
Penfluridol as a Candidate of Drug Repurposing for Anticancer Agent



Abstract
1. Introduction
2. What is the Penfluridol?
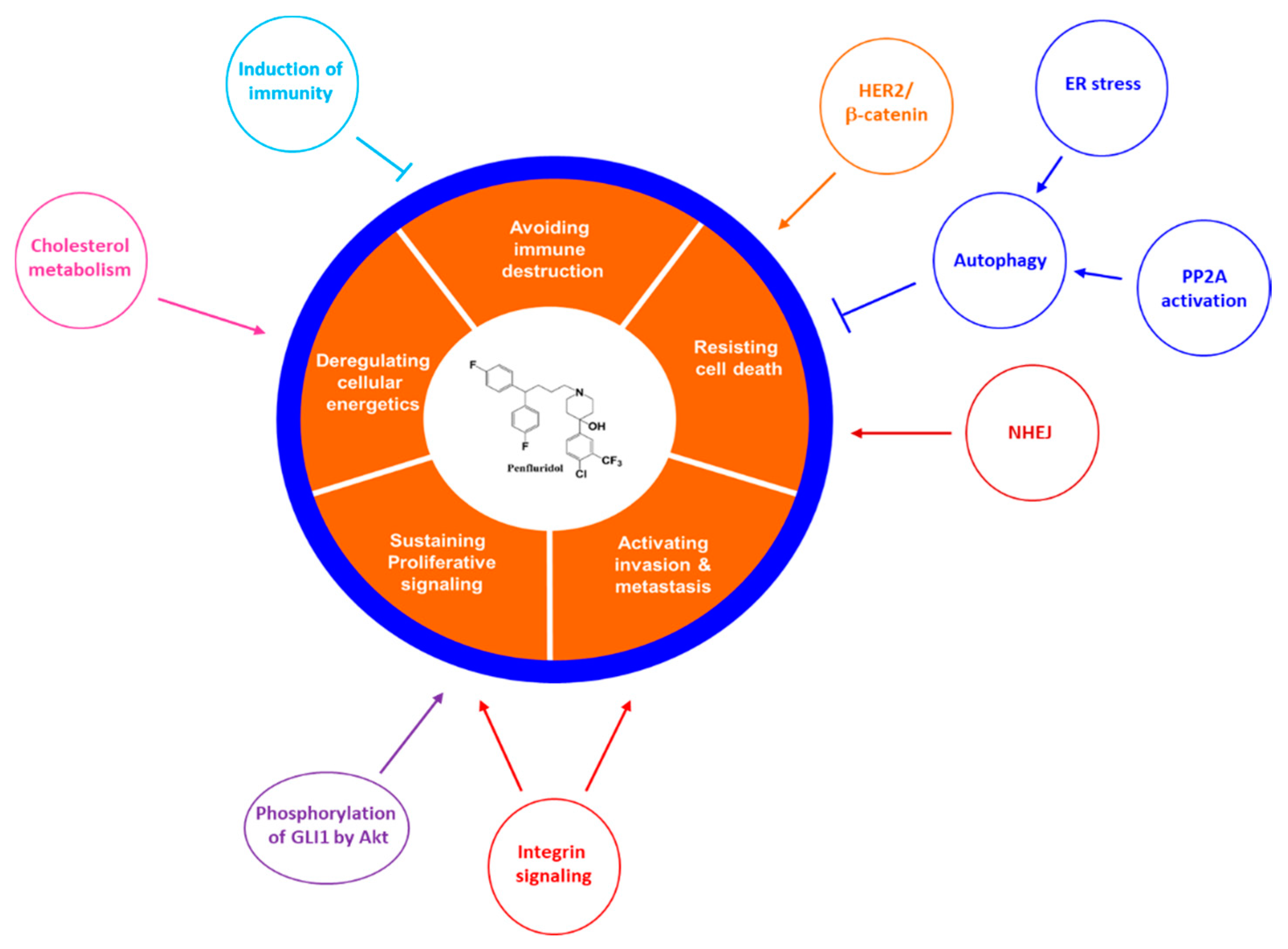
3. Anti-Cancer Effects of Penfluridol
3.1. Penfluridol Suppresses Cell Proliferation
3.2. Penfluridol Induces Cell Death
3.3. Penfluridol Impedes Metastasis and Invasion
3.4. Penfluridol Hinders Angiogenesis
3.5. Penfluridol and Evading Immune Destruction
3.6. Penfluridol and Inflammation
3.7. Penfluridol and Replicative Immortality
3.8. Penfluridol Increases the Efficiency of Growth Suppressors
3.9. Penfluridol and Genome Instability and Mutation
3.10. Penfluridol and Deregulating Cellular Energetics
4. Mechanism of Action of Penfluridol on Cancer
4.1. The Antipsychotic-Related Mechanism of Action of Penfluridol on Cancer
4.1.1. The Relationship between Dopamine Receptor D2 and Cancer
4.1.2. The Relationship between T-type Calcium Channels and Cancer
4.2. The Novel Molecular Mechanism of Action of Penfluridol on Cancer
4.2.1. Inhibition of Integrin Signalling Pathway
4.2.2. Inhibition of Akt-Mediated Phosphorylation of Glioma-Associated Oncogene 1 (GLI1)
4.2.3. Induction of Autophagy
4.2.4. Inhibition of Cholesterol Metabolism
4.2.5. Enhancement of Protein Phosphatase 2A (PP2A) Activity
4.2.6. Induction of Immunity
4.2.7. Miscelleneous Mechanisms Involved in Overcoming Resistance
5. Needs for Penfluridol Derivatives
6. Perspectives
Author Contributions
Funding
Conflicts of Interest
Ethical Approval
Informed Consent
Open Access
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N.; Stemmer, S.M.; Greenberg, D.; Goldstein, D.A. Trajectories of injectable cancer drug costs after launch in the United States. J. Clin. Oncol. 2018, 36, 319–325. [Google Scholar] [CrossRef]
- Howard, D.H.; Bach, P.B.; Berndt, E.R.; Conti, R.M. Pricing in the market for anticancer drugs. J. Econ. Perspect. 2015, 29, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.A.; Niemegeers, C.J.; Schellekens, K.H.; Lenaerts, F.M.; Verbruggen, F.J.; Van Nueten, J.M.; Schaper, W.K. The pharmacology of penfluridol (R 16341) a new potent and orally long-acting neuroleptic drug. Eur. J. Pharmacol. 1970, 11, 139–154. [Google Scholar] [CrossRef]
- Soares, B.G.; Lima, M.S. Penfluridol for schizophrenia. Cochrane Database Syst. Rev. 2006, 2, CD002923. [Google Scholar] [CrossRef] [PubMed]
- Shintomi, K.; Yamamura, M. Effects of penfluridol and other drugs on apomorphine-induced stereotyped behavior in monkeys. Eur. J. Pharmacol. 1975, 31, 273–280. [Google Scholar] [CrossRef]
- Kline, C.L.B.; Ralff, M.D.; Lulla, A.R.; Wagner, J.M.; Abbosh, P.H.; Dicker, D.T.; Allen, J.E.; El-Deiry, W.S. Role of Dopamine receptors in the anticancer activity of ONC201. Neoplasia 2018, 20, 80–91. [Google Scholar] [CrossRef]
- Santi, C.M.; Cayabyab, F.S.; Sutton, K.G.; McRory, J.E.; Mezeyova, J.; Hamming, K.S.; Parker, D.; Stea, A.; Snutch, T.P. Differential inhibition of T-type calcium channels by neuroleptics. J. Neurosci. 2002, 22, 396–403. [Google Scholar] [CrossRef]
- Ashraf-Uz-Zaman, M.; Sajib, M.S.; Cucullo, L.; Mikelis, C.M.; German, N.A. Analogs of penfluridol as chemotherapeutic agents with reduced central nervous system activity. Bioorg. Med. Chem. Lett. 2018, 28, 3652–3657. [Google Scholar] [CrossRef]
