
Development of Targeted Alpha Particle Therapy for Solid Tumors

 , ,
, ,  and
and
Abstract
1. Introduction
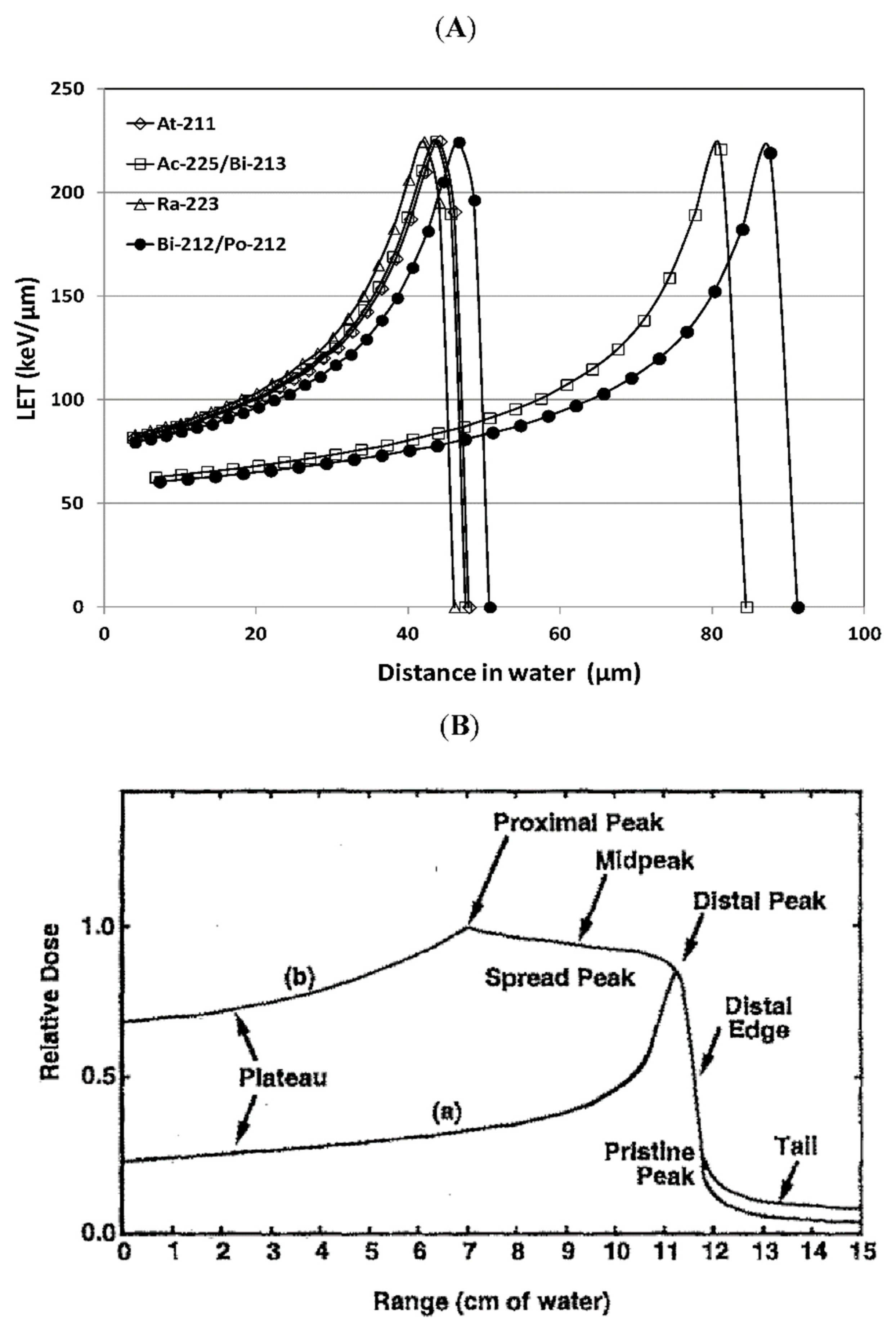
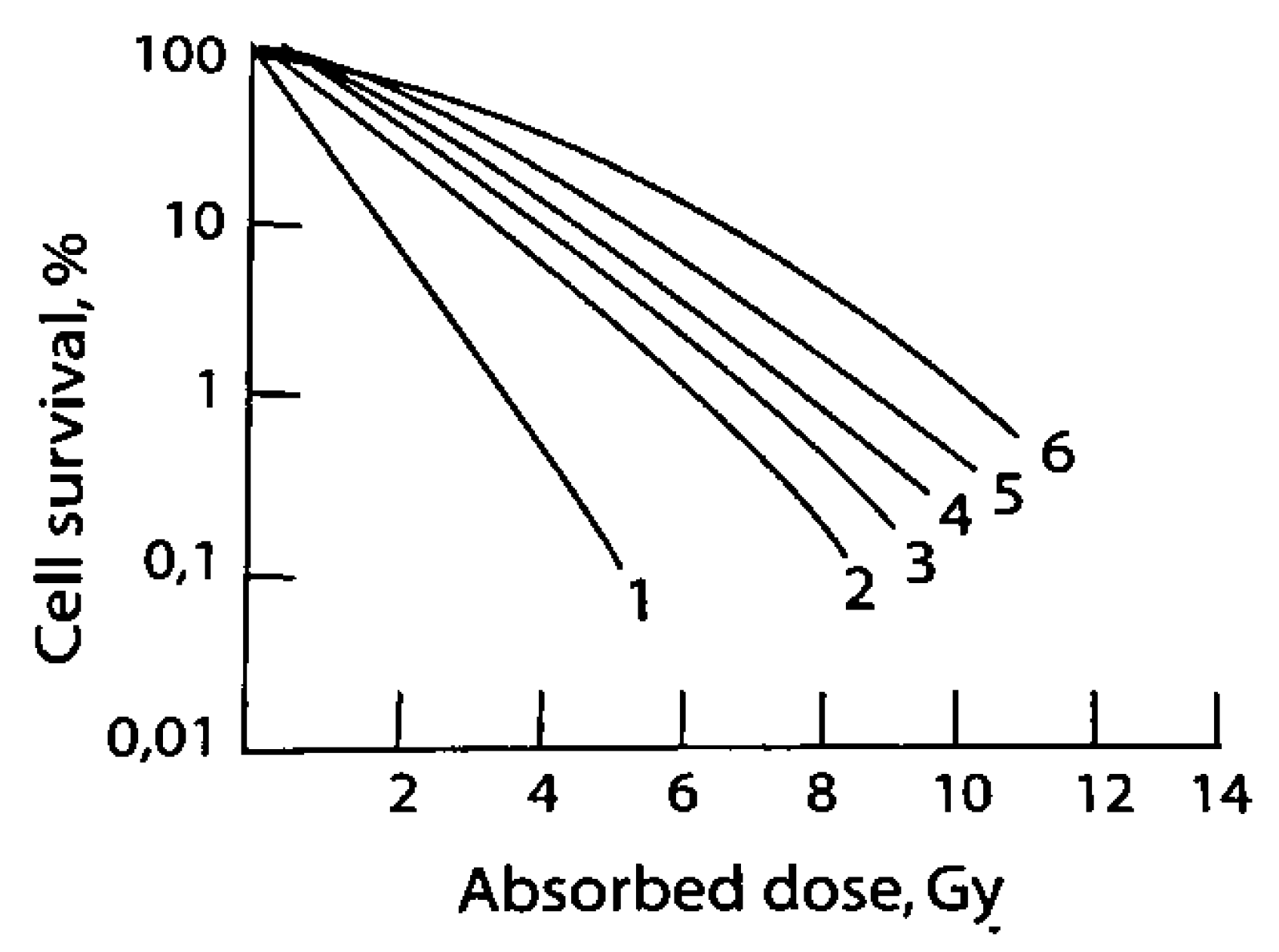
2. Mechanism of Action/Tumor Cell Killing
3. Alpha-Particle Emitting Radionuclides
4. Targeting Moieties
4.1. Small Molecules
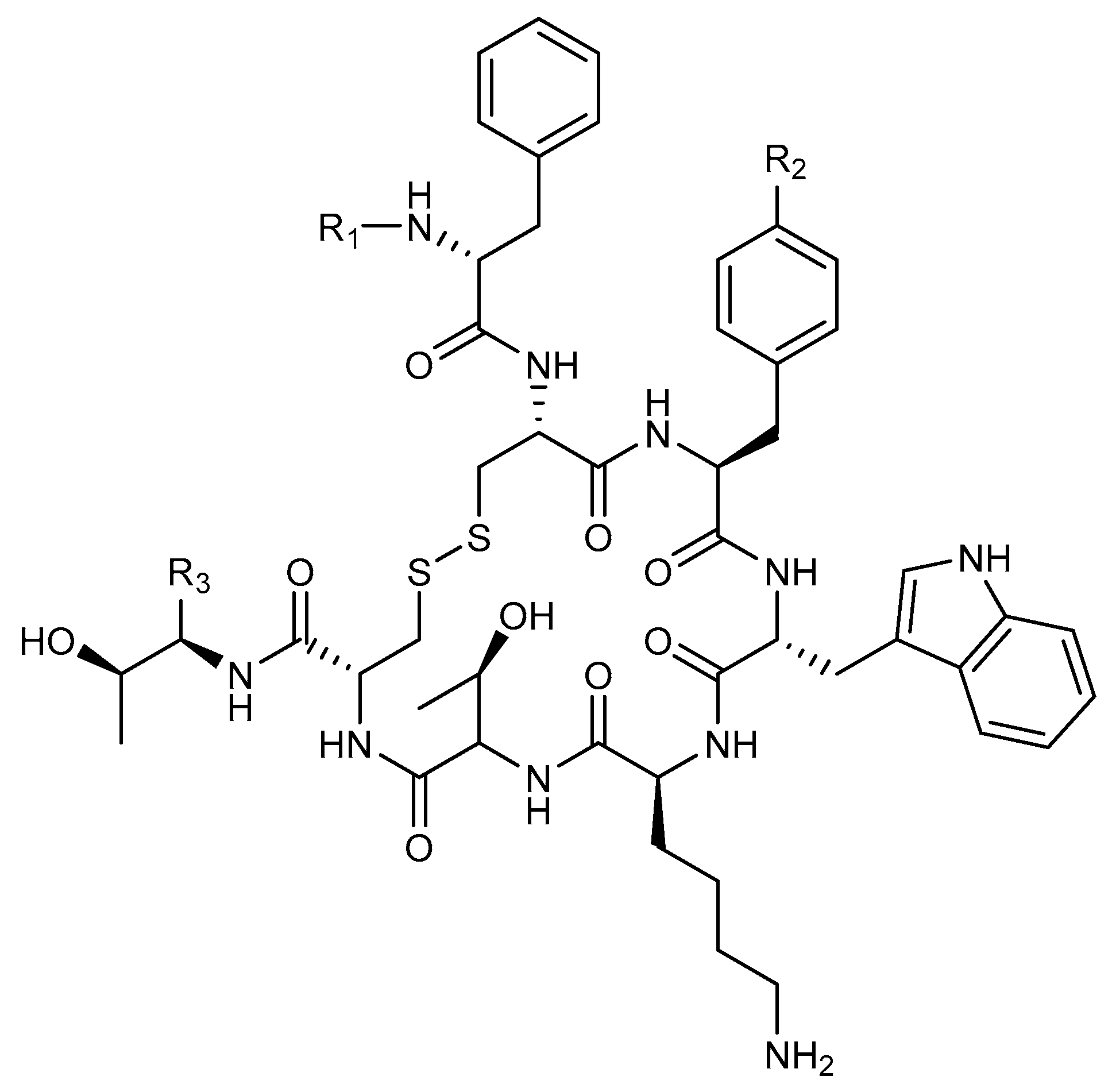
4.2. Peptides
4.3. Antibodies
4.4. Antibody Fragments
4.5. Passive Targeting
5. Chelation/Attachment
5.1. Radiosynthesis
5.2. Linkers/Rational Design
6. Radiation Dosimetry
7. Pre-Clinical Studies
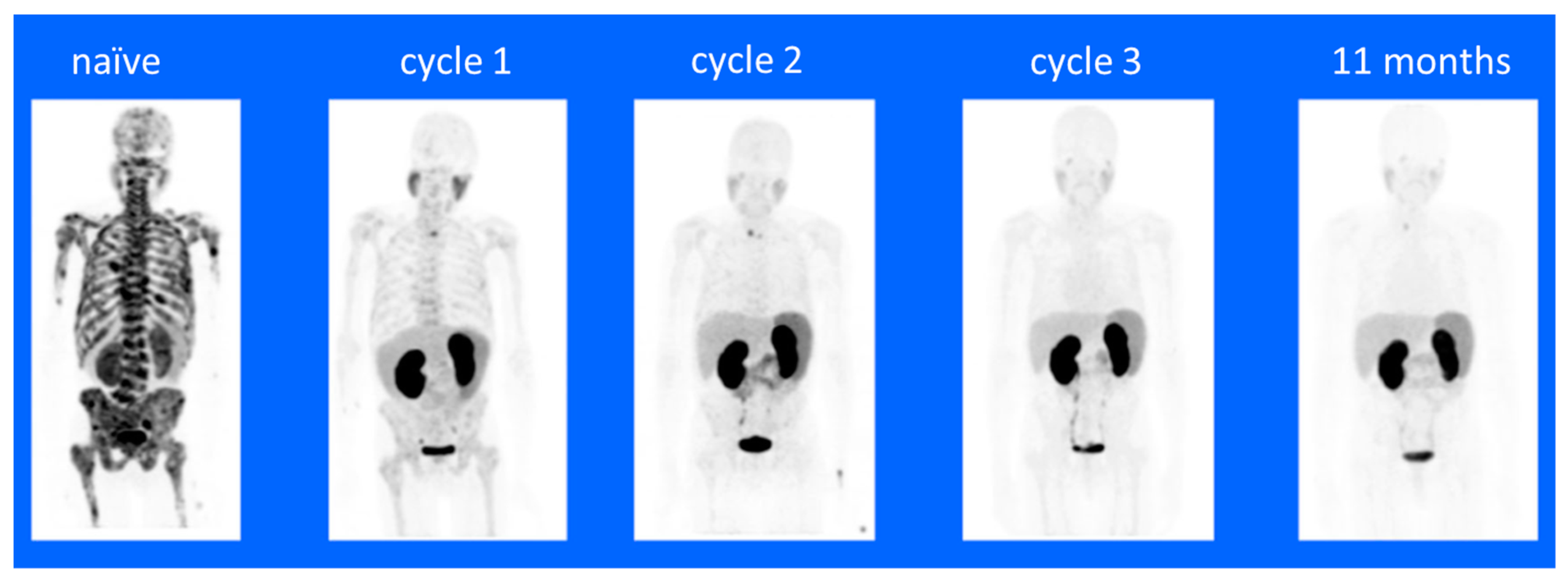
7.1. Preclinical Therapeutics Studies
7.2. Preclinical Imaging
8. Medicinal Chemistry
8.1. Lead Optimization
8.2. cGMP Production
9. Clinical Studies
9.1. Recent TAT Clinical Trials
9.2. Radium-223 Dichloride (223RaCl2) Trials
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pouget, J.P.; Navarro-Teulon, I.; Bardies, M.; Chouin, N.; Cartron, G.; Pelegrin, A.; Azria, D. Clinical radioimmunotherapy--the role of radiobiology. Nat. Rev. Clin. Oncol. 2011, 8, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Bodet-Milin, C.; Kraeber-Bodere, F.; Eugene, T.; Guerard, F.; Gaschet, J.; Bailly, C.; Mougin, M.; Bourgeois, M.; Faivre-Chauvet, A.; Cherel, M.; et al. Radioimmunotherapy for Treatment of Acute Leukemia. Semin. Nucl. Med. 2016, 46, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, G.; Roeske, J.C.; McDevitt, M.R.; Palm, S.; Allen, B.J.; Fisher, D.R.; Brill, A.B.; Song, H.; Howell, R.W.; Akabani, G.; et al. MIRD Pamphlet No. 22 (abridged): Radiobiology and dosimetry of alpha-particle emitters for targeted radionuclide therapy. J. Nucl. Med. 2010, 51, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Dizdarevic, S.; Jessop, M.; Begley, P.; Main, S.; Robinson, A. (223)Ra-Dichloride in castration-resistant metastatic prostate cancer: Improving outcomes and identifying predictors of survival in clinical practice. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Hohenfellner, M.; Giesel, F.L.; Haberkorn, U.; Morgenstern, A. Targeted alpha-Therapy of Metastatic Castration-Resistant Prostate Cancer with (225)Ac-PSMA-617: Swimmer-Plot Analysis Suggests Efficacy Regarding Duration of Tumor Control. J. Nucl. Med. 2018, 59, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Sathekge, M.; Bruchertseifer, F.; Knoesen, O.; Reyneke, F.; Lawal, I.; Lengana, T.; Davis, C.; Mahapane, J.; Corbett, C.; Vorster, M.; et al. (225)Ac-PSMA-617 in chemotherapy-naive patients with advanced prostate cancer: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 129–138. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- Sartor, O.; Sharma, D. Radium and other alpha emitters in prostate cancer. Transl. Urol. 2018, 7, 436–444. [Google Scholar] [CrossRef]
- Franchet-Beuzit, J.; Spotheim-Maurizot, M.; Sabattier, R.; Blazy-Baudras, B.; Charlier, M. Radiolytic footprinting. Beta rays, gamma photons, and fast neutrons probe DNA-protein interactions. Biochemistry 1993, 32, 2104–2110. [Google Scholar] [CrossRef]
- Schulte-Frohlinde, D. Mechanism of radiation-induced strand break formation in DNA and polynucleotides. Adv. Space Res. 1986, 6, 89–96. [Google Scholar] [CrossRef]
- Cox, R.; Masson, W.K. Mutation and inactivation of cultured mammalian cells exposed to beams of accelerated heavy ions. III. Human diploid fibroblasts. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1979, 36, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T.; Munson, R.J.; Thacker, J.; Cox, R. Mutation and inactivation of cultured mammalian cells exposed to beams of accelerated heavy ions. IV. Biophysical interpretation. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1980, 37, 135–167. [Google Scholar] [CrossRef] [PubMed]
- Munson, R.J.; Bance, D.A.; Stretch, A.; Goodhead, D.T. Mutation and inactivation of cultured mammalian cells exposed to beams of accelerated heavy ions. I. Irradiation facilities and methods. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1979, 36, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Thacker, J.; Stretch, A.; Stephens, M.A. Mutation and inactivation of cultured mammalian cells exposed to beams of accelerated heavy ions. II. Chinese hamster V79 cells. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1979, 36, 137–148. [Google Scholar] [CrossRef]
- Pouget, J.P.; Mather, S.J. General aspects of the cellular response to low- and high-LET radiation. Eur. J. Nucl. Med. 2001, 28, 541–561. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Sgouros, G. Alpha-particles for targeted therapy. Adv. Drug Deliv. Rev. 2008, 60, 1402–1406. [Google Scholar] [CrossRef]
- Song, H.; Senthamizhchelvan, S.; Hobbs, R.F.; Sgouros, G. Alpha Particle Emitter Radiolabeled Antibody for Metastatic Cancer: What Can We Learn from Heavy Ion Beam Radiobiology? Antibodies 2012, 1, 124–148. [Google Scholar] [CrossRef]
- Lawrence, J.H.; Tobias, C.A.; Born, J.L.; Linfoot, J.A.; Kling, R.P.; Gottschalk, A. Alpha and Proton Heavy Particles and the Bragg Peak in Therapy. Trans. Am. Clin. Clim. Assoc. 1964, 75, 111–116. [Google Scholar]
- Kudryashow, Y.B. Radiation Biophysics (Ionization Radiation); Nova Science Publishers Inc.: Hauppauge, NY, USA, 2006. [Google Scholar]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Elgqvist, J.; Andersson, H.; Back, T.; Hultborn, R.; Jensen, H.; Karlsson, B.; Lindegren, S.; Palm, S.; Warnhammar, E.; Jacobsson, L. Therapeutic efficacy and tumor dose estimations in radioimmunotherapy of intraperitoneally growing OVCAR-3 cells in nude mice with (211)At-labeled monoclonal antibody MX35. J. Nucl. Med. 2005, 46, 1907–1915. [Google Scholar]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Seymour, C. Radiation-induced bystander effects: Evidence for an adaptive response to low dose exposures? Dose Response 2006, 4, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Sawal, H.A.; Asghar, K.; Bureik, M.; Jalal, N. Bystander signaling via oxidative metabolism. Onco. Target 2017, 10, 3925–3940. [Google Scholar] [CrossRef] [PubMed]
- Ludgate, C.M. Optimizing cancer treatments to induce an acute immune response: Radiation Abscopal effects, PAMPs, and DAMPs. Clin. Cancer Res. 2012, 18, 4522–4525. [Google Scholar] [CrossRef]
- Williams, L.E.; DeNardo, G.L.; Meredith, R.F. Targeted radionuclide therapy. Med. Phys. 2008, 35, 3062–3068. [Google Scholar] [CrossRef]
- Jadvar, H.; Quinn, D.I. Targeted alpha-particle therapy of bone metastases in prostate cancer. Clin. Nucl. Med. 2013, 38, 966–971. [Google Scholar] [CrossRef]
- Miederer, M.; Scheinberg, D.A.; McDevitt, M.R. Realizing the potential of the Actinium-225 radionuclide generator in targeted alpha particle therapy applications. Adv. Drug Deliv. Rev. 2008, 60, 1371–1382. [Google Scholar] [CrossRef]
- Allen, B.J.; Huang, C.Y.; Clarke, R.A. Targeted alpha anticancer therapies: Update and future prospects. Biologics 2014, 8, 255–267. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef]
- Imam, S.K. Advancements in cancer therapy with alpha-emitters: A review. Int. J. Radiat Oncol. Biol. Phys. 2001, 51, 271–278. [Google Scholar] [CrossRef]
- Elgqvist, J.; Frost, S.; Pouget, J.P.; Albertsson, P. The potential and hurdles of targeted alpha therapy—Clinical trials and beyond. Front. Oncol. 2014, 3, 324. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.R.; Ma, D.; Lai, L.T.; Simon, J.; Borchardt, P.; Frank, R.K.; Wu, K.; Pellegrini, V.; Curcio, M.J.; Miederer, M.; et al. Tumor therapy with targeted atomic nanogenerators. Science 2001, 294, 1537–1540. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, O.; Guatelli, S.; Allen, B.; Barbet, J.; Cherel, M.; Bardies, M.; Bruchertseifer, F.; Seidl, C.; Bombardieri, E.; Bilewicz, A.; et al. Report: Technical Meeting on “Alpha emitting radionuclides and radiopharmaceuticals for therapy”. Available online: http://www-naweb.iaea.org/napc/iachem/working_materials/TM-44815-report-Alpha-Therapy.pdf (accessed on 25 September 2019).
- Kim, Y.S.; Brechbiel, M.W. An overview of targeted alpha therapy. Tumour Biol. 2012, 33, 573–590. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.R.; Finn, R.D.; Ma, D.; Larson, S.M.; Scheinberg, D.A. Preparation of alpha-emitting 213Bi-labeled antibody constructs for clinical use. J. Nucl. Med. 1999, 40, 1722–1727. [Google Scholar]
- Couturier, O.; Supiot, S.; Degraef-Mougin, M.; Faivre-Chauvet, A.; Carlier, T.; Chatal, J.F.; Davodeau, F.; Cherel, M. Cancer radioimmunotherapy with alpha-emitting nuclides. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 601–614. [Google Scholar] [CrossRef]
- Corson, D.R.; MacKenzie, K.R.; Segre, E. Artificially Radioactive Element 85. Phys. Rev. J. 1940, 58, 672. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Pruszynski, M. Astatine-211: Production and availability. Curr. Radiopharm. 2011, 4, 177–185. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Vaidyanathan, G. Astatine-211-labeled radiotherapeutics: An emerging approach to targeted alpha-particle radiotherapy. Curr. Pharm. Des. 2000, 6, 1433–1455. [Google Scholar] [CrossRef]
- Guerard, F.; Gestin, J.F.; Brechbiel, M.W. Production of [(211)At]-astatinated radiopharmaceuticals and applications in targeted alpha-particle therapy. Cancer Biother. Radiopharm. 2013, 28, 1–20. [Google Scholar] [CrossRef]
- Wadas, T.J.; Pandya, D.N.; Solingapuram Sai, K.K.; Mintz, A. Molecular targeted alpha-particle therapy for oncologic applications. AJR Am. J. Roentgenol. 2014, 203, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Zhuikov, B.L.; Kalmykov, S.N.; Ermolaev, R.A.; Aliev, V.M.; Kokhanyuk, V.L.; Matushko, I.G.; Tananaev, B.F.; Myasoedov, B.F. Production of 225Ac and 223Ra by irradiation of Th with accelerated protons. Radiochemistry 2011, 53, 77–80. [Google Scholar] [CrossRef]
- Griswold, J.R.; Medvedev, D.G.; Engle, J.W.; Copping, R.; Fitzsimmons, J.M.; Radchenko, V.; Cooley, J.C.; Fassbender, M.E.; Denton, D.L.; Murphy, K.E.; et al. Large scale accelerator production of (225)Ac: Effective cross sections for 78–192 MeV protons incident on (232)Th targets. Appl. Radiat Isot. 2016, 118, 366–374. [Google Scholar] [CrossRef] [PubMed]
- John, K. Targeted Alpha Therapy: The US DOE Tri-Lab (ORNL, BNL, LANL) Research Effort to Provide Accelerator-Produced 225Ac for Radiotherapy. In Proceedings of the American Physical Society Annual Meeting, New Orleans, LA, USA, 28–31 January 2017. [Google Scholar]
- Khabibullin, A.R.; Karolak, A.; Budzevich, M.M.; McLaughlin, M.L.; Morse, D.L.; Woods, L.M. Structure and properties of DOTA-chelated radiopharmaceuticals within the (225)Ac decay pathway. Medchemcomm 2018, 9, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Butterworth-Heinemann: Oxford, UK, 1997; p. 946. ISBN 0-08-037941-9. [Google Scholar]
- Bruland, O.S.; Nilsson, S.; Fisher, D.R.; Larsen, R.H. High-linear energy transfer irradiation targeted to skeletal metastases by the alpha-emitter 223Ra: Adjuvant or alternative to conventional modalities? Clin. Cancer Res. 2006, 12, 6250s–6257s. [Google Scholar] [CrossRef]
- Henriksen, G.; Bruland, O.S.; Larsen, R.H. Thorium and actinium polyphosphonate compounds as bone-seeking alpha particle-emitting agents. Anticancer Res. 2004, 24, 101–105. [Google Scholar]
- Mausner, L.F.; Straub, R.F.; Srivastava, S.C. The in vivo generator for radioimmunotherapy. J. Label. Compd. Radiopharm. 1989, 26, 498–500. [Google Scholar] [CrossRef]
- Baidoo, K.E.; Milenic, D.E.; Brechbiel, M.W. Methodology for Labeling Proteins and Peptides with Lead-212 ((212)Pb). Nucl. Med. Biol. 2013, 40, 592–599. [Google Scholar] [CrossRef]
