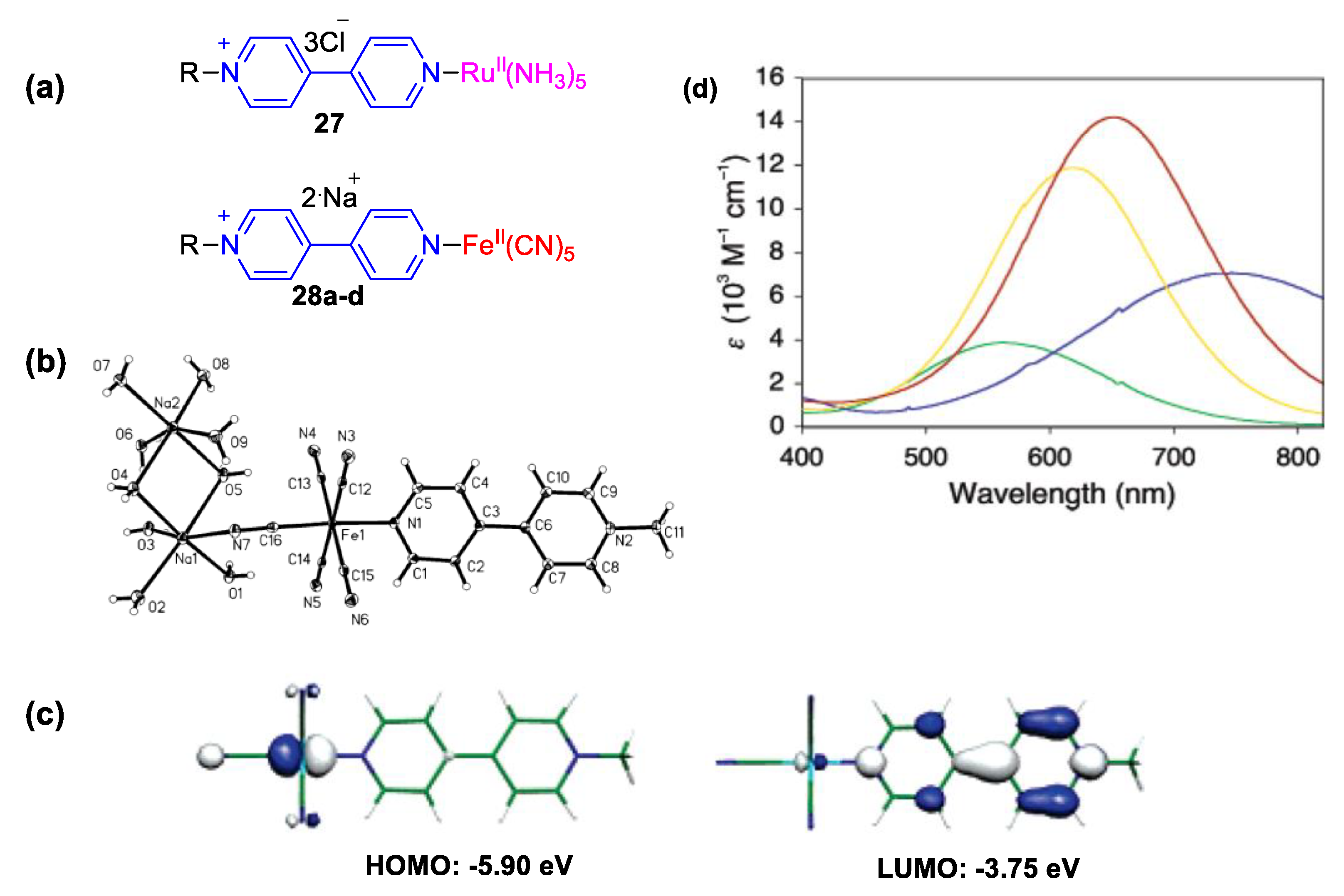
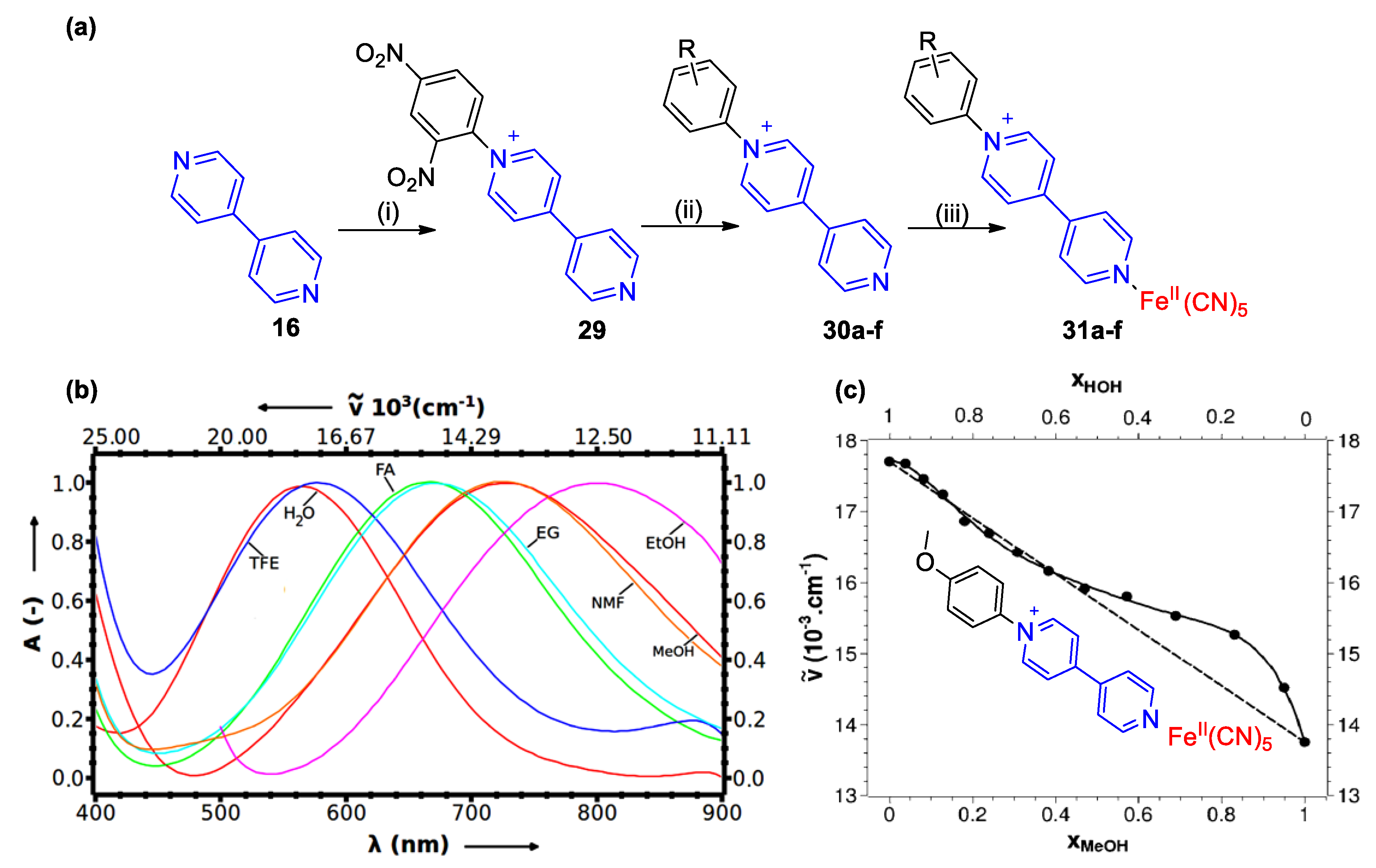
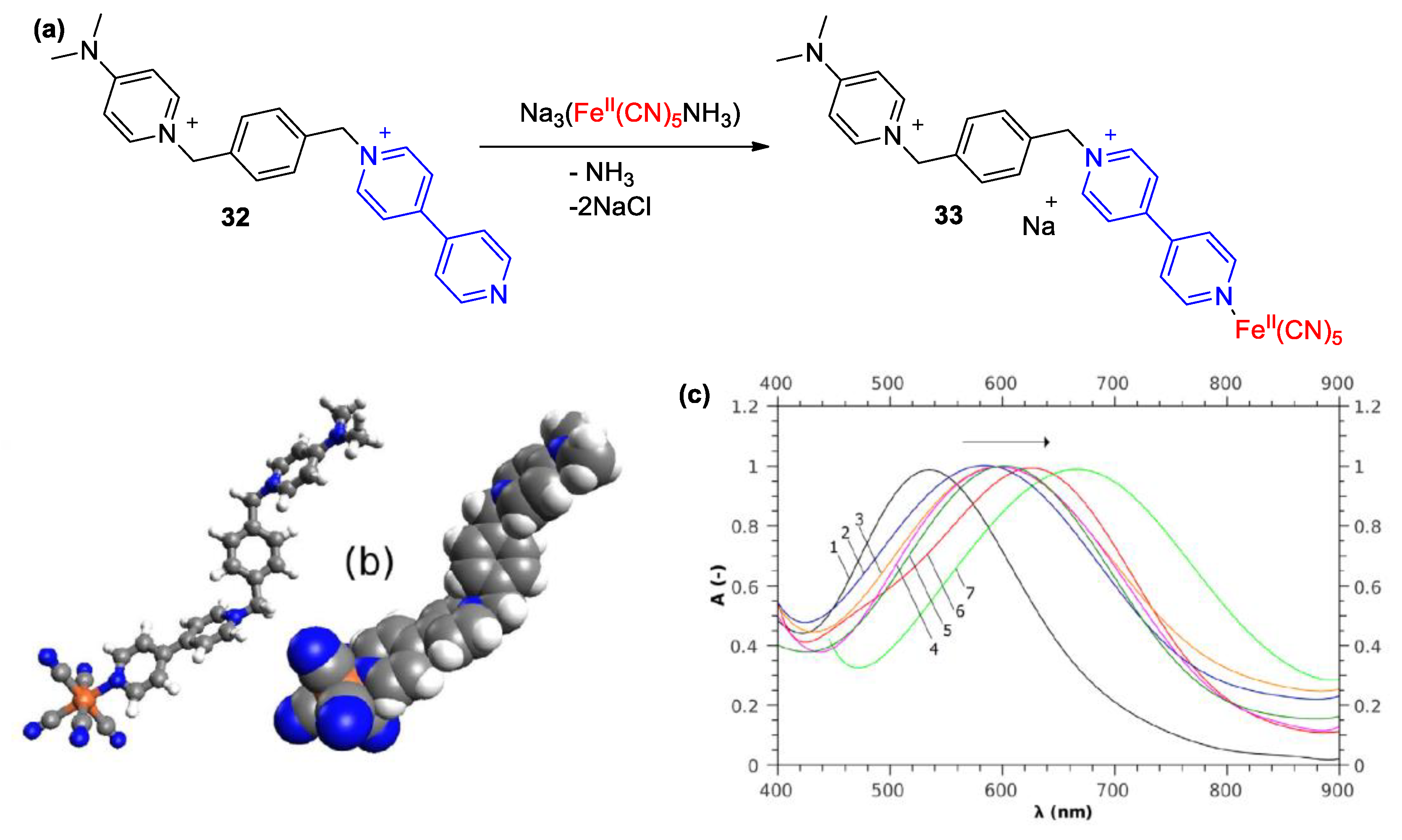
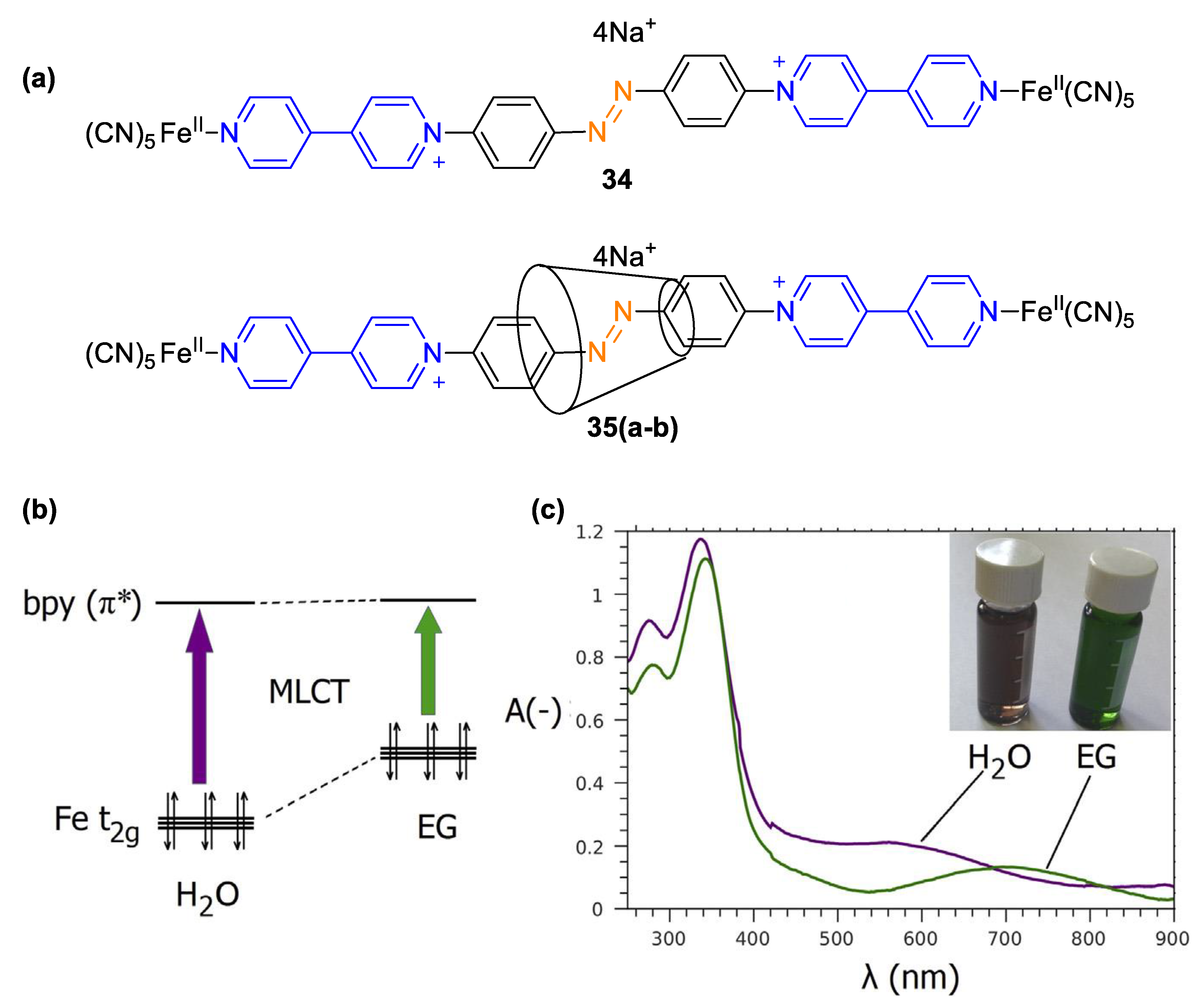
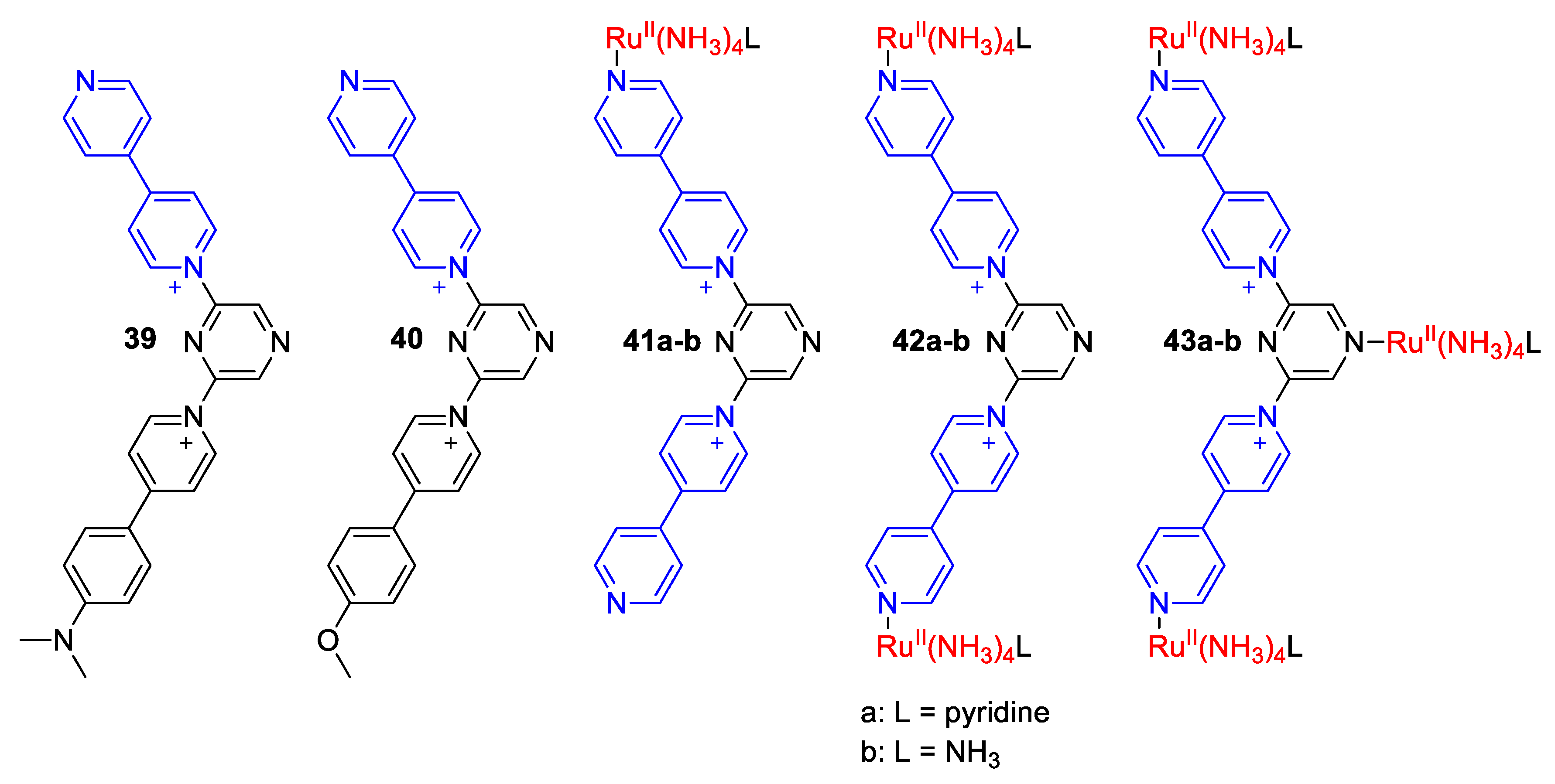
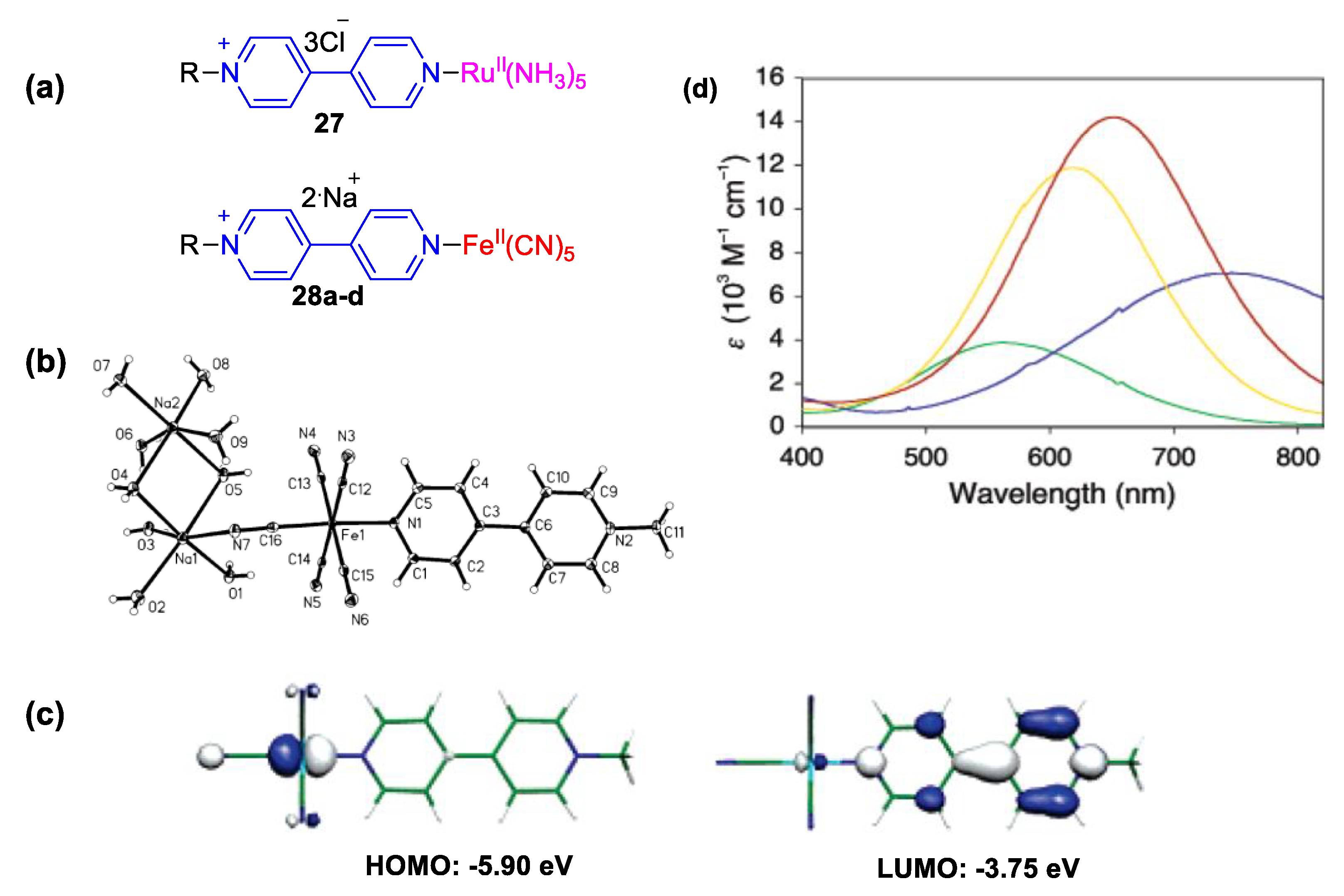
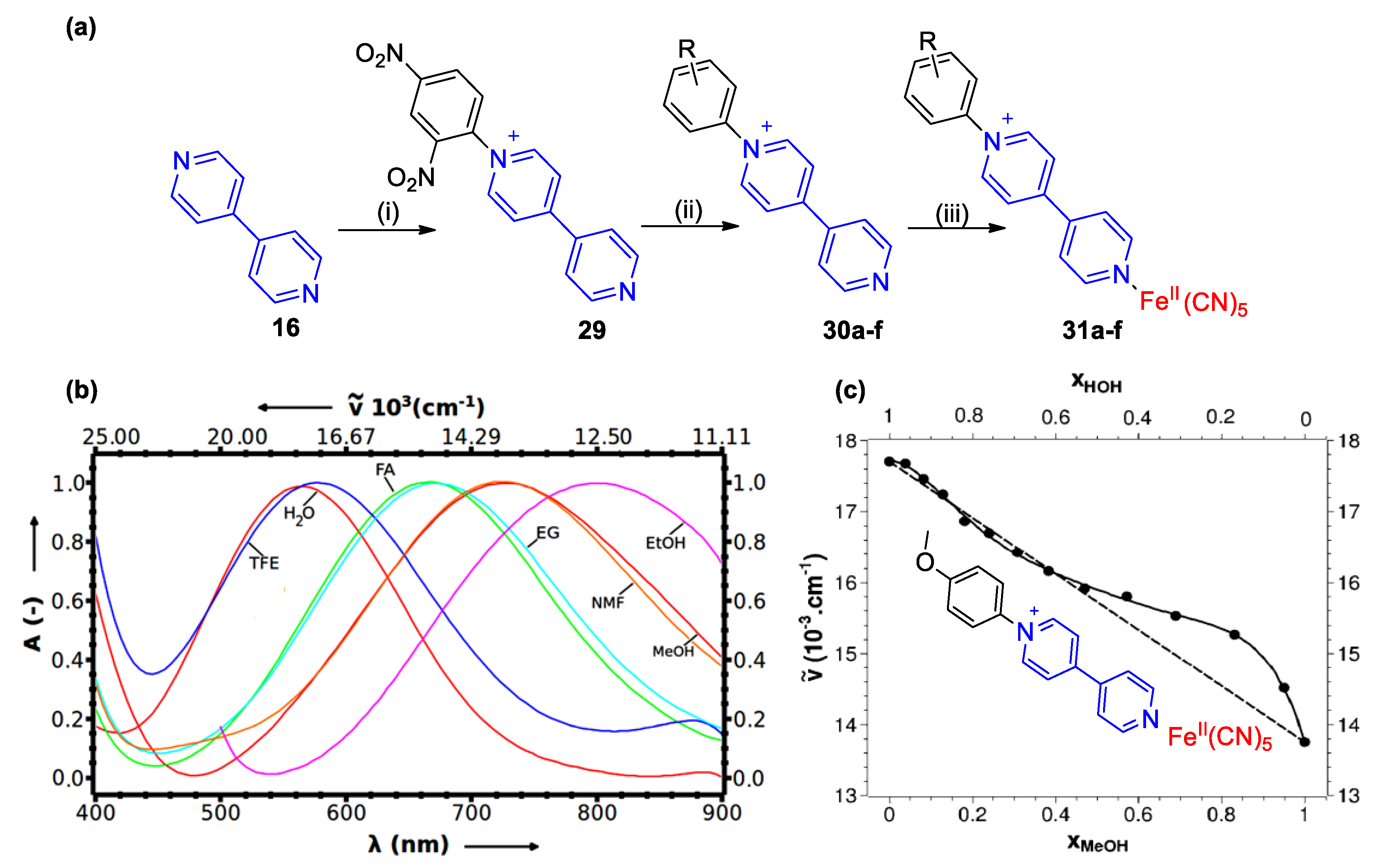
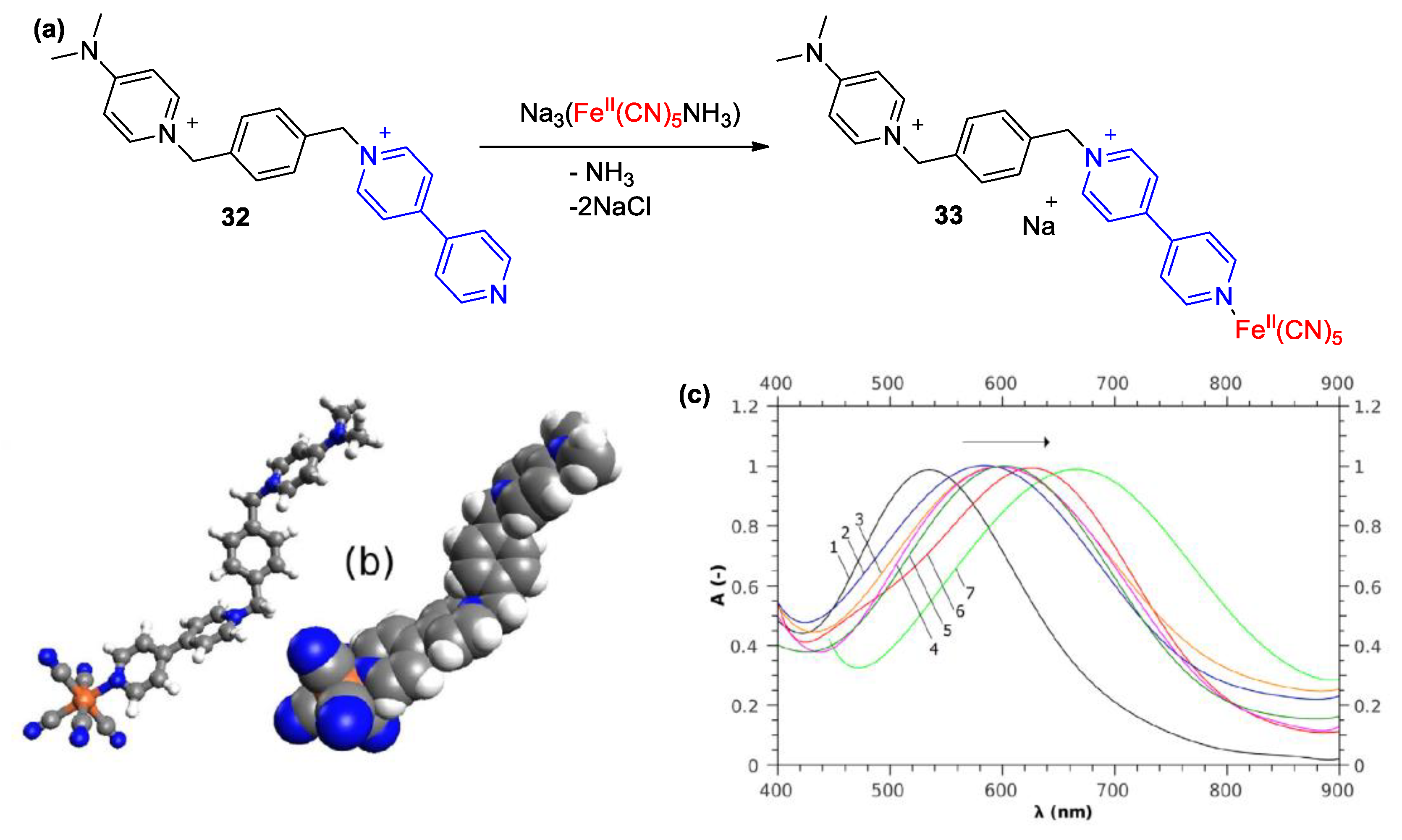
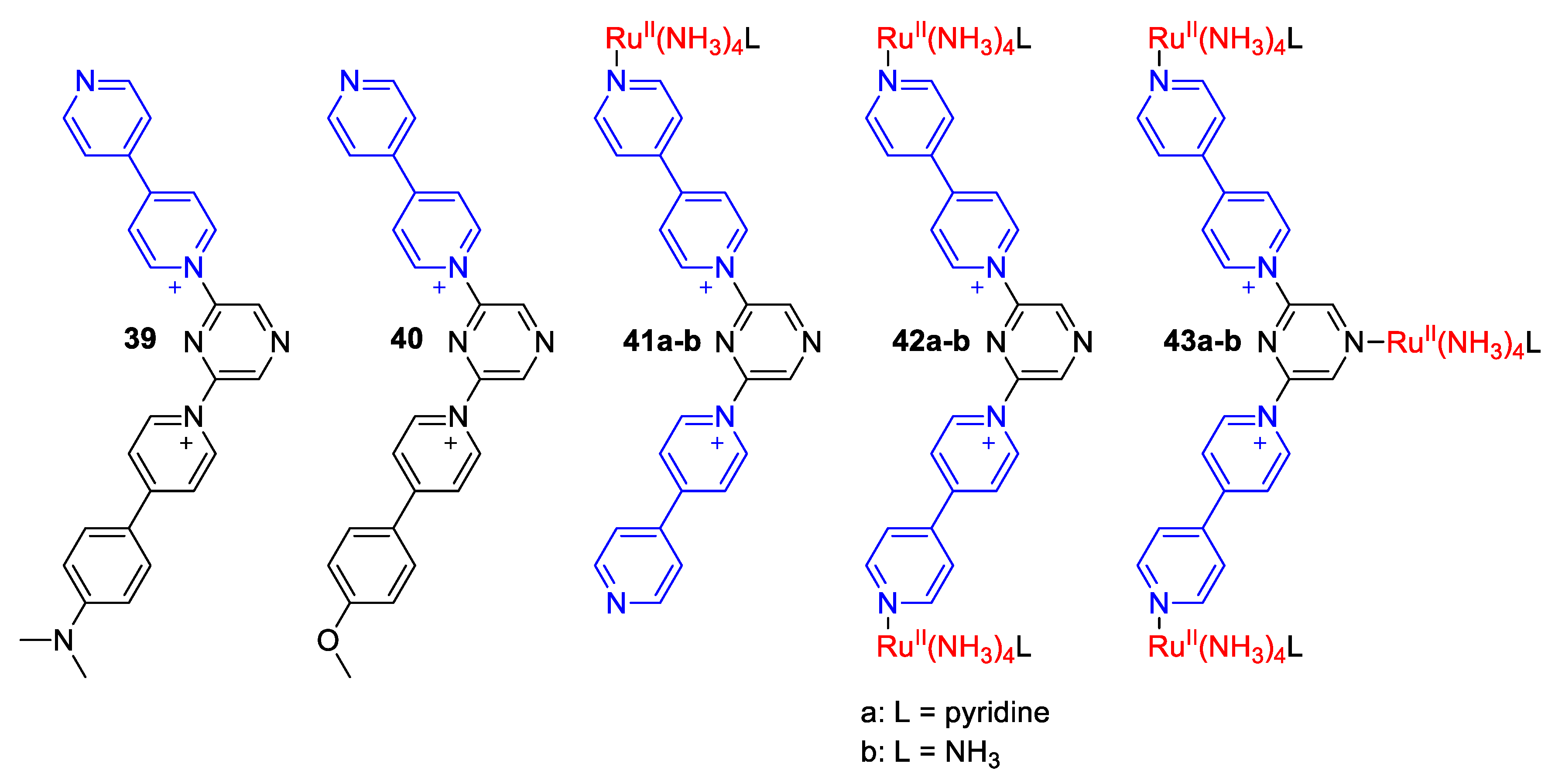
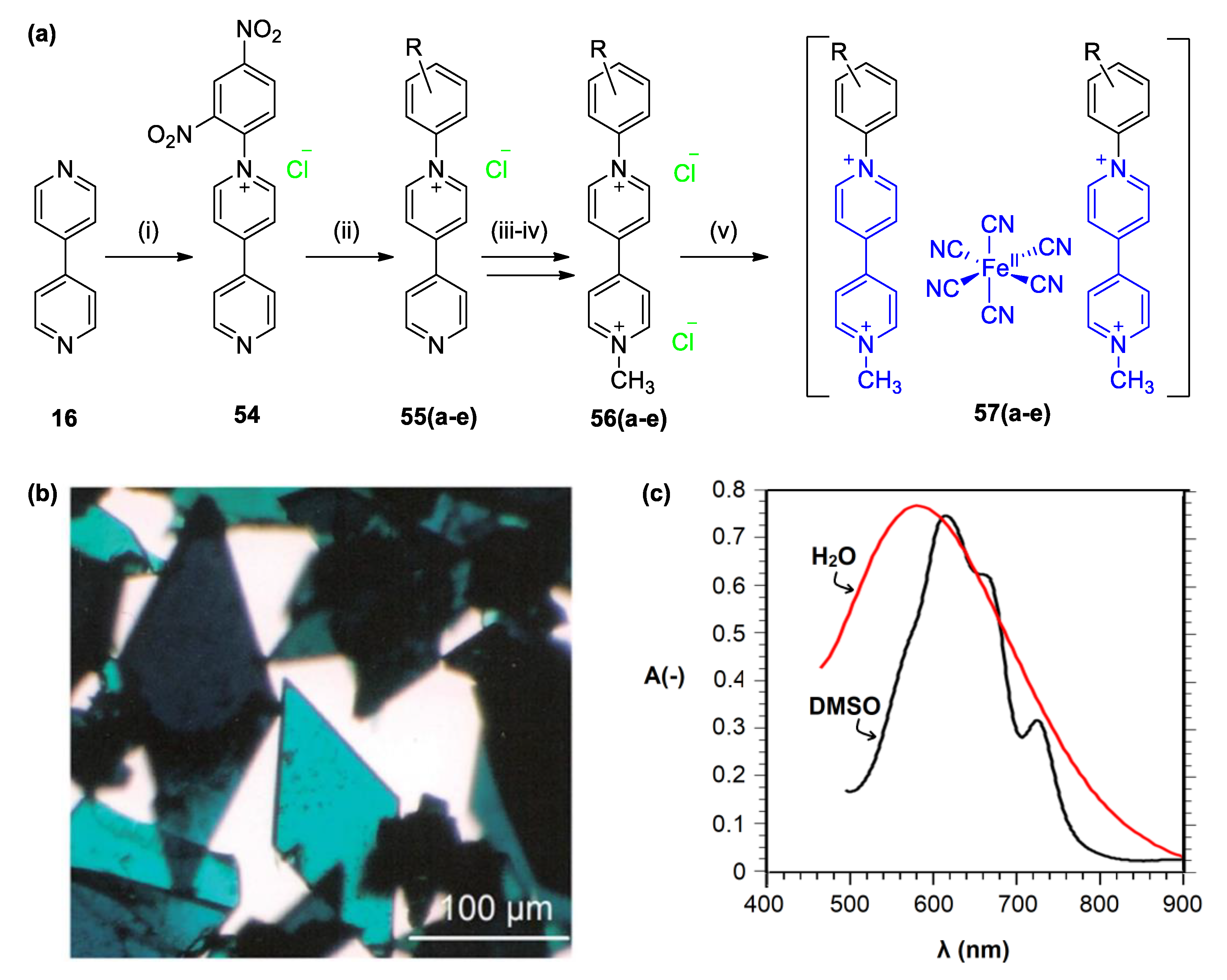
4. Solvatochromic Complexes Involving Monoquats as Ligands
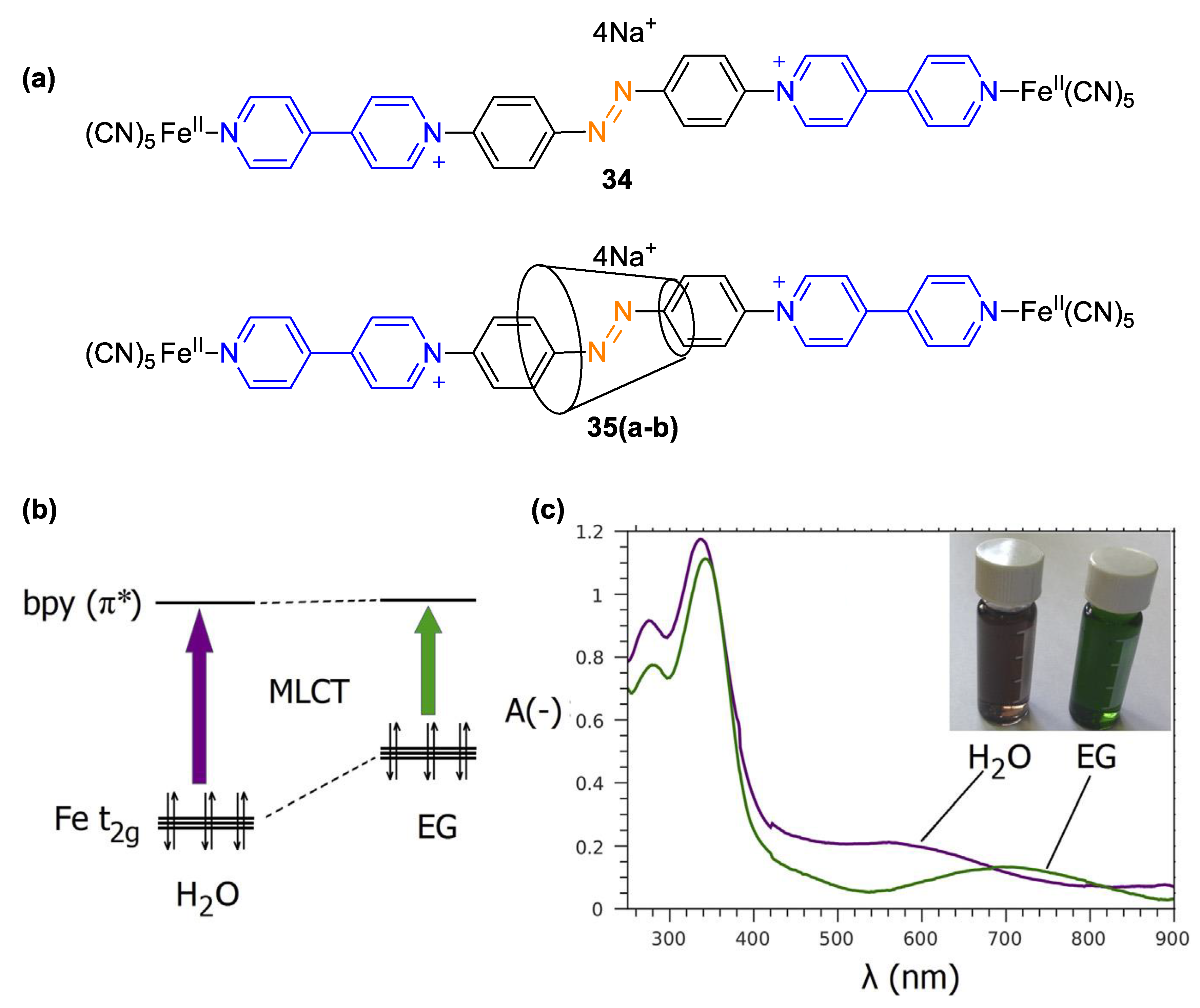
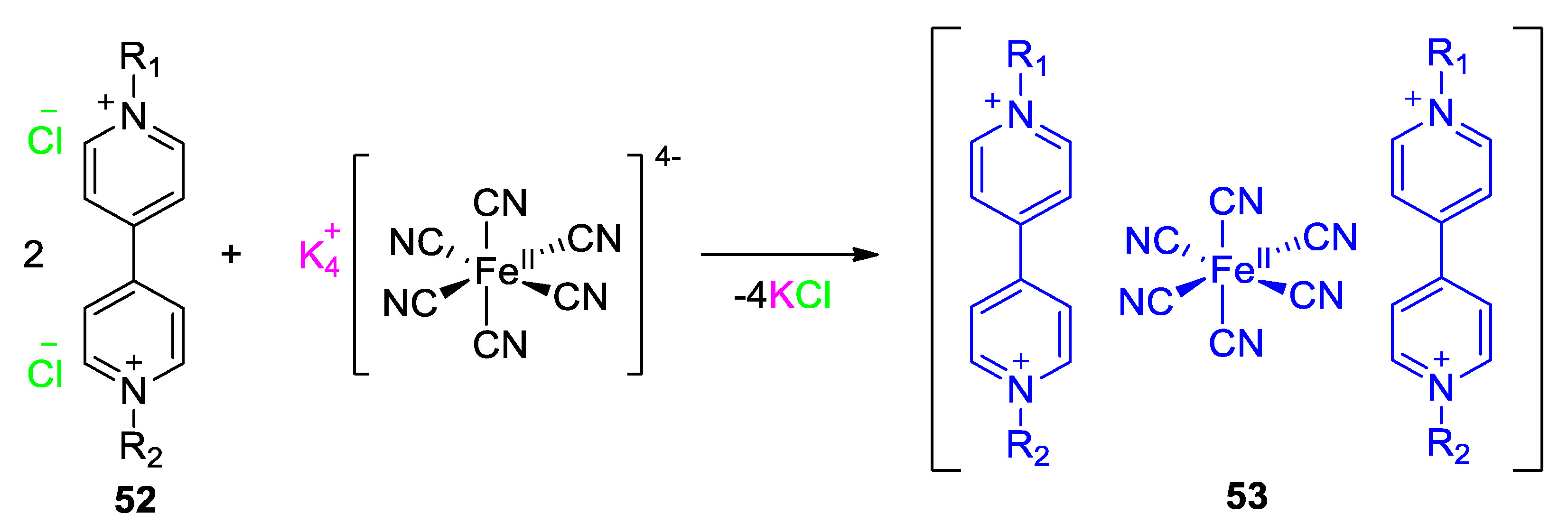
5. Multicomponent Chromic Systems Involving Viologens
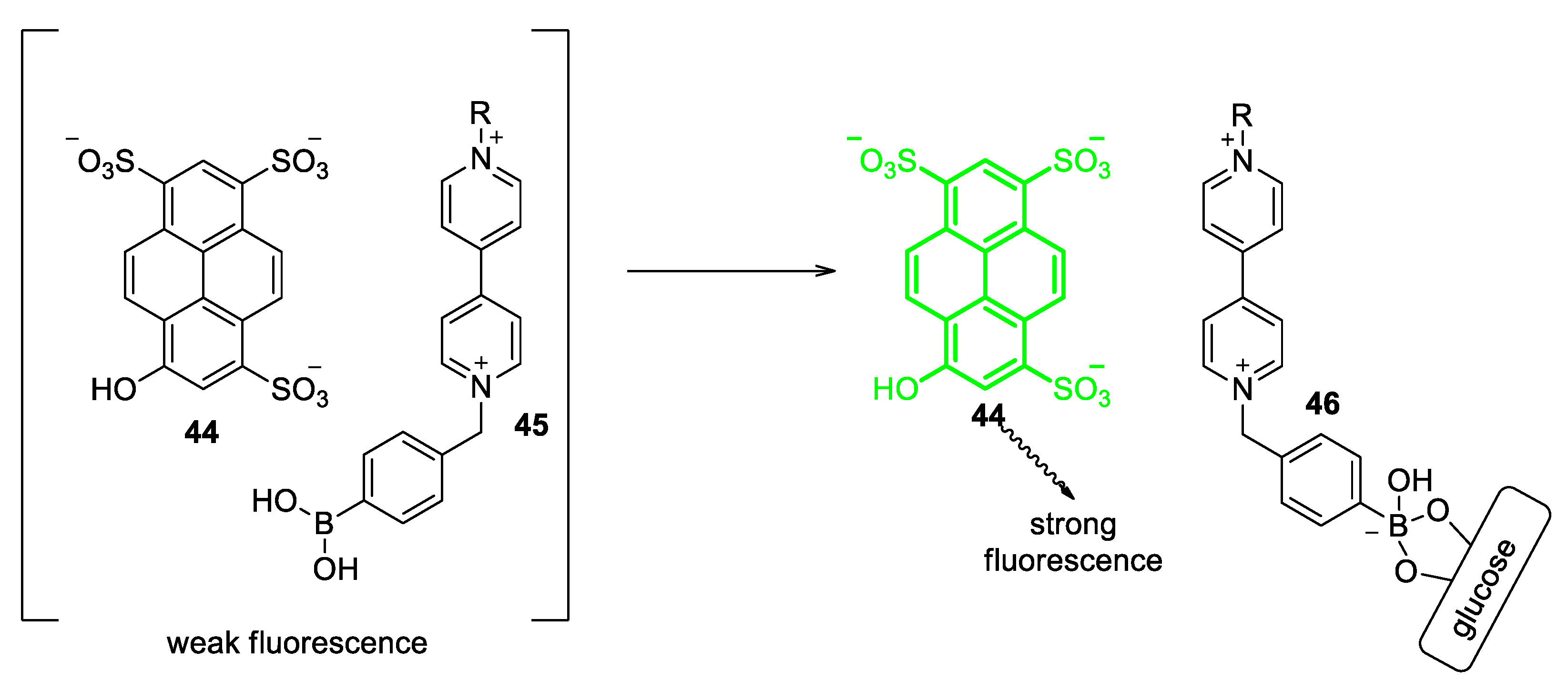
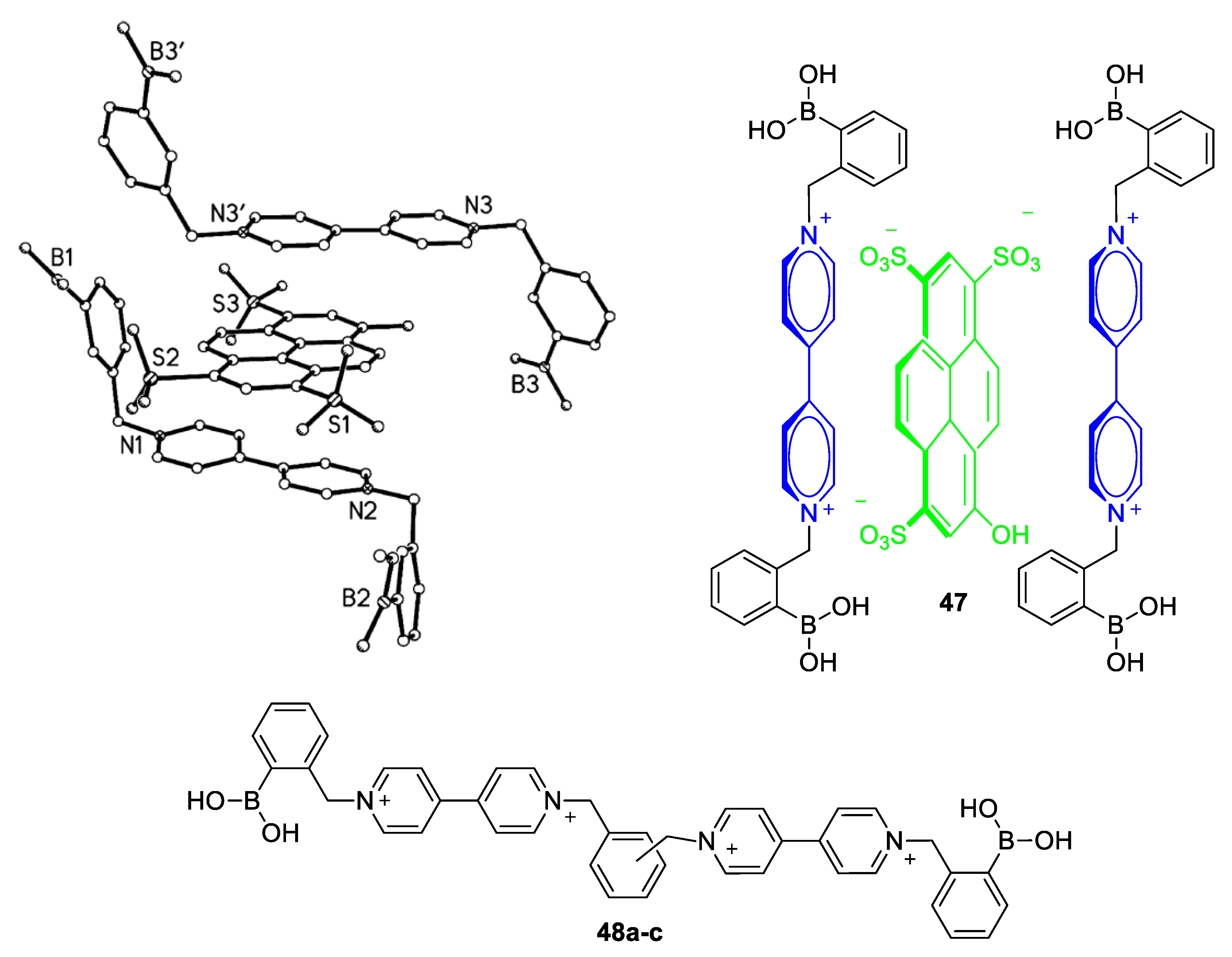
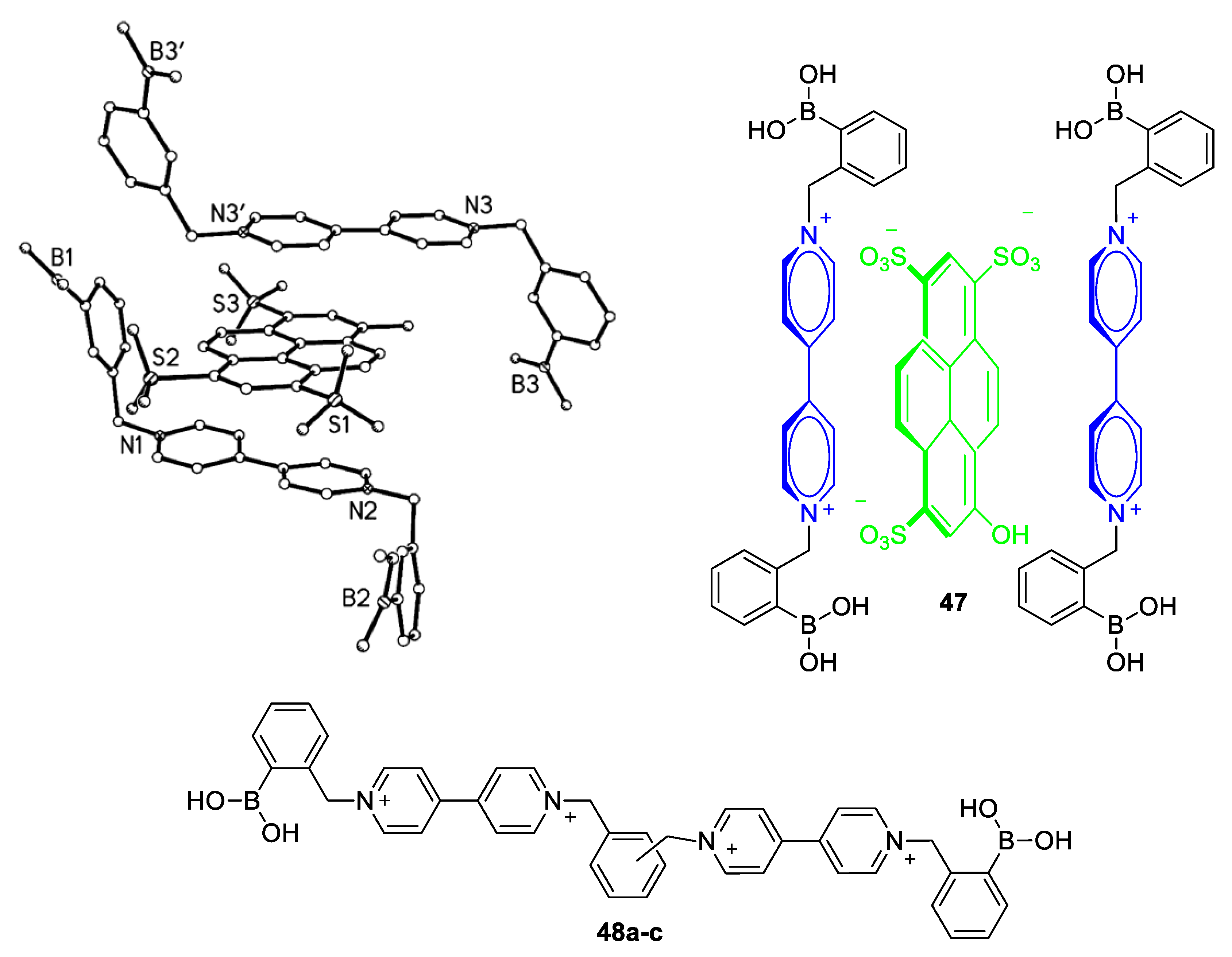
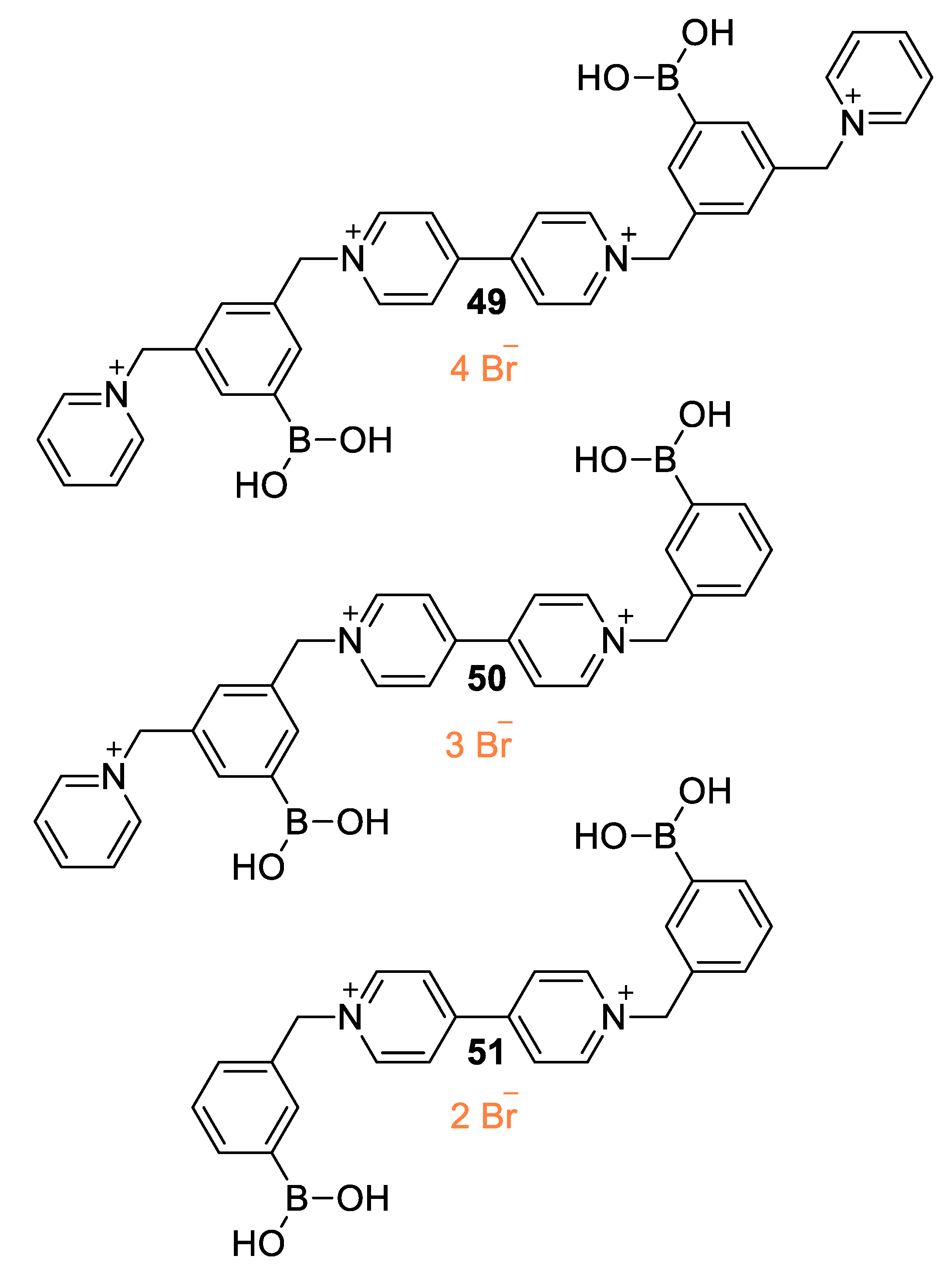
5.1. Glucose-Sensing Systems Involving Viologens
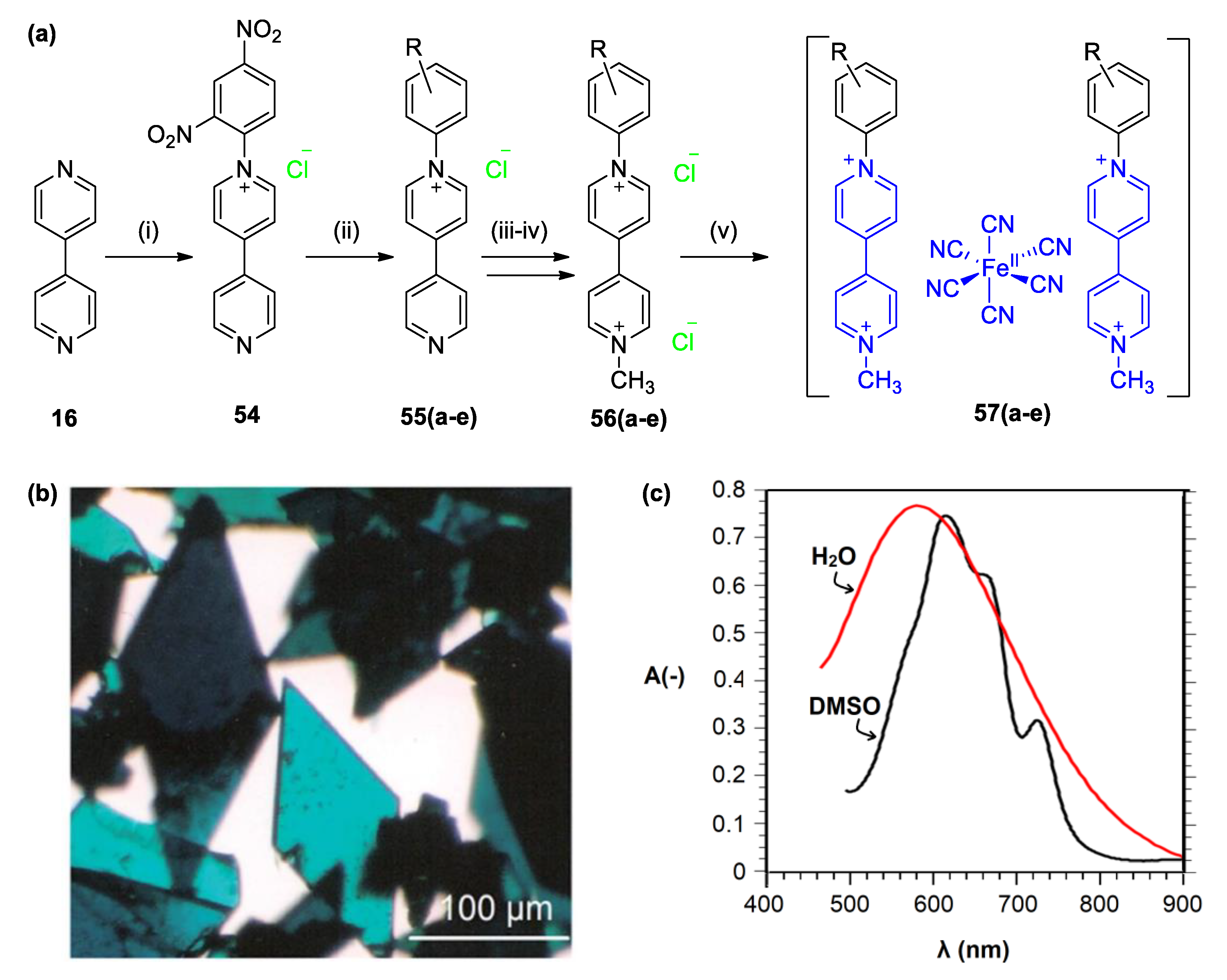
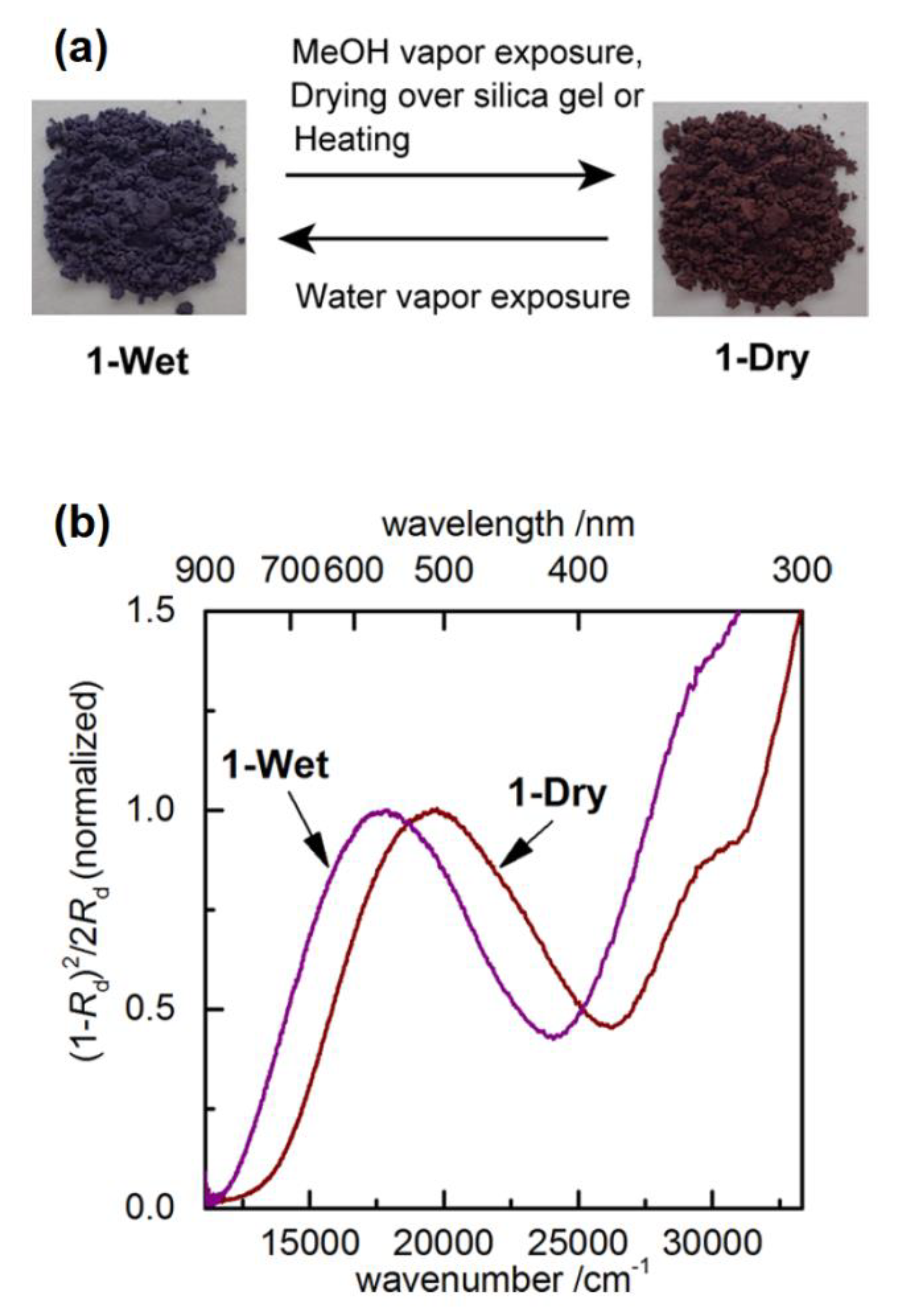
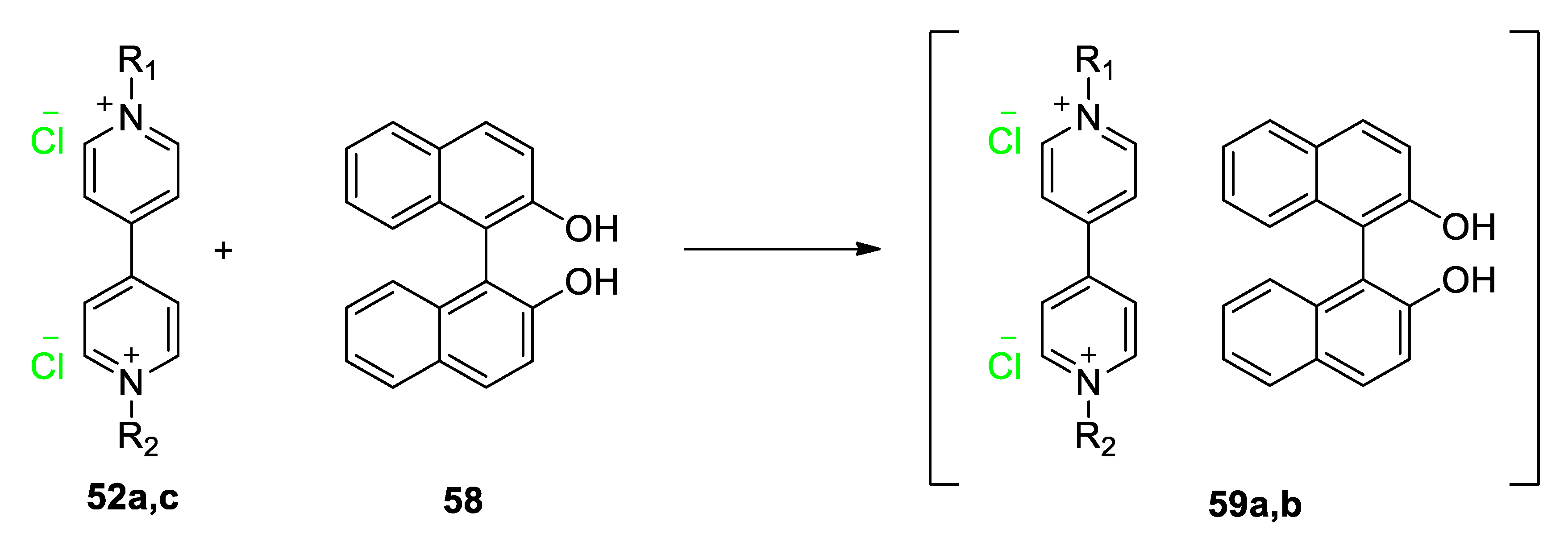
5.2. Charge Transfer Complexes of Viologens
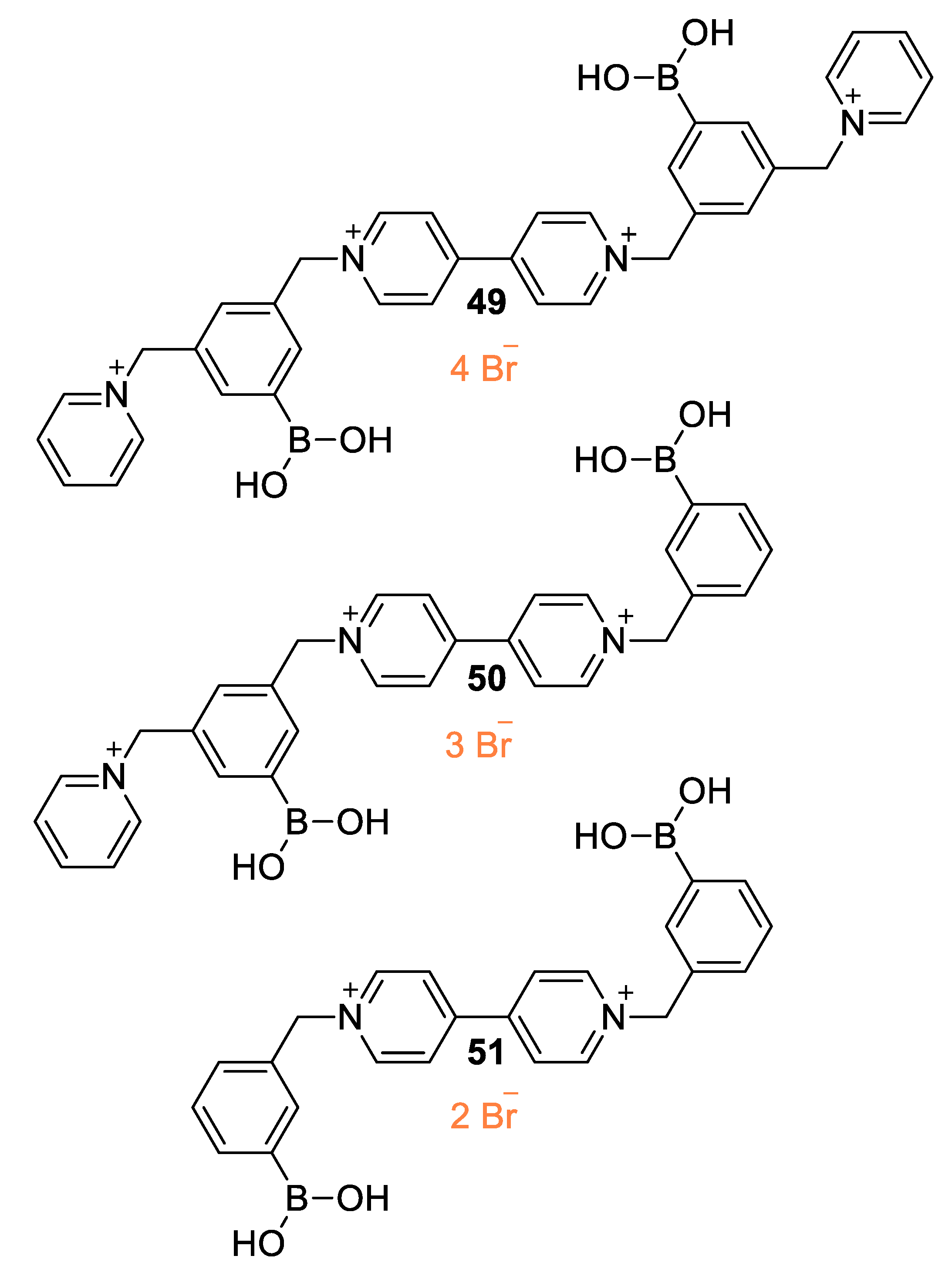
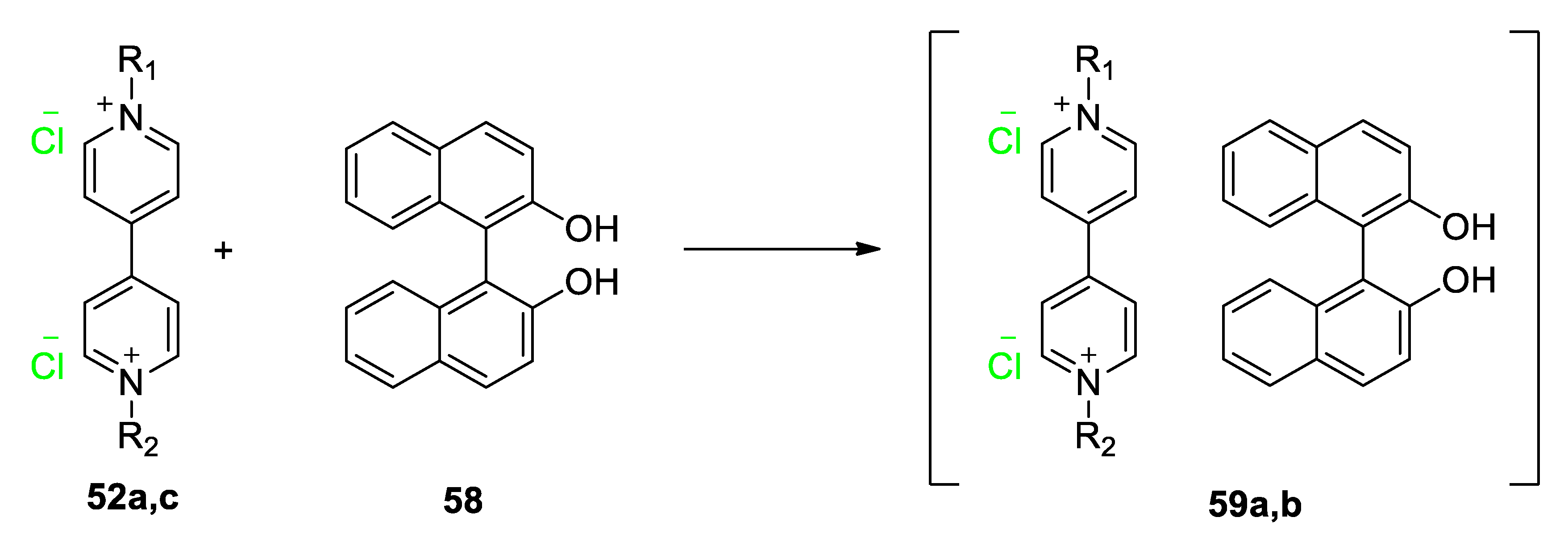
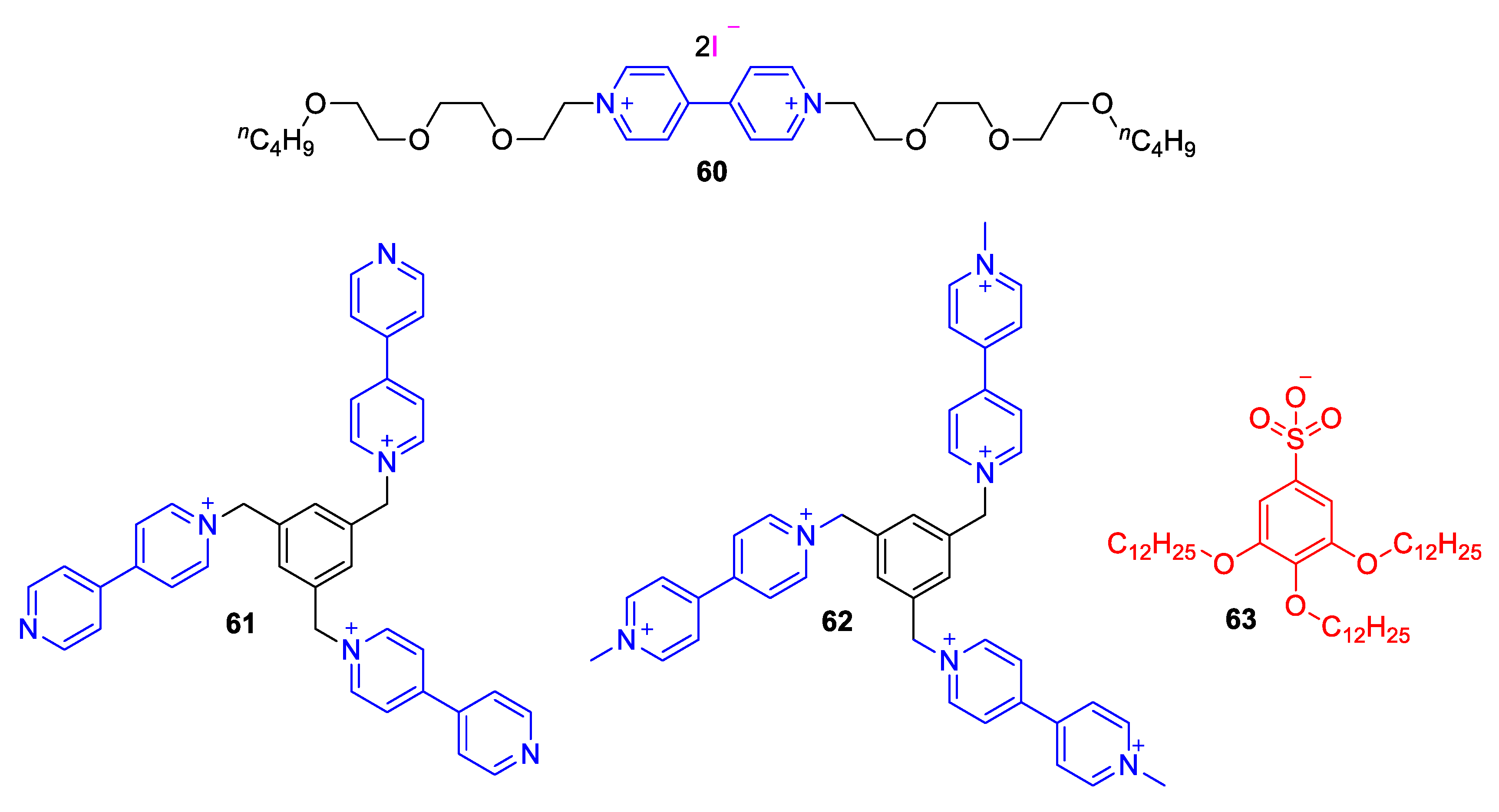
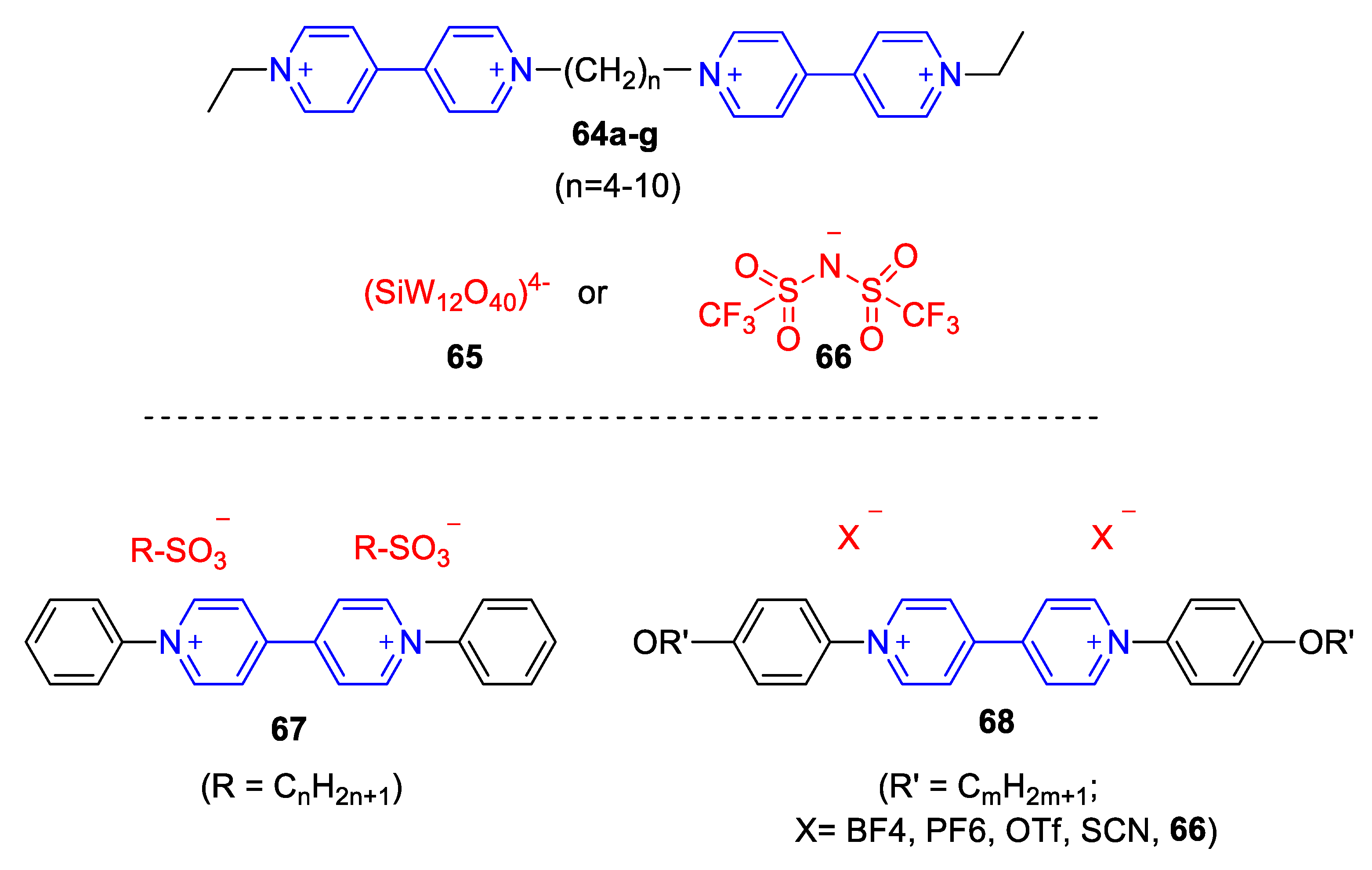
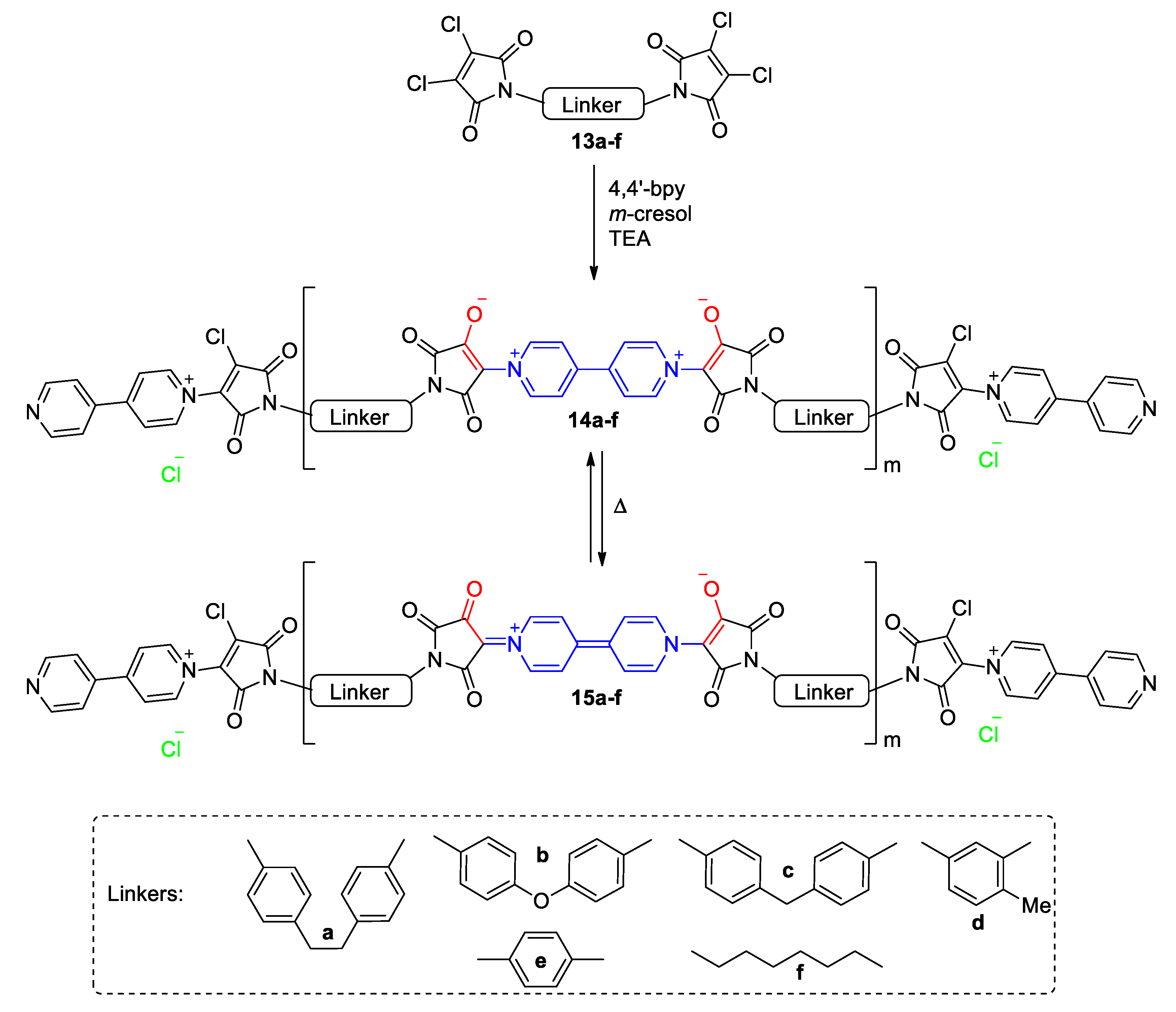
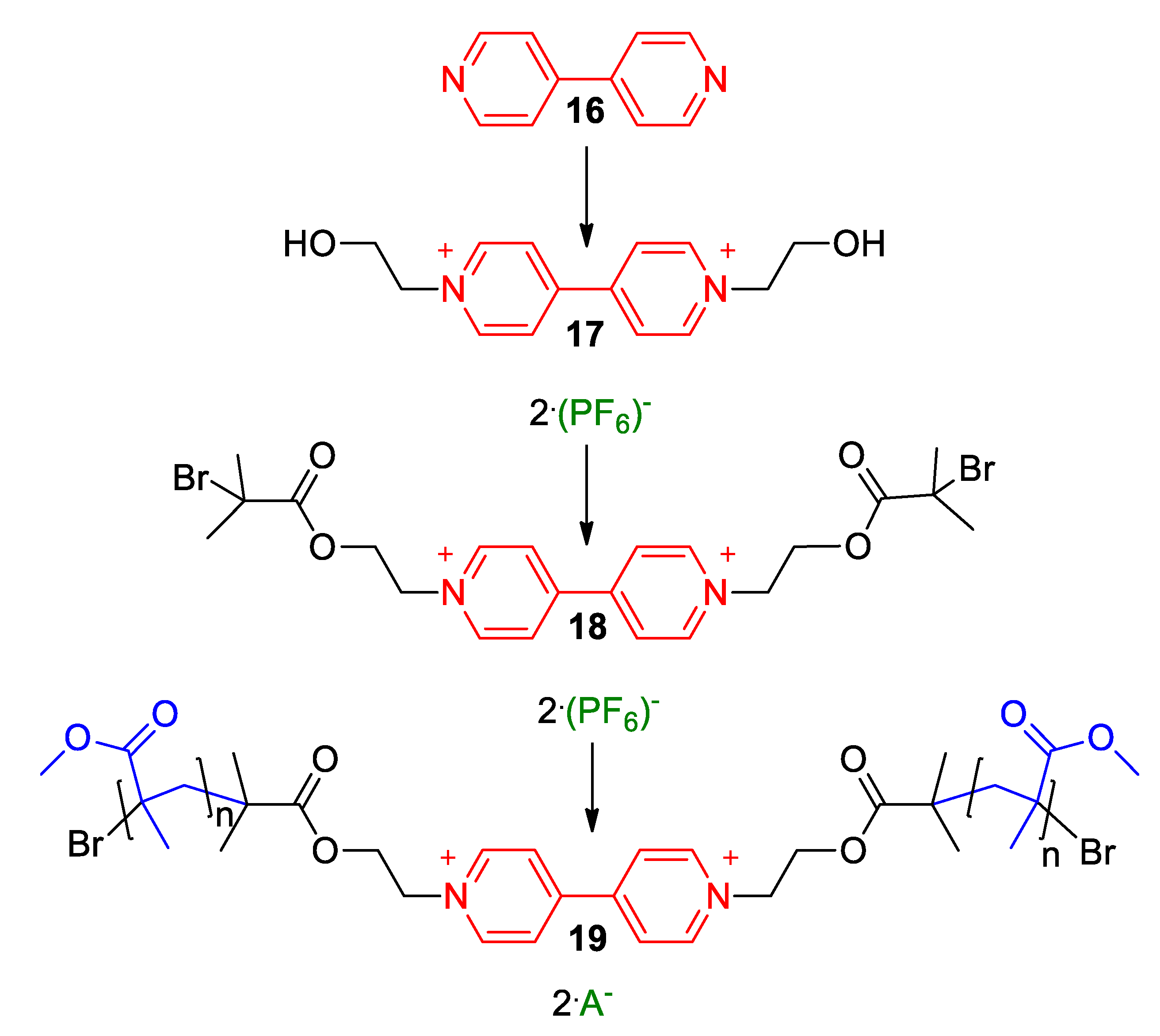
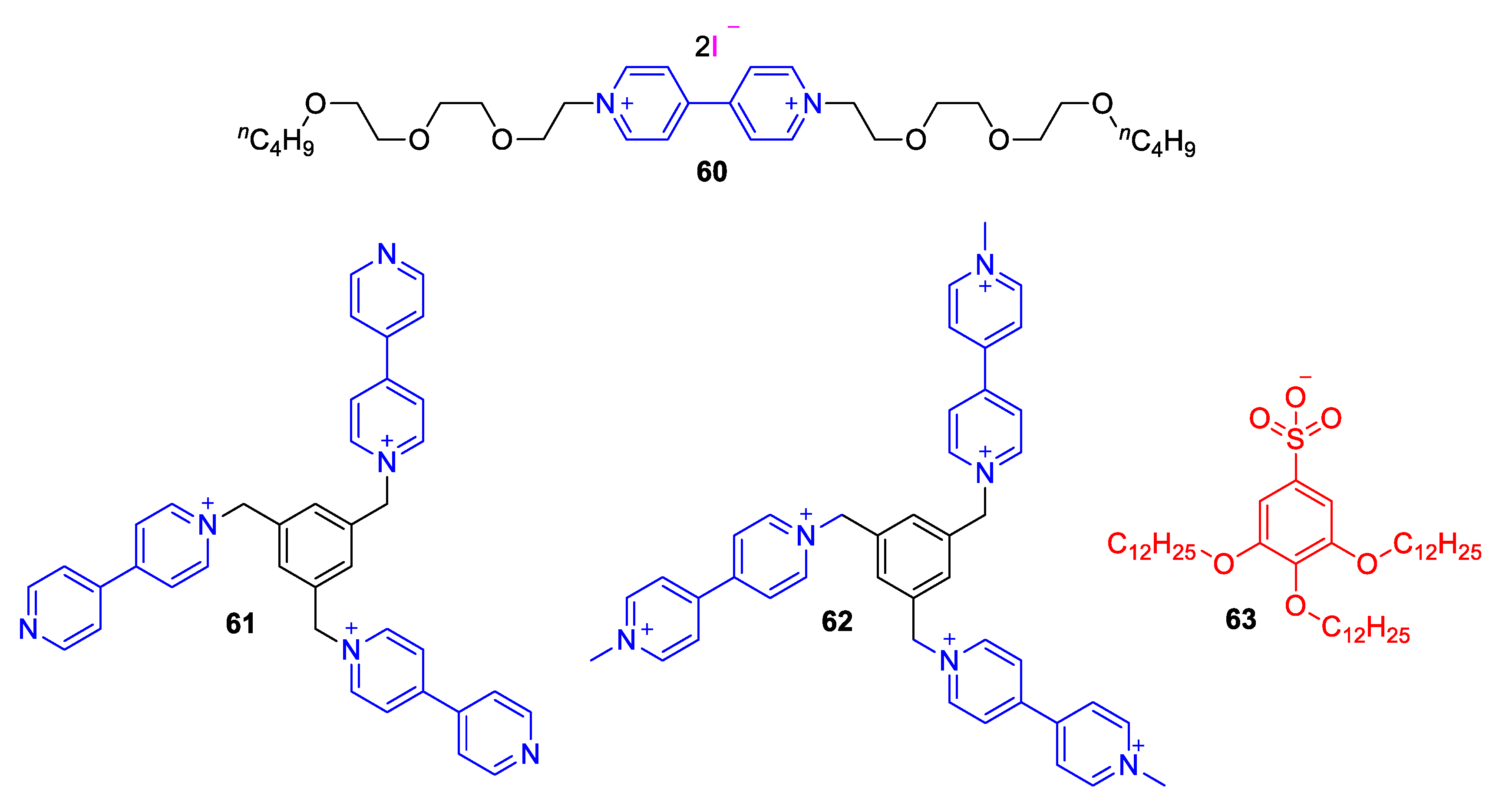
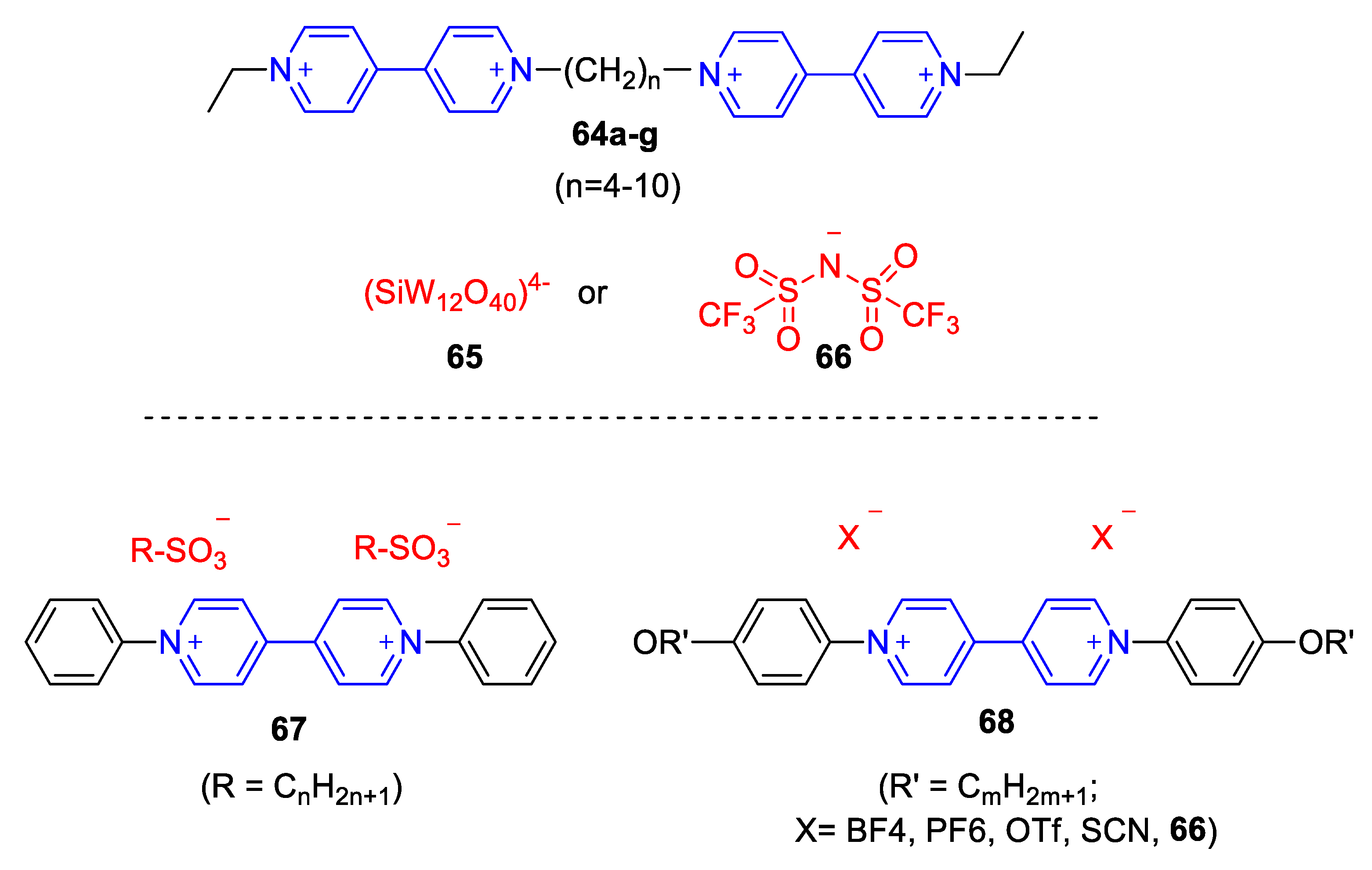
5.3. Ionic Liquid Crystalline Systems Involving Viologens
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Monk, P.M.S. The Viologens: Physicochemical Properties, Synthesis and Applications of the Salts of 4,4-Bipyridine; Wiley: Chichester, UK, 1998. [Google Scholar]
- Baroncini, M.; Silvi, S.; Credi, A. Photo- and Redox-Driven Artificial Molecular Motors. Chem. Rev. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Frasconi, M.; Stoddart, J.F. Introducing Stable Radicals into Molecular Machines. ACS Cent. Sci. 2017, 3, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Erbas-Cakmak, S.; Leigh, D.A.; McTernan, C.T.; Nussbaumer, A.L. Artificial Molecular Machines. Chem. Rev. 2015, 115, 10081–10206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrenbach, A.C.; Zhu, Z.; Cao, D.; Liu, W.