
Quantitative Analysis of Terpenic Compounds in Microsamples of Resins by Capillary Liquid Chromatography



Abstract
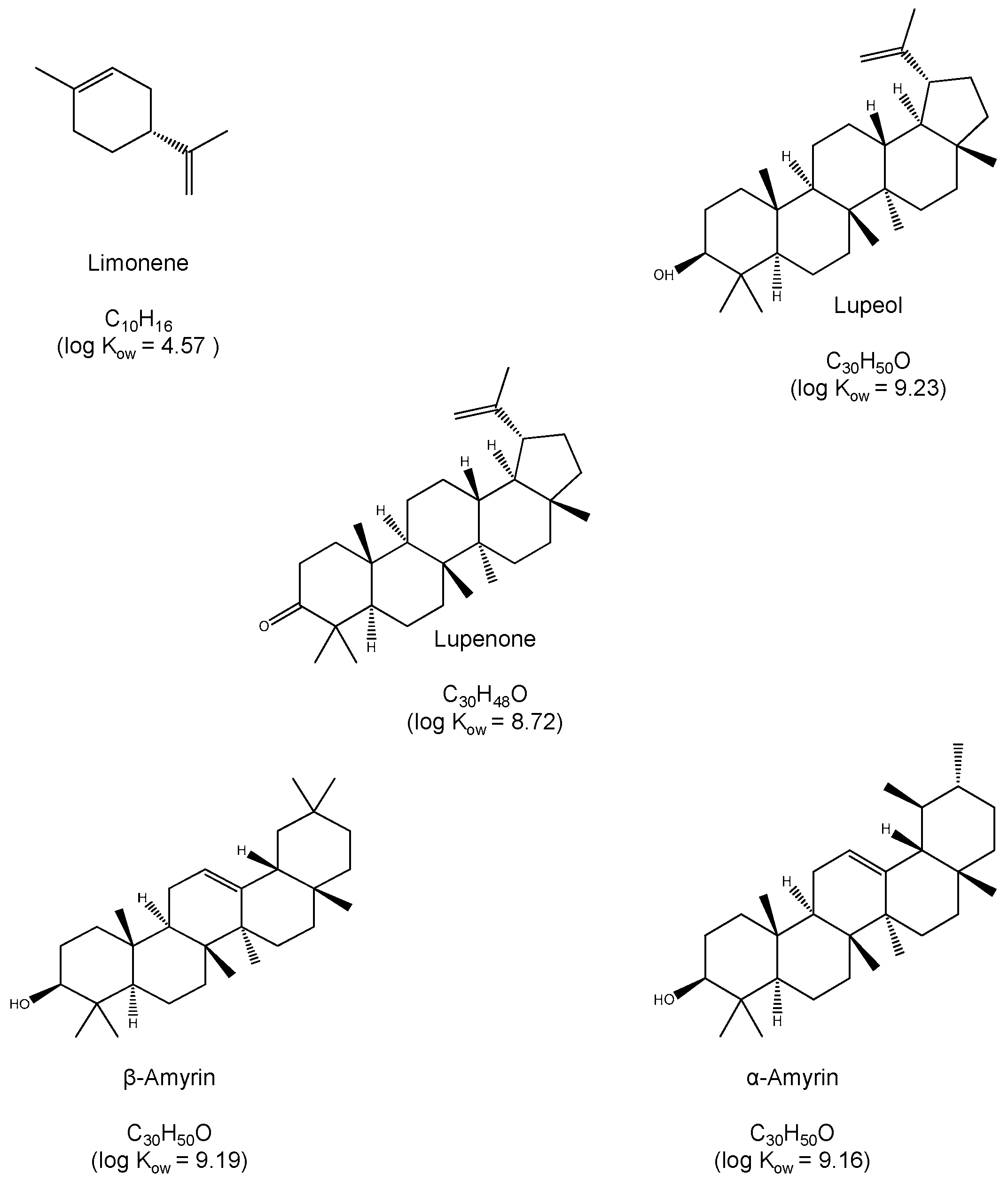
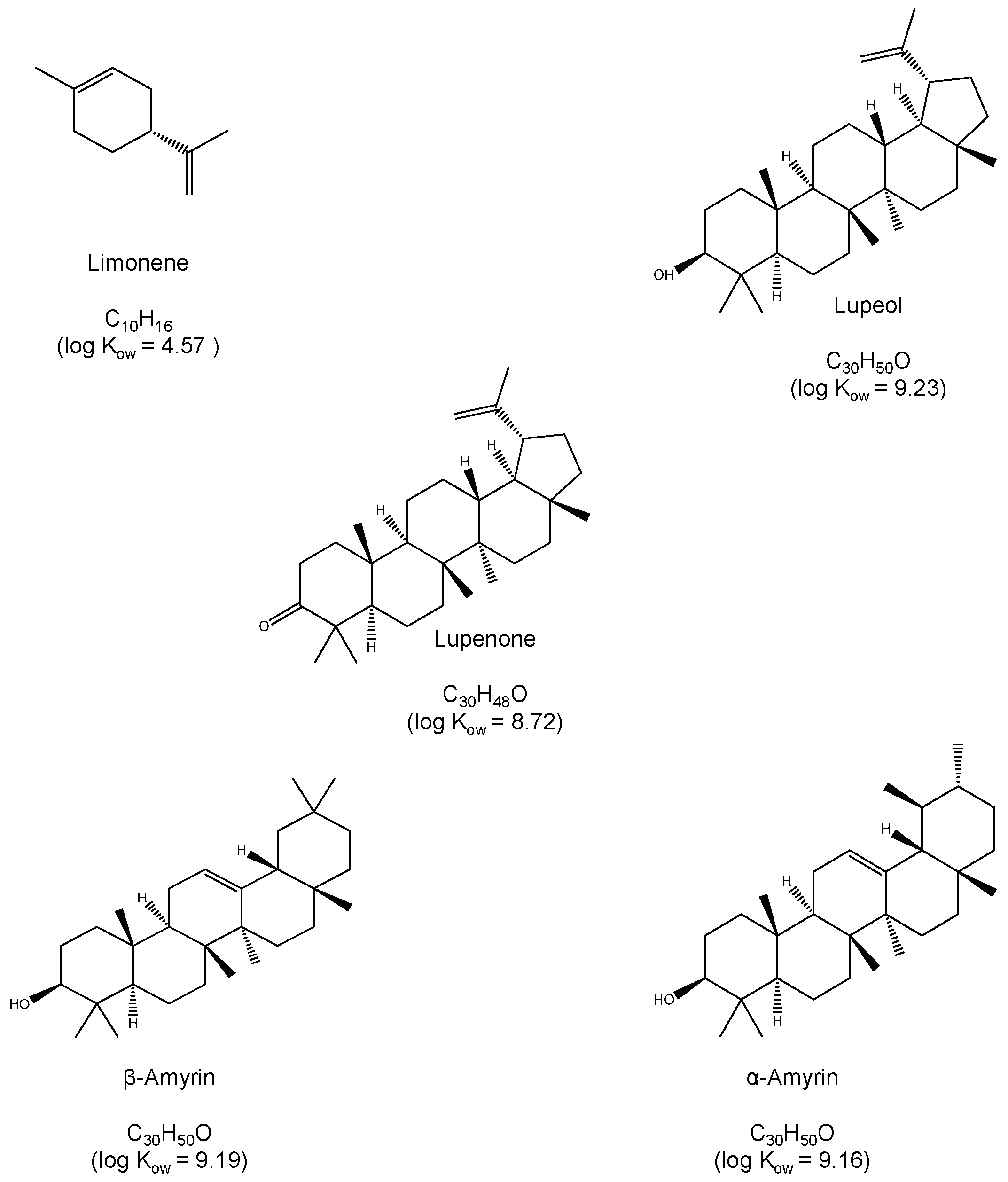
:1. Introduction
2. Results
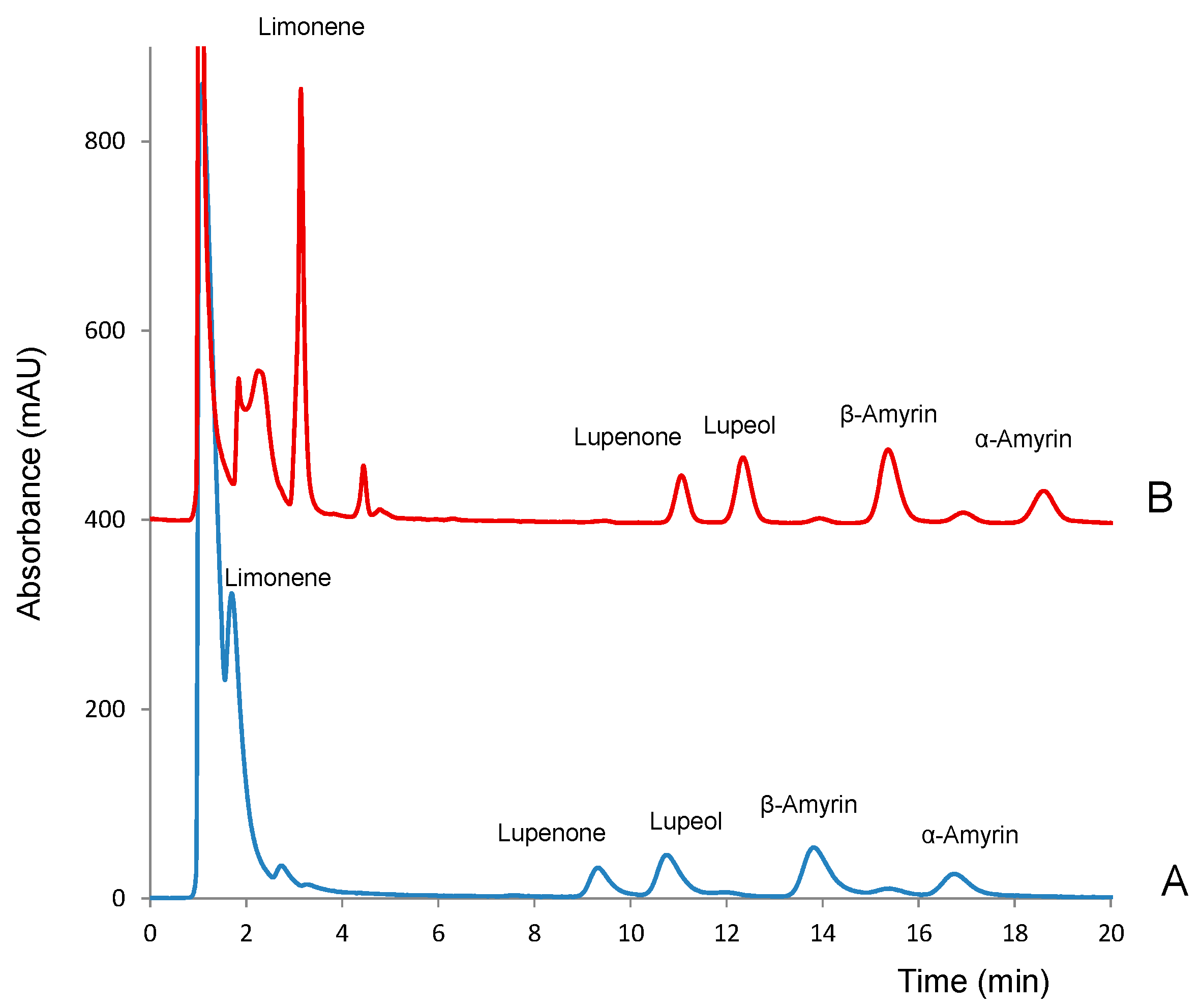
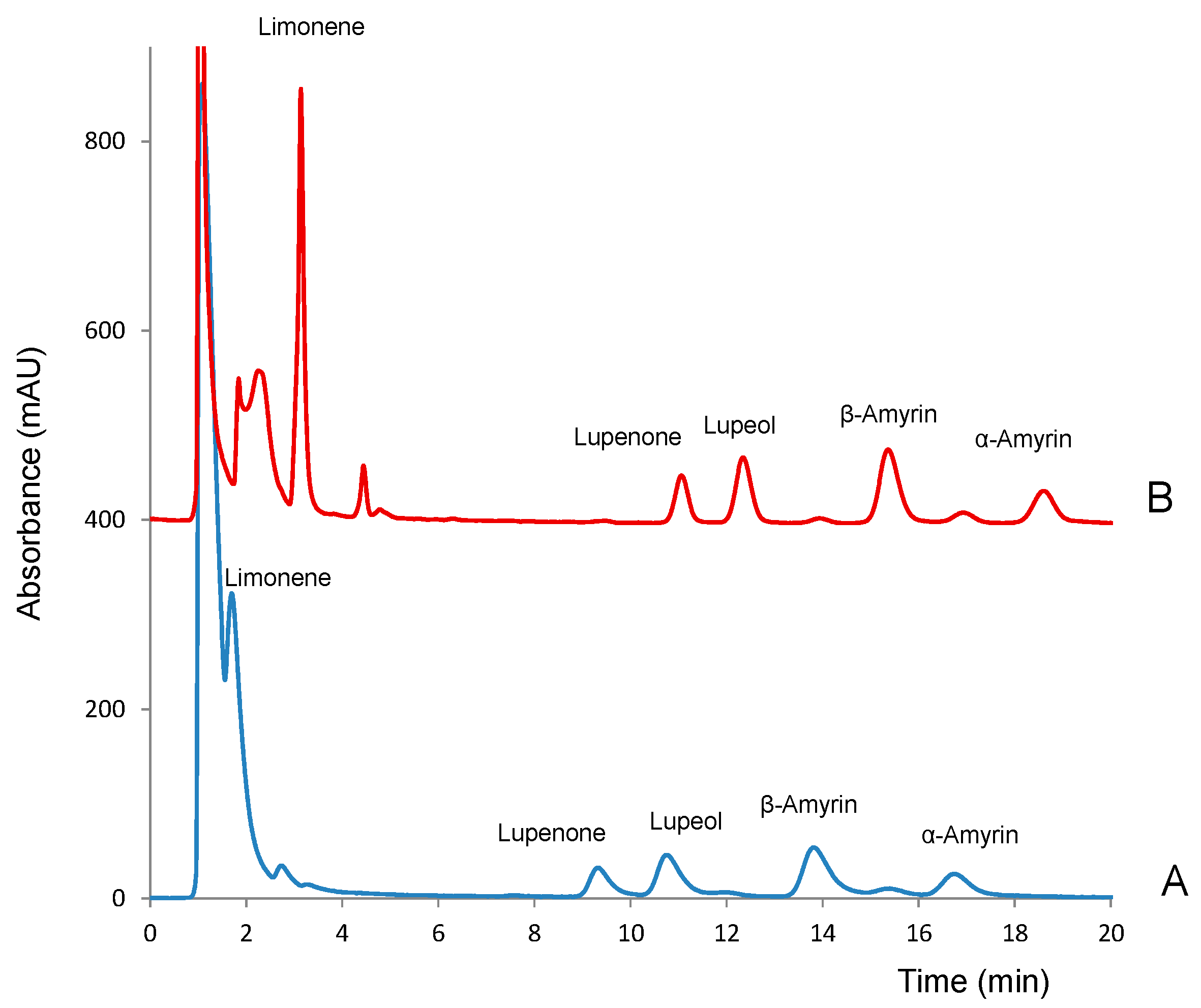
2.1. Chromatographic Conditions
2.2. Method Validation
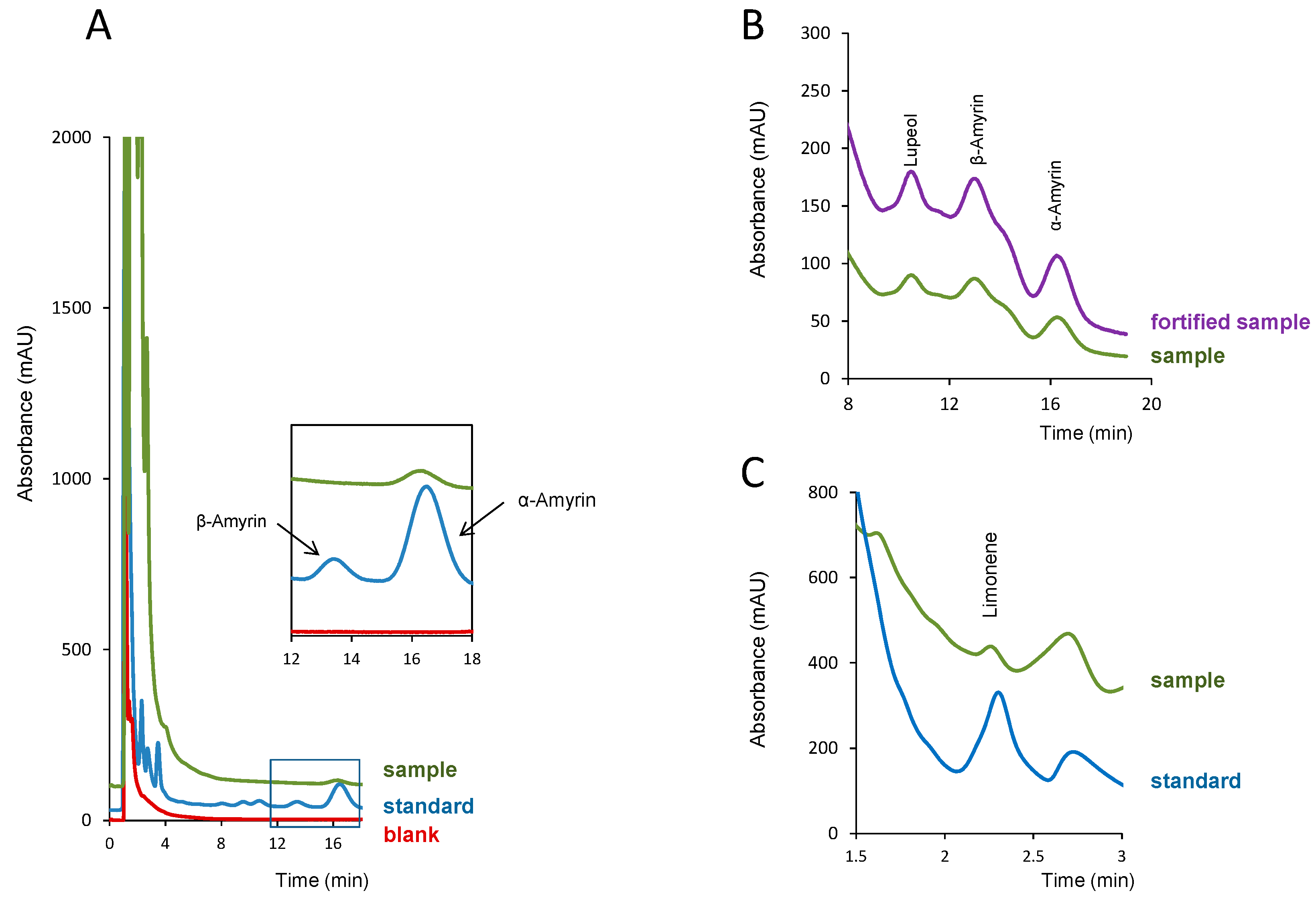
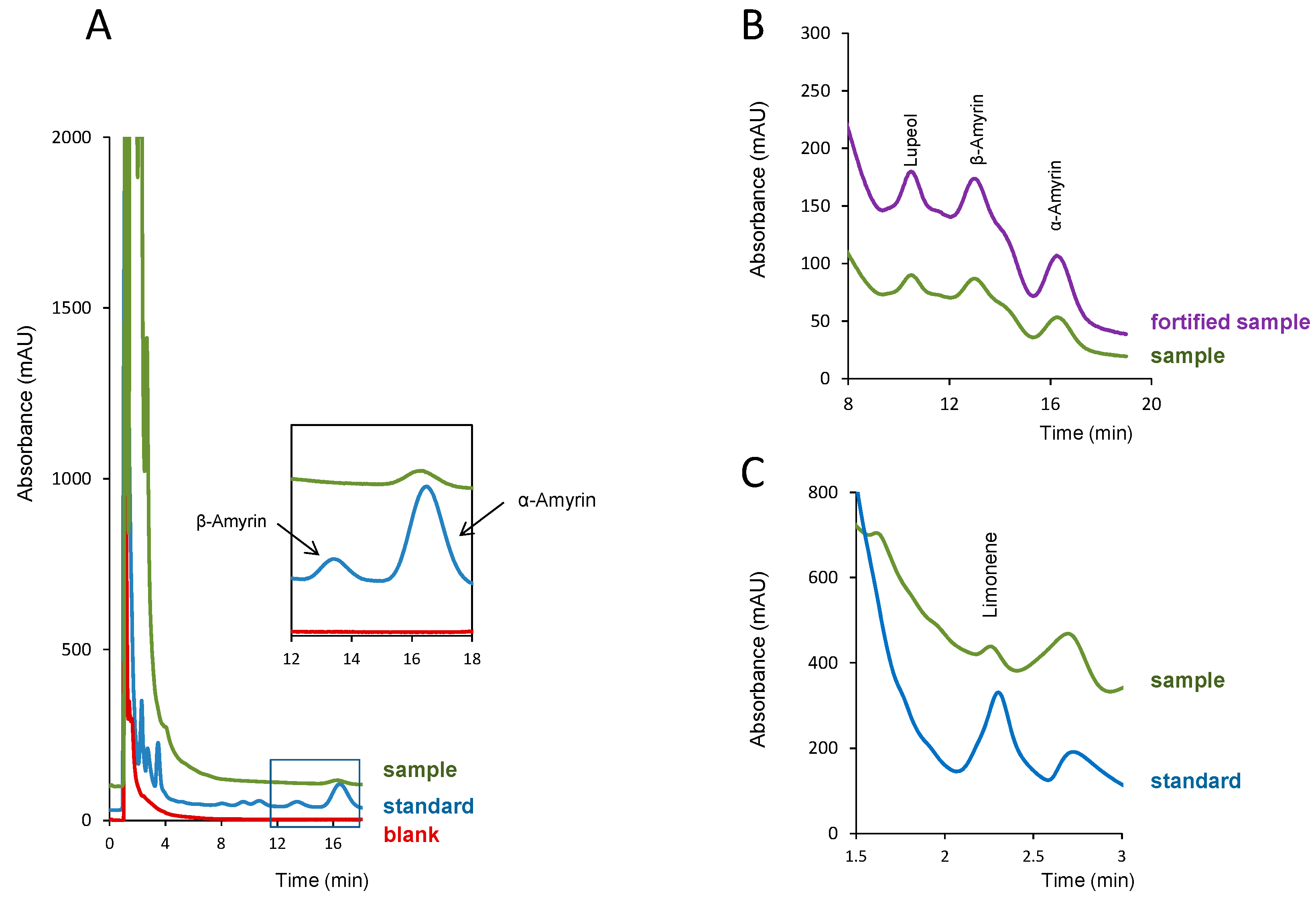
2.3. Analysis of Resins
2.3.1. Sample Preparation
2.3.2. Quantification Studies
3. Discussion
4. Materials and Methods
4.1. Chemicals and Solutions
4.2. Instrumentation and Analytical Conditions
4.3. Analysis of Resins
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gigliarelli, G.; Becerra, J.X.; Curini, M.; Marcotullio, M.C. Chemical Composition and Biological Activities of Fragrant Mexican Copal (Bursera spp.). Molecules 2015, 20, 22383–22394. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, A.L.; Siani, A.C.; Veiga Junior, V.V. The chemistry and pharmacology of the South America genus Protium Burm. f. (Burseraceae). Phcog. Rev. 2007, 1, 93–104. [Google Scholar]
- Hernández Vázquez, L.; Palazon, J.; Navarro-Ocaña, A. The Pentacyclic Triterpenes α, β-amyrins: A Review of Sources and Biological Activities. In Phytochemicals—A Global Perspective of Their Role in Nutrition and Health; Venketeshwer-Estrada, R., Ed.; IntechOpen: Rijeka, Croatia, 2012; pp. 487–502. ISBN 978-953-51-0296-0. [Google Scholar]
- Romero-Estrada, A.; Maldonado-Magaña, A.; González-Christen, J.; Bahena, S.M.; Garduño-Ramírez, M.L.; Rodríguez-López, V.; Alvarez, L. Anti-inflammatory and antioxidative effects of six pentacyclic triterpenes isolated from the mexican copal resin of bursera copallifera. BMC Complement. Altern. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.E.P.; Pires, F.B.; Bressan, L.P.; da Silva, F.B., Jr.; Lameira, O.; da Rosa, M.B. Some triterpenic compounds in extracts of Cecropia and Bauhinia species for different sampling years. Rev. Bras. Farmacogn. 2018, 28, 21–26. [Google Scholar] [CrossRef]
- Villa-Ruano, N.; Pacheco-Hernández, Y.; Becerra-Martínez, Y.; Zárate-Reyes, J.A.; Cruz-Durán, R. Chemical profile and pharmacological effects of the resin and essential oil from Bursera slechtendalii: A medicinal “copal tree” of southern Mexico. Fitoterapia 2018, 128, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Stacey, R.J.; Cartwright, C.R.; McEwan, C.R. Chemical characterization of ancient Mesoamerican “copal” resins: Preliminary results. Archaeometry 2006, 48, 323–340. [Google Scholar] [CrossRef]
