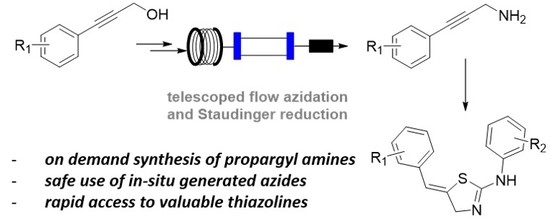
Development of a Telescoped Flow Process for the Safe and Effective Generation of Propargylic Amines


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
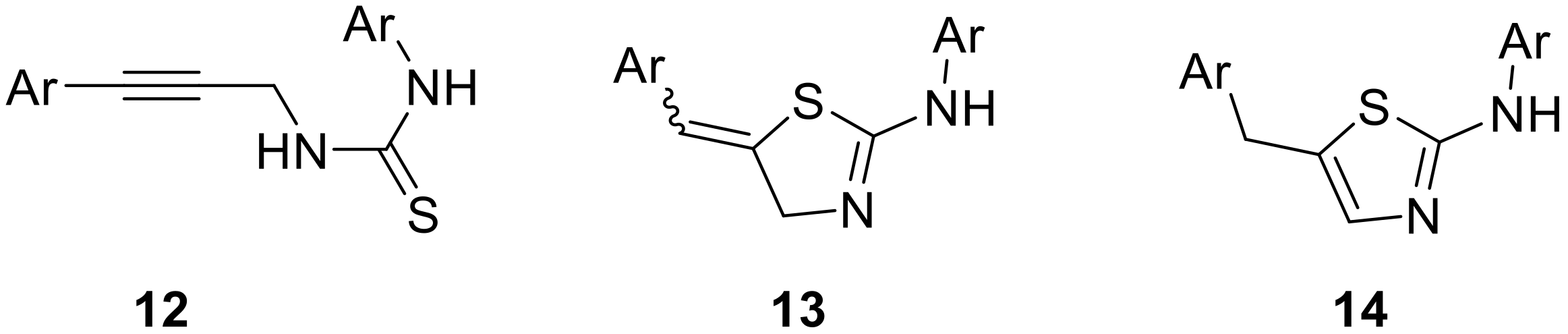
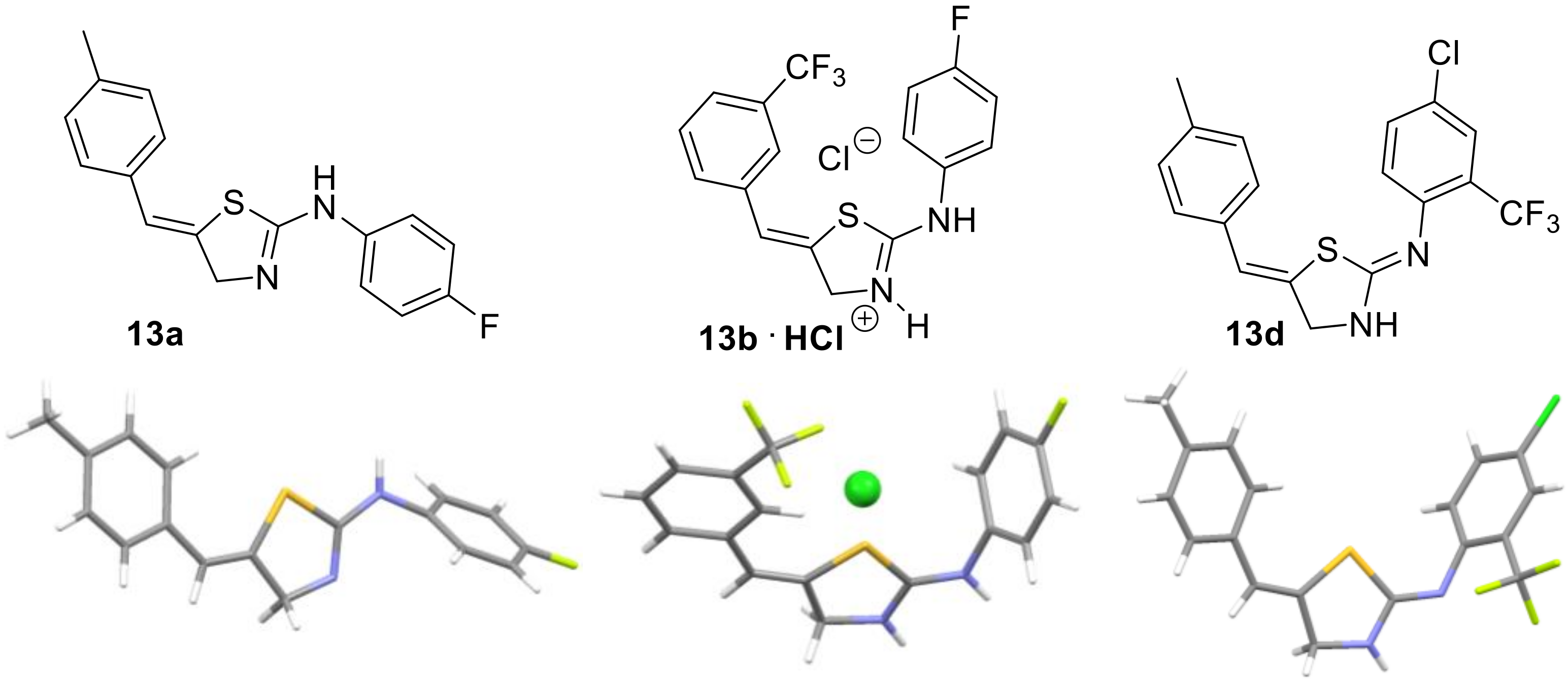
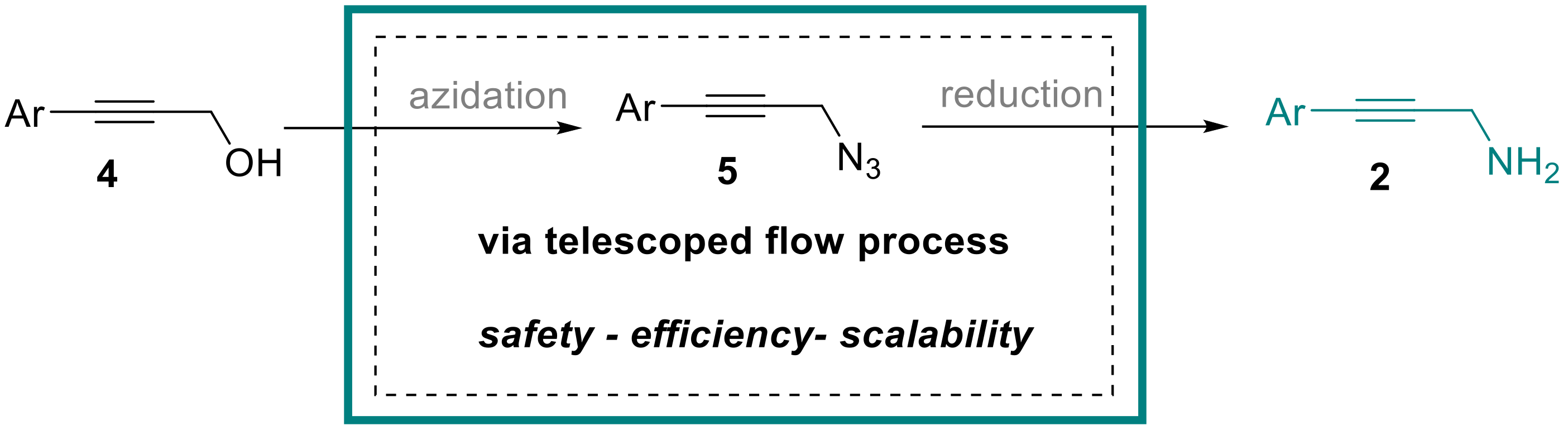
2. Results
3. Materials and Methods
3.1. General Information
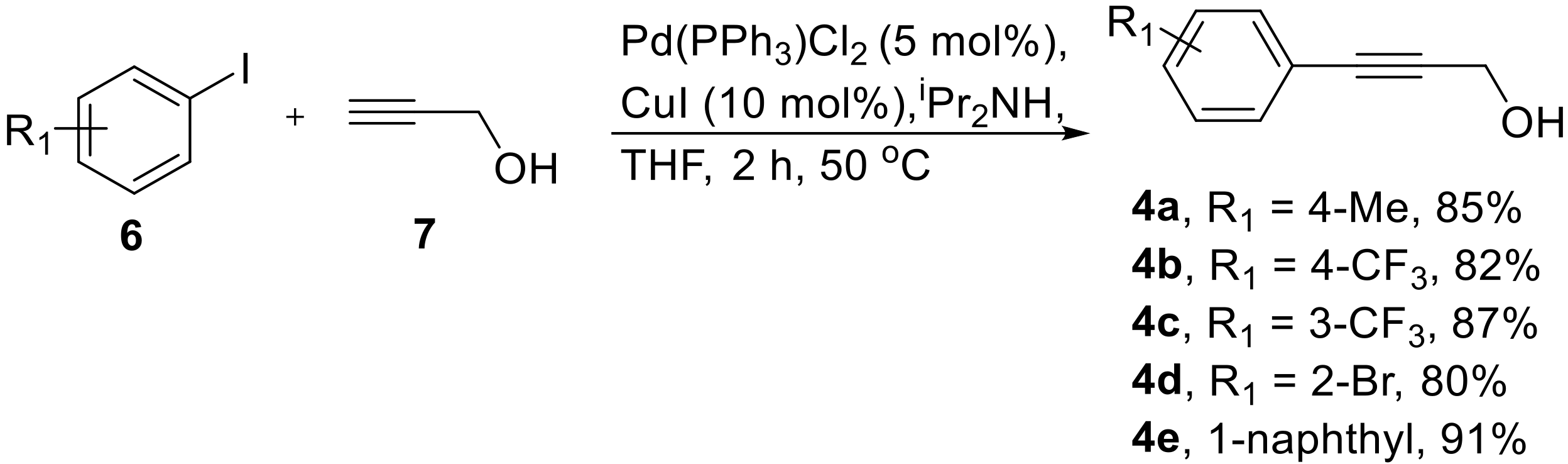
3.2. General Procedure for the Synthesis of Propargyl Alcohols 4a–e
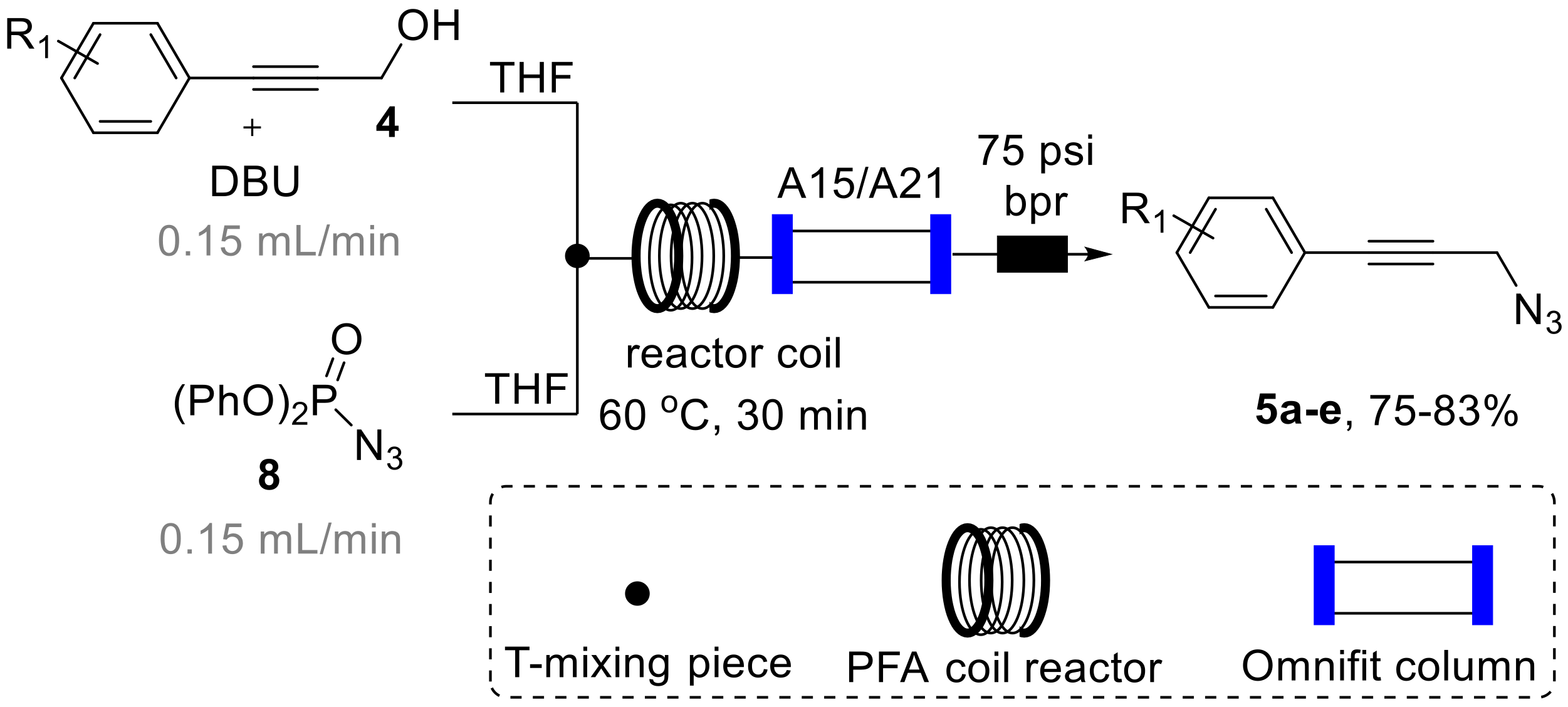
3.3. General Procedure for the Synthesis of Propargyl Azides 5a–e