-G.; Li, H.; Dey, S.K.; Basu, S.; Trabolsi, A.; Botros, Y.Y.; Goddard III, W.A.; et al. Radically Enhanced Molecular Switches. J. Am. Chem. Soc. 2012, 134, 16275–16288. [Google Scholar] [CrossRef] [Green Version]
- Feringa, B.L.; Browne, W.R. Molecular Switches; Wiley VCH: Weinheim, Germany, 2011; ISBN 978-3-527-63441-5. [Google Scholar]
- Škorjanc, T.; Shetty, D.; Olson, M.A.; Trabolsi, A. Design Strategies and Redox-Dependent Applications of Insoluble Viologen-Based Covalent Organic Polymers. ACS App. Mater. Inter. 2019, 11, 6705–6716. [Google Scholar] [CrossRef]
- Aulakh, D.; Varghese, J.R.; Wriedt, M. A New Design Strategy to Access Zwitterionic Metal–Organic Frameworks from Anionic Viologen Derivates. Inorg. Chem. 2015, 54, 1756–1764. [Google Scholar] [CrossRef]
- Li, P.; Zhou, L.-J.; Yang, N.-N.; Sui, Q.; Gong, T.; Gao, E.-Q. Metal–Organic Frameworks with Extended Viologen Units: Metal-Dependent Photochromism, Photomodulable Fluorescence, and Sensing Properties. Cryst. Growth Des. 2018, 18, 7191–7198. [Google Scholar] [CrossRef]
- Striepe, L.; Baumgartner, T. Viologens and Their Application as Functional Materials. Chem. Eur. J. 2017, 23, 16924–16940. [Google Scholar] [CrossRef]
- Ding, J.; Zheng, C.; Wang, L.; Lu, C.; Zhang, B.; Chen, Y.; Li, M.; Zhai, G.; Zhuang, X. Viologen-inspired functional materials: Synthetic strategies and applications. J. Mater. Chem. A 2019, 7, 23337–23360. [Google Scholar] [CrossRef]
- Shah, K.W.; Wang, S.-X.; Xiang, D.; Soo, Y.; Xu, J. Viologen-Based Electrochromic Materials: From Small Molecules, Polymers and Composites to Their Applications. Polymers 2019, 11, 1839. [Google Scholar] [CrossRef] [Green Version]
