1. Introduction
Ginseng is a well-known traditional herbal medicine that has been used for its pharmacological and therapeutic effects in Korea, China, and Japan for thousands of years [
1,
2]. It has been studied extensively and has been shown to have effects on neurological disorders, such as Alzheimer’s disease (AD), stroke, Parkinson’s disease, and multiple sclerosis [
3,
4,
5,
6,
7]. There have been several researches on ginseng itself, as well as on individual ingredients in ginseng, such as ginsenosides (a group of active ingredients in ginseng), most of which have focused on Rb1, Rg1, Rg3, Re, and Rd [
8,
9]. However, many studies on the pharmacological actions of ginseng have been limited to ginsenosides, even though ginsenosides alone cannot explain the wide variety of ginseng effects [
7,
10].
Gintonin, a component of ginseng and a novel exogenous ligand of G protein-coupled lysophosphatidic acid (LPA) receptors, has been identified as a non-saponin and non-acidic carbohydrate polymer [
11,
12]. The gintonin-enriched fraction (GEF) of ginseng extract contains a large amount of gintonin, as the name implies, and also linoleic acid (C18:2), phosphatidic acid (PA) 16:0-18:2, LPA C18:2, as well as other bioactive lysophospholipids in addition to LPAs [
12]. GEF transiently induces [Ca
2+]
i and stimulates diverse Ca
2+-dependent cellular effects, ranging from neurotransmitter release to neuronal cell proliferation, through G protein-coupled LPA receptors [
11,
13]. Although the relationship between gintonin and the nervous system has been well-investigated, relatively little is known about the effects of gintonin on skin, specifically on fibroblasts that are closely related to skin biology.
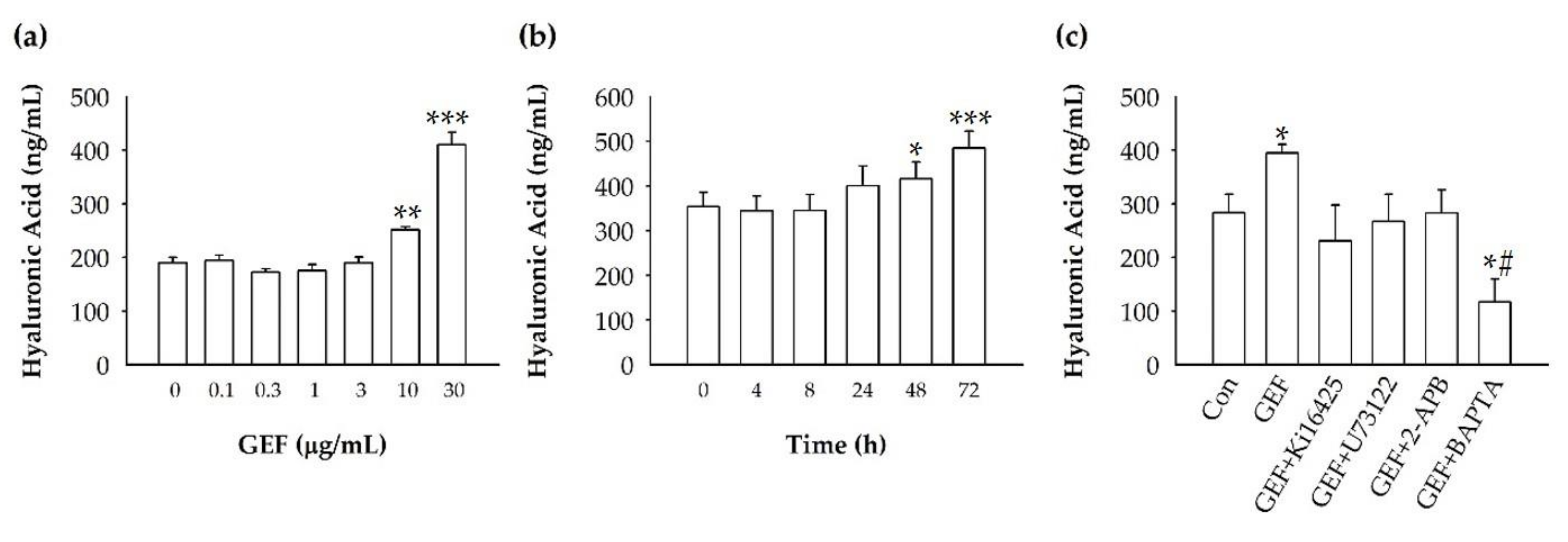
Fibroblasts are one of the most important skin cell types, not only for proliferating, but also for maintaining skin homeostasis. LPA has been known to induce the proliferation and migration of fibroblasts [
14]. Therefore, we hypothesized that GEF could be useful for revitalizing the skin by inducing the secretion of skin-hydrating or -protecting materials by stimulating fibroblast activities. Two major and well-known factors affecting skin hydration and protection are hyaluronic acid (HA) and collagen. HA is an extracellular matrix (ECM) component that hydrates skin, lubricates joints, and has a space-filling capacity. HA synthesis increases during tissue injury and wound healing. HA also regulates the activation of inflammatory cells to induce the cellular immune response upon fibroblast cell injury [
14]. Collagen is required for deposition in fibroblasts and tissue regeneration [
15,
16].