- Ranjan, A.; Gupta, P.; Srivastava, S.K. Penfluridol: An antipsychotic agent suppresses metastatic tumor growth in triple-negative breast cancer by inhibiting Integrin signaling axis. Cancer Res. 2016, 76, 877–890. [Google Scholar] [CrossRef]
- Ranjan, A.; Srivastava, S.K. Penfluridol suppresses glioblastoma tumor growth by Akt-mediated inhibition of GLI1. Oncotarget 2017, 8, 32960. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Srivastava, S.K. Penfluridol suppresses pancreatic tumor growth by autophagy-mediated apoptosis. Sci. Rep. 2016, 6, 26165. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, Y.Y.; Li, Z.X.; Zhao, Q.; Wang, X.; Yu, Y.; Wang, Y.Y.; Wang, Y.Q.; Luo, F. Anti-tumor effects of Penfluridol through dysregulation of Cholesterol homeostasis. Asian Pac. J. Cancer Prev. 2014, 15, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.; Sun, Q.-Y.; Lee, K.L.; Ding, L.-W.; Wuensche, P.; Torres-Fernandez, L.A.; Tan, S.Z.; Tokatly, I.; Zaiden, N.; Poellinger, L. Activation of protein phosphatase 2A tumor suppressor as potential treatment of pancreatic cancer. Mol. Oncol. 2015, 9, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Schonthal, A.H. Role of serine/threonine protein phosphatase 2A in cancer. Cancer Lett. 2001, 170, 1–13. [Google Scholar] [CrossRef]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef]
- Gupta, N.; Gupta, P.; Srivastava, S.K. Penfluridol overcomes paclitaxel resistance in metastatic breast cancer. Sci. Rep. 2019, 9, 5066. [Google Scholar] [CrossRef] [PubMed]
- Wittekind, C.; Neid, M. Cancer invasion and metastasis. Oncology 2005, 69, 14–16. [Google Scholar] [CrossRef]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Dakir, E.H.; Pickard, A.; Srivastava, K.; McCrudden, C.M.; Gross, S.R.; Lloyd, S.; Zhang, S.D.; Margariti, A.; Morgan, R.; Rudland, P.S.; et al. The anti-psychotic drug pimozide is a novel chemotherapeutic for breast cancer. Oncotarget 2018, 9, 34889–34910. [Google Scholar] [CrossRef] [PubMed]
- Schluter, A.; Weller, P.; Kanaan, O.; Nel, I.; Heusgen, L.; Hoing, B.; Hasskamp, P.; Zander, S.; Mandapathil, M.; Dominas, N.; et al. CD31 and VEGF are prognostic biomarkers in early-stage, but not in late-stage, laryngeal squamous cell carcinoma. BMC Cancer 2018, 18, 272. [Google Scholar] [CrossRef]
- DeLisser, H.M.; Christofidou-Solomidou, M.; Strieter, R.M.; Burdick, M.D.; Robinson, C.S.; Wexler, R.S.; Kerr, J.S.; Garlanda, C.; Merwin, J.R.; Madri, J.A.; et al. Involvement of endothelial PECAM-1/CD31 in angiogenesis. Am. J. Pathol. 1997, 151, 671–677. [Google Scholar] [PubMed]
- Gorgun, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Cai, Z.; Zhang, Y.; Yutzy, W.H.; Roby, K.F.; Roden, R.B. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res. 2006, 66, 6807–6815. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Wright, S.; Srivastava, S.K. Immune consequences of penfluridol treatment associated with inhibition of glioblastoma tumor growth. Oncotarget 2017, 8, 47632–47641. [Google Scholar] [CrossRef]