- Bird, C.L.; Kuhn, A.T. Electrochemistry of the viologens. Chem. Soc. Rev. 1981, 10, 49–82. [Google Scholar] [CrossRef]
- Škorjanc, T.; Shetty, D.; Olson, M.A.; Trabolsi, A. Viologen-based electrochromic materials and devices. J. Mater. Chem. C 2019, 7, 4622–4637. [Google Scholar] [CrossRef]
- Murugavel, K. Benzylic viologen dendrimers: A review of their synthesis, properties and applications. Polym. Chem. 2014, 5, 5873–5884. [Google Scholar] [CrossRef]
- Monk, P.; Mortimer, R.; Rosseinsky, D. Electrochromism and Electrochromic Devices; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Benniston, A.C.; Harriman, A.; Li, P.; Rostron, J.P.; Harrington, R.W.; Clegg, W. A Spectroscopic Study of the Reduction of Geometrically Restrained Viologens. Chem. Eur. J. 2007, 13, 7838–7851. [Google Scholar] [CrossRef] [PubMed]
- Crano, J.C.; Guglielmetti, R.J. Organic photochromic and thermochromic compounds. In Main Photochromic Families; Kluwer Akademic Publishers: New York, NY, USA, 2002; Volume 1, pp. 341–367. [Google Scholar]
- Tsukahara, K.; Wilkins, R.G. Kinetics of reduction of eight viologens by dithionite ion. J. Am. Chem. Soc. 1985, 107, 2632–2635. [Google Scholar] [CrossRef]
- Fiala, T.; Ludvíková, L.; Heger, D.; Švec, J.; Slanina, T.; Vetráková, L.; Babiak, M.; Nečas, M.; Kulhánek, P.; Klán, P.; et al. Bambusuril as a One-Electron Donor for Photoinduced Electron Transfer to Methyl Viologen in Mixed Crystals. J. Am. Chem. Soc. 2017, 139, 2597–2603. [Google Scholar] [CrossRef]
- Simonsen, K.B.; Becher, J.; Thorup, N.; Cava, M.P. Synthesis and X-ray crystal structure of the first tetrathiafulvalene-based acceptor–donor–acceptor sandwich. Chem. Commun. 1998, 901–902. [Google Scholar] [CrossRef]
- Ponnu, A.; Sung, J.; Spears, K.G. Ultrafast Electron-Transfer and Solvent Adiabaticity Effects in Viologen Charge-Transfer Complexes. J. Phys. Chem. A 2006, 110, 12372–12384. [Google Scholar] [CrossRef]