Panax ginseng extract has wound-healing properties and has, thus, been used to maximize collagen synthesis for anti-wrinkle effects; however, its active ingredient(s) have not been clearly identified [
17]. A large amount of research is, therefore, still needed to understand the effects of GEF binding to LPA receptors, as well as the relationship between GEF and skin biology.
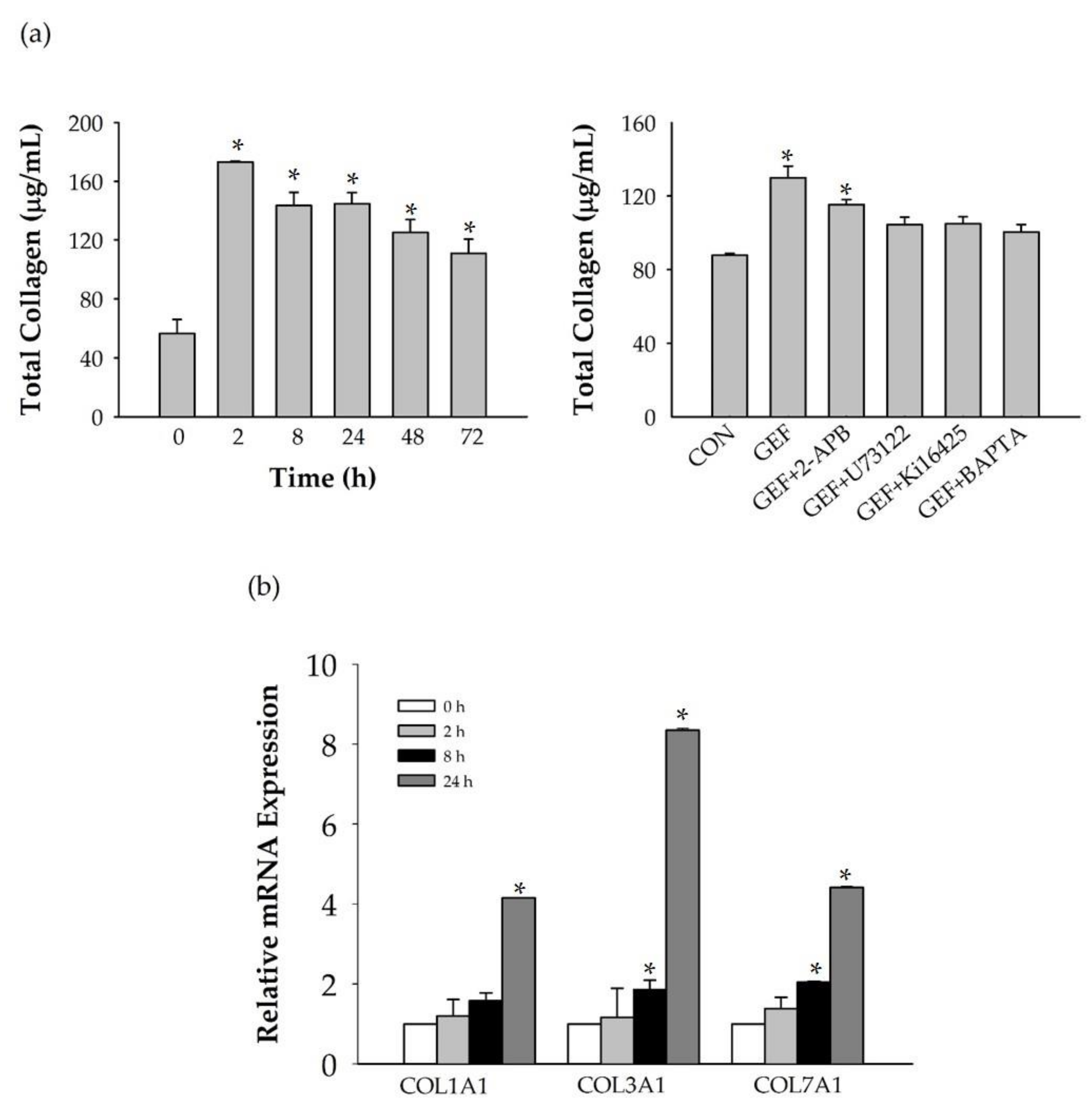
In this study, we investigated how GEF promotes HA and collagen release. We found that GEF-mediated LPA receptor activation is coupled to Ca2+ signaling and is followed by HA and collagen release, which are closely associated with skin aging and skin protection. Thus, GEF can potentially be used as a cosmetic ingredient.