- Lee, C.H. Epithelial-mesenchymal transition: Initiation by cues from chronic inflammatory tumor microenvironment and termination by anti-inflammatory compounds and specialized pro-resolving lipids. Biochem. Pharmacol. 2018, 158, 261–273. [Google Scholar] [CrossRef]
- Alfonso-De Matte, M.Y.; Moses-Soto, H.; Kruk, P.A. Calcium-mediated telomerase activity in ovarian epithelial cells. Arch. Biochem. Biophys. 2002, 399, 239–244. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Yao, Y.; Dai, W. Genomic instability and cancer. J. Carcinog. Mutagenes. 2014, 5, 1000165. [Google Scholar] [PubMed]
- Hung, W.Y.; Chang, J.H.; Cheng, Y.; Cheng, G.Z.; Huang, H.C.; Hsiao, M.; Chung, C.L.; Lee, W.J.; Chien, M.H. Autophagosome accumulation-mediated ATP energy deprivation induced by penfluridol triggers nonapoptotic cell death of lung cancer via activating unfolded protein response. Cell Death Dis. 2019, 10, 538. [Google Scholar] [CrossRef] [PubMed]
- Weissenrieder, J.S.; Neighbors, J.D.; Mailman, R.B.; Hohl, R.J. Cancer and the Dopamine D2 receptor: A pharmacological perspective. J. Pharmacol. Exp. Ther. 2019, 370, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Brami-Cherrier, K.; Valjent, E.; Garcia, M.; Pages, C.; Hipskind, R.A.; Caboche, J. Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: A new route to cAMP response element-binding protein phosphorylation. J. Neurosci. 2002, 22, 8911–8921. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Dong, S.M.; Kim, B.R.; Park, M.S.; Trink, B.; Byun, H.J.; Rho, S.B. Thioridazine induces apoptosis by targeting the PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells. Apoptosis 2012, 17, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Yu, T.; Hu, J.; Hu, L. Dopamine D2 receptor blocker thioridazine induces cell death in human uterine cervical carcinoma cell line SiHa. J. Obstet. Gynaecol. Res. 2015, 41, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Chung, Y.M.; Ma, J.; Yang, Q.; Berek, J.S.; Hu, M.C. Pharmacological activation of FOXO3 suppresses triple-negative breast cancer in vitro and in vivo. Oncotarget 2016, 7, 42110–42125. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, M.K.; Yu, H.T.; Zhong, Z.H.; Cai, N.; Chen, G.Z.; Zhang, P.; Chen, J.J. The antipsychotic drug pimozide inhibits cell growth in prostate cancer through suppression of STAT3 activation. Int. J. Oncol. 2016, 48, 322–328. [Google Scholar] [CrossRef]
- Das, A.; Pushparaj, C.; Bahi, N.; Sorolla, A.; Herreros, J.; Pamplona, R.; Vilella, R.; Matias-Guiu, X.; Marti, R.M.; Canti, C. Functional expression of voltage-gated calcium channels in human melanoma. Pigment Cell Melanoma Res. 2012, 25, 200–212. [Google Scholar] [CrossRef]
- Antal, L.; Martin-Caraballo, M. T-type Calcium channels in cancer. Cancers (Basel) 2019, 11, 134. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Gray, L.S.; Dziegielewski, J. T-type calcium channels blockers as new tools in cancer therapies. Pflugers Arch. 2014, 466, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Seagar, M.J.; Jones, J.F.; Reber, B.F.; Catterall, W.A. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 5478–5482. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Ertel, S.I.; Ertel, E.A.; Clozel, J.P. T-type Ca2+ channels and pharmacological blockade: Potential pathophysiological relevance. Cardiovasc. Drugs Ther. 1997, 11, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Enyeart, J.J.; Biagi, B.A.; Day, R.N.; Sheu, S.S.; Maurer, R.A. Blockade of low and high threshold Ca2+ channels by diphenylbutylpiperidine antipsychotics linked to inhibition of prolactin gene expression. J. Biol. Chem. 1990, 265, 16373–16379. [Google Scholar] [PubMed]