- Hwang, H.J.; Lee, S.K.; Lee, S.; Park, J.W. An NMR study on the conformation of naphthalene–viologen linked compounds: Effect of flexible spacer length. J. Chem. Soc. Perkin Trans. 1999, 2, 1081–1086. [Google Scholar] [CrossRef]
- Santos, W.G.; Budkina, D.S.; Deflon, V.M.; Tarnovsky, A.N.; Cardoso, D.R.; Forbes, M.D.E. Photoinduced Charge Shifts and Electron Transfer in Viologen–Tetraphenylborate Complexes: Push–Pull Character of the Exciplex. J. Am. Chem. Soc. 2017, 139, 7681–7684. [Google Scholar] [CrossRef]
- Abouelwafa, A.S.; Mereacre, V.; Balaban, T.S.; Anson, C.E.; Powell, A.K. Photo- and thermally-enhanced charge separation in supramolecular viologen–hexacyanoferrate complexes. Cryst. Eng. Comm. 2010, 12, 94–99. [Google Scholar] [CrossRef]
- Mau, A.W.-H.; Overbeek, J.M.; Loder, J.W.; Sasse, W.H.F. On the fluorescence and photoreduction of methyl viologens. J. Chem. Soc. Faraday Trans. 1986, 82, 869–876. [Google Scholar] [CrossRef]
- Alvaro, M.; Facey, G.A.; García, H.; García, S.; Scaiano, J.C. Intrazeolite Photochemistry. 16. Fluorescence of Methylviologen Adsorbed within Medium- and Large-Pore Zeolites. J. Phys. Chem. 1996, 100, 18173–18176. [Google Scholar] [CrossRef]
- Peon, J.; Tan, X.; Hoerner, J.D.; Xia, C.; Luk, Y.F.; Kohler, B. Excited State Dynamics of Methyl Viologen. Ultrafast Photoreduction in Methanol and Fluorescence in Acetonitrile. J. Phys. Chem. A 2001, 105, 5768–5777. [Google Scholar] [CrossRef]
- Clennan, E.L. Viologen embedded zeolites. Coord. Chem. Rev. 2004, 248, 477–492. [Google Scholar] [CrossRef]
- Freitag, M.; Gundlach, L.; Piotrowiak, P.; Galoppini, E. Fluorescence Enhancement of Di-p-tolyl Viologen by Complexation in Cucurbit uril. J. Am. Chem. Soc. 2012, 134, 3358. [Google Scholar] [CrossRef] [PubMed]
- Villemure, G.; Detellier, C.; Szabo, A.G. Fluorescence of clay-intercalated methylviologen. J. Am. Chem. Soc. 1986, 108, 4658–4659. [Google Scholar] [CrossRef]