- Mirzadeh, S.; Kumar, K.; Gansow, O.A. The Chemical Fate of 212Bi-DOTA Formed by β− Decay of 212Pb(DOTA)2−. Radiochem. Acta 1993, 60, 1–10. [Google Scholar] [CrossRef]
- Ruble, G.; Wu, C.; Squire, R.A.; Ganswo, O.A.; Strand, M. The use of 212Pb-labeled monoclonal antibody in the treatment of murine erythroleukemia. Int. J. Radiat. Oncol. Biol. Phys. 1996, 34, 609–616. [Google Scholar] [CrossRef]
- Su, F.M.; Beaumier, P.; Axworthy, D.; Atcher, R.; Fritzberg, A. Pretargeted radioimmunotherapy in tumored mice using an in vivo 212Pb/212Bi generator. Nucl. Med. Biol. 2005, 32, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Fendler, W.P.; Cutler, C. More alpha Than beta for Prostate Cancer? J. Nucl. Med. 2017, 58, 1709–1710. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, A.; Apostolidis, C.; Kratochwil, C.; Sathekge, M.; Krolicki, L.; Bruchertseifer, F. An Overview of Targeted Alpha Therapy with (225)Actinium and (213)Bismuth. Curr. Radiopharm. 2018, 11, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-Targeted alpha-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Bronzel, M.; Apostolidis, C.; Weichert, W.; Haberkorn, U.; Giesel, F.L.; Morgenstern, A. Targeted alpha-Therapy of Metastatic Castration-Resistant Prostate Cancer with (225)Ac-PSMA-617: Dosimetry Estimate and Empiric Dose Finding. J. Nucl. Med. 2017, 58, 1624–1631. [Google Scholar] [CrossRef]
- Bauer, W.; Briner, U.; Doepfner, W.; Haller, R.; Huguenin, R.; Marbach, P.; Petcher, T.J.; Pless, J. SMS 201–995: A very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sci. 1982, 31, 1133–1140. [Google Scholar] [CrossRef]
- Maack, T.; Johnson, V.; Kau, S.T.; Figueiredo, J.; Sigulem, D. Renal filtration, transport, and metabolism of low-molecular-weight proteins: A review. Kidney Int. 1979, 16, 251–270. [Google Scholar] [CrossRef]
- Sabet, A.; Ezziddin, K.; Pape, U.-F.; Reichman, K.; Haslerud, T.; Ahmadzadehfar, H.; Biersack, H.-J.; Nagarajah, J.; Ezziddin, S. Accurate assessment of long-term nephrotoxicity after peptide receptor radionuclide therapy with 177Lu-octreotate. Eur. J. Nucl. Med. Mol. Imaging 2013, 41, 505–510. [Google Scholar] [CrossRef]
- Norenberg, J.P.; Krenning, B.J.; Konings, I.R.; Kusewitt, D.F.; Nayak, T.K.; Anderson, T.L.; de Jong, M.; Garmestani, K.; Brechbiel, M.W.; Kvols, L.K. 213Bi-[DOTA0, Tyr3]octreotide peptide receptor radionuclide therapy of pancreatic tumors in a preclinical animal model. Clin. Cancer Res. 2006, 12, 897–903. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. (213)Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: A first-in-human experience. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2106–2119. [Google Scholar] [CrossRef]
- Chan, H.S.; Konijnenberg, M.W.; de Blois, E.; Koelewijn, S.; Baum, R.P.; Morgenstern, A.; Bruchertseifer, F.; Breeman, W.A.; de Jong, M. Influence of tumour size on the efficacy of targeted alpha therapy with (213)Bi-[DOTA(0),Tyr(3)]-octreotate. EJNMMI Res. 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Tafreshi, N.K.; Tichacek, C.J.; Pandya, D.N.; Doligalski, M.L.; Budzevich, M.M.; Kil, H.; Bhatt, N.B.; Kock, N.D.; Messina, J.L.; Ruiz, E.E.; et al. Melanocortin 1 Receptor-Targeted alpha-Particle Therapy for Metastatic Uveal Melanoma. J. Nucl. Med. 2019, 60, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Figueroa, S.D.; Fisher, D.R.; Moore, H.A.; Testa, R.F.; Hoffman, T.J.; Quinn, T.P. 203Pb-labeled alpha-melanocyte-stimulating hormone peptide as an imaging probe for melanoma detection. J. Nucl. Med. 2008, 49, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, D.; Lee, D.; Kapoor, S.; Gibson-Corley, K.N.; Quinn, T.P.; Sagastume, E.A.; Mott, S.L.; Walsh, S.A.; Acevedo, M.R.; et al. Enhancing the Efficacy of Melanocortin 1 Receptor-Targeted Radiotherapy by Pharmacologically Upregulating the Receptor in Metastatic Melanoma. Mol. Pharm. 2019. [Google Scholar] [CrossRef]
- Chang, M.-Y.; Seideman, J.; Sofou, S. Enhanced Loading Efficiency and Retention of 225Ac in Rigid Liposomes for Potential Targeted Therapy of Micrometastases. Bioconjug. Chem. 2008, 19, 1274–1282. [Google Scholar] [CrossRef]
- Woodward, J.; Kennel, S.J.; Stuckey, A.; Osborne, D.; Wall, J.; Rondinone, A.J.; Standaert, R.F.; Mirzadeh, S. LaPO4 Nanoparticles Doped with Actinium-225 that Partially Sequester Daughter Radionuclides. Bioconjug. Chem. 2011, 22, 766–776. [Google Scholar] [CrossRef]
- Zhu, C.; Bandekar, A.; Sempkowski, M.; Banerjee, S.R.; Pomper, M.G.; Bruchertseifer, F.; Morgenstern, A.; Sofou, S. Nanoconjugation of PSMA-Targeting Ligands Enhances Perinuclear Localization and Improves Efficacy of Delivered Alpha-Particle Emitters against Tumor Endothelial Analogues. Mol. Cancer Ther. 2016, 15, 106–113. [Google Scholar] [CrossRef]
- Ballangrud, A.M.; Yang, W.H.; Palm, S.; Enmon, R.; Borchardt, P.E.; Pellegrini, V.A.; McDevitt, M.R.; Scheinberg, D.A.; Sgouros, G. Alpha-particle emitting atomic generator (Actinium-225)-labeled trastuzumab (herceptin) targeting of breast cancer spheroids: Efficacy versus HER2/neu expression. Clin. Cancer Res. 2004, 10, 4489–4497. [Google Scholar] [CrossRef]
- Jaggi, J.S.; Seshan, S.V.; McDevitt, M.R.; LaPerle, K.; Sgouros, G.; Scheinberg, D.A. Renal Tubulointerstitial Changes after Internal Irradiation with α-Particle–Emitting Actinium Daughters. J. Am. Soc. Nephrol. 2005, 16, 2677–2689. [Google Scholar] [CrossRef]
- Singh Jaggi, J.; Kappel, B.J.; McDevitt, M.R.; Sgouros, G.; Flombaum, C.D.; Cabassa, C.; Scheinberg, D.A. Efforts to Control the Errant Products of a Targeted In vivo Generator. Cancer Res. 2005, 65, 4888–4895. [Google Scholar] [CrossRef]
- Orozco, J.J.; Bäck, T.; Kenoyer, A.; Balkin, E.R.; Hamlin, D.K.; Wilbur, D.S.; Fisher, D.R.; Frayo, S.L.; Hylarides, M.D.; Green, D.J.; et al. Anti-CD45 radioimmunotherapy using (211)At with bone marrow transplantation prolongs survival in a disseminated murine leukemia model. Blood 2013, 121, 3759–3767. [Google Scholar] [CrossRef][Green Version]
- Green, D.J.; Shadman, M.; Jones, J.C.; Frayo, S.L.; Kenoyer, A.L.; Hylarides, M.D.; Hamlin, D.K.; Wilbur, D.S.; Balkan, E.R.; Lin, Y.; et al. Astatine-211 conjugated to an anti-CD20 monoclonal antibody eradicates disseminated B-cell lymphoma in a mouse model. Blood 2015, 125, 2111–2119. [Google Scholar] [CrossRef]
- Yokota, T.; Milenic, D.E.; Whitlow, M.; Schlom, J. Rapid Tumor Penetration of a Single-Chain Fv and Comparison with Other Immunoglobulin Forms. Cancer Res. 1992, 52, 3402–3408. [Google Scholar]
- Hudson, P.J.; Souriau, C. Engineered antibodies. Nat. Med. 2003, 9, 129–134. [Google Scholar] [CrossRef]
- Steffen, A.C.; Almqvist, Y.; Chyan, M.K.; Lundqvist, H.; Tolmachev, V.; Wilbur, D.S.; Carlsson, J. Biodistribution of 211At labeled HER-2 binding affibody molecules in mice. Oncol. Rep. 2007, 17, 1141–1147. [Google Scholar] [CrossRef]
- D’Huyvetter, M.; Xavier, C.; Caveliers, V.; Lahoutte, T.; Muyldermans, S.; Devoogdt, N. Radiolabeled nanobodies as theranostic tools in targeted radionuclide therapy of cancer. Expert. Opin. Drug Deliv. 2014, 11, 1939–1954. [Google Scholar] [CrossRef]
- Jadvar, H.; Challa, S.; Quinn, D.I.; Conti, P.S. One-Year Postapproval Clinical Experience with Radium-223 Dichloride in Patients with Metastatic Castrate-Resistant Prostate Cancer. Cancer Biother. Radiopharm. 2015, 30, 195–199. [Google Scholar] [CrossRef]
- Takalkar, A.; Adams, S.; Subbiah, V. Radium-223 dichloride bone-targeted alpha particle therapy for hormone-refractory breast cancer metastatic to bone. Exp. Hematol. Oncol. 2014, 3, 1–7. [Google Scholar] [CrossRef]
- Kozempel, J.; Vlk, M.; Málková, E.; Bajzíková, A.; Bárta, J.; Santos-Oliveira, R.; Malta Rossi, A. Prospective carriers of 223Ra for targeted alpha particle therapy. J. Radioanal. Nucl. Chem. 2014, 304, 443–447. [Google Scholar] [CrossRef]
- Schwartz, J.; Jaggi, J.S.; O’Donoghue, J.A.; Ruan, S.; McDevitt, M.; Larson, S.M.; Scheinberg, D.A.; Humm, J.L. Renal uptake of bismuth-213 and its contribution to kidney radiation dose following administration of actinium-225-labeled antibody. Phys. Med. Biol. 2011, 56, 721–733. [Google Scholar] [CrossRef]
- McLaughlin, M.F.; Woodward, J.; Boll, R.A.; Wall, J.S.; Rondinone, A.J.; Kennel, S.J.; Mirzadeh, S.; Robertson, J.D. Gold Coated Lanthanide Phosphate Nanoparticles for Targeted Alpha Generator Radiotherapy. PLoS ONE 2013, 8, e54531. [Google Scholar] [CrossRef]
- Harrison, M.R.; Wong, T.Z.; Armstrong, A.J.; George, D.J. Radium-223 chloride: A potential new treatment for castration-resistant prostate cancer patients with metastatic bone disease. Cancer Manag. Res. 2013, 5, 1–14. [Google Scholar] [CrossRef]
- Wilbur, D.S. Chemical and radiochemical considerations in radiolabeling with alpha-emitting radionuclides. Curr. Radiopharm. 2011, 4, 214–247. [Google Scholar] [CrossRef]
- de Kruijff, R.M.; Wolterbeek, H.T.; Denkova, A.G. A Critical Review of Alpha Radionuclide Therapy-How to Deal with Recoiling Daughters? Pharmaceuticals (Basel) 2015, 8, 321–336. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Reardon, D.A.; Pozzi, O.R.; Vaidyanathan, G.; Bigner, D.D. Targeted alpha-particle radiotherapy with 211At-labeled monoclonal antibodies. Nucl. Med. Biol. 2007, 34, 779–785. [Google Scholar] [CrossRef]
- Hassfjell, S.; Brechbiel, M.W. The development of the alpha-particle emitting radionuclides 212Bi and 213Bi, and their decay chain related radionuclides, for therapeutic applications. Chem. Rev. 2001, 101, 2019–2036. [Google Scholar] [CrossRef]
- Morgenstern, A.; Bruchertseifer, F.; Apostolidis, C. Targeted alpha therapy with 213Bi. Curr. Radiopharm. 2011, 4, 295–305. [Google Scholar] [CrossRef]
- Raes, F. Description of the properties of unattached 218Po and 212Pb particles by means of the classical theory of cluster formation. Health Phys. 1985, 49, 1177–1187. [Google Scholar] [CrossRef]
- Yong, K.; Brechbiel, M. Application of Pb for Targeted alpha-particle Therapy (TAT): Pre-clinical and Mechanistic Understanding through to Clinical Translation. AIMS Med. Sci. 2015, 2, 228–245. [Google Scholar] [CrossRef]
- Gott, M.; Steinbach, J.; Mamat, C. The Radiochemical and Radiopharmaceutical Applications of Radium. Open Chem. 2016, 14, 118–129. [Google Scholar] [CrossRef]
- Scheinberg, D.A.; McDevitt, M.R. Actinium-225 in targeted alpha-particle therapeutic applications. Curr. Radiopharm. 2011, 4, 306–320. [Google Scholar] [CrossRef]
- Moss, L.; Edelstein, N.; Fuger, J. Actinium. In The Chemistry of the Actinide and TRANSACTINIDE Elements; Springer: Amsterdam, The Netherlands, 2006; pp. 18–51. [Google Scholar] [CrossRef]
- Diamond, R.; Street Jr, K.; Seaborg, G. An ion-exchange study of possible hybridized 5f bonding in the actinides. J. Am. Chem. Soc. 1954, 76, 1461–1469. [Google Scholar] [CrossRef]
- Summary and Comparison of the Properties of the Actinide and Transactinide Elements; Moss, L., Edelstein, N., Fuger, J., Eds.; Springer: Amsterdam, The Netherlands, 2006; pp. 1753–1835. [Google Scholar]
- Gorden, A.E.; DeVore, M.A., 2nd; Maynard, B.A. Coordination chemistry with f-element complexes for an improved understanding of factors that contribute to extraction selectivity. Inorg. Chem. 2013, 52, 3445–3458. [Google Scholar] [CrossRef]
- Chappell, L.L.; Ma, D.; Milenic, D.E.; Garmestani, K.; Venditto, V.; Beitzel, M.P.; Brechbiel, M.W. Synthesis and evaluation of novel bifunctional chelating agents based on 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid for radiolabeling proteins. Nucl. Med. Biol. 2003, 30, 581–595. [Google Scholar] [CrossRef]
- Chappell, L.L.; Deal, K.A.; Dadachova, E.; Brechbiel, M.W. Synthesis, conjugation, and radiolabeling of a novel bifunctional chelating agent for (225)Ac radioimmunotherapy applications. Bioconjug. Chem. 2000, 11, 510–519. [Google Scholar] [CrossRef]