3. Discussion
Skin is comprised of the epidermis and the dermis. The dermis is an inner layer that is comprised of fibroblasts. Skin acts as the front line of protection against bacterial infections, water loss, and physical injury [
18,
19,
20]. It is well known that the fibroblast layer of skin is easily damaged by various factors, ranging from UV exposure to physical hazards [
20]. Although previous reports have demonstrated that several ginsenosides and ginsenoside metabolites, such as compound K, exert skin-hydrating effects by regulating HA synthases and prevent skin aging and degradation of collagen by regulating collagenases under UVB irradiation conditions [
20,
21], other ingredients of ginseng, such as gintonin, have not been examined, and relatively little is known about how GEF affects the molecular mechanisms of skin homeostasis. To understand the mechanisms, we studied the beneficial effects of GEF on human skin fibroblasts.
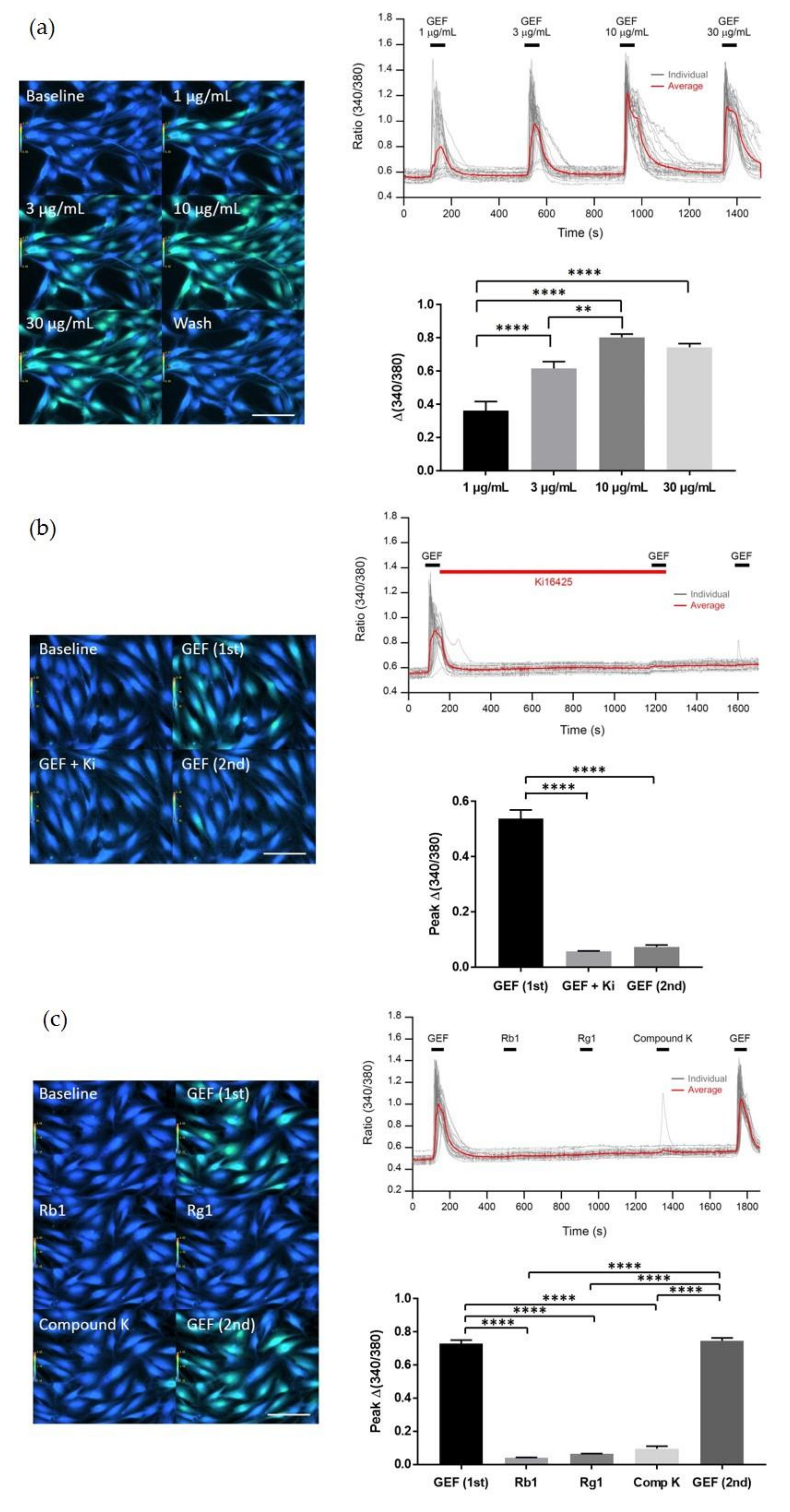
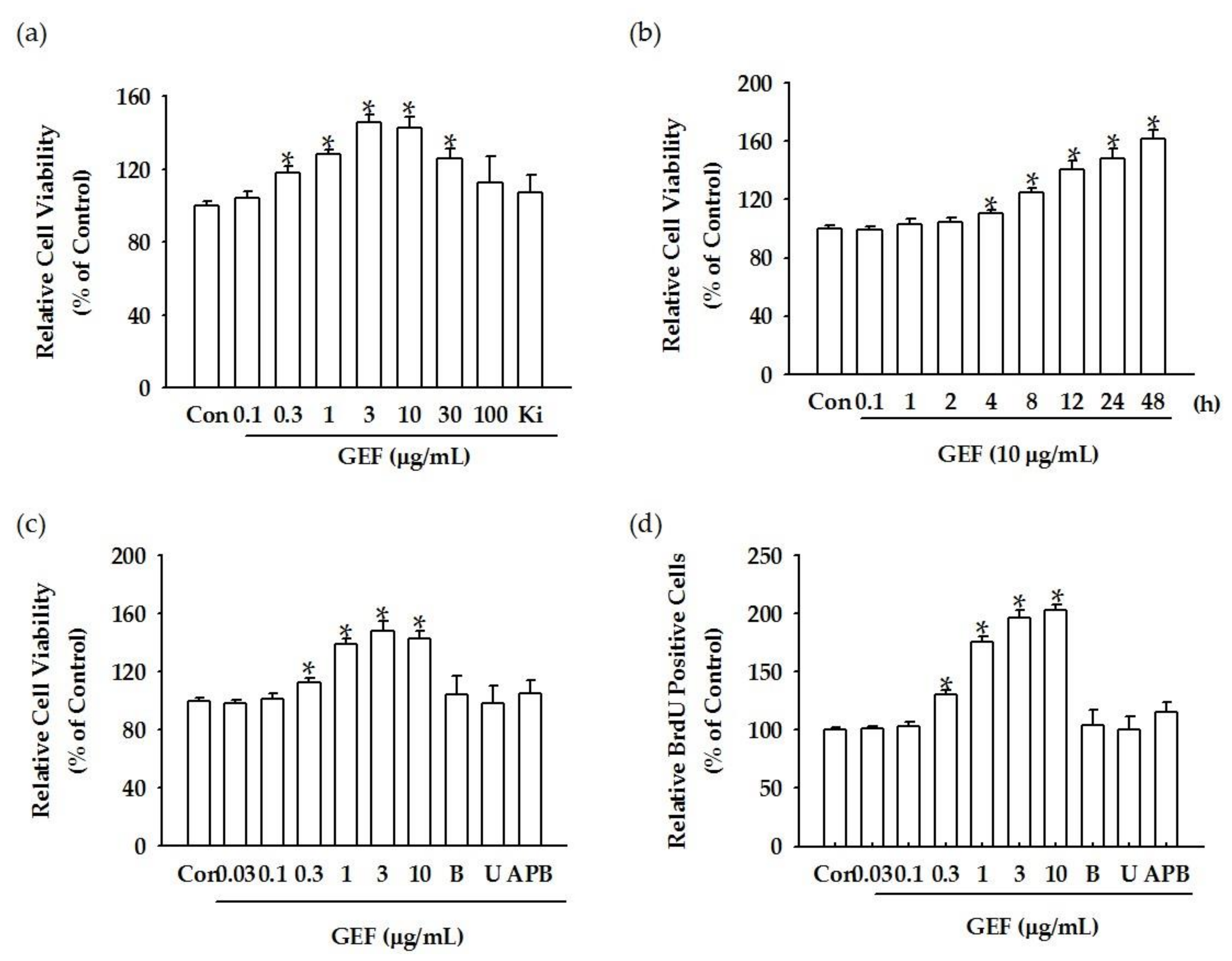
We found that GEF treatment of HDF cells can induce [Ca
2+]
i transiently via regulation of LPA receptor signal transduction and further induce in vitro cell proliferation via the LPA receptor signaling pathway. Interestingly, GEF, but not ginsenosides, such as ginsenoside Rb1, ginsenoside Rg1, and compound K, induced [Ca
2+]
i transiently, indicating that GEF, but not ginsenosides, is an active ingredient of ginseng that promotes HDF cell regulation via transient induction of [Ca
2+]
i. Next, we examined whether GEF-mediated transient induction of [Ca
2+]
i via LPA receptor regulation could be coupled to HA and collagen release via LPA receptor activation. We found that GEF stimulates HA and collagen release via the LPA receptor signaling pathway and that GEF treatment of HDF cells enhanced HA and collagen synthase gene expression. Finally, we found that GEF treatment increased LPA receptor subtype expression, at both the gene and protein levels. The present study, thus, shows that GEF-mediated cellular activity changes via LPA receptor subtype regulation are linked induction of [Ca
2+]
i, transient to cell proliferation, HA and collagen release, and changes in LPA receptor expression in HDF cells. These results are similar to those obtained in our previous studies on gintonin-mediated wound healing in corneal epithelium cells through in vitro, as well as in vivo experiments using a rabbit corneal injury model [
22].