- Alvaro, M.; García, H.; García, S.; Márquez, F.; Scaiano, J.C. Intrazeolite Photochemistry. 17. Zeolites as Electron Donors: Photolysis of Methylviologen Incorporated within Zeolites. J. Phys. Chem. B 1997, 101, 3043–3051. [Google Scholar] [CrossRef]
- Bâldea, I.; Köppel, H.; Wenzel, W. (4,4′)-Bipyridine in vacuo and in solvents: A quantum chemical study of a prototypical floppy molecule from a molecular transport perspective. Phys. Chem. Chem. Phys. 2013, 15, 1918–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Papadakis, R.; Deligkiozi, I.; Tsolomitis, A. Spectroscopic investigation of the solvatochromic behavior of a new synthesized non symmetric viologen dye: Study of the solvent-solute interactions. Anal. Bioanal. Chem. 2010, 397, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, R.; Deligkiozi, I.; Tsolomitis, A. Synthesis and characterization of a group of new medium responsive non-symmetric viologens. Chromotropism and structural effects. Dyes Pigment. 2012, 95, 478–484. [Google Scholar] [CrossRef]
- Gutmann, V. Lecture Notes on Solution Chemistry; World Scientific: Singapore, 1995. [Google Scholar]
- Kamlet, M.J.; Abboud, J.L.M.; Abraham, M.H.; Taft, R.W. Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters, Pi.*, Alpha., and Beta., and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar] [CrossRef]
- Shi, W.; Xing, F.; Bai, Y.-L.; Hu, M.; Zhao, Y.; Li, M.-X.; Zhu, S. High Sensitivity Viologen for a Facile and Versatile Sensor of Base and Solvent Polarity in Solution and Solid State in Air Atmosphere. ACS Appl. Mater. Inter. 2015, 7, 14493–14500. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Xing, F.; Zhao, Y.; Bai, Y.-L.; Li, M.-X.; Zhu, S. Phenolacetyl Viologen as Multifunctional Chromic Material for Fast and Reversible Sensor of Solvents, Base, Temperature, Metal Ions, NH3 Vapor, and Grind in Solution and Solid State. ACS Omega 2017, 2, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Gaina, C.; Gaina, V.; Airinei, A.; Avram, E. Polyimides containing 4,4′-bipyridinium units. Appl. Polym. Sci. 2004, 94, 2091–2100. [Google Scholar] [CrossRef]