- Davis, I.A.; Glowienka, K.A.; Boll, R.A.; Deal, K.A.; Brechbiel, M.W.; Stabin, M.; Bochsler, P.N.; Mirzadeh, S.; Kennel, S.J. Comparison of 225actinium chelates: Tissue distribution and radiotoxicity. Nucl. Med. Biol. 1999, 26, 581–589. [Google Scholar] [CrossRef]
- McDevitt, M.R.; Ma, D.; Simon, J.; Frank, R.K.; Scheinberg, D.A. Design and synthesis of 225Ac radioimmunopharmaceuticals. Appl. Radiat. Isot. 2002, 57, 841–847. [Google Scholar] [CrossRef]
- Gouin, S.G.; Gestin, J.F.; Monrandeau, L.; Segat-Dioury, F.; Meslin, J.C.; Deniaud, D. Synthesis and metal complexation properties of Ph-DTPA and Ph-TTHA: Novel radionuclide chelating agents for use in nuclear medicine. Org. Biomol. Chem. 2005, 3, 454–461. [Google Scholar] [CrossRef]
- Thiele, N.A.; Brown, V.; Kelly, J.M.; Amor-Coarasa, A.; Jermilova, U.; MacMillan, S.N.; Nikolopoulou, A.; Ponnala, S.; Ramogida, C.F.; Robertson, A.K.H.; et al. An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy. Angew. Chem. Int. Ed. Engl. 2017, 56, 14712–14717. [Google Scholar] [CrossRef]
- Escorcia, F.E.; Henke, E.; McDevitt, M.R.; Villa, C.H.; Smith-Jones, P.; Blasberg, R.G.; Benezra, R.; Scheinberg, D.A. Selective killing of tumor neovasculature paradoxically improves chemotherapy delivery to tumors. Cancer Res. 2010, 70, 9277–9286. [Google Scholar] [CrossRef]
- Maguire, W.F.; McDevitt, M.R.; Smith-Jones, P.M.; Scheinberg, D.A. Efficient 1-step radiolabeling of monoclonal antibodies to high specific activity with 225Ac for alpha-particle radioimmunotherapy of cancer. J. Nucl. Med. 2014, 55, 1492–1498. [Google Scholar] [CrossRef]
- Poty, S.; Membreno, R.; Glaser, J.M.; Ragupathi, A.; Scholz, W.W.; Zeglis, B.M.; Lewis, J.S. The inverse electron-demand Diels-Alder reaction as a new methodology for the synthesis of (225)Ac-labelled radioimmunoconjugates. Chem. Commun. (Camb) 2018, 54, 2599–2602. [Google Scholar] [CrossRef]
- Sofou, S.; Kappel, B.J.; Jaggi, J.S.; McDevitt, M.R.; Scheinberg, D.A.; Sgouros, G. Enhanced retention of the alpha-particle-emitting daughters of Actinium-225 by liposome carriers. Bioconjug. Chem. 2007, 18, 2061–2067. [Google Scholar] [CrossRef]
- Sofou, S.; Thomas, J.L.; Lin, H.Y.; McDevitt, M.R.; Scheinberg, D.A.; Sgouros, G. Engineered liposomes for potential alpha-particle therapy of metastatic cancer. J. Nucl. Med. 2004, 45, 253–260. [Google Scholar]
- Matson, M.L.; Villa, C.H.; Ananta, J.S.; Law, J.J.; Scheinberg, D.A.; Wilson, L.J. Encapsulation of alpha-Particle-Emitting 225Ac3+ Ions Within Carbon Nanotubes. J. Nucl. Med. 2015, 56, 897–900. [Google Scholar] [CrossRef]
- McDevitt, M.R.; Chattopadhyay, D.; Kappel, B.J.; Jaggi, J.S.; Schiffman, S.R.; Antczak, C.; Njardarson, J.T.; Brentjens, R.; Scheinberg, D.A. Tumor targeting with antibody-functionalized, radiolabeled carbon nanotubes. J. Nucl. Med. 2007, 48, 1180–1189. [Google Scholar] [CrossRef]
- Mulvey, J.J.; Villa, C.H.; McDevitt, M.R.; Escorcia, F.E.; Casey, E.; Scheinberg, D.A. Self-assembly of carbon nanotubes and antibodies on tumours for targeted amplified delivery. Nat. Nanotechnol. 2013, 8, 763–771. [Google Scholar] [CrossRef]
- Ruggiero, A.; Villa, C.H.; Holland, J.P.; Sprinkle, S.R.; May, C.; Lewis, J.S.; Scheinberg, D.A.; McDevitt, M.R. Imaging and treating tumor vasculature with targeted radiolabeled carbon nanotubes. Int. J. Nanomed. 2010, 5, 783–802. [Google Scholar] [CrossRef]
- Gouard, S.; Pallardy, A.; Gaschet, J.; Faivre-Chauvet, A.; Bruchertseifer, F.; Morgenstern, A.; Maurel, C.; Matous, E.; Kraeber-Bodere, F.; Davodeau, F.; et al. Comparative analysis of multiple myeloma treatment by CD138 antigen targeting with bismuth-213 and Melphalan chemotherapy. Nucl. Med. Biol. 2014, 41, e30–e35. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques, 3rd ed.; Hermanson, G.T., Ed.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Tavaré, R.; Wu, W.H.; Zettlitz, K.A.; Salazar, F.B.; McCabe, K.E.; Marks, J.D.; Wu, A.M. Enhanced immunoPET of ALCAM-positive colorectal carcinoma using site-specific 64Cu-DOTA conjugation. Protein Eng. Des. Sel. 2014, 27, 317–324. [Google Scholar] [CrossRef][Green Version]
- Li, L.; Olafsen, T.; Anderson, A.-L.; Wu, A.; Raubitschek, A.A.; Shively, J.E. Reduction of Kidney Uptake in Radiometal Labeled Peptide Linkers Conjugated to Recombinant Antibody Fragments. Site-Specific Conjugation of DOTA-Peptides to a Cys-Diabody. Bioconjug. Chem. 2002, 13, 985–995. [Google Scholar] [CrossRef]
- Akizawa, H.; Imajima, M.; Hanaoka, H.; Uehara, T.; Satake, S.; Arano, Y. Renal Brush Border Enzyme-Cleavable Linkages for Low Renal Radioactivity Levels of Radiolabeled Antibody Fragments. Bioconjug. Chem. 2013, 24, 291–299. [Google Scholar] [CrossRef]
- Loevinger, R.; Budinger, T.F.; Watson, E.E. MIRD Primer for Absorbed Dose Calculations, Revised Edition; The Society of Nuclear Medicine, Inc: New York, NY, USA, 1991. [Google Scholar]
- Kellerer, A.M.; Chmelevsky, D. Criteria for the applicability of LET. Radiat. Res. 1975, 63, 226–234. [Google Scholar] [CrossRef]
- Fisher, K.J.; Jooss, K.; Alston, J.; Yang, Y.; Haecker, S.E.; High, K.; Pathak, R.; Raper, S.E.; Wilson, J.M. Recombinant adeno-associated virus for muscle directed gene therapy. Nat. Med. 1997, 3, 306–312. [Google Scholar] [CrossRef]
- Zubal, I.G.; Harrell, C.R.; Smith, E.O.; Rattner, Z.; Gindi, G.; Hoffer, P.B. Computerized three-dimensional segmented human anatomy. Med. Phys. 1994, 21, 299–302. [Google Scholar] [CrossRef]
- Bolch, W.E.; Bouchet, L.G.; Robertson, J.S.; Wessels, B.W.; Siegel, J.A.; Howell, R.W.; Erdi, A.K.; Aydogan, B.; Costes, S.; Watson, E.E.; et al. MIRD pamphlet No. 17: The dosimetry of nonuniform activity distributions--radionuclide S values at the voxel level. Medical Internal Radiation Dose Committee. J. Nucl. Med. 1999, 40, 11S–36S. [Google Scholar]
- Stabin, M.G.; Sparks, R.B.; Crowe, E. OLINDA/EXM: The second-generation personal computer software for internal dose assessment in nuclear medicine. J. Nucl. Med. 2005, 46, 1023–1027. [Google Scholar]
- Sgouros, G.; Frey, E.; Wahl, R.; He, B.; Prideaux, A.; Hobbs, R. Three-dimensional imaging-based radiobiological dosimetry. Semin. Nucl. Med. 2008, 38, 321–334. [Google Scholar] [CrossRef][Green Version]
- Gil, A.V.; Pérez, M.C.; Aroche, L.T.; Pacilio, M.; Botta, F.; Cremonesi, M. MCID: A personalized dosimetric tool associating voxel-based models with MCNP5. In Proceedings of the IAEA International Conference on Radiation Protection in Medicine, Setting the Scene for the Next Decade, Bonn, Germany, 3–7 December 2012. [Google Scholar]
- Chiavassa, S.; Bardies, M.; Guiraud-Vitaux, F.; Bruel, D.; Jourdain, J.R.; Franck, D.; Aubineau-Laniece, I. OEDIPE: A personalized dosimetric tool associating voxel-based models with MCNPX. Cancer Biother. Radiopharm. 2005, 20, 325–332. [Google Scholar] [CrossRef]
- Yoriyaz, H.; Stabin, M.G.; dos Santos, A. Monte Carlo MCNP-4B-based absorbed dose distribution estimates for patient-specific dosimetry. J. Nucl. Med. 2001, 42, 662–669. [Google Scholar]
- Prideaux, A.R.; Song, H.; Hobbs, R.F.; He, B.; Frey, E.C.; Ladenson, P.W.; Wahl, R.L.; Sgouros, G. Three-dimensional radiobiologic dosimetry: Application of radiobiologic modeling to patient-specific 3-dimensional imaging-based internal dosimetry. J. Nucl. Med. 2007, 48, 1008–1016. [Google Scholar] [CrossRef]
- Liu, X.; Ljungberg, M.; Strand, S.E. DOSIMG: A 3D voxel-based Monte Carlo program for absorbed dose calculations. J. Nucl. Med. 2001, 42, 243P. [Google Scholar]
- Nelson, W.R.; Hirayama, H.; Rogers, D.W.O. The EGS4 Code System; National Technical Information Service, U.S. Department of Commerce: Springfield, VA, USA; Stanford University Linear Accelerator Center: Stanford, CA, USA, 1985. [Google Scholar]
- Marcatili, S.; Pettinato, C.; Daniels, S.; Lewis, G.; Edwards, P.; Fanti, S.; Spezi, E. Development and validation of RAYDOSE: A Geant4-based application for molecular radiotherapy. Phys. Med. Biol. 2013, 58, 2491–2508. [Google Scholar] [CrossRef]
- Besemer, A.E.; Yang, Y.M.; Grudzinski, J.J.; Hall, L.T.; Bednarz, B.P. Development and Validation of RAPID: A Patient-Specific Monte Carlo Three-Dimensional Internal Dosimetry Platform. Cancer Biother. Radiopharm. 2018, 33, 155–165. [Google Scholar] [CrossRef]
- Agostinelli, S.; Allison, J.; Amako, K.; Apostolakis, J.; Araujo, H.; Arce, P.; Asai, M.; Axen, D.; Banerjee, S.; Barrand, G.; et al. GEANT4-a simulation toolkit. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2003, 506, 250–303. [Google Scholar] [CrossRef]
- Sempau, J.; Wilderman, S.J.; Bielajew, A.F. DPM, a fast, accurate Monte Carlo code optimized for photon and electron radiotherapy treatment planning dose calculations. Phys. Med. Biol. 2000, 45, 2263–2291. [Google Scholar] [CrossRef]
- Miller, B.W.; Gregory, S.J.; Fuller, E.S.; Barrett, H.H.; Barber, B.; Furenlid, L.R. The iQID camera: An ionizing-radiation quantum imaging detector. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2014, 767, 146–152. [Google Scholar] [CrossRef]
- Back, T.; Jacobsson, L. The alpha-camera: A quantitative digital autoradiography technique using a charge-coupled device for ex vivo high-resolution bioimaging of alpha-particles. J. Nucl. Med. 2010, 51, 1616–1623. [Google Scholar] [CrossRef]
- Demartis, S.; Tarli, L.; Borsi, L.; Zardi, L.; Neri, D. Selective targeting of tumour neovasculature by a radiohalogenated human antibody fragment specific for the ED-B domain of fibronectin. Eur. J. Nucl. Med. 2001, 28, 534–539. [Google Scholar] [CrossRef]
- Park, S.I.; Shenoi, J.; Pagel, J.M.; Hamlin, D.K.; Wilbur, D.S.; Orgun, N.; Kenoyer, A.L.; Frayo, S.; Axtman, A.; Back, T.; et al. Conventional and pretargeted radioimmunotherapy using bismuth-213 to target and treat non-Hodgkin lymphomas expressing CD20: A preclinical model toward optimal consolidation therapy to eradicate minimal residual disease. Blood 2010, 116, 4231–4239. [Google Scholar] [CrossRef]
- Robinson, M.K.; Shaller, C.; Garmestani, K.; Plascjak, P.S.; Hodge, K.M.; Yuan, Q.A.; Marks, J.D.; Waldmann, T.A.; Brechbiel, M.W.; Adams, G.P. Effective treatment of established human breast tumor xenografts in immunodeficient mice with a single dose of the alpha-emitting radioisotope astatine-211 conjugated to anti-HER2/neu diabodies. Clin. Cancer Res. 2008, 14, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yao, Z.; Garmestani, K.; Axworthy, D.B.; Zhang, Z.; Mallett, R.W.; Theodore, L.J.; Goldman, C.K.; Brechbiel, M.W.; Carrasquillo, J.A.; et al. Pretargeting radioimmunotherapy of a murine model of adult T-cell leukemia with the alpha-emitting radionuclide, bismuth 213. Blood 2002, 100, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Poty, S.; Carter, L.M.; Mandleywala, K.; Membreno, R.; Abdel-Atti, D.; Ragupathi, A.; Scholz, W.W.; Zeglis, B.M.; Lewis, J.S. Leveraging Bioorthogonal Click Chemistry to Improve (225)Ac-Radioimmunotherapy of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2019, 25, 868–880. [Google Scholar] [CrossRef]
- Li, Y.; Song, E.; Abbas Rizvi, S.M.; Power, C.A.; Beretov, J.; Raja, C.; Cozzi, P.J.; Morgenstern, A.; Apostolidis, C.; Allen, B.J.; et al. Inhibition of micrometastatic prostate cancer cell spread in animal models by 213Bilabeled multiple targeted alpha radioimmunoconjugates. Clin. Cancer Res. 2009, 15, 865–875. [Google Scholar] [CrossRef]
- Makvandi, M.; Lieberman, B.P.; LeGeyt, B.; Hou, C.; Mankoff, D.A.; Mach, R.H.; Pryma, D.A. The pre-clinical characterization of an alpha-emitting sigma-2 receptor targeted radiotherapeutic. Nucl. Med. Biol. 2016, 43, 35–41. [Google Scholar] [CrossRef]