LPA, a lysophospholipid, acts as a ligand for at least six G protein-coupled LPA receptor subtypes (LPA1-6). LPA receptors are expressed in a variety of cells and tissues, including fibroblasts [
14,
15,
22]. The present study provides further evidence that GEF treatment in normal human dermal fibroblast can help promote HA and collagen release by stimulating the LPA receptor–Ca
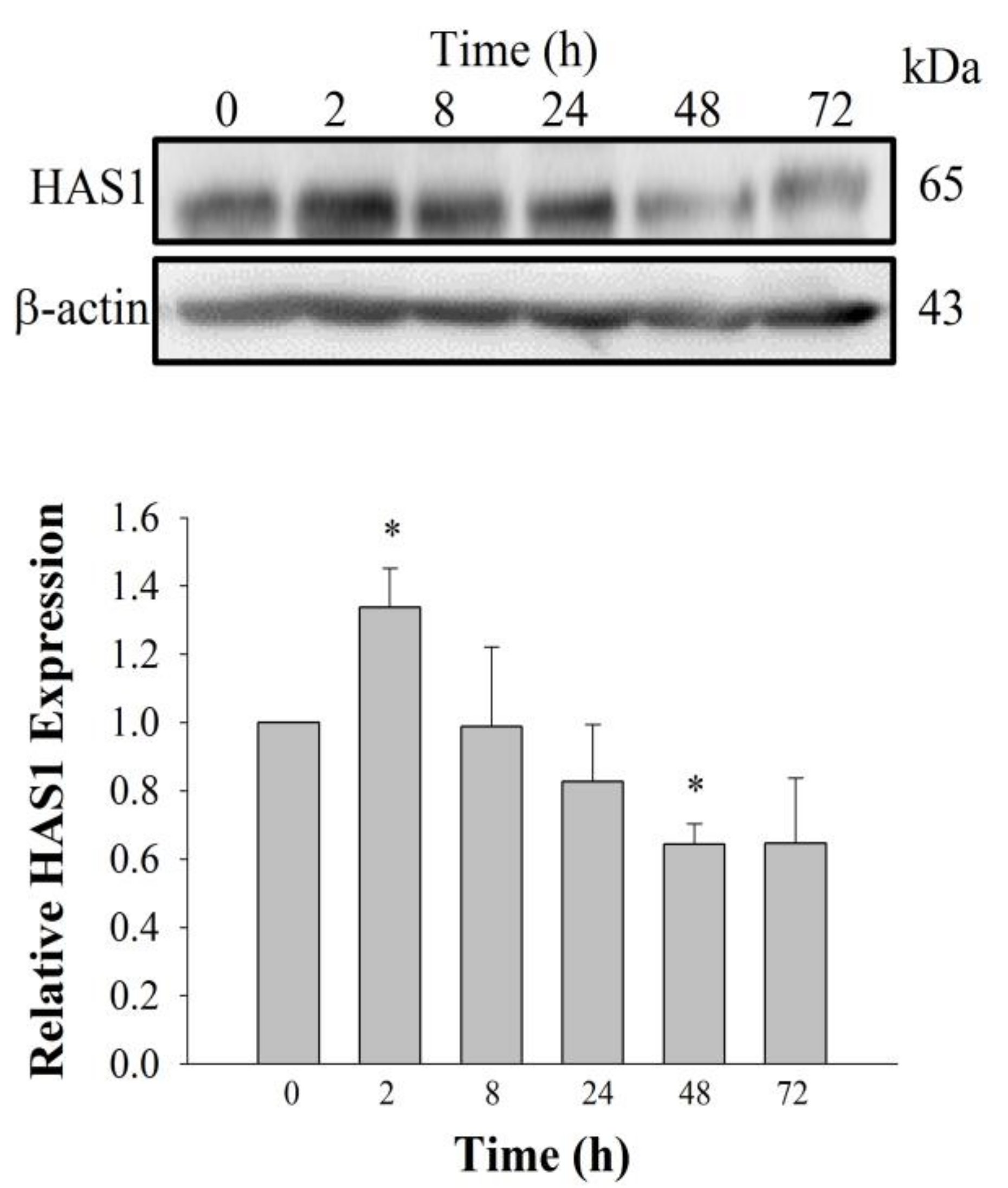
2+ signaling pathway. Using cyclic phosphatidic acid, Maeda-Sano et al. revealed that HA synthesis takes place via the CREB transcription factor signaling pathway through the activation of HAS-2, and not HAS-1; this finding is not consistent with our results [
15]. This discrepancy could be explained by our use of GEF, which is different from the compound used by Maeda-Sano et al. Moreover, we used a HAS-1 antibody, not HAS-2 antibody utilized by Maeda-Sano et al. to test protein expression levels (
Figure 4) [
15]. Further studies are needed to understand the genes and proteins that are responsible for induction of HA synthesis upon treatment of fibroblasts with GEF. These results also raise the possibility that GEF, an exogenous LPA receptor ligand, can play a key role in fibroblast hydration, as well as anti-aging if used as an externally applicable ingredient on the skin. Thus, GEF could be used as a base material for topical skin treatments. In the future, GEF could be used as an herb-derived cosmetic ingredient for daily skin treatment due to its skin-hydrating effect.