- Lucero-Gómez, P.; Mathe, C.; Vieillescazes, C.; Bucio-Galindo, L.; Belio-Reyes, I.; Vega-Aviña, R. Archeobotanic: HPLC molecular profiles for the discrimination of copals in Mesoamerica Application to the study of resins materials from objects of Aztec offerings. ArcheoSciences, revue d’archéométrie 2014, 38, 119–133. [Google Scholar] [CrossRef]
- Lucero-Gómez, P.; Mathe, C.; Vieillescazes, C.; Bucio-Galindo, L.; Belio-Reyes, I.; Vega-Aviña, R. Analysis of Mexican reference standards for Bursera spp. Resins by gas-chromatography-mass spectrometry and application to archaeological objects. J. Archaeol. Sci. 2014, 41, 679–690. [Google Scholar] [CrossRef]
- Rhourrhi-Frih, B.; West, C.; Pasquier, L.; André, P.; Chaimbault, P.; Lafosse, M. Classification of natural resins by liquid chromatography-mass spectrometry and gas chromatography-mass spectrometry using chemometric analysis. J. Chromatogr. A 2012, 1256, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Vázquez, L.; Mangas, S.; Palazón, J.; Navarro-Ocaña, A. Valuable medicinal plants and resins: Commercial phytochemicals with bioactive properties. Ind. Crops. Prod. 2010, 31, 476–480. [Google Scholar] [CrossRef]
- Merali, Z.; Cayer, C.; Kent, P.; Liu, R.; Cal, V.R.; Harris, C.S.; Arnason, J.T. Sacred Maya incense, copal (Protium copal - Burseraceae), has antianxiety effects in animal models. J. Ethnopharmacol. 2018, 216, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Drzewicz, P.; Natkaniec-Nowak, L.; Czapla, D. Analytical approaches for studies of fossil resins. Trends Anal. Chem. 2016, 85C, 75–84. [Google Scholar] [CrossRef]
- Martelanc, M.; Vovk, I.; Simonovska, B. Separation and identification of some common isomeric plant triterpenoids by thin-layer chromatography and high-performance liquid chromatography. J. Chromatogr. A 2009, 1216, 6662–6670. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Montañez, G.; Ragazzo-Sánchez, J.A.; Calderón-Santoyo, M.; Velázquez-de la Cruz, G.; Ramírez de León, J.A.; Navarro-Ocaña, A. Evaluation of extraction methods for preparative scale obtention of mangiferin and lupeol from mango peels (Mangifera indica L.). Food. Chem. 2014, 159, 267–272. [Google Scholar] [CrossRef]
- Bahadir-Acıkara, Ö.; Özbilgin, S.; Saltan-Iscan, G.; Dall’Acqua, S.; Rjašková, V.; Özgökçe, F.; Suchý, V.; Šmejkal, K. Phytochemical analysis of Podospermum and Scorzonera n-hexane extracts and the HPLC quantitation of triterpenes. Molecules 2018, 23, 1813. [Google Scholar] [CrossRef] [PubMed]
- Herrera-López, M.G.; Rubio-Hernández, E.I.; Leyte-Lugo, M.A.; Schinkovitz, A.; Richomme, P.; Calvo-Irabién, L.M.; Peña-Rodríguez, L.M. Botanical Origin of Triterpenoids from Yucatecan Propolis. Phytochem. Lett. 2019, 29, 25–29. [Google Scholar] [CrossRef]
- Nazario, C.E.D.; Silva, M.R.; Franco, M.S.; Lanças, F.M. Evolution in Miniaturized Column Liquid Chromatography Instrumentation and Applications: An Overview. J. Chromatogr. A 2015, 1421, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Jornet-Martínez, N.; Ortega-Sierra, A.; Verdú-Andrés, J.; Herráez-Hernández, R.; Campíns-Falcó, P. Analysis of Contact Traces of Cannabis by In-Tube Solid-Phase Microextraction Coupled to Nanoliquid Chromatography. Molecules 2018, 23, 2359. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, B.; Örnemark, U. (Eds.) Eurachem Guide: The Fitness for Purpose of Analytical Methods: A Laboratory Guide to Method Validation and Related Topics, 2nd ed. 2014, p. 57. Available online: https://www.eurachem.org/images/stories/Guides/pdf/MV_guide_2nd_ed_EN.pdf (accessed on 10 November 2019).