- Willhauck, M.J.; Samani, B.R.; Wolf, I.; Senekowitsch-Schmidtke, R.; Stark, H.J.; Meyer, G.J.; Knapp, W.H.; Goke, B.; Morris, J.C.; Spitzweg, C. The potential of 211Astatine for NIS-mediated radionuclide therapy in prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1272–1281. [Google Scholar] [CrossRef]
- Borchardt, P.E.; Yuan, R.R.; Miederer, M.; McDevitt, M.R.; Scheinberg, D.A. Targeted actinium-225 in vivo generators for therapy of ovarian cancer. Cancer Res. 2003, 63, 5084–5090. [Google Scholar]
- Miederer, M.; McDevitt, M.R.; Sgouros, G.; Kramer, K.; Cheung, N.K.; Scheinberg, D.A. Pharmacokinetics, dosimetry, and toxicity of the targetable atomic generator, 225Ac-HuM195, in nonhuman primates. J. Nucl. Med. 2004, 45, 129–137. [Google Scholar]
- Miederer, M.; McDevitt, M.R.; Borchardt, P.; Bergman, I.; Kramer, K.; Cheung, N.K.; Scheinberg, D.A. Treatment of neuroblastoma meningeal carcinomatosis with intrathecal application of alpha-emitting atomic nanogenerators targeting disialo-ganglioside GD2. Clin. Cancer Res. 2004, 10, 6985–6992. [Google Scholar] [CrossRef]
- Miederer, M.; Henriksen, G.; Alke, A.; Mossbrugger, I.; Quintanilla-Martinez, L.; Senekowitsch-Schmidtke, R.; Essler, M. Preclinical evaluation of the alpha-particle generator nuclide 225Ac for somatostatin receptor radiotherapy of neuroendocrine tumors. Clin. Cancer Res. 2008, 14, 3555–3561. [Google Scholar] [CrossRef]
- Song, H.; Hobbs, R.F.; Vajravelu, R.; Huso, D.L.; Esaias, C.; Apostolidis, C.; Morgenstern, A.; Sgouros, G. Radioimmunotherapy of breast cancer metastases with alpha-particle emitter 225Ac: Comparing efficacy with 213Bi and 90Y. Cancer Res. 2009, 69, 8941–8948. [Google Scholar] [CrossRef]
- Essler, M.; Gartner, F.C.; Neff, F.; Blechert, B.; Senekowitsch-Schmidtke, R.; Bruchertseifer, F.; Morgenstern, A.; Seidl, C. Therapeutic efficacy and toxicity of 225Ac-labelled vs. 213Bi-labelled tumour-homing peptides in a preclinical mouse model of peritoneal carcinomatosis. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 602–612. [Google Scholar] [CrossRef]
- Drecoll, E.; Gaertner, F.C.; Miederer, M.; Blechert, B.; Vallon, M.; Muller, J.M.; Alke, A.; Seidl, C.; Bruchertseifer, F.; Morgenstern, A.; et al. Treatment of peritoneal carcinomatosis by targeted delivery of the radio-labeled tumor homing peptide bi-DTPA-[F3]2 into the nucleus of tumor cells. PLoS ONE 2009, 4, e5715. [Google Scholar] [CrossRef]
- Sattiraju, A.; Solingapuram Sai, K.K.; Xuan, A.; Pandya, D.N.; Almaguel, F.G.; Wadas, T.J.; Herpai, D.M.; Debinski, W.; Mintz, A. IL13RA2 targeted alpha particle therapy against glioblastomas. Oncotarget 2017, 8, 42997–43007. [Google Scholar] [CrossRef]
- Sattiraju, A.; Xiong, X.; Pandya, D.N.; Wadas, T.J.; Xuan, A.; Sun, Y.; Jung, Y.; Sai, K.K.S.; Dorsey, J.F.; Li, K.C.; et al. Alpha Particle Enhanced Blood Brain/Tumor Barrier Permeabilization in Glioblastomas Using Integrin Alpha-v Beta-3-Targeted Liposomes. Mol. Cancer 2017, 16, 2191–2200. [Google Scholar] [CrossRef]
- McLaughlin, M.F.; Robertson, D.; Pevsner, P.H.; Wall, J.S.; Mirzadeh, S.; Kennel, S.J. LnPO4 nanoparticles doped with Ac-225 and sequestered daughters for targeted alpha therapy. Cancer Biother. Radiopharm. 2014, 29, 34–41. [Google Scholar] [CrossRef]
- Nedrow, J.R.; Josefsson, A.; Park, S.; Back, T.; Hobbs, R.F.; Brayton, C.; Bruchertseifer, F.; Morgenstern, A.; Sgouros, G. Pharmacokinetics, microscale distribution, and dosimetry of alpha-emitter-labeled anti-PD-L1 antibodies in an immune competent transgenic breast cancer model. EJNMMI Res. 2017, 7, 57. [Google Scholar] [CrossRef]
- Pfannkuchen, N.; Bausbacher, N.; Pektor, S.; Miederer, M.; Rosch, F. In vivo Evaluation of [(225)Ac]Ac-DOTA(ZOL) for alpha-Therapy of Bone Metastases. Curr. Radiopharm. 2018, 11, 223–230. [Google Scholar] [CrossRef]
- Kelly, J.M.; Amor-Coarasa, A.; Ponnala, S.; Nikolopoulou, A.; Williams, C., Jr.; Thiele, N.A.; Schlyer, D.; Wilson, J.J.; DiMagno, S.G.; Babich, J.W. A Single Dose of (225)Ac-RPS-074 Induces a Complete Tumor Response in a LNCaP Xenograft Model. J. Nucl. Med. 2018. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; McLendon, R.E.; Garg, P.K.; Archer, G.E.; Schuster, J.M.; Bigner, D.D. Radioimmunotherapy of neoplastic meningitis in rats using an alpha-particle-emitting immunoconjugate. Cancer Res. 1994, 54, 4719–4725. [Google Scholar]
- Zalutsky, M.R.; Stabin, M.G.; Larsen, R.H.; Bigner, D.D. Tissue distribution and radiation dosimetry of astatine-211-labeled chimeric 81C6, an alpha-particle-emitting immunoconjugate. Nucl. Med. Biol. 1997, 24, 255–261. [Google Scholar] [CrossRef]
- Andersson, H.; Elgqvist, J.; Horvath, G.; Hultborn, R.; Jacobsson, L.; Jensen, H.; Karlsson, B.; Lindegren, S.; Palm, S. Astatine-211-labeled antibodies for treatment of disseminated ovarian cancer: An overview of results in an ovarian tumor model. Clin. Cancer Res. 2003, 9, 3914S–3921S. [Google Scholar] [PubMed]
- Andersson, H.; Lindegren, S.; Back, T.; Jacobsson, L.; Leser, G.; Horvath, G. Radioimmunotherapy of nude mice with intraperitoneally growing ovarian cancer xenograft utilizing 211At-labelled monoclonal antibody MOv18. Anticancer Res. 2000, 20, 459–462. [Google Scholar] [PubMed]
- Andersson, H.; Lindegren, S.; Back, T.; Jacobsson, L.; Leser, G.; Horvath, G. The curative and palliative potential of the monoclonal antibody MOv18 labelled with 211At in nude mice with intraperitoneally growing ovarian cancer xenografts--a long-term study. Acta Oncol. 2000, 39, 741–745. [Google Scholar] [CrossRef]
- Andersson, H.; Palm, S.; Lindegren, S.; Back, T.; Jacobsson, L.; Leser, G.; Horvath, G. Comparison of the therapeutic efficacy of 211At- and 131I-labelled monoclonal antibody MOv18 in nude mice with intraperitoneal growth of human ovarian cancer. Anticancer Res. 2001, 21, 409–412. [Google Scholar]
- Elgqvist, J.; Andersson, H.; Jensen, H.; Kahu, H.; Lindegren, S.; Warnhammar, E.; Hultborn, R. Repeated Intraperitoneal alpha-Radioimmunotherapy of Ovarian Cancer in Mice. J. Oncol. 2010, 2010, 394913. [Google Scholar] [CrossRef][Green Version]
- Elgqvist, J.; Bernhardt, P.; Hultborn, R.; Jensen, H.; Karlsson, B.; Lindegren, S.; Warnhammar, E.; Jacobsson, L. Myelotoxicity and RBE of 211At-conjugated monoclonal antibodies compared with 99mTc-conjugated monoclonal antibodies and 60Co irradiation in nude mice. J. Nucl. Med. 2005, 46, 464–471. [Google Scholar]
- Zhang, M.; Yao, Z.; Patel, H.; Garmestani, K.; Zhang, Z.; Talanov, V.S.; Plascjak, P.S.; Goldman, C.K.; Janik, J.E.; Brechbiel, M.W.; et al. Effective therapy of murine models of human leukemia and lymphoma with radiolabeled anti-CD30 antibody, HeFi-1. Proc. Natl. Acad. Sci. USA 2007, 104, 8444–8448. [Google Scholar] [CrossRef]
- Cheng, J.; Ekberg, T.; Engstrom, M.; Nestor, M.; Jensen, H.J.; Tolmachev, V.; Anniko, M. Radioimmunotherapy with astatine-211 using chimeric monoclonal antibody U36 in head and neck squamous cell carcinoma. Laryngoscope 2007, 117, 1013–1018. [Google Scholar] [CrossRef]
- Nakamae, H.; Wilbur, D.S.; Hamlin, D.K.; Thakar, M.S.; Santos, E.B.; Fisher, D.R.; Kenoyer, A.L.; Pagel, J.M.; Press, O.W.; Storb, R.; et al. Biodistributions, myelosuppression, and toxicities in mice treated with an anti-CD45 antibody labeled with the alpha-emitting radionuclides bismuth-213 or astatine-211. Cancer Res. 2009, 69, 2408–2415. [Google Scholar] [CrossRef]
- Palm, S.; Back, T.; Claesson, I.; Danielsson, A.; Elgqvist, J.; Frost, S.; Hultborn, R.; Jensen, H.; Lindegren, S.; Jacobsson, L. Therapeutic efficacy of astatine-211-labeled trastuzumab on radioresistant SKOV-3 tumors in nude mice. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.E.; Back, T.; Elgstrom, E.; Jensen, H.; Nilsson, R.; Lindegren, S.; Tennvall, J. Successful radioimmunotherapy of established syngeneic rat colon carcinoma with 211At-mAb. EJNMMI Res. 2013, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, Y.; Sudo, H.; Watanabe, S.; Nagatsu, K.; Tsuji, A.B.; Sakashita, T.; Ito, Y.M.; Yoshinaga, K.; Higashi, T.; Ishioka, N.S. Antitumor effects of radionuclide treatment using alpha-emitting meta-(211)At-astato-benzylguanidine in a PC12 pheochromocytoma model. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Li, H.K.; Hasegawa, S.; Nakajima, N.I.; Morokoshi, Y.; Minegishi, K.; Nagatsu, K. Targeted cancer cell ablation in mice by an alpha-particle-emitting astatine-211-labeled antibody against major histocompatibility complex class I chain-related protein A and B. Biochem. Biophys. Res. Commun. 2018, 506, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Behr, T.M.; Behe, M.; Stabin, M.G.; Wehrmann, E.; Apostolidis, C.; Molinet, R.; Strutz, F.; Fayyazi, A.; Wieland, E.; Gratz, S.; et al. High-linear energy transfer (LET) alpha versus low-LET beta emitters in radioimmunotherapy of solid tumors: Therapeutic efficacy and dose-limiting toxicity of 213Bi- versus 90Y-labeled CO17-1A Fab’ fragments in a human colonic cancer model. Cancer Res. 1999, 59, 2635–2643. [Google Scholar]
- Huneke, R.B.; Pippin, C.G.; Squire, R.A.; Brechbiel, M.W.; Gansow, O.A.; Strand, M. Effective alpha-particle-mediated radioimmunotherapy of murine leukemia. Cancer Res. 1992, 52, 5818–5820. [Google Scholar] [PubMed]
- Hartmann, F.; Horak, E.M.; Garmestani, K.; Wu, C.; Brechbiel, M.W.; Kozak, R.W.; Tso, J.; Kosteiny, S.A.; Gansow, O.A.; Nelson, D.L.; et al. Radioimmunotherapy of nude mice bearing a human interleukin 2 receptor alpha-expressing lymphoma utilizing the alpha-emitting radionuclide-conjugated monoclonal antibody 212Bi-anti-Tac. Cancer Res. 1994, 54, 4362–4370. [Google Scholar]
- Nikula, T.K.; McDevitt, M.R.; Finn, R.D.; Wu, C.; Kozak, R.W.; Garmestani, K.; Brechbiel, M.W.; Curcio, M.J.; Pippin, C.G.; Tiffany-Jones, L.; et al. Alpha-emitting bismuth cyclohexylbenzyl DTPA constructs of recombinant humanized anti-CD33 antibodies: Pharmacokinetics, bioactivity, toxicity and chemistry. J. Nucl. Med. 1999, 40, 166–176. [Google Scholar]
- Milenic, D.; Garmestani, K.; Dadachova, E.; Chappell, L.; Albert, P.; Hill, D.; Schlom, J.; Brechbiel, M. Radioimmunotherapy of human colon carcinoma xenografts using a 213Bi-labeled domain-deleted humanized monoclonal antibody. Cancer Biother. Radiopharm. 2004, 19, 135–147. [Google Scholar] [CrossRef]
- Bloechl, S.; Beck, R.; Seidl, C.; Morgenstern, A.; Schwaiger, M.; Senekowitsch-Schmidtke, R. Fractionated locoregional low-dose radioimmunotherapy improves survival in a mouse model of diffuse-type gastric cancer using a 213Bi-conjugated monoclonal antibody. Clin. Cancer Res. 2005, 11, 7070s–7074s. [Google Scholar] [CrossRef]
- Knor, S.; Sato, S.; Huber, T.; Morgenstern, A.; Bruchertseifer, F.; Schmitt, M.; Kessler, H.; Senekowitsch-Schmidtke, R.; Magdolen, V.; Seidl, C. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Pfost, B.; Seidl, C.; Autenrieth, M.; Saur, D.; Bruchertseifer, F.; Morgenstern, A.; Schwaiger, M.; Senekowitsch-Schmidtke, R. Intravesical alpha-radioimmunotherapy with 213Bi-anti-EGFR-mAb defeats human bladder carcinoma in xenografted nude mice. J. Nucl. Med. 2009, 50, 1700–1708. [Google Scholar] [CrossRef] [PubMed]