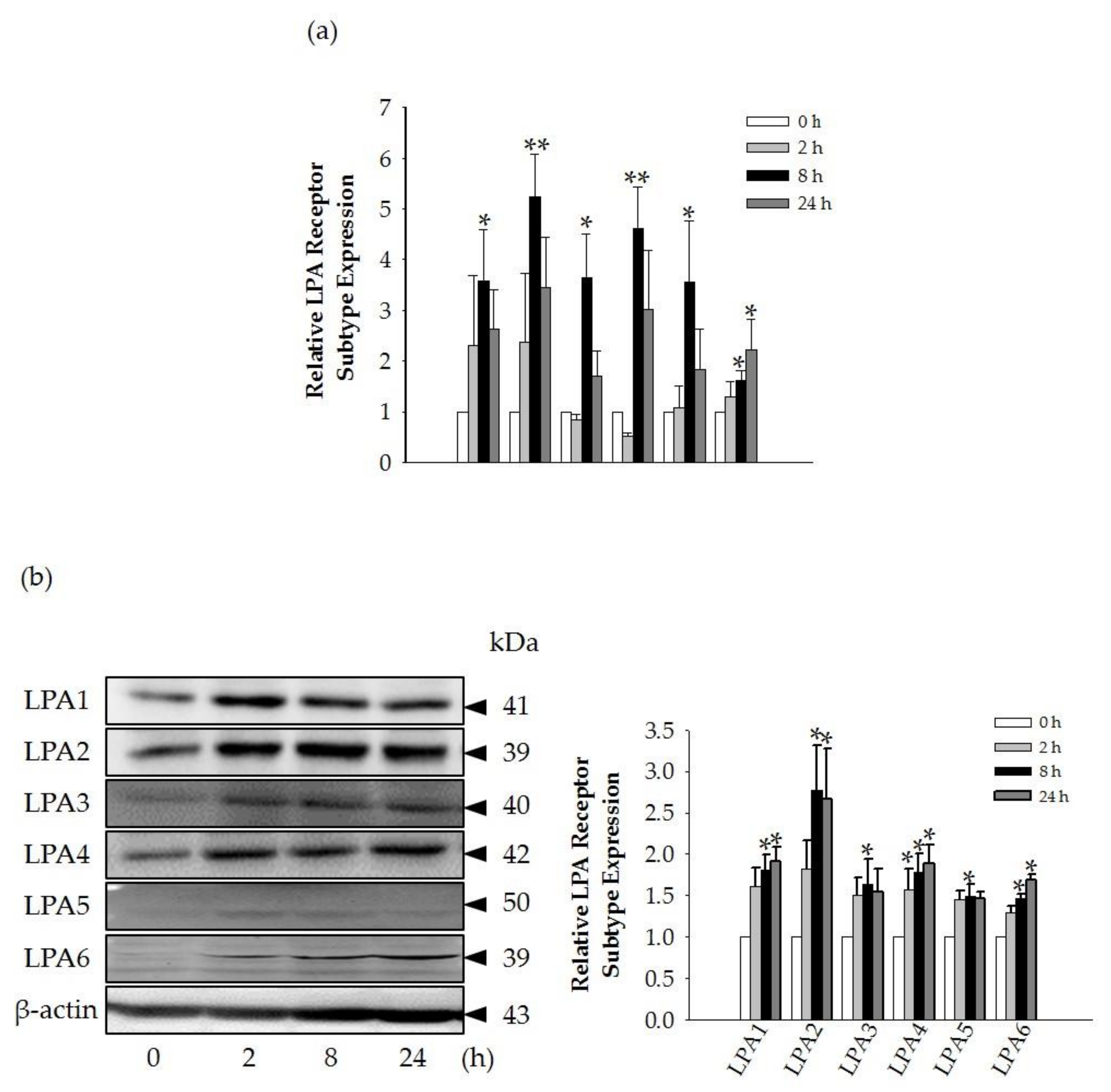
Interestingly, we found that the GEF-mediated transient induction of [Ca
2+]
i, cell proliferation, and HA and collagen release were attenuated by LPA1/3 receptor antagonists. However, as shown in
Figure 6, we found that GEF treatment of HDFs increased the expression levels of nearly all of the six LPA receptor subtypes with different degrees in HDFs, indicating the possibility that GEF exerts its effects through all six LPA receptors. However, in the present study, we could not confirm the involvement of other LPA receptor subtypes except LPA1/3 receptors, due to the limited availability of selective and specific LPA receptor subtype antagonists. In future studies, the development of selective LPA receptor subtype antagonists will allow us to identify which LPA receptor subtypes are more involved in affecting HDF physiology with respect to HA and collagen release.
There was a report that matrix metallopeptidases (MMPs) may play an important role by LPA upregulating procollagen expression in normal skin conditions and downregulating MMP-1 under ultraviolet (UV)-induced conditions [
23]. It is also known that UV irradiation causes up-regulation of several MMPs that can impair extracellular matrix (ECM) proteins that can lead to skin aging [
24]. In future study, it will be interesting to examine the effects of GEF on MMPs related with skin biology.