- Costello, L.C. The suppression of Prolactin is required for the treatment of advanced Prostate cancer. Oncogen (Westerville) 2019, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Valerie, N.C.; Dziegielewska, B.; Hosing, A.S.; Augustin, E.; Gray, L.S.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. Inhibition of T-type calcium channels disrupts Akt signaling and promotes apoptosis in glioblastoma cells. Biochem. Pharmacol. 2013, 85, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chong, K.; Ryu, B.-K.; Park, K.-J.; Yu, M.O.; Lee, J.; Chung, S.; Choi, S.; Park, M.-J.; Chung, Y.-G. Repurposing Penfluridol in combination with Temozolomide for the treatment of Glioblastoma. Cancers 2019, 11, 1310. [Google Scholar] [CrossRef]
- Levite, M.; Chowers, Y.; Ganor, Y.; Besser, M.; Hershkovits, R.; Cahalon, L. Dopamine interacts directly with its D3 and D2 receptors on normal human T cells, and activates β1 integrin function. Eur. J. Immunol. 2001, 31, 3504–3512. [Google Scholar] [CrossRef]
- Lambert, A.W.; Ozturk, S.; Thiagalingam, S. Integrin signaling in mammary epithelial cells and breast cancer. ISRN Oncol. 2012, 2012, 493283. [Google Scholar] [CrossRef]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Gobira, P.H.; Ropke, J.; Aguiar, D.C.; Crippa, J.A.; Moreira, F.A. Animal models for predicting the efficacy and side effects of antipsychotic drugs. Braz. J. Psychiatry 2013, 35, S132–S139. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, E.; Li, X.; Safe, S. Penfluridol represses integrin expression in breast cancer through induction of reactive oxygen species and downregulation of Sp transcription factors. Mol. Cancer Ther. 2017, 16, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Abdelrahim, M. Sp transcription factor family and its role in cancer. Eur. J. Cancer 2005, 41, 2438–2448. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; De Tribolet, N.; Radovanovic, I.; Ruiz, I.; Altaba, A. HEDGEHOG-GLI1 signaling regulates human Glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Beaulieu, J.-M.; Tirotta, E.; Sotnikova, T.D.; Masri, B.; Salahpour, A.; Gainetdinov, R.R.; Borrelli, E.; Caron, M.G. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J. Neurosci. 2007, 27, 881–885. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Ktistakis, N.T.; Tooze, S.A. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016, 26, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ji, X.; Liu, J.; Li, Z.; Zhang, X. Dopamine receptor subtypes differentially regulate autophagy. Int. J. Mol. Sci. 2018, 19, 1540. [Google Scholar] [CrossRef] [PubMed]