- Wang, Z.; Tsarevsky, N.V. Well-defined polymers containing a single mid-chain viologen group: Synthesis, environment-sensitive fluorescence, and redox activity. Polym. Chem. 2016, 7, 4402–4410. [Google Scholar] [CrossRef]
- Sui, Q.; Ren, X.-T.; Dai, Y.-X.; Wang, K.; Li, W.-T.; Gong, T.; Fang, J.-J.; Zou, B.; Gao, E.-Q.; Wang, L. Piezochromism and hydrochromism through electron transfer: New stories for viologen. Mater. Chem. Sci. 2017, 8, 2758–2768. [Google Scholar] [CrossRef] [Green Version]
- Papadakis, R.; Deligkiozi, I. Solvent Effects in Supramolecular Systems [Online First]; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Pramanik, B.; Mondal, J.H.; Singha, N.; Ahmed, S.; Mohanty, J.; Das, D. A Viologen–Perylenediimide Conjugate as an Efficient Base Sensor with Solvatochromic Property. Chem. Phys. Chem. 2017, 18, 245–252. [Google Scholar] [CrossRef]
- Gong, T.; Yang, X.; Fang, J.-J.; Sui, Q.; Xi, F.-G.; Gao, E.-Q. Distinct Chromic and Magnetic Properties of Metal–Organic Frameworks with a Redox Ligand. ACS Appl. Mater. Interfaces 2017, 9, 5503–5512. [Google Scholar] [CrossRef]
- Li, S.-L.; Han, M.; Zhang, Y.; Li, G.-P.; Li, M.; He, G.; Zhang, X.-M. X-ray and UV Dual Photochromism, Thermochromism, Electrochromism, and Amine-Selective Chemochromism in an Anderson-like Zn7Cluster-Based 7-Fold Interpenetrated Framework. J. Am. Chem. Soc. 2019, 141, 12663–12672. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.-X.; Ju, Z.-F.; Jin, X.-H.; Zhang, J. Novel Polythreaded Coordination Polymer: From an armed-Polyrotaxane Sheet to a 3D Polypseudorotaxane Array, Photo- and Thermochromic Behaviors. Inorg. Chem. 2009, 48, 1266–1268. [Google Scholar] [CrossRef] [PubMed]