In conclusion, we have demonstrated in this study that LPA receptor activation induced by GEF treatment of HDFs makes GEF an effective candidate for skin hydration and for possible anti-aging in HDFs. GEF is, therefore, a good candidate cosmetic ingredient to promote skin hydration, and possibly prevent or treat skin aging.
4. Materials and Methods
4.1. GEF Preparation and Reagents
GEFs were prepared as previously described [
7]. Briefly, 4-year-old Korean white ginseng (Korea Ginseng Cooperation, Daejon, Korea), purchased locally from a ginseng market, was ground into small pieces (>3 mm) and refluxed with 70% fermentation ethanol. The ethanol extracts were concentrated and dissolved in distilled water. After ethanol extraction, the supernatant and precipitate produced by water fractionation were separated by centrifugation (1977×
g, 20 min). The precipitate was lyophilized after being centrifuged. This fraction was designated GEF and had a yield of 1.3%. 2-Aminoethoxydiphenyl borate (2-APB), phospholipase C inhibitor (U73122), a membrane permeable Ca
2+ chelator to chelate free cytosolic Ca
2+ (calcium-specific aminoplolycarboxilic acid, BAPTA-AM), Ki16425 (LPA1/3 inhibitor), and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). HAS1, LPA1-4, and anti-β-actin–HRP-conjugated antibodies were purchased from Abcam (Cambridge, MA, USA). LPA5 was obtained from Biorbyt (San Francisco, CA, USA), LPA6 was obtained from OriGene Technologies, Inc. (Rockville, MD, USA), and goat anti-rabbit IgG antibody was obtained from GeneTex (Irvine, CA, USA). For all molecular studies, the reagents and kits used were purchased from BIORAD (Hercules, CA, USA) and Invitrogen (Walthamm, MA, USA).
4.2. Cell Culture
For all experiments, human dermal fibroblasts (HDFs), from passages 3 to 10, were used (NB1RGB, Biosolution, Catalog No. MC1233). All cells were cultured in 95% Minimum Essential Medium (MEM) α with 5% fetal bovine serum and maintained at 37 °C in humidified conditions under 5% CO2. For testing dose dependence, cells were incubated in a 100-mm culture dish, followed by serum starvation for 24 h, and treated with vehicle or GEF (final concentration of 0, 1, 3, 10, 30, and 100 μg/mL for LPA subtype detection and cell viability test) for 24 h. Cells were incubated in a 100-mm culture dish, moved to fresh serum-free media for another 24 h, and treated with vehicle or GEF (final conc. of 10 μg/mL for HAS1 and LPA subtype detection and cell) for time dependence tests (0, 4, 8, 24, 48, and 72 h) or pre-treated with antagonists for 30 min followed by GEF treatment (final conc. of 10 μg/mL) for 24 h. Cell culture supernatants were used for detection of hyaluronic acid and collagen secretion into media.
4.3. Primer Designing and mRNA Analysis
To investigate mRNA expression of LPA receptors (LPA1-6), collagen type I, alpha 1 (COL1A1), collagen, type III, alpha 1 (COL3A1), and collagen, type VII, alpha 1 (COL7A1), β-actin expression was used as a normalization control in HDFs. Total RNA was isolated from HDFs with TRIzol (Invitrogen) according to the manufacturer’s instructions, with minor modifications. qRT-PCR (real-time PCR) was performed with individually reverse-transcribed cDNAs. The CFX96 real-time PCR system (BIORAD, U.S.A) was used for analysis. iQ SYBR Green Supermix (BIORAD) was utilized with the following steps: Pre-denaturation at 95 °C for 30 s, 35 repeats of denaturation at 95 °C for 10 s, 60 °C for 10 s, 72 °C for 25 s, final elongation at 72 °C for 30 s, and detection of melting curve from 72 °C to 97 °C. The following primers were used for this experiment: LPA1-5’gtcttctgggccattttcaa3´, 5´tcatagtcctctggcgaaca3´, 91 bp; LPA2-5´gaggccaactcactggtca3´, ggcgcatctcagcatctc3´, 58 bp; LPA3-5´gaagctaatgaagacggtgatga3´, 5´agcaggaaccaccttttcac3´, 135bp; LPA4-5´tctggatcctagtcctcagtgg3´, 5´ccagacacgtttggagaagc3´, 107 bp; LPA5-5´cgccatcttccagatgaac3´, 5´tagcggtccacgttgatg3´, 66 bp; LPA6-5´tctggcaattgtctacccatt3´, 5´tcaaagcaggcttctgagg3´, 165 bp; β-actin-5´ttctacaatgagctgcgtgtg3´, 5´ggggtgttgaaggtctcaaa3´, 122 bp; COL1A1-5´gccaagacgaagacatccca3´, 5´cagatcacgtcatcgcacaac3´, 136bp; COL3A1-5´cccactattattttggcacaacag3´, 5´aacggatcctgagtcacagaca3´, 129 bp; COL7A1-5´ctcagcagctatcacctggac3´, 5´tgtccaccacacgtagttcaa3´, 154 bp.