- Visa, A.; Sallán, M.C.; Maiques, O.; Alza, L.; Talavera, E.; López-Ortega, R.; Santacana, M.; Herreros, J.; Cantí, C. T-type Cav3. 1 channels mediate progression and chemotherapeutic resistance in glioblastoma. Cancer Res. 2019, 79, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Pushparaj, C.; Herreros, J.; Nager, M.; Vilella, R.; Portero, M.; Pamplona, R.; Matias-Guiu, X.; Martí, R.M.; Cantí, C. T-type calcium channel blockers inhibit autophagy and promote apoptosis of malignant melanoma cells. Pigment Cell Melanoma Res. 2013, 26, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Ranjan, A.; German, N.; Mikelis, C.; Srivenugopal, K.; Srivastava, S.K. Penfluridol induces endoplasmic reticulum stress leading to autophagy in pancreatic cancer. Tumour Biol. 2017, 39, 1010428317705517. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Wen, Y.-C.; Ku, C.-C.; Yang, Y.-C.; Chow, J.-M.; Yang, S.-F.; Lee, W.-J.; Chien, M.-H. Penfluridol triggers cytoprotective autophagy and cellular apoptosis through ROS induction and activation of the PP2A-modulated MAPK pathway in acute myeloid leukemia with different FLT3 statuses. J. Biomed. Sci. 2019, 26, 1–13. [Google Scholar] [CrossRef]
- Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. J. Cell. Biochem. 2004, 91, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol. 2011, 178, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, E.D.; Catts, V.S.; Catts, S.V.; Ng, T.F.; Whitaker, N.J.; Brown, A.J.; Lutze-Mann, L.H. Cytotoxic effects of antipsychotic drugs implicate cholesterol homeostasis as a novel chemotherapeutic target. Int. J. Cancer 2010, 126, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Bánréti, Á.; Lukácsovich, T.; Csikós, G.; Erdélyi, M.; Sass, M. PP2A regulates autophagy in two alternative ways in Drosophila. Autophagy 2012, 8, 623–636. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, X.; Qin, C.; Cuevas, S.; Jose, P.A.; Armando, I. Dopamine D2 receptors’ effects on renal inflammation are mediated by regulation of PP2A function. Am. J. Physiol.Renal Physiol. 2015, 310, F128–F134. [Google Scholar] [CrossRef]
- Clerkin, J.; Naughton, R.; Quiney, C.; Cotter, T. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett 2008, 266, 30–36. [Google Scholar] [CrossRef]
- Wainszelbaum, M.J.; Liu, J.; Kong, C.; Srikanth, P.; Samovski, D.; Su, X.; Stahl, P.D. TBC1D3, a hominoid-specific gene, delays IRS-1 degradation and promotes insulin signaling by modulating p70 S6 kinase activity. PLoS ONE 2012, 7, e31225. [Google Scholar] [CrossRef]
- Figueroa, C.; Gálvez-Cancino, F.; Oyarce, C.; Contreras, F.; Prado, C.; Valeria, C.; Cruz, S.; Lladser, A.; Pacheco, R. Inhibition of dopamine receptor D3 signaling in dendritic cells increases antigen cross-presentation to CD8+ T-cells favoring anti-tumor immunity. J. Neuroimmunol. 2017, 303, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.-I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Okada, H. Myeloid-derived suppressor cells (MDSCs) in gliomas and glioma-development. Immunol. Investig. 2012, 41, 658–679. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Edwards, C.M.; Mundy, G.R. Gr-1+ CD11b+ myeloid-derived suppressor cells: Formidable partners in tumor metastasis. J. Bone Miner. Res. 2010, 25, 1701–1706. [Google Scholar] [CrossRef]