- Blandamer, M.J.; Burgess, J.; Haines, R.I. Kinetic and equilibrium properties of pentacyano(3,5-dimethylpyridine)-iron(II) and related anions in mixed aqueous solvents. J. Chem. Soc. Dalton Trans. 1976, 5, 1293–1298. [Google Scholar] [CrossRef]
- Toma, H.E.; Takasugi, M.S. Spectroscopic studies of preferential and asymmetric solvation in substituted cyanoiron(II) complexes. J. Solution Chem. 1983, 12, 547–561. [Google Scholar] [CrossRef]
- Pinheiro, C.; Lima, J.C.; Parola, A.J. Using hydrogen bonding-specific interactions to detect water in aprotic solvents at concentrations below 50 ppm. Sens. Actuators B 2006, 114, 978–983. [Google Scholar] [CrossRef]
- Papadakis, R.; Tsolomitis, A. Study of the correlations of the MLCT Vis absorption maxima of 4-pentacyanoferrate-40′-arylsubstituted bispyridinium complexes with the Hammett substituent parameters and the solvent polarity parameters ETN and AN. J. Phys. Org. Chem. 2009, 22, 515–521. [Google Scholar] [CrossRef]
- Papadakis, R.; Tsolomitis, A. Solvatochromism and preferential solvation of 4-pentacyanoferrate 4′-aryl substituted bipyridinium complexes in binary mixtures of hydroxylic and non-hydroxylic solvents. J. Solution Chem. 2011, 40, 1108–1125. [Google Scholar] [CrossRef]
- Coe, B.J.; Harries, J.L.; Helliwell, M.; Jones, L.A.; Asselberghs, I.; Clays, K.; Brunschwig, B.S.; Harris, J.A.; Garín, J.; Orduna, J. Pentacyanoiron(II) as an electron donor group for nonlinear optics: Medium-responsive properties and comparisons with related pentaammineruthenium(II) complexes. J. Am. Chem. Soc. 2006, 128, 12192–12204. [Google Scholar] [CrossRef] [Green Version]
- The term ”solvatochromic intensity” is used here by the author to express the solvatochromic response (CT wavelength, wavenumber, or energy change) induced by a certain input (change in solvent polarity expressed by a polarity parameter like Reichardt’s polarity scale ET(30) [34]).