- AOAC Official Methods of Analysis. Appendix K: Guidelines for Dietary Supplements and Botanicals; AOAC, International: Gaithersburg, MD, USA, 2013; p. 8. [Google Scholar]
- Sanchez, J. Estimating detection limits in chromatography from calibration data: Ordinary least squares regression vs. weighted least squares. Separations 2018, 5, 49. [Google Scholar] [CrossRef]
- Piña-Torres, C.; Lucero-Gómez, P.; Nieto, S.; Vázquez, A.; Bucio, L.; Belio, I.; Vega, R.; Mathe, C.; Vieillescazes, C. An analytical strategy based on Fourier transform infrared spectroscopy, principal component analysis and linear discriminant analysis to suggest the botanical origin of resins from Bursera. Application to archaeological Aztec Samples. J. Cult. Herit. 2018, 33, 48–59. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Linearity *, ** (n = 15) | Mean Found Concentration ** (n = 3) | Precision, rsd (%) (n = 3) | LOD (µg mL−1) | LOQ (µg mL−1) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| y = (a ± sa) + (b ± sb) x | R2 | 2.5 µg mL−1 | 7.5 µg mL−1 | Intraday | Interday | |||||
| 2.5 µg mL−1 | 7.5 µg mL−1 | 2.5 µg mL−1 | 7.5 µg mL−1 | |||||||
| Limonene | y = (−77 ± 2) + (433 ± 6)x | 0.997 | 2.3 ± 0.1 | 6.5 ± 0.4 | 2 | 0.6 | 3 | 4 | 0.1 | 0.4 |
| Lupenone | y = (−22 ± 9) + (63.9 ± 1.6)x | 0.994 | 2.4 ± 0.1 | 7.0 ± 0.1 | 4 | 0.8 | 7 | 7 | 0.25 | 0.8 |
| Lupeol | y = (−42 ± 12) + (111 ± 2)x | 0.996 | 2.6 ± 0.1 | 7.4 ± 0.2 | 1.4 | 2 | 7 | 8 | 0.25 | 0.8 |
| β-Amyrin | y = (−20 ± 17) + (135 ± 3)x | 0.995 | 2.3 ± 0.1 | 7.5 ± 0.6 | 3 | 8 | 8 | 8 | 0.25 | 0.8 |
| α-Amyrin | y = (72 ± 42) + (313 ± 8)x | 0.994 | 2.9 ± 0.2 | 8.4 ± 0.1 | 9 | 17 | 16 | 17 | 0.25 | 0.8 |
| Compound | Recovery (%) |
|---|---|
| Limonene | 103 ± 4 |
| Lupenone | 101 ± 1 |
| Lupeol | 79 ± 9 |
| β-Amyrin | 52 ± 5 |
| α-Amyrin | 75 ± 3 |
| Sample | Percentage a (%), (n = 3) | |||||
|---|---|---|---|---|---|---|
| Limonene | Lupenone | Lupeol | β-Amyrin | α-Amyrin | ||
| White copal | 1 mg | 0.9 ± 0.2 | <LOD | <LOD | <LOD | <LOD |
| 15 mg | 1.2 ± 0.2 | <LOD | <LOD | <LOD | 0.020 ± 0.002 | |
| Copal in tears | 10 mg | <LOD | <LOD | 0.034 ± 0.001 | 0.069 ± 0.002 | 0.011 ± 0.001 |
| 10 mg b | <LOD | <LOD | 0.033 ± 0.001 | 0.074 ± 0.001 | 0.010 ± 0.003 | |
| 10 mg c | <LOD | <LOD | 0.035 ± 0.002 | 0.082 ± 0.005 | 0.010 ± 0.004 | |
| Ocote | 1 mg | 9.3 ± 0.2 | <LOD | <LOD | <LOD | <LOQ |
| 10 mg | 9.3 ± 0.1 | <LOD | <LOD | <LOD | 0.093 ± 0.003 | |
| 10 mg b | 7.2 ± 0.1 | <LOD | <LOD | <LOD | 0.16 ± 0.01 | |
| 10 mg c | 7.3 ± 0.3 | <LOD | <LOD | <LOD | 0.16 ± 0.02 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponce-Rodríguez, H.D.; Herráez-Hernández, R.; Verdú-Andrés, J.; Campíns-Falcó, P. Quantitative Analysis of Terpenic Compounds in Microsamples of Resins by Capillary Liquid Chromatography. Molecules 2019, 24, 4068. https://doi.org/10.3390/molecules24224068
Ponce-Rodríguez HD, Herráez-Hernández R, Verdú-Andrés J, Campíns-Falcó P. Quantitative Analysis of Terpenic Compounds in Microsamples of Resins by Capillary Liquid Chromatography. Molecules. 2019; 24(22):4068. https://doi.org/10.3390/molecules24224068
Chicago/Turabian StylePonce-Rodríguez, H. D., R. Herráez-Hernández, J. Verdú-Andrés, and P. Campíns-Falcó. 2019. "Quantitative Analysis of Terpenic Compounds in Microsamples of Resins by Capillary Liquid Chromatography" Molecules 24, no. 22: 4068. https://doi.org/10.3390/molecules24224068
APA StylePonce-Rodríguez, H. D., Herráez-Hernández, R., Verdú-Andrés, J., & Campíns-Falcó, P. (2019). Quantitative Analysis of Terpenic Compounds in Microsamples of Resins by Capillary Liquid Chromatography. Molecules, 24(22), 4068. https://doi.org/10.3390/molecules24224068