- Milenic, D.E.; Brady, E.D.; Garmestani, K.; Albert, P.S.; Abdulla, A.; Brechbiel, M.W. Improved efficacy of alpha-particle-targeted radiation therapy: Dual targeting of human epidermal growth factor receptor-2 and tumor-associated glycoprotein 72. Cancer 2010, 116, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Frischknecht, M.; Zhang, H.; Morgenstern, A.; Bruchertseifer, F.; Boisclair, J.; Provencher-Bolliger, A.; Reubi, J.C.; Maecke, H.R. Alpha- versus beta-particle radiopeptide therapy in a human prostate cancer model (213Bi-DOTA-PESIN and 213Bi-AMBA versus 177Lu-DOTA-PESIN). Cancer Res. 2011, 71, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Fichou, N.; Gouard, S.; Maurel, C.; Barbet, J.; Ferrer, L.; Morgenstern, A.; Bruchertseifer, F.; Faivre-Chauvet, A.; Bigot-Corbel, E.; Davodeau, F.; et al. Single-Dose Anti-CD138 Radioimmunotherapy: Bismuth-213 is More Efficient than Lutetium-177 for Treatment of Multiple Myeloma in a Preclinical Model. Front. Med. (Lausanne) 2015, 2, 76. [Google Scholar] [CrossRef]
- Seidl, C.; Zockler, C.; Beck, R.; Quintanilla-Martinez, L.; Bruchertseifer, F.; Senekowitsch-Schmidtke, R. 177Lu-immunotherapy of experimental peritoneal carcinomatosis shows comparable effectiveness to 213Bi-immunotherapy, but causes toxicity not observed with 213Bi. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 312–322. [Google Scholar] [CrossRef]
- Cherel, M.; Gouard, S.; Gaschet, J.; Sai-Maurel, C.; Bruchertseifer, F.; Morgenstern, A.; Bourgeois, M.; Gestin, J.F.; Bodere, F.K.; Barbet, J.; et al. 213Bi radioimmunotherapy with an anti-mCD138 monoclonal antibody in a murine model of multiple myeloma. J. Nucl. Med. 2013, 54, 1597–1604. [Google Scholar] [CrossRef]
- Fazel, J.; Rotzer, S.; Seidl, C.; Feuerecker, B.; Autenrieth, M.; Weirich, G.; Bruchertseifer, F.; Morgenstern, A.; Senekowitsch-Schmidtke, R. Fractionated intravesical radioimmunotherapy with (213)Bi-anti-EGFR-MAb is effective without toxic side-effects in a nude mouse model of advanced human bladder carcinoma. Cancer Biol. 2015, 16, 1526–1534. [Google Scholar] [CrossRef]
- Milenic, D.E.; Baidoo, K.E.; Kim, Y.S.; Brechbiel, M.W. Evaluation of cetuximab as a candidate for targeted alpha-particle radiation therapy of HER1-positive disseminated intraperitoneal disease. MAbs 2015, 7, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Hylarides, M.; Fisher, D.R.; Shelton, T.; Moore, H.; Wester, D.W.; Fritzberg, A.R.; Winkelmann, C.T.; Hoffman, T.; Quinn, T.P. Melanoma therapy via peptide-targeted {alpha}-radiation. Clin. Cancer Res. 2005, 11, 5616–5621. [Google Scholar] [CrossRef]
- Boudousq, V.; Bobyk, L.; Busson, M.; Garambois, V.; Jarlier, M.; Charalambatou, P.; Pelegrin, A.; Paillas, S.; Chouin, N.; Quenet, F.; et al. Comparison between internalizing anti-HER2 mAbs and non-internalizing anti-CEA mAbs in alpha-radioimmunotherapy of small volume peritoneal carcinomatosis using 212Pb. PLoS ONE 2013, 8, e69613. [Google Scholar] [CrossRef] [PubMed]
- Milenic, D.E.; Garmestani, K.; Brady, E.D.; Albert, P.S.; Ma, D.; Abdulla, A.; Brechbiel, M.W. Alpha-particle radioimmunotherapy of disseminated peritoneal disease using a (212)Pb-labeled radioimmunoconjugate targeting HER2. Cancer Biother. Radiopharm. 2005, 20, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Milenic, D.E.; Garmestani, K.; Brady, E.D.; Albert, P.S.; Abdulla, A.; Flynn, J.; Brechbiel, M.W. Potentiation of high-LET radiation by gemcitabine: Targeting HER2 with trastuzumab to treat disseminated peritoneal disease. Clin. Cancer Res. 2007, 13, 1926–1935. [Google Scholar] [CrossRef] [PubMed]
- Kasten, B.B.; Arend, R.C.; Katre, A.A.; Kim, H.; Fan, J.; Ferrone, S.; Zinn, K.R.; Buchsbaum, D.J. B7-H3-targeted (212)Pb radioimmunotherapy of ovarian cancer in preclinical models. Nucl. Med. Biol. 2017, 47, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Kasten, B.B.; Gangrade, A.; Kim, H.; Fan, J.; Ferrone, S.; Ferrone, C.R.; Zinn, K.R.; Buchsbaum, D.J. (212)Pb-labeled B7-H3-targeting antibody for pancreatic cancer therapy in mouse models. Nucl. Med. Biol. 2018, 58, 67–73. [Google Scholar] [CrossRef]
- Kasten, B.B.; Oliver, P.G.; Kim, H.; Fan, J.; Ferrone, S.; Zinn, K.R.; Buchsbaum, D.J. (212)Pb-Labeled Antibody 225.28 Targeted to Chondroitin Sulfate Proteoglycan 4 for Triple-Negative Breast Cancer Therapy in Mouse Models. Int. J. Mol. Sci. 2018, 19, 925. [Google Scholar] [CrossRef]
- Milenic, D.E.; Kim, Y.S.; Baidoo, K.E.; Wong, K.J.; Barkley, R.; Delgado, J.; Brechbiel, M.W. Exploration of a F(ab′)2 Fragment as the Targeting Agent of alpha-Radiation Therapy: A Comparison of the Therapeutic Benefit of Intraperitoneal and Intravenous Administered Radioimmunotherapy. Cancer Biother. Radiopharm. 2018, 33, 182–193. [Google Scholar] [CrossRef]
- Dahle, J.; Jonasdottir, T.J.; Heyerdahl, H.; Nesland, J.M.; Borrebaek, J.; Hjelmerud, A.K.; Larsen, R.H. Assessment of long-term radiotoxicity after treatment with the low-dose-rate alpha-particle-emitting radioimmunoconjugate (227)Th-rituximab. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 93–102. [Google Scholar] [CrossRef]
- Abbas, N.; Heyerdahl, H.; Bruland, O.S.; Borrebaek, J.; Nesland, J.; Dahle, J. Experimental alpha-particle radioimmunotherapy of breast cancer using 227Th-labeled p-benzyl-DOTA-trastuzumab. EJNMMI Res. 2011, 1, 18. [Google Scholar] [CrossRef]
- Hagemann, U.B.; Mihaylova, D.; Uran, S.R.; Borrebaek, J.; Grant, D.; Bjerke, R.M.; Karlsson, J.; Cuthbertson, A.S. Targeted alpha therapy using a novel CD70 targeted thorium-227 conjugate in in vitro and in vivo models of renal cell carcinoma. Oncotarget 2017, 8, 56311–56326. [Google Scholar] [CrossRef]
- Westrom, S.; Bonsdorff, T.B.; Bruland, O.S.; Larsen, R.H. Therapeutic Effect of alpha-Emitting (224)Ra-Labeled Calcium Carbonate Microparticles in Mice with Intraperitoneal Ovarian Cancer. Transl. Oncol. 2018, 11, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Briel, A. Innovative diagnostics enhances and advances the impact of in vivo small-animal imaging in drug discovery and pharmaceutical development. Mod. Biopharm. 2013, 183–209. [Google Scholar] [CrossRef]
- Chaudhury, S.; Thakur, B.; Chatterjee, S.; Ray, P. Molecular Imaging Aided Improvement in Drug Discovery and Development. Curr. Biotechnol. (SharjahUnited Arab Emir.) 2014, 3, 218–237. [Google Scholar] [CrossRef]
- Medhi, B.; Misra, S.; Avti Pramod, K.; Kumar, P.; Kumar, H.; Singh, B. Role of neuroimaging in drug development. Rev. Neurosci. 2014, 25, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Poels, E.M.P.; Kegeles, L.S.; Kantrowitz, J.T.; Slifstein, M.; Javitt, D.C.; Lieberman, J.A.; Abi-Dargham, A.; Girgis, R.R. Imaging glutamate in schizophrenia: Review of findings and implications for drug discovery. Mol. Psychiatry 2014, 19, 20–29. [Google Scholar] [CrossRef]
- Pecking, A.P.; Bellet, D.; Alberini, J.L. Immuno-SPET/CT and immuno-PET/CT: A step ahead to translational imaging. Clin. Exp. Metastasis 2012, 29, 847–852. [Google Scholar] [CrossRef]
- Chappell, L.L.; Dadachova, E.; Milenic, D.E.; Garmestani, K.; Wu, C.; Brechbiel, M.W. Synthesis, characterization, and evaluation of a novel bifunctional chelating agent for the lead isotopes 203Pb and 212Pb. Nucl. Med. Biol. 2000, 27, 93–100. [Google Scholar] [CrossRef]
- Sgouros, G.; Ballangrud, A.M.; Jurcic, J.G.; McDevitt, M.R.; Humm, J.L.; Erdi, Y.E.; Mehta, B.M.; Finn, R.D.; Larson, S.M.; Scheinberg, D.A. Pharmacokinetics and dosimetry of an alpha-particle emitter labeled antibody: 213Bi-HuM195 (anti-CD33) in patients with leukemia. J. Nucl. Med. 1999, 40, 1935–1946. [Google Scholar]
- Lohrmann, C.; Zhang, H.; Thorek, D.L.; Desai, P.; Zanzonico, P.B.; O’Donoghue, J.; Irwin, C.P.; Reiner, T.; Grimm, J.; Weber, W.A. Cerenkov Luminescence Imaging for Radiation Dose Calculation of a 90Y-Labeled Gastrin-Releasing Peptide Receptor Antagonist. J. Nucl. Med. 2015, 56, 805–811. [Google Scholar] [CrossRef]
- Ruggiero, A.; Holland, J.P.; Lewis, J.S.; Grimm, J. Cerenkov luminescence imaging of medical isotopes. J. Nucl. Med. 2010, 51, 1123–1130. [Google Scholar] [CrossRef]
- Das, S.; Thorek, D.L.; Grimm, J. Cerenkov imaging. Adv. Cancer Res. 2014, 124, 213–234. [Google Scholar] [CrossRef] [PubMed]
- Thorek, D.; Robertson, R.; Bacchus, W.A.; Hahn, J.; Rothberg, J.; Beattie, B.J.; Grimm, J. Cerenkov imaging—A new modality for molecular imaging. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 163–173. [Google Scholar]
- Beattie, B.J.; Thorek, D.L.; Schmidtlein, C.R.; Pentlow, K.S.; Humm, J.L.; Hielscher, A.H. Quantitative modeling of Cerenkov light production efficiency from medical radionuclides. PLoS ONE 2012, 7, e31402. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, N.L.; Graves, E.E. The potential for Cerenkov luminescence imaging of alpha-emitting radionuclides. Phys. Med. Biol. 2012, 57, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, G. Long-lived alpha emitters in radioimmunotherapy: The mischievous progeny. Cancer Biother. Radiopharm. 2000, 15, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Pandya, D.N.; Hantgan, R.; Budzevich, M.M.; Kock, N.D.; Morse, D.L.; Batista, I.; Mintz, A.; Li, K.C.; Wadas, T.J. Preliminary Therapy Evaluation of 225Ac-DOTA-c(RGDyK) Demonstrates that Cerenkov Radiation Derived from 225Ac Daughter Decay Can be Detected by Optical Imaging for In vivo Tumor Visualization. Theranostics 2016, 6, 698–709. [Google Scholar] [CrossRef]
- Chen, X.; Hou, Y.; Tohme, M.; Park, R.; Khankaldyyan, V.; Gonzales-Gomez, I.; Bading, J.R.; Laug, W.E.; Conti, P.S. Pegylated Arg-Gly-Asp peptide: 64Cu labeling and PET imaging of brain tumor alphavbeta3-integrin expression. J. Nucl. Med. 2004, 45, 1776–1783. [Google Scholar]
- Chen, X.; Liu, S.; Hou, Y.; Tohme, M.; Park, R.; Bading, J.R.; Conti, P.S. MicroPET imaging of breast cancer alphav-integrin expression with 64Cu-labeled dimeric RGD peptides. Mol. Imaging Biol. 2004, 6, 350–359. [Google Scholar] [CrossRef]
- Chen, X.; Park, R.; Tohme, M.; Shahinian, A.H.; Bading, J.R.; Conti, P.S. MicroPET and autoradiographic imaging of breast cancer alpha v-integrin expression using 18F- and 64Cu-labeled RGD peptide. Bioconjug. Chem. 2004, 15, 41–49. [Google Scholar] [CrossRef]
- Chen, X. Integrin Targeted Imaging and Therapy. Theranostics 2011, 2011, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Patterson, J.T.; Sarkar, M.; Pedzisa, L.; Kodadek, T.; Roush, W.R.; Rader, C. Site-Specific Dual Antibody Conjugation via Engineered Cysteine and Selenocysteine Residues. Bioconjug. Chem. 2015, 26, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- van Rij, C.M.; Frielink, C.; Goldenberg, D.M.; Sharkey, R.M.; Lütje, S.; McBride, W.J.; Oyen, W.J.G.; Boerman, O.C. Pretargeted Radioimmunotherapy of Prostate Cancer with an Anti-TROP-2×Anti-HSG Bispecific Antibody and a (177)Lu-Labeled Peptide. Cancer Biother. Radiopharm. 2014, 29, 323–329. [Google Scholar] [CrossRef]
- Frampas, E.; Rousseau, C.; Bodet-Milin, C.; Barbet, J.; Chatal, J.-F.; Kraeber-Bodéré, F. Improvement of Radioimmunotherapy Using Pretargeting. Front. Oncol. 2013, 3, 159. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, M.; Ferlin, M.; Legouffe, E.; Horvathova, M.; Liautard, J.; Rossi, J.F.; Wijdenes, J.; Brochier, J.; Klein, B. The myeloma cell antigen syndecan-1 is lost by apoptotic myeloma cells. Br. J. Haematol. 1998, 100, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Adumeau, P.; Sharma, S.K.; Brent, C.; Zeglis, B.M. Site-Specifically Labeled Immunoconjugates for Molecular Imaging--Part 2: Peptide Tags and Unnatural Amino Acids. Mol. Imaging Biol. 2016, 18, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Adumeau, P.; Sharma, S.K.; Brent, C.; Zeglis, B.M. Site-Specifically Labeled Immunoconjugates for Molecular Imaging--Part 1: Cysteine Residues and Glycans. Mol. Imaging Biol. 2016, 18, 1–17. [Google Scholar] [CrossRef]
- Schwarz, S.W.; Decristoforo, C.; Goodbody, A.E.; Singhal, N.; Saliba, S.; Ruddock, R.S.; Zukotynski, K.; Ross, A.A. Harmonization of U.S., European Union, and Canadian First-in-Human Regulatory Requirements for Radiopharmaceuticals: Is This Possible? J. Nucl. Med. 2019, 60, 158–166. [Google Scholar] [CrossRef]
- Krolicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Krolicki, B.; Jakucinski, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Prolonged survival in secondary glioblastoma following local injection of targeted alpha therapy with (213)Bi-substance P analogue. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1636–1644. [Google Scholar] [CrossRef]
- Krolicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Krolicki, B.; Jakucinski, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Safety and efficacy of targeted alpha therapy with (213)Bi-DOTA-substance P in recurrent glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2018. [Google Scholar] [CrossRef]
- Autenrieth, M.E.; Seidl, C.; Bruchertseifer, F.; Horn, T.; Kurtz, F.; Feuerecker, B.; D’Alessandria, C.; Pfob, C.; Nekolla, S.; Apostolidis, C.; et al. Treatment of carcinoma in situ of the urinary bladder with an alpha-emitter immunoconjugate targeting the epidermal growth factor receptor: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1364–1371. [Google Scholar] [CrossRef]
- Autenrieth, M.E.; Horn, T.; Kurtz, F.; Nguyen, K.; Morgenstern, A.; Bruchertseifer, F.; Schwaiger, M.; Blechert, M.; Seidl, C.; Senekowitsch-Schmidtke, R.; et al. [Intravesical radioimmunotherapy of carcinoma in situ of the urinary bladder after BCG failure]. Urol. A 2017, 56, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Hadaschik, B. Re: 225Ac-PSMA-617 for PSMA-Targeting Alpha-radiation Therapy of Patients with Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2016, 70, 1080–1081. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. Ac-225-DOTATOCan empiric dose finding for alpha particle emitter based radionuclide therapy of neuroendocrine tumors. J. Nucl. Med. 2015, 56, 1232. [Google Scholar]
- Jurcic, J.G.; Levy, M.Y.; Park, J.H.; Ravandi, F.; Perl, A.E.; Pagel, M.J.; Smith, D.B.; Estey, E.H.; Kantarjian, H.; Cicic, D.; et al. Phase I Trial of Targeted Alpha-Particle Therapy with Actinium-225 (225Ac)-Lintuzumab and Low-Dose Cytarabine (LDAC) in Patients Age 60 or Older with Untreated Acute Myeloid Leukemia (AML). In Proceedings of the ASH Annual Meeting, Atlanta, GA, USA, 9–12 December 2017; p. 4050. [Google Scholar]
- Rathke, H.; Kratochwil, C.; Hohenberger, R.; Giesel, F.L.; Bruchertseifer, F.; Flechsig, P.; Morgenstern, A.; Hein, M.; Plinkert, P.; Haberkorn, U.; et al. Initial clinical experience performing sialendoscopy for salivary gland protection in patients undergoing (225)Ac-PSMA-617 RLT. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Sathekge, M.; Bruchertseifer, F.; Vorster, M.; Lawal, I.; Knoesen, O.; Mahapane, J.; Davis, C.; Reyneke, F.; Maes, A.; Kratochwil, C.; et al. PREDICTORS OF OVERALL AND DISEASE FREE SURVIVAL IN METASTATIC CASTRATION-RESISTANT PROSTATE CANCER PATIENTS RECEIVING (225)Ac-PSMA-617 RADIOLIGAND THERAPY. J. Nucl. Med. 2019. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Reardon, D.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; McLendon, R.E.; Wong, T.Z.; Bigner, D.D. Clinical experience with alpha-particle emitting 211At: Treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J. Nucl. Med. 2008, 49, 30–38. [Google Scholar] [CrossRef]
- Andersson, H.; Cederkrantz, E.; Back, T.; Divgi, C.; Elgqvist, J.; Himmelman, J.; Horvath, G.; Jacobsson, L.; Jensen, H.; Lindegren, S.; et al. Intraperitoneal alpha-particle radioimmunotherapy of ovarian cancer patients: Pharmacokinetics and dosimetry of (211)At-MX35 F(ab′)2—A phase I study. J. Nucl. Med. 2009, 50, 1153–1160. [Google Scholar] [CrossRef]
- Jurcic, J.G.; Larson, S.M.; Sgouros, G.; McDevitt, M.R.; Finn, R.D.; Divgi, C.R.; Ballangrud, A.M.; Hamacher, K.A.; Ma, D.; Humm, J.L.; et al. Targeted alpha particle immunotherapy for myeloid leukemia. Blood 2002, 100, 1233–1239. [Google Scholar] [CrossRef]
- Rosenblat, T.L.; McDevitt, M.R.; Mulford, D.A.; Pandit-Taskar, N.; Divgi, C.R.; Panageas, K.S.; Heaney, M.L.; Chanel, S.; Morgenstern, A.; Sgouros, G.; et al. Sequential cytarabine and alpha-particle immunotherapy with bismuth-213-lintuzumab (HuM195) for acute myeloid leukemia. Clin. Cancer Res. 2010, 16, 5303–5311. [Google Scholar] [CrossRef]
- Heeger, S.; Moldenhauer, G.; Egerer, G.; Wesch, H.; Martin, S.; Nikula, T.K.; Apostolidis, C.; Brechbiel, M.; Ho, A.; Haas, R. Alpha radioimmunotherapy of B-lineage non-Hodgkin’s lymphoma using 213Bi-labeled anti-CD19- and anti-CD20-CHX-A”-DTPA conjugates. J. Clin. Oncol. 2004, 22 (Suppl. S14), 2625. [Google Scholar] [CrossRef]
- Kneifel, S.; Cordier, D.; Good, S.; Ionescu, M.C.; Ghaffari, A.; Hofer, S.; Kretzschmar, M.; Tolnay, M.; Apostolidis, C.; Waser, B.; et al. Local targeting of malignant gliomas by the diffusible peptidic vector 1,4,7,10-tetraazacyclododecane-1-glutaric acid-4,7,10-triacetic acid-substance p. Clin. Cancer Res. 2006, 12, 3843–3850. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.J.; Singla, A.A.; Rizvi, S.M.; Graham, P.; Bruchertseifer, F.; Apostolidis, C.; Morgenstern, A. Analysis of patient survival in a Phase I trial of systemic targeted alpha-therapy for metastatic melanoma. Immunotherapy 2011, 3, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Raja, C.; Graham, P.; Abbas Rizvi, S.M.; Song, E.; Goldsmith, H.; Thompson, J.; Bosserhoff, A.; Morgenstern, A.; Apostolidis, C.; Kearsley, J.; et al. Interim analysis of toxicity and response in phase 1 trial of systemic targeted alpha therapy for metastatic melanoma. Cancer Biol. 2007, 6, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Meredith, R.; Torgue, J.; Shen, S.; Fisher, D.R.; Banaga, E.; Bunch, P.; Morgan, D.; Fan, J.; Straughn, J.M., Jr. Dose escalation and dosimetry of first-in-human alpha radioimmunotherapy with 212Pb-TCMC-trastuzumab. J. Nucl. Med. 2014, 55, 1636–1642. [Google Scholar] [CrossRef]
- Meredith, R.F.; Torgue, J.; Azure, M.T.; Shen, S.; Saddekni, S.; Banaga, E.; Carlise, R.; Bunch, P.; Yoder, D.; Alvarez, R. Pharmacokinetics and imaging of 212Pb-TCMC-trastuzumab after intraperitoneal administration in ovarian cancer patients. Cancer Biother. Radiopharm. 2014, 29, 12–17. [Google Scholar] [CrossRef]
- Castello, A.; Macapinlac, H.A.; Lopci, E.; Santos, E.B. Prostate-specific antigen flare induced by (223)RaCl2 in patients with metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2256–2263. [Google Scholar] [CrossRef]
- Dizdarevic, S.; Petersen, P.M.; Essler, M.; Versari, A.; Bourre, J.C.; la Fougere, C.; Valdagni, R.; Paganelli, G.; Ezziddin, S.; Kalinovsky, J.; et al. Interim analysis of the REASSURE (Radium-223 alpha Emitter Agent in non-intervention Safety Study in mCRPC popUlation for long-teRm Evaluation) study: Patient characteristics and safety according to prior use of chemotherapy in routine clinical practice. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1102–1110. [Google Scholar] [CrossRef]
- Hoskin, P.; Sartor, O.; O’Sullivan, J.M.; Johannessen, D.C.; Helle, S.I.; Logue, J.; Bottomley, D.; Nilsson, S.; Vogelzang, N.J.; Fang, F.; et al. Efficacy and safety of radium-223 dichloride in patients with castration-resistant prostate cancer and symptomatic bone metastases, with or without previous docetaxel use: A prespecified subgroup analysis from the randomised, double-blind, phase 3 ALSYMPCA trial. Lancet Oncol. 2014, 15, 1397–1406. [Google Scholar] [CrossRef]
- Parker, C.; Finkelstein, S.E.; Michalski, J.M.; O’Sullivan, J.M.; Bruland, O.; Vogelzang, N.J.; Coleman, R.E.; Nilsson, S.; Sartor, O.; Li, R.; et al. Efficacy and Safety of Radium-223 Dichloride in Symptomatic Castration-resistant Prostate Cancer Patients With or Without Baseline Opioid Use From the Phase 3 ALSYMPCA Trial. Eur. Urol. 2016, 70, 875–883. [Google Scholar] [CrossRef]
- Jo, K.I.; Kim, M.S.; Yeon, J.Y.; Kim, J.S.; Hong, S.C. Recurrent Bleeding in Hemorrhagic Moyamoya Disease: Prognostic Implications of the Perfusion Status. J. Korean Neurosurg. Soc. 2016, 59, 117–121. [Google Scholar] [CrossRef]
- Falkmer, U.; Jarhult, J.; Wersall, P.; Cavallin-Stahl, E. A systematic overview of radiation therapy effects in skeletal metastases. Acta Oncol 2003, 42, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, E.B. Dosage and response in radiopharmaceutical therapy of painful osseous metastases. J. Nucl. Med. 1996, 37, 249–252. [Google Scholar] [PubMed]
- Pandit-Taskar, N.; Batraki, M.; Divgi, C.R. Radiopharmaceutical therapy for palliation of bone pain from osseous metastases. J. Nucl. Med. 2004, 45, 1358–1365. [Google Scholar] [PubMed]
- Henriksen, G.; Fisher, D.R.; Roeske, J.C.; Bruland, O.S.; Larsen, R.H. Targeting of osseous sites with alpha-emitting 223Ra: Comparison with the beta-emitter 89Sr in mice. J. Nucl. Med. 2003, 44, 252–259. [Google Scholar] [PubMed]
- Nilsson, S.; Larsen, R.H.; Fossa, S.D.; Balteskard, L.; Borch, K.W.; Westlin, J.E.; Salberg, G.; Bruland, O.S. First clinical experience with alpha-emitting radium-223 in the treatment of skeletal metastases. Clin. Cancer Res. 2005, 11, 4451–4459. [Google Scholar] [CrossRef] [PubMed]
- Lewington, V.J. Bone-seeking radionuclides for therapy. J. Nucl. Med. 2005, 46 (Suppl. 1), 38S–47S. [Google Scholar]
- Dauer, L.T.; Williamson, M.J.; Humm, J.; O’Donoghue, J.; Ghani, R.; Awadallah, R.; Carrasquillo, J.; Pandit-Taskar, N.; Aksnes, A.K.; Biggin, C.; et al. Radiation safety considerations for the use of (2)(2)(3)RaCl(2) DE in men with castration-resistant prostate cancer. Health Phys. 2014, 106, 494–504. [Google Scholar] [CrossRef]
- Nilsson, S.; Strang, P.; Aksnes, A.K.; Franzen, L.; Olivier, P.; Pecking, A.; Staffurth, J.; Vasanthan, S.; Andersson, C.; Bruland, O.S. A randomized, dose-response, multicenter phase II study of radium-223 chloride for the palliation of painful bone metastases in patients with castration-resistant prostate cancer. Eur. J. Cancer 2012, 48, 678–686. [Google Scholar] [CrossRef]
- Sartor, O.; Coleman, R.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: Results from a phase 3, double-blind, randomised trial. Lancet. Oncol. 2014, 15, 738–746. [Google Scholar] [CrossRef]
- Nilsson, S.; Cislo, P.; Sartor, O.; Vogelzang, N.J.; Coleman, R.E.; O’Sullivan, J.M.; Reuning-Scherer, J.; Shan, M.; Zhan, L.; Parker, C. Patient-reported quality of life analysis of radium-223 dichloride from the phase 3 ALSYMPCA study. Ann. Oncol. 2016. [Google Scholar] [CrossRef]
- Crawford, E.D.; Higano, C.S.; Shore, N.D.; Hussain, M.; Petrylak, D.P. Treating Patients with Metastatic Castration Resistant Prostate Cancer: A Comprehensive Review of Available Therapies. J. Urol. 2015, 194, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Loriot, Y.; Sweeney, C.J.; Fizazi, K.; Ryan, C.J.; Shevrin, D.H.; Antonarakis, E.S.; Pandit-Taskar, N.; Deandreis, D.; Jacene, H.A.; et al. Radium-223 in combination with docetaxel in patients with castration-resistant prostate cancer and bone metastases: A phase 1 dose escalation/randomised phase 2a trial. Eur. J. Cancer 2019, 114, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Shore, N.D.; Tutrone, R.F.; Mariados, N.F.; Nordquist, L.T.; Mehlhaff, B.A.; Steere, K.J.; Harrelson, S.S. eRADicAte: A Prospective Evaluation Combining Radium-223 Dichloride and Abiraterone Acetate Plus Prednisone in Patients With Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2018, 16, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Parker, C.C.; Saad, F.; Miller, K.; Tombal, B.; Ng, Q.S.; Bogemann, M.; Matveev, V.; Piulats, J.M.; Zucca, L.E.; et al. ERA 223: A phase 3 trial of radium-223 dichloride (Ra-223) in combination with abiraterone acetate (abiraterone) and prednisone in the treatment of asymptomatic or mildly symptomatic chemotherapy-naive patients (pts) with bone predominant metastatic castration-resistant prostate cancer (mCRPC). In Proceedings of the ESMO 2018 Congress, Munich, Germany, 19 October 2018. [Google Scholar]