4.4. Immunoblotting
First, 20 μg of total protein from lysed HDF cells was used to detect LPA receptor subtypes and HAS1. The cells were lysed using modified radio immunoprecipitation assay buffer (RIPA buffer) with a protease inhibitor cocktail, and LPA receptor expression was detected using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and semi-dry blotting of the protein onto a 0.45-μm PVDF membrane using rabbit anti-LPA1 polyclonal antibody (1:2000), rabbit anti-LPA2 polyclonal antibody (1:1000), rabbit anti-LPA3 polyclonal antibody (1:500), rabbit anti-LPA4 polyclonal antibody (1:2000), rabbit anti-LPA5 polyclonal antibody (1:2000), rabbit anti-LPA6 polyclonal antibody (1:2000), or rabbit anti-HAS1 polyclonal antibody (1:1000). The blotted membrane was stripped and re-probed with mouse anti-β-actin monoclonal antibody conjugated to HRP (1:30,000). Images were visualized using Clarity Western ECL Substrate Bio-Rad (Hercules, CA, USA) using the iBright CL1000 (Thermo Fischer Scientific, Waltham, MA, United States). Chemi-documentation was conducted for further documentation preservation. Densitometry analysis of the blots was performed using ImageJ.
4.5. Measuring Hyaluronic Acid Using Enzyme-Linked Immunosorbent Assay (ELISA)
Hyaluronic acid measurement was performed using the Hyaluronan Enzyme-Linked Immunosorbent Assay Kit (HA-ELISA, K-1201, Echelon, Salt Lake City, UT, USA) according to the manufacturer’s instructions. Briefly, cells were plated in a 100-mm dish (1 × 105 cells/plate) for 24 h, serum-starved for another 24 h, then pre-treated with antagonists (2-APB, U73122, Ki16425, and BAPTA-AM) for 30 min, followed by GEF treatment. The collected supernatants were analyzed by the detection plate (K-1201, Echelon, Salt Lake City, UT, USA) and measured at 405 nm at T = 45 min using a scanning multi-well spectrophotometer.
4.6. Measuring Collagen Using Enzyme-Linked Immunosorbent Assay (ELISA)
Collagen analysis from cell media was conducted according to the manufacturer’s instructions. Briefly, cells were processed as described above, and the supernatants were collected, freeze-dried to yield powder, and re-suspended with 1× PBS (pH 7.2) to obtain 10× concentrated media. The samples were diluted at a 1:1 ratio with 12 M HCl, boiled at 95 °C for 20 h, and centrifuged for 10 min at 13,000× g. The prepared samples were treated with detection reagents for another 60 min and the plate was read at 570 nm by a scanning multi-well spectrophotometer (Spectra Max 190, Molecular Devices, Sunnyvale, CA, USA).
4.7. Cell Viability Assay
The cell viability after each treatment was determined using a water-soluble tetrazolium salt-1(WST-1) assay. The WST-1 cell proliferation assay (Catalog No. ab155902, Abcam, Cambridge, UK) was performed using a plate reader (Synergy 2, BioTek, Winooski, VT, USA). Cells were plated in 96-well plates (2 × 104 cells/well) overnight and were subjected to GEF treatment for the indicated time. Then, the culture plate was incubated with the WST-1 reagent for 0.5–4 h, according to the manufacturer’s instructions. After incubation, the resulting formazan dye was quantified using a scanning multi-well spectrophotometer (Spectra Max 190, Molecular Devices, Sunnyvale, CA, USA). The optical density (OD) of each well was measured by a plate reader with a filter setting at 570 nm.
4.8. Intracellular Ca2+ Measurement
Intracellular Ca
2+ levels in HDFs were measured after exposure to GEF. HDFs from each group were incubated for 40–60 min at room temperature with 5 μM Fura-2/AM (Molecular Probes, Eugene, OR, USA) and 0.001% pluronic F-127 (Molecular Probes) in a 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered solution composed of the following: 150 mM NaCl, 5 mM KCl, 1 mM MgCl
2, 2 mM CaCl
2, 10 mM HEPES, and 10 mM glucose, with the pH adjusted to 7.4 with NaOH. Cells were illuminated using a xenon arc lamp, and the excitation wavelengths (340 and 380 nm) were selected using a computer-controlled filter wheel (Sutter Instruments, Novato, CA, USA). The emitted fluorescence was reflected through a 515-nm-long pass filter to a frame transfer–cooled CCD camera (Olympus, Shinjuku, Tokyo, Japan), and the ratios of the emitted fluorescence were calculated using a digital fluorescence analyzer and then converted to intracellular free Ca
2+ concentrations [Ca
2+]
i. All imaging data were collected and analyzed using the Universal Imaging software (Bedford Hills, New York, USA) [
25].
4.9. Statistical Analysis
All values are presented as either mean ± standard error of the mean (S.E.M.) or % of control. A p-value under 0.05 was considered to be statistically significant. Differences between the means of the control and treatment groups were compared using unpaired Student’s t-test or one-way ANOVA.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