- Du, J.; Shang, J.; Chen, F.; Zhang, Y.; Yin, N.; Xie, T.; Zhang, H.; Yu, J.; Liu, F. A CRISPR/Cas9–based screening for non-homologous end joining inhibitors reveals Ouabain and Penfluridol as Radiosensitizers. Mol. Cancer Ther. 2018, 17, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009, 417, 639–650. [Google Scholar] [CrossRef]
- Li, Y.-H.; Wang, X.; Pan, Y.; Lee, D.-H.; Chowdhury, D.; Kimmelman, A.C. Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PloS ONE 2012, 7, e39588. [Google Scholar] [CrossRef] [PubMed]
- Hait, W.; Gesmonde, J.; Lazo, J. Effect of anti-calmodulin drugs on the growth and sensitivity of C6 rat glioma cells to bleomycin. Anticancer Res. 1994, 14, 1711–1721. [Google Scholar]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: an unmet medical need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef]
- O’Toole, S.A.; Beith, J.M.; Millar, E.K.; West, R.; McLean, A.; Cazet, A.; Swarbrick, A.; Oakes, S.R. Therapeutic targets in triple negative breast cancer. J. Clin. Pathol. 2013, 66, 530–542. [Google Scholar] [CrossRef]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.; Morizono, K.; Karanikolas, B.D.; Wu, L. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef] [PubMed]
- Reiche, E.M.V.; Nunes, S.O.V.; Morimoto, H.K. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004, 5, 617–625. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.-B.; Luo, C.; Mao, X.-Y.; Li, X.; Yin, J.-Y.; Zhang, W.; Zhou, H.-H.; Liu, Z.-Q. The prospective value of dopamine receptors on Bio-behavior of tumor. J. Cancer 2019, 10, 1622. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Yu, C.Y.; Li, X.X.; Zhang, P.; Tang, J.; Yang, Q.; Fu, T.; Zhang, X.; Cui, X.; Tu, G. Therapeutic target database update 2018: Enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2017, 46, D1121–D1127. [Google Scholar]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood brain barrier: A challenge for effectual therapy of brain tumors. Biomed Res. Int. 2015, 2015, 320941. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of Cancer | Tested Cell Lines | IC50 (µmol/L) | References |
|---|---|---|---|
| Breast cancer | MDA-MB-231, HCC 1806, 4 T1 | (5.75–7.5)/24h | [10] |
| Glioblastoma | GBM 43, GBM 10, GBM 44, GBM 28, GBM 14, T98G, U251 MG, U87MG, SJ-GBM2, CHLA-200 | (4.5–10)/24h | [11] |
| Pancreatic cancer | Panc-1, AsPC-1, BxPC-3 | (6.0–6.5)/24h | [12] |
| Lung Cancer | LCC | 4.3/24h | [9] |
| LL/2 | 2.45/48h | [13] | |
| Colon Cancer | CT26 | 2.74/48h | [13] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuan, N.M.; Lee, C.H. Penfluridol as a Candidate of Drug Repurposing for Anticancer Agent. Molecules 2019, 24, 3659. https://doi.org/10.3390/molecules24203659
Tuan NM, Lee CH. Penfluridol as a Candidate of Drug Repurposing for Anticancer Agent. Molecules. 2019; 24(20):3659. https://doi.org/10.3390/molecules24203659
Chicago/Turabian StyleTuan, Nguyen Minh, and Chang Hoon Lee. 2019. "Penfluridol as a Candidate of Drug Repurposing for Anticancer Agent" Molecules 24, no. 20: 3659. https://doi.org/10.3390/molecules24203659
APA StyleTuan, N. M., & Lee, C. H. (2019). Penfluridol as a Candidate of Drug Repurposing for Anticancer Agent. Molecules, 24(20), 3659. https://doi.org/10.3390/molecules24203659