- van der Doelen, M.J.; Mehra, N.; Hermsen, R.; Janssen, M.J.R.; Gerritsen, W.R.; van Oort, I.M. Patient Selection for Radium-223 Therapy in Patients With Bone Metastatic Castration-Resistant Prostate Cancer: New Recommendations and Future Perspectives. Clin. Genitourin. Cancer 2019, 17, 79–87. [Google Scholar] [CrossRef]
- Malamas, A.S.; Gameiro, S.R.; Knudson, K.M.; Hodge, J.W. Sublethal exposure to alpha radiation (223Ra dichloride) enhances various carcinomas’ sensitivity to lysis by antigen-specific cytotoxic T lymphocytes through calreticulin-mediated immunogenic modulation. Oncotarget 2016, 7, 86937–86947. [Google Scholar] [CrossRef]
- Nilsson, S.; Franzen, L.; Parker, C.; Tyrrell, C.; Blom, R.; Tennvall, J.; Lennernas, B.; Petersson, U.; Johannessen, D.C.; Sokal, M.; et al. Two-year survival follow-up of the randomized, double-blind, placebo-controlled phase II study of radium-223 chloride in patients with castration-resistant prostate cancer and bone metastases. Clin. Genitourin. Cancer 2013, 11, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Aksnes, A.K.; Naume, B.; Garcia, C.; Jerusalem, G.; Piccart, M.; Vobecky, N.; Thuresson, M.; Flamen, P. A phase IIa, nonrandomized study of radium-223 dichloride in advanced breast cancer patients with bone-dominant disease. Breast Cancer Res. Treat. 2014, 145, 411–418. [Google Scholar] [CrossRef]
- Berenson, J.R.; Yellin, O.; Patel, R.; Duvivier, H.; Nassir, Y.; Mapes, R.; Abaya, C.D.; Swift, R.A. A phase I study of samarium lexidronam/bortezomib combination therapy for the treatment of relapsed or refractory multiple myeloma. Clin. Cancer Res. 2009, 15, 1069–1075. [Google Scholar] [CrossRef]
- Anderson, P.M.; Subbiah, V.; Rohren, E. Bone-seeking radiopharmaceuticals as targeted agents of osteosarcoma: Samarium-153-EDTMP and radium-223. Adv. Exp. Med. Biol. 2014, 804, 291–304. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
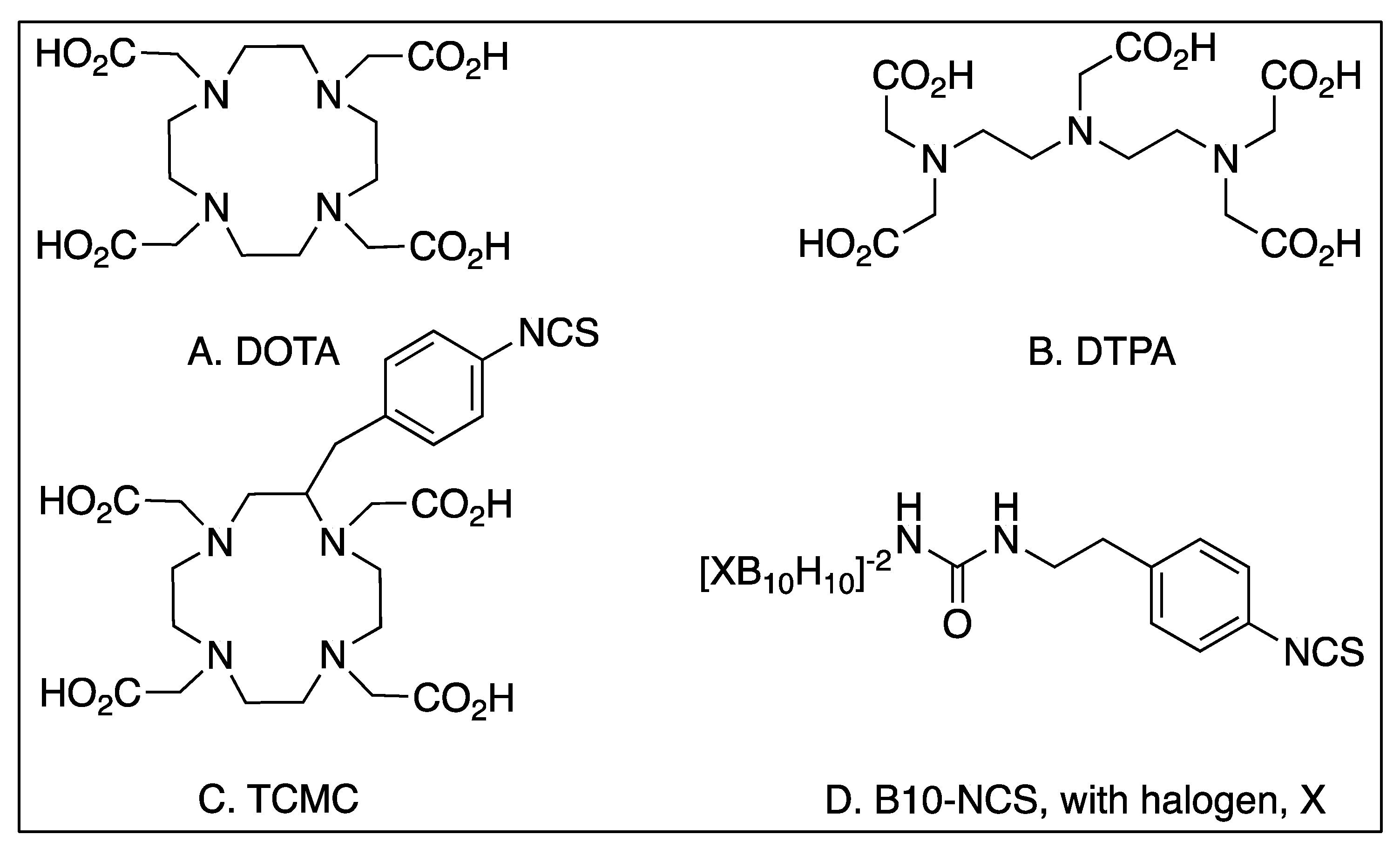{kind=link}
{kind=link}
| Isotope | Half-Life | Max Energy | Emissions Per Decay |
|---|---|---|---|
| 225Ac | 10.1 d | 5.83 | 4 α, 2β- |
| 211At | 7.2 h | 5.87 | 1 α, 1 EC |
| 212Bi | 1.01 h | 6.09 | 1 α, 1β- |
| 213Bi | 45.6 min | 5.87 | 1 α, 2β- |
| 212Pb | 10.6 h | 6.09 | 1 α, 2β- |
| 223Ra | 11.4 d | 5.87 | 4 α, 2β- |
| 224Ra | 3.6 d | 8.8 | 5 α, 2 β- |
| 149Tb | 4.1 h | 3.96 | 1 α, 1 β+ |
| 227Th | 18.7 d | 6.04 | 5 α, 2β- |
| Isotope | Study | Molecular Target | Targeting Moiety | Drug(s) & Route | Cancer Type & Animal Model | Key Results | Ref |
|---|---|---|---|---|---|---|---|
| 225Ac | Efficacy, toxicity | PSMA, CD19 | J591 & B4 mAbs | 225Ac–DOTA–J591, 225Ac-B4, i.v. | Human LNCaP prostate s.c. xenografts & disseminated Daudi lymphoma in male nude mice. | Both effective without toxicity. | [35] |
| 225Ac | Efficacy, toxicity | HER-2/neu | Trastuzumab | 225Ac–DOTA–trastuzumab, i.p. | SKOV3 human ovarian cancer s.c. xenografts in female nude mice. | Effective with no toxicity. | [148] |
| 225Ac | PK, RD, toxicity | CD33 | HuM195 Ab | 225Ac–DOTA–HuM195, i.v. | Cynomolgus monkey leukemia (does not express the human CD33 target). | 12 d blood T1/2, dosimetry kinetics estimated, efficacy without renal toxicity. | [149] |
| 225Ac | BD, efficacy, toxicity | Ganglioside GD2 | 3F8 Ab | 225Ac–DOTA–3F8, i.v. | NMB-7 human neuroblastoma xenografts in nude mice (BD), meningeal carcinomatosis xenografts in nude rats (efficacy) & cynomolgus monkeys (toxicity). | Tumor specificity, increased survival, no toxicity. | [150] |
| 225Ac, 177Lu | BD, efficacy, toxicity | Somatostatin receptors | DOTATOC peptide | 225Ac and 177Lu–DOTATOC, i.v. | AR42J rat pancreatic exocrine s.c. xenografts in nude mice. | 225Ac-TAT had greater efficacy relative to 177Lu-TBT with low toxicity. | [151] |
| 225Ac, 213Bi, 90Y | BD, dosimetery, efficacy, toxicity | HER-2/neu | 7.16.4 mAb | 225Ac, 213Bi and 90Y-7.16.4, i.v. | neu-N transgenic mouse model with rat HER-2/neu expression and spontaneous lung metastases & NT2.5 mouse mammary fat pad xenografts with rat HER-2/neu. | 225Ac-TAT had greater efficacy but with renal toxicity relative to 213Bi-TAT & 90Y-TBT. | [152] |
| 225Ac, 213Bi | BD, efficacy, toxicity | nucleolin | F3 peptide | 225Ac–DOTA–F3, 213Bi–DTPA–F3, i.p. | MDA-MB-435 human peritoneal carcinomatosis in SCID mice. | 225Ac-TAT had greater efficacy relative to 213Bi-TAT with specific tumor uptake and minor renal toxicity. | [153,154] |
| 225Ac | Vascular normalization & efficacy | Vascular endothelial (VE)-cadherin | E4G10 Ab | 225Ac–DOTA–E4G10, i.v. | LS174T human colon s.c. xenografts in female nude mice. | Improved tumor vascular architecture & increased efficacy when combined with chemotherapy. | [107] |
| 225Ac | Safety and efficacy | IL13RA2 | Pep-1L peptide | [225Ac]Pep-1L, stereotactic intracranial injection | U8251 human glioblastoma orthotopic xenografts in male nude mice. | Efficacy with no significant toxicity. | [155] |
| 225Ac | BBB and BTB permeabili-zation | Integrin αvβ3 | small-molecule antagonist | 225Ac-labeled targeted liposomes (225Ac-TL), intracranial injection | U87 MG human glioblastoma orthotopic xenografts in male nude mice. | Enhanced blood-brain barrier (BBB) and bood-tumor barrier (BTB) permeability. | [156] |
| 225Ac | BD, Efficacy | Thrombo-modulin | 201b mAb | LnPO4 nanoparticles (NPs) doped with 225Ac-201b, i.v. | Syngeneic EMT6 mouse breast epithelial cell metastases in BALB/c mouse lung following i.v. injection of cells | Retention of 225Ac and daughters in lung tissue, metastasis burden reduced. | [157] |
| 225Ac | Micro BD, RD | PD-L1 | anti-PD-L1-BC Ab | 225Ac–DOTA–anti-PD-L1-BC, i.v. | NT2.5 mouse mammary xenografts in female nude mice. | Uniform distribution in liver, non-uniform in kidney and tumor, liver RD was limiting. | [158] |
| 225Ac | BD and toxicity | Bone metastasis | Zoledronic acid (ZOL) | 225Ac–DOTAZOL, i.v. | Wistar rats. | High bone:blood ratio. Kidney toxicity. | [159] |
| 225Ac | BD, RD and dose response | PSMA | PSMA ligands with albumin-binding moiety | 225Ac-RPS-074, i.v. | LNCaP human prostate cancer s.c. xenografts in BALB/c mice. | Decreased clearance rate, single administration had complete response in 86% of tumors. | [160] |
| 225Ac | PK, BD, specificity, RD, toxicity, efficacy | MC1R | MC1RL peptide | 225Ac–DOTA–MC1RL, i.v. | PK (Sprague-Dawley rats), BALB/c mice (toxicity and BD) and MEL270 human uveal melanoma s.c. xenografts in SCID mice (BD and efficacy). | Renal and hepatobiliary excretion, rapid blood clearance, low toxicity, prolonged survival and decreased metastasis after single injection. | [67] |
| 225Ac | Efficacy, toxicity | CA19.9 | 5B1 human mAb | 225Ac-labeled tetrazine radioligand and a transcyclooctene5B1 for pretargeting, i.v. | Bilateral MIAPaCa-2 (CA19.9-negative) and BxPC3 (CA19.9-positive) pancreatic cancer s.c. xenografts, and BxPC3 orthotopic xenografts in nude mice. | Pretargeting has similar efficacy compared to conventional TAT with reduced hematotoxicity. | [144] |
| 211At | BD, RD, specificity, efficacy, toxicity | Tenascin glycoprotein | 81C6 mAb | 211At-81C6, subarachnoid catheter or i.v. | Female athymic rat model of neoplastic meningitis by inoculation of human rhabdomyosarcoma cells via subarachnoid catheter. | Efficacy without significant toxicity. RD estimates. | [161,162] |
| 211At | PK, BD, efficacy, toxicity | gp38 | MOv18 mAb | 211At- & 131I-MOv18, i.p. or i.v. | Peritoneal OVCAR-3 human ovarian xenografts in BALB/c ν/ν or nude mice following IP injection of cells. | 211At-TAT had greater efficacy relative to 131I-TBT. | [163,164,165,166] |
| 211At | Tumor neo-vasculature targeting | Fibronectin ED-B domain | Human scFv(L19) | 211At-scFv(L19), i.v. | Murine F9 teratocarcinoma & rat FE8 sarcoma in female nude mice. | Retained at tumor blood vessels resulting in increased tumor to blood ratios. | [140] |
| 211At | BD, tumor dosimetry, efficacy, toxicity | 95-kDa glycoprotein | MX35 mAb | 211At-MX35, i.p. or i.v. | OVCAR-3 human ovarian cancer micrometastases in nude mice. | Fractionated treatment increased efficacy without significant toxicity. | [23,167,168] |
| 211A, 90Y | Efficacy | CD30 | HeFi-1 mAb | 211At-, 90Y HeFi-1, i.v. | Human anaplastic large cell lymphoma cells in SCID/NOD mice. Karpas 299 cell i.v. injection for leukemia & SUDHL-1 xenografts for lymphoma. | 211At-HeFi-1 increased survival in the leukemia model & combination with unlabeled HeFI-1 further improved efficacy. 90Y-HeFi-1 TBT increased survival in the lymphoma model. | [169] |
| 211At | BD, efficacy, toxicity | CD44v6 | U36 chimeric mAb | 211At-U36, i.v. | UT-SCC7 human head and neck squamous cell carcinoma s.c. xenografts in nude mice. | Reduced tumor growth with no significant toxicity. BD consistent with targeting. | [170] |
| 211At | BD, efficacy, toxicity | HER2/neu | C6.5 diabody | 211At-SAPS-C6.5, i.v. | HER2/neu-positive MDA-MB-361/DYT2 breast xenografts in nude mice. | Tumor growth delay with low renal toxicity. | [142] |
| 211At | Efficacy | NIS-transduced tumor cells | Astatide (HAt) peptide | 211At-astatide, i.p. | NIS transduced LNCaP human prostate (NP-1) and parental (P-1) s.c. xenografts in male nude mice. | Accumulation similar to iodine with efficacy against NP-1 tumors relative to control P-1 tumors. | [147] |
| 211At, 213Bi | BD, myelo suppression, toxicity | CD45 | 30F11 Ab | 211At-30F11-ADTM, 213Bi-30F11-CHX-A″, i.v. | Female BALB/c mice. | 211At-TAT induced myeloablation in haematopoietic tissues with greater efficacy and less toxicity relative to the 213Bi conjugate. | [171] |
| 211At | Efficacy | HER-2/neu | Trastuzumab | 211At-trastuzumab, i.p. | SKOV3 human ovarian i.p. xenografts in nude mice. | Combination of trastuzumab and 211At-trastuzumab resulted in complete tumor eradication. | [172] |
| 211At | Dosimetry, toxicity, efficacy | Lewis Y epitope | BR96, chimeric IgG1 mAb | 211At-BR96, i.v. | BN7005-H1D2 rat syngeneic sub-peritoneal colon engraftments. | Resulted in undetectable tumors with tolerable toxicity. | [173] |
| 211At | Efficacy | CD20 | 1F5 mAb | 211At-1F5, i.v. | Human Ramos (Burkitt lymphoma) s.c. xenografts in nude mice and i.v. injection of Ramos cells in SCID mice for disseminated lymphoma. | Highly effective in minimal residual disease mouse model. | [77] |
| 211At | BD, dosimetry | Sigma-2 receptor | MM3 ligand | 211At-MM3, i.v. | EMT6 murine breast syngeneic tumor in female BALB/c mice. | Tumor specific targeting. | [146] |
| 211At | Efficacy | Norepineph-erine transporter | Benzyl-guanidine | meta-[211At]-astatobenzyl-guanidine, i.v. | PC12 rat pheochromocytoma s.c. xenograft in nude mice. | Reduced tumor size without weight loss. | [174] |
| 211At | BD, efficacy | MICA/B | anti MICA/B Ab | 211At-anti MICA/B Ab, i.v. | HCT116 (p53-/- & MICA/B positive) human colon cancer s.c. xenograft in nude mice. | Significant reduction in tumor growth, no weight loss, erythrocytopenia with recovery in 3-4 wks. | [175] |
| 213Bi, 90Y | Toxicity and efficacy | CO17-1A | CO17-1A Fab’ | 213Bi-Fab’ and 90Y-Fab’, i.v. | GW-39 human colon cancer s.c. xenograft in nude mice. | TAT had greater efficacy and lower toxicity than TBT. | [176] |
| 212Bi | Specificity, efficacy, toxicity | gp70 | 103A mAb | 212Bi–CHX-A-DTPA–103A, i.v. | RLV induced erythroleukemia in BALB/c mice. | Clinical and histological remission of erythroleukemia and prolonged survival with low toxicity. | [177] |