- Papadakis, R. Preferential solvation of a highly medium responsive pentacyanoferrate(II) complex in binary solvent mixtures: Understanding the role of dielectric enrichment and the specificity of solute–solvent interactions. J. Phys. Chem. B. 2016, 120, 9422–9433. [Google Scholar] [CrossRef]
- Papadakis, R. The solvatochromic behavior and degree of ionicity of a synthesized pentacyano (N-substituted-4, 4′-bipyridinium) ferrate (II) complex in different media. Tuning the solvatochromic intensity in aqueous glucose solutions. Chem. Phys. 2014, 430, 29–39. [Google Scholar] [CrossRef]
- Deligkiozi, I.; Voyiatzis, E.; Tsolomitis, A.; Papadakis, R. Synthesis and characterization of new azobenzenecontaining bis pentacyanoferrate(II) stoppered push-pull [2]rotaxanes, with alpha- and beta-cyclodextrin. Towards highly medium responsive dyes. Dyes Pigment. 2015, 113, 709–722. [Google Scholar] [CrossRef]
- Papadakis, R.; Deligkiozi, I.; Nowak, K.E. Study of the preferential solvation effects in binary solvent mixtures with the use of intensely solvatochromic azobenzene involving [2] rotaxane solutes. J. Mol. Liq. 2019, 274, 715–723. [Google Scholar] [CrossRef]
- Deligkiozi, I.; Papadakis, R.; Tsolomitis, A. Synthesis, characterisation and photoswitchability of a new [2]rotaxane of α-cyclodextrin with a diazobenzene containing π-conjugated molecular dumbbell. Supramol. Chem. 2012, 24, 333–343. [Google Scholar] [CrossRef]
- Baer, A.J.; Macartney, D.H. α- and β-cyclodextrin rotaxanes of μ-Bis(4-pyridyl)bis[pentacyanoferrate(II)] Complexes. Inorg. Chem. 2000, 39, 1410–1417. [Google Scholar] [CrossRef]
- Deligkiozi, I.; Papadakis, R.; Tsolomitis, A. Photoconductive properties of a p-conjugated a-cyclodextrin containing [2]rotaxane and its corresponding molecular dumbbell. Phys. Chem. Chem. Phys. 2013, 15, 3497–3503. [Google Scholar] [CrossRef]
- Papadakis, R.; Deligkiozi, I.; Li, H. Photoconductive Interlocked Molecules and Macromolecules. In Photodetectors; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-D.; Zhu, R.; Yin, J.-P.; Sun, L.; Guo, R.-Y.; Zhan, J. Bipyridinium-Bearing Multi-stimuli Responsive Chromic Material with High Stability. Cryst. Growth Des. 2018, 18, 3236–3243. [Google Scholar] [CrossRef]
- Coe, B.J.; Fielden, J.; Foxon, S.P.; Helliwell, M.; Asselberghs, I.; Clays, K.; De Mey, K.; Brunschwig, B.S. Syntheses and Properties of Two-Dimensional, Dicationic Nonlinear Optical Chromophores Based on Pyrazinyl Cores. J. Org. Chem. 2010, 75, 8550–8563. [Google Scholar] [CrossRef] [Green Version]
- Coe, B.J.; Pilkington, R.A. Theoretical Studies on Two-Dimensional Nonlinear Optical Chromophores with Pyrazinyl Cores and Organic or Ruthenium(II) Ammine Electron Donors. J. Phys. Chem. A 2014, 118, 2253–2268. [Google Scholar] [CrossRef]
- Sun, X.; James, T.D. Glucose Sensing in Supramolecular Chemistry. Chem. Rev. 2015, 115, 8001–8037. [Google Scholar] [CrossRef] [Green Version]
- De Borba, E.B.; Amaral, C.L.C.; Politi, M.J.; Villalobos, R.; Baptista, M.S. Photophysical and Photochemical Properties of Pyranine/Methyl Viologen Complexes in Solution and in Supramolecular Aggregates: A Switchable Complex. Langmuir 2000, 16, 5900–5907. [Google Scholar] [CrossRef]
- Brooks, W.L.A.; Sumerlin, B.S. Synthesis and Applications of Boronic Acid-Containing Polymers: From Materials to Medicine. Chem. Rev. 2016, 116, 1375–1397. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.G. Boronic Acids: Preparation and Applications in Organic Synthesis. In Medicine and Materials, 2nd ed.; Wiley-VCH Verlag: Weinheim, Germany, 2011; ISBN 9783527639328. [Google Scholar]
- Gamsey, S.; Miller, A.; Olmstead, M.M.; Beavers, M.; Hirayama, L.C.; Pradhan, S.; Wessling, R.A.; Singaram, B. Boronic Acid-Based Bipyridinium Salts as Tunable Receptors for Monosaccharides and α-Hydroxycarboxylates. J. Am. Chem. Soc. 2007, 129, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Sharrett, Z.; Gamsey, S.; Levine, P.; Cunningham-Bryant, D.; Vilozny, B.; Schiller, A.; Wessling, R.A.; Singaram, B. Boronic acid-appended bis-viologens as a new family of viologen quenchers for glucose sensing. Tetrahedron Lett. 2008, 49, 300–304. [Google Scholar] [CrossRef]
- Sharrett, Z.; Gamsey, S.; Fat, J.; Cunningham-Bryant, D.; Wessling, R.A.; Singaram, B. The effect of boronic acid acidity on performance of viologen-based boronic acids in a two-component optical glucose-sensing system. Tetrahedron Lett. 2007, 48, 5125–5129. [Google Scholar] [CrossRef]
- Cordes, D.B.; Gamsey, S.; Sharrett, Z.; Miller, A.; Thoniyot, P.; Wessling, R.A.; Singaram, B. The Interact ion of Boronic Acid-Substituted Viologens with Pyranine: The Effects of Quencher Charge on Fluorescence Quenching and Glucose Response. Langmuir 2005, 21, 6540–6547. [Google Scholar] [CrossRef]
- Gamsey, S.; Suri, J.T.; Wessling, R.A.; Singaram, B. Continuous Glucose Detection Using Boronic Acid-Substituted Viologens in Fluorescent Hydrogels: Linker Effects and Extension to Fiber Optics. Langmuir 2006, 22, 9067–9074. [Google Scholar] [CrossRef]
- Singaram, B.; Wessling, R.A. Polyhydroxyl-substituted organic molecule sensing optical in vivo method utilizing a boronic acid adduct and the device thereof. US Patent 6,627,177,B2, 30 September 2003. [Google Scholar]
- Foster, R. Organic Charge-Transfer Complexes; Academic Press: London, UK, 1969. [Google Scholar]
- Gutmann, F.; Johnson, C.; Keyzer, H.; Molnar, J. Charge Transfer Complexes in Biological Systems, 1st ed.; Marcel Dekker Inc.: New York, NY, USA, 1997. [Google Scholar]
- Saielli, G. Ion-Pairing of Octyl Viologen Diiodide in Low-Polar Solvents: An Experimental and Computational Study. J. Phys. Chem. A 2008, 112, 7987–7995. [Google Scholar] [CrossRef]
- Nakahara, A.; Wang, J.H. Charge transfer complexes of methylviologen. J. Phys. Chem. 1963, 67, 496–498. [Google Scholar] [CrossRef]
- Kosower, E.M.; Cotter, J.L. Stable Free Radicals. II. The Reduction of 1-Methyl-4-cyanopyridinium Ion to Methylviologen Cation Radical. J. Am. Chem. Soc. 1964, 86, 5524–5527. [Google Scholar] [CrossRef]
- Curtis, J.C.; Sullivan, B.P.; Meyer, T.J. Calculation of electron-transfer rate constants from the properties of charge-transfer absorption bands. The PQ2+, Fe(CN)64- system. Inorg. Chem. 1980, 19, 3833–3839. [Google Scholar] [CrossRef]
- Watanabe, T.; Honda, K. Measurement of the extinction coefficient of the methyl viologen cation radical and the efficiency of its formation by semiconductor photocatalysis. J. Phys. Chem. 1982, 86, 2617–2619. [Google Scholar] [CrossRef]
- Bockman, T.M.; Kochi, J.K. Isolation and oxidation-reduction of methylviologen cation radicals. Novel disproportionation in charge-transfer salts by X-ray crystallography. J. Org. Chem. 1990, 55, 4127–4135. [Google Scholar] [CrossRef]
- Hammack, W.S.; Drickamer, H.G.; Hendrickson, D.N. Effect of pressure on the charge-transfer band of the [Fe(CN)6]4−·dimethyl viologen ion pair. Chem. Phys. Lett. 1988, 151, 469–473. [Google Scholar] [CrossRef]
- Monk, P.M.S.; Hodgkinson, N.M.; Partridge, R.D. The colours of charge-transfer complexes of methyl viologen: Effects of donor, ionic strength and solvent. Dyes Pigment. 1999, 43, 241–251. [Google Scholar] [CrossRef]
- Papadakis, R.; Deligkiozi, I.; Giorgi, M.; Faure, B.; Tsolomitis, A. Supramolecular complexes involving nonsymmetric viologen cations and hexacyanoferrate (II) anions. A spectroscopic, crystallographic and computational study. RSC Adv. 2016, 6, 575–585. [Google Scholar] [CrossRef]
- Tanaka, R.; Matsushi, N. A charge-transfer salt composed of methyl viologen and hexacyanidoferrate(II). Acta Cryst. C 2017, 73, 476–480. [Google Scholar] [CrossRef]
- Antipin, M.Y.; Ilyukhin, A.B.; Kotov, V.Y. Ice-like (H2O)12 and (H2O)14 clusters in the crystal structures of alkali metal–ethyl viologen hexacyanometallates. Mendeleev Commun. 2001, 11, 210–211. [Google Scholar] [CrossRef]
- Tanaka, R.; Okazawa, A.; Konaka, H.; Sasaki, A.; Kojima, N.; Matsushita, N. Unique Hydration/Dehydration-Induced Vapochromic Behavior of a Charge-Transfer Salt Comprising Viologen and Hexacyanidoferrate (II). Inorg. Chem. 2018, 57, 2209–2217. [Google Scholar] [CrossRef]
- Kinuta, T.; Sato, T.; Tajima, N.; Kuroda, R.; Matsubara, Y.; Imai, Y. Solid-state thermochromism observed in charge-transfer complex composed of binaphthol and viologen. J. Mol. Struct. 2010, 982, 45–49. [Google Scholar] [CrossRef]
- Saielli, G. Special Issue Editorial: Ionic Liquid Crystals. Crystals 2019, 9, 274. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, S.; Weiner, J. Ueber einige Abkömmlinge des Propylamins. Ber. Dtsch. Chem. Ges. 1888, 21, 2669–2679. [Google Scholar] [CrossRef] [Green Version]
- Reinitzer, F. Beiträge zur Kenntniss des Cholesterins. Monatsh. für Chem. (Wien) 1888, 9, 421–441. [Google Scholar] [CrossRef]
- Cîrcu, V. Ionic Liquid C rystals Based on Pyridinium Salts, Progress and Developments in Ionic Liquids. In Progress and Developments in Ionic Liquids; Handy, S., Ed.; IntechOpen: Basel, Switzerland, 2019. [Google Scholar] [CrossRef] [Green Version]
- Saielli, G. Ionic Liquid Crystals. In Crystals 2019; MDPI: Basel, Switzerland, 2019; ISBN 978-3-03921-086-2. [Google Scholar] [CrossRef]
- Tabushi, I.; Yamamura, K.; Kominami, K. Electric stimulus-response behavior of liquid-crystalline viologen. J. Am. Chem. Soc. 1986, 108, 6409–6410. [Google Scholar] [CrossRef]
- Asaftei, S.; Ciobanu, M.; Lepadatu, A.M.; Song, E.; Beginn, U. Thermotropic ionic liquid crystals by molecular assembly and ion pairing of 4,4′-bipyridinium derivatives and tris(dodecyloxy)benzenesulfonates in a non-polar solvent. J. Mater. Chem. 2012, 22, 14426–14437. [Google Scholar] [CrossRef]
- Bonchio, M.; Carraro, M.; Casella, G.; Causin, V.; Rastrelli, F.; Saielli, G. Thermal behaviour and electrochemical properties of bis(trifluoromethanesulfonyl)amide and dodecatungstosilicate viologen dimers. Phys. Chem. Chem. Phys. 2012, 14, 2710–2717. [Google Scholar] [CrossRef]
- Gunaratne, H.Q.N.; Nockemann, P.; Olejarz, S.; Reid, S.M.; Seddon, K.R.; Srinivasan, G. Ionic Liquids with Solvatochromatic and Charge-Transfer Functionalities Incorporating the Viologen Moiety. Austr. J. Chem. 2013, 66, 607–611. [Google Scholar] [CrossRef]
- Wang, R.-T.; Lee, G.-H.; Lai, C.K. Anion-induced ionic liquid crystals of Diphenylviologens. J. Mater. Chem. C 2018, 6, 9430–9444. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ichikawa, T. Design of Viologen-Based Liquid Crystals Exhibiting Bicontinuous Cubic Phases and Their Redox-Active Behavior. Materials 2017, 10, 1243. [Google Scholar] [CrossRef] [Green Version]
- Mortimer, R.J.; Rosseinsky, D.R.; Monk, P.M.S. Electrochromic Materials and Devices; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; ISBN 9783527679850. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | λmax (EMLCT) 1 in H2O | λmax (EMLCT) 1 in MeOH | Δλmax (ΔΕMLCT) 1 | Ref. |
|---|---|---|---|---|---|
| 28a | Me | 534 (2.32) | 686 (1.81) | 152 (0.51) | [55] |
| 28b | Ph | 566 (2.19) | 738 (1.68) | 172 (0.51) | [55] |
| 28c | 4-AcPh | 584 (2.12) | 771 (1.61) | 187 (0.51) | [55] |
| 28d | 2-Pym | 618 (2.00) | 854 (1.45) | 236 (0.55) | [55] |
| 31a | 4-OMePh | 565 (2.19) | 727 (1.71) | 162 (0.48) | [53] |
| 31b | 4-MePh | 563 (2.20) | 730 (1.70) | 167 (0.50) | [53] |
| 31c | 3-MePh | 563 (2.20) | 731 (1.70) | 168 (0.50) | [53] |
| 31d | 4-ClPh | 572 (2.17) | 741 (1.67) | 169 (0.50) | [53] |
| 31e | 4-BrPh | 573 (2.16) | 741 (1.67) | 168 (0.49) | [53] |
| 31f | 4-CNPh | 586 (2.12) | 755 (1.64) | 169 (0.48) | [53] |
| CTC | R1, R2 1 | φ (ο) 2 | RCC (Å) 3 | RpyFem (Å) 4 | λmax (nm) 5 | Ref. |
|---|---|---|---|---|---|---|
| 53a | Me, Me | 37.4 | 1.481 | 5.603 | 540 | [89] |
| 53b | Et, Et | 1.30 | 1.481 | 6.321 | – | [90] |
| 53b | Et, Et | 15.6 | 1.476 | 6.137 | 507,560 | [91] |
| 57a | p-tolyl, Me (1) | 10.3 | 1.434 | 5.767 | 574 | [88] |
| 57a | p-tolyl, Me (2) | 27.6 | 1.480 | 5.604 | 574 | [88] |
| 53c | 2,4-(NO2)2Ph, 2,4-(NO2)2Ph | 13.8 | 1.480 | 5.520 | 420, 532 | [25] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadakis, R. Mono- and Di-Quaternized 4,4′-Bipyridine Derivatives as Key Building Blocks for Medium- and Environment-Responsive Compounds and Materials. Molecules 2020, 25, 1. https://doi.org/10.3390/molecules25010001
Papadakis R. Mono- and Di-Quaternized 4,4′-Bipyridine Derivatives as Key Building Blocks for Medium- and Environment-Responsive Compounds and Materials. Molecules. 2020; 25(1):1. https://doi.org/10.3390/molecules25010001
Chicago/Turabian StylePapadakis, Raffaello. 2020. "Mono- and Di-Quaternized 4,4′-Bipyridine Derivatives as Key Building Blocks for Medium- and Environment-Responsive Compounds and Materials" Molecules 25, no. 1: 1. https://doi.org/10.3390/molecules25010001
APA StylePapadakis, R. (2020). Mono- and Di-Quaternized 4,4′-Bipyridine Derivatives as Key Building Blocks for Medium- and Environment-Responsive Compounds and Materials. Molecules, 25(1), 1. https://doi.org/10.3390/molecules25010001