| 212Bi | BD, efficacy, toxicity | CD25 | Anti-Tac, humanized mAb | 2l2Bi–CHX-A-anti-Tac, i.v. | SP2 and SP2/Tac syngeneic murine lymphoma in nude mice. | Effective in treatment of bulky solid tumors. | [178] |
| 213Bi | Stability, PK, toxicity | CD33 | HuM195 mAb | 213-Bi-CHX-A-DTPA–HuM195, i.p. or i.v. | Normal BALB/c mice without leukemia. | Favorable stability, PK and toxicity. | [179] |
| 213Bi, 90Y | Pretargeting efficacy | CD25 (Tac) | Humanized anti-Tac mAb (HAT) | 213Bi- & 90Y-DOTA–HAT; & HAT–streptavidin & 213Bi–DOTA–biotin or 90Y-DOTA–biotin, i.v. | Intraperitoneal MET-1 human adult T-cell leukemia in SCID/NOD mice. | Pre-targeted 213Bi TAT increased survival relative to 213Bi–DOTA–HAT, 90Y TBT & pre-targeted TBT. | [143] |
| 213Bi, 131I | Efficacy | TAG-72 | Humanized, domain-deleted CC49 mAb (HuCC49ΔCH2) | 213Bi- or 131I-HuCC49ΔCH2, i.p. | TAG-72+ LS-174T & TAG-72 negative MIP human colon i.p. xenografts in nude mice. | 213Bi-TAT had greater growth inhibition or regression relative to 131I-TBT. | [180] |
| 213Bi | Efficacy, toxicity | d9-E-cad | d9-E-cad mAb | 213Bi-d9-E-cad mAb, i.p. | HSC45-M2 human gastric i.p. xenografts with d9-E-cad mutation in female nude mice. | Double administration had greater efficacy relative to single administration, with no toxicity. | [181] |
| 213Bi | BD, efficacy, toxicity | Somatostatin receptors | DOTATOC peptide | 213Bi–DOTATOC, i.v. | CA20948 rat pancreatic adenocarcinoma tumors in Lewis rats. | Antitumor efficacy with low toxicity. | [64] |
| 213Bi | Specificity, BD | CD87 | P-P4D peptide | 213Bi-P-P4D, i.p. | OV-MZ-6 human ovarian i.p. xenografts in female nude mice. | Specific tumor uptake, kidney uptake reduced by co-injection of gelofusine. | [182] |
| 213Bi | Efficacy, toxicity | MUC1, uPAR and BLCA-38 | C595 & BLCA-38 mAbs, & PAI2 protein | 213Bi-C595, -BLCA-38 & -PAI2, i.p. | PC-3 human prostate orthotopic, intratibial and s.c. xenograft tumors in NOD SCID mice. | Multiple TAT can overcome heterogeneous antigen expression with efficacy against micrometastases. | [145] |
| 213Bi | Efficacy, toxicity | EGFR | Matuzumab | 213Bi-matuzumab, intravesical | EJ28 human orthotopic bladder xenografts in nude mice. | Increased survival without toxicity. Combination with mitomycin C increased efficacy with nephrotoxicity. | [183] |
| 213Bi | Efficacy | TAG-72 | Humanized CC49 mAb (HuCC49DCH2) | 213Bi- HuCC49DCH2, i.p. | LS-174T human colon i.p. xenografts in female nude mice. | Combination trastuzumab and i.p. TAT increased efficacy and was well tolerated. | [184] |
| 213Bi | Pretargeting efficacy | CD20 | scFv-1F5-SA (streptavidin fusion protein) | 1F5-SA & 213Bi–DOTA–biotin, i.v. | Ramos human lymphoma xenografts in nude mice. | Tumor regression and increased survival in mice with small tumors via pretargeting. | [141] |
| 213Bi, 177Lu | BD, dosimetry, efficacy, toxicity | GRP | PESIN and AMBA peptides | 177Lu–DOTA–PESIN, 213Bi–DOTA–PESIN, or 213Bi-AMBA, i.v. | PC-3 human prostate s.c. xenografts in female nude mice. | 213Bi-TAT had greater efficacy compared to 177Lu-TBT. 213Bi–DOTA–PESIN had lower renal toxicity relative to 213Bi-AMBA. | [185] |
| 213Bi, 177Lu | Efficacy | CD138 | 9E7.4 mAb | 213Bi-9E7.4 and 177Lu-9E7.4, i.v. | 5T33 murine multiple myeloma cell syngeneic i.v. injection into C57/BL6 mice. | 213Bi-9E7.4 increased survival and cured 45%, 177Lu-9E7.4′ increased survival, no cures. | [186] |
| 213Bi, 177Lu | Efficacy, toxicity | Mutant d9-E-cadherin | d9MAb | 213Bi-d9Mab & 177Lu-d9Mab, i.p. | HSC45-M2 human gastric cancer cell i.p. injection in nude mice. | 213Bi had comparable efficacy with lower toxicity. | [187] |
| 213Bi | BD, efficacy, toxicity | CD138 | Anti-mouse CD138 Ab | 213Bi-CD138, i.v. | 5T33 mouse multiple myeloma cell engraftment into syngeneic C57BL/KaLwRij mice. | Increased survival with only moderate and transient toxicity. | [188] |
| 213Bi | Efficacy, toxicity | EGFR | Matuzumab | 213Bi-matuzumab, intravesical. | EJ28 human orthotopic bladder xenografts in nude mice. | Increased survival with low toxicity. | [189] |
| 213Bi | PK, efficacy, dosimetry, toxicity | SSTR2 | DOTATATE peptide | 213Bi–DOTATATE, i.v. | Neuroendocrine H69 human small cell lung carcinoma and CA20948 rat pancreatic s.c. xenografts in nude mice. | Effective in small and large tumors (both types), with dose limiting renal toxicity. | [66] |
| 212Pb | Efficacy | HER-1 | Cetuximab | 212Pb-cetuximab, i.p. | ILS174T human colon i.p. xenografts in nude mice. | Extended survival and combined with gemcitabine & carboplatin increased efficacy. | [190] |
| 212Pb | Efficacy | MC1R | DOTA-Re (Arg11)CCMSH peptide | 212Pb[DOTA]–Re (Arg11)CCMSH, i.v. | B16/F1 murine melanoma syngeneic s.c. engraftments in C57BL/6 mice. | Tumor eradication at higher activities. | [191] |
| 212Pb | Efficacy | HER-2 and CEA | Trastuzumab & 35A7 | 212Pb-trastuzumab & 212Pb-35A7, i.p. | A-431 HER-2 positive and CEA transfected vulvar squamous carcinoma cells i.p in nude mice. | Internalizing anti-HER2 labeled Ab had greater efficacy than non-internalizing anti-CEA labeled Ab. | [192] |
| 212Pb | Efficacy, toxicity | HER-2/neu | Trastuzumab | 212Pb-trastuzumab, i.p. | LS174T human colon & Shaw human pancreatic i.p. xenografts in nude mice. | Increased survival with low toxicity. | [193] |
| 212Pb | Efficacy | HER-2/neu | Trastuzumab | 212Pb-trastuzumab, i.p. | LS-174T human colon i.p. xenografts in nude mice. | Combination with gemcitabine increased survival. | [194] |
| 212Pb | BD, efficacy | B7-H3 | 376.96 mAb | 212Pb-376.96, i.p. | ES-2 or A2780cp20 human ovarian cancer cells i.p. into nude mice. | High peritoneal retention, tumor tissue accumulation & increased survival. | [195] |
| 212Pb | BD, efficacy | B7-H3 | 376.96 mAb | 212Pb-376.96, i.v. | Panc039 pancreatic cancer orthotopic xenografts in nude mice. | High tumor uptake & tumor growth inhibition. | [196] |
| 212Pb | BD, efficacy | CSPG4 | 225.28 mAb | 212Pb-225.28, i.v. | SUM159 & 2LMP human triple negative breast cancer (TNBC) orthotopic mammary fat pad xenografts in nude mice. | Dose-dependent growth inhibition. | [197] |
| 212Pb | Administration route, toxicity, efficacy | EGFR | Panitumumab F(ab’)2 fragment | 212Pb-panitumumab F(ab’)2, i.p. & i.v. | ILS-174T human colon i.p. xenografts in nude mice. | Increased survival with tolerated toxicity via i.p. or i.v. injection. | [198] |
| 212Pb | Efficacy, combination therapy | MC1R | ee-cyclized α-MSH peptide | 212Pb–DOTA–MC1L, BRAFi & HDACi | A2058 & MEWO human melanoma xenografts in nude mice. | Improved tumor response by combination therapy. | [69] |
| 227Th | BD, efficacy, toxicity | CD20 | Rituximab | 227Th–DOTA–p-benzyl-rituximab, i.v. | BALB/c mice & Raji human B-cell lymphoma s.c. xenografts in nude mice. | Increased efficacy with managable toxicity. | [199] |
| 227Th | BD, efficacy, toxicity | HER-2/neu | Trastuzumab | 227Th–DOTA–trastuzumab, i.v. | SKBR-3 human breast cancer xenografts in nude mice. | Tumor growth inhibition with no toxicity. | [200] |
| 227Th | BD, efficacy, toxicity | CD70 | Anti-human CD70 mAb | CD70-TTC, i.v. | 786-O human renal cancer s.c. xenografts in nude mice | Well tolerated with inhibition of tumor growth. | [201] |
| 224Ra | Efficacy, toxicity | peritoneal metastases | Injection into peritoneum | 224Ra-labeled calcium carbonate microparticles, i.p. | ES-2 and SKOV3 human ovarian cancer i.p. xenografts in nude mice. | Well tolerated with antitumor effect. | [202] |
| Isotope | Molecular Target | Targeting Moiety | Drug | Cancer Type | Trial/# of Patients | Administration Route | Key Results | Ref |
|---|---|---|---|---|---|---|---|---|
| 225Ac | CD33 | HuM195 | 225Ac–DOTA–HuM195 | AML | Phase I/20, Ongoing multicentric phase I, II | Intravenous | Safe at doses ≤ 3 µCi/kg, anti-leukemic activity across all dose levels studied, no acute toxicities, myelosuppression | [235] |
| 225Ac | PSMA | PSMA-617 ligand | 225Ac–PSMA-617 | Prostate cancer | NA/40 | Intravenous | Remarkable anti-tumor response was observed in the patients. Xerostomia in salivary gland was the main side effect. | [5,6,60,233,236,237] |
| 225Ac | Somatostatin receptors | DOTATOC peptide | 225Ac–DOTATOC | Neuroendocrine tumors | NA/34 | Not mentioned | Well-tolerated with promising treatment efficacy | [234] |
| 211At | Tenascin-C | chimeric 816 antibody | 211At-ch81C6 | Glioblastoma | Phase I/18 | surgically created resection cavity | Increased Median survival (54 weeks), No dose-limiting toxicity, No-grade 3 toxicity | [238] |
| 211At | NaPi2b | MX35 F(ab′)2 | 211At-MX35 F(ab′)2 | Ovarian carcinoma | Phase I/9 | Intraperitoneal | No adverse effects, grade I toxicity, no bone marrow toxicity | [239] |
| 213Bi | CD33 | HuM195 | 213Bi–CHX-A-DTPA–HuM1 95 | AML | Phase I/18 | Intravenous | 14 patients had reductions in marrow blasts | [240] |
| 213Bi | CD33 | HuM195 | 213Bi–CHX-A-DTPA–HuM1 95 | AML | Phase I, II/31 | Intravenous | dose-response relationship with remission at the highest doses | [241] |
| 213Bi | CD20 | Rituximab | 213Bi-CHX-A”- Rituximab | Non-Hodgkin lymphoma | Phase I/9 | Intravenous | Myelosuppression and no other toxic side, two patients responded | [242] |
| 213Bi | Neurokinin type-1 receptor | Substance P | 213Bi–DOTA–[Thi8, Met (O2) 11]–substance P | Glioblastoma | NA/2, 9, 20 | Intrathecal | Well-tolerated, favorable response | [229,230,243] |
| 213Bi | NG2 proteoglycan | 9.2.27 antibody | 213Bi–cDTPA–9.2.27 | Melanoma | Phase I/38 | Intralesional | TAT was safe up to 450 mCi and effective at 200 mCi | [244,245] |
| 213Bi | Somatostatin receptors | DOTATOC peptide | 213Bi–DOTATOC | Neuroendocrine tumors | NA/7 | Intraarterial infusion | responses were observed in all patients | [65] |
| 213Bi | EGFR | Cetuximab | 213Bi–CHX-A-DTPA–anti-EGFR | carcinoma in situ (CIS) of the bladder | Pilot studies/9 and 12 | Intravesical | TAT well tolerated and showed therapeutic efficacy | [231,232] |
| 212Pb | HER2 | Trastuzumab | 212Pb–TCMC–trastuzumab | Ovarian Cancer | Phase I/3 | Intraperitoneal | Dose escalation showed a little agent-related toxicity, consistent with the dosimetry data | [246,247] |
| 223Ra | Hydroxy-apatite | NA | 223Ra–chloride | Prostate cancer mets | Phase I-III/921 | Intravenous | radium-223 improved overall survival | [7,8,82,248,249,250,251] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tafreshi, N.K.; Doligalski, M.L.; Tichacek, C.J.; Pandya, D.N.; Budzevich, M.M.; El-Haddad, G.; Khushalani, N.I.; Moros, E.G.; McLaughlin, M.L.; Wadas, T.J.; et al. Development of Targeted Alpha Particle Therapy for Solid Tumors. Molecules 2019, 24, 4314. https://doi.org/10.3390/molecules24234314
Tafreshi NK, Doligalski ML, Tichacek CJ, Pandya DN, Budzevich MM, El-Haddad G, Khushalani NI, Moros EG, McLaughlin ML, Wadas TJ, et al. Development of Targeted Alpha Particle Therapy for Solid Tumors. Molecules. 2019; 24(23):4314. https://doi.org/10.3390/molecules24234314
Chicago/Turabian StyleTafreshi, Narges K., Michael L. Doligalski, Christopher J. Tichacek, Darpan N. Pandya, Mikalai M. Budzevich, Ghassan El-Haddad, Nikhil I. Khushalani, Eduardo G. Moros, Mark L. McLaughlin, Thaddeus J. Wadas, and et al. 2019. "Development of Targeted Alpha Particle Therapy for Solid Tumors" Molecules 24, no. 23: 4314. https://doi.org/10.3390/molecules24234314
APA StyleTafreshi, N. K., Doligalski, M. L., Tichacek, C. J., Pandya, D. N., Budzevich, M. M., El-Haddad, G., Khushalani, N. I., Moros, E. G., McLaughlin, M. L., Wadas, T. J., & Morse, D. L. (2019). Development of Targeted Alpha Particle Therapy for Solid Tumors. Molecules, 24(23), 4314. https://doi.org/10.3390/molecules24234314