3.4. General Procedure for the Synthesis of Propargyl Amines 2a–e
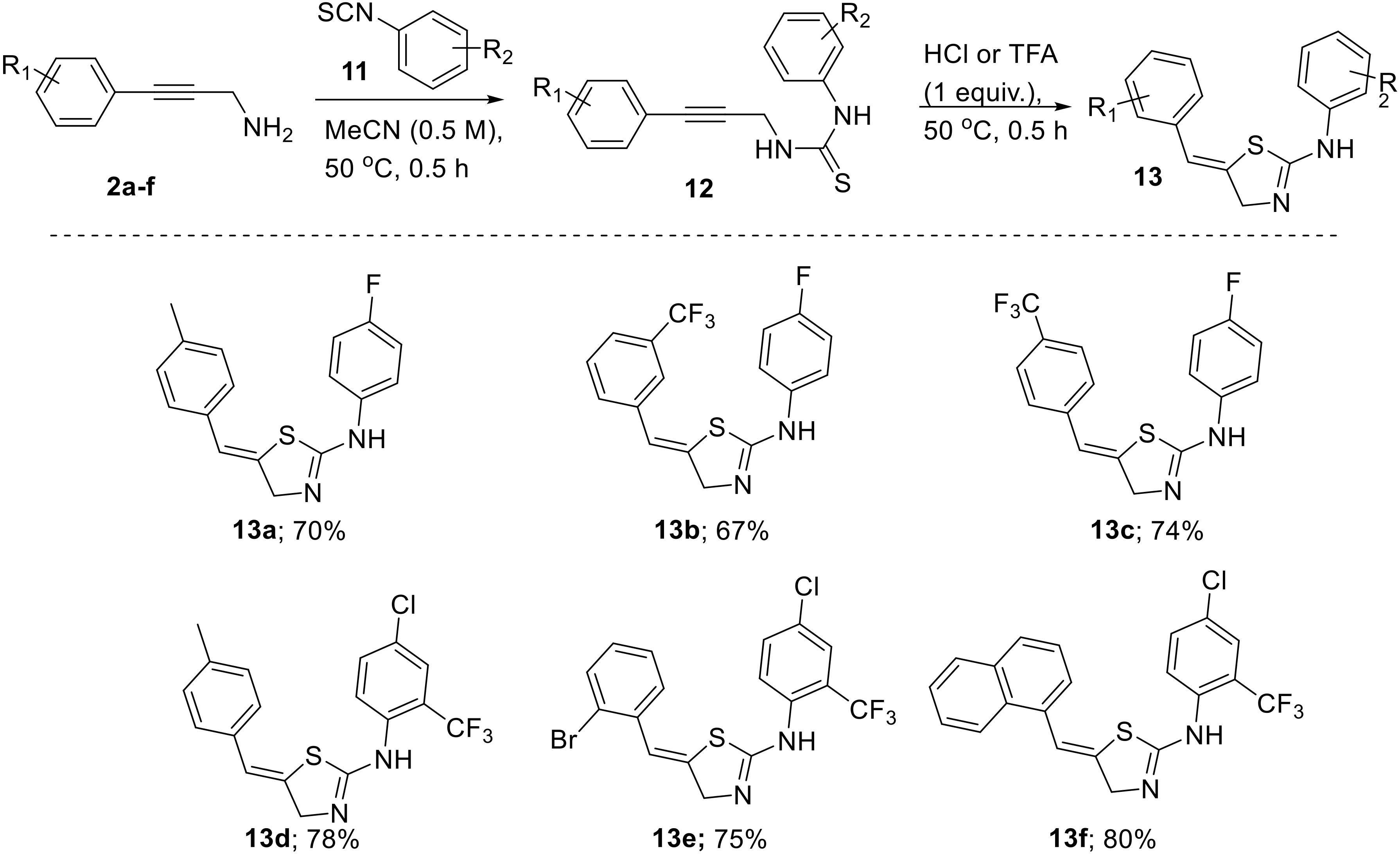
3.5. General Procedure for the Synthesis of Thiazolines 13a–e
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Lauder, K.; Toscani, A.; Scalacci, N.; Castagnolo, D. Synthesis and Reactivity of Propargylamines in Organic Chemistry. Chem. Rev. 2017, 117, 14091–14200. [Google Scholar] [CrossRef]
- Uredi, D.; Motati, D.R.; Watkins, E.B. A Unified Strategy for the Synthesis of β-Carbolines, γ-Carbolines, and Other Fused Azaheteroaromatics under Mild, Metal-Free Conditions. Org. Lett. 2018, 20, 6336–6339. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Xie, Z. Atom-Economical Synthesis of N-Heterocycles via Cascade Inter-/Intramolecular C-N Bond-Forming Reactions Catalyzed by Ti-Amides. J. Am. Chem. Soc. 2010, 132, 11473–11480. [Google Scholar] [CrossRef] [PubMed]
- Kamouka, S.; Moran, W.J. Iodoarene-Catalyzed Cyclizations of N-Propargylamides and β-Amidoketones: Synthesis of 2-Oxazolines. Beilstein J. Org. Chem. 2017, 13, 1823–1827. [Google Scholar] [CrossRef] [PubMed]
- Peshkov, V.A.; Pereshivko, O.P.; Van der Eycken, E.V. A Walk Around the A3-coupling. Chem. Soc. Rev. 2012, 41, 3790–3807. [Google Scholar] [CrossRef]
- Wei, C.; Li, Z.; Li, C.-J. The Development of A3-Coupling (Aldehyde-Alkyne-Amine) and AA3-Coupling (Asymmetric Aldehyde-Alkyne-Amine). Synlett 2004, 9, 1472–1483. [Google Scholar] [CrossRef]
- Shi, L.; Tu, Y.-Q.; Wang, M.; Zhang, F.-M.; Fan, C.-A. Microwave-Promoted Three-Component Coupling of Aldehyde, Alkyne, and Amine via C−H Activation Catalyzed by Copper in Water. Org. Lett. 2004, 6, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Gommermann, N.; Konradin, C.; Polborn, K.; Knochel, P. Enantioselective, Copper(I)-Catalyzed Three-Component Reaction for the Preparation of Propargylamines. Angew. Chem. Int. Ed. 2003, 42, 5763–5766. [Google Scholar] [CrossRef] [PubMed]
- Bisai, A.; Singh, V.K. Enantioselective One-Pot Three-Component Synthesis of Propargylamines. Org. Lett. 2006, 8, 2405–2408. [Google Scholar] [CrossRef] [PubMed]
- Kidwai, M.; Bansal, V.; Kumar, A.; Mozumdar, S. The First Au-Nanoparticles Catalyzed Green Synthesis of Propargylamines via a Three-Component Coupling Reaction of Aldehyde, Alkyne and Amine. Green Chem. 2007, 9, 742–745. [Google Scholar] [CrossRef]
- Purkait, N.; Okumura, S.; Souto, J.A.; Muniz, K. Hypervalent Iodine Mediated Oxidative Amination of Allenes. Org. Lett. 2014, 16, 4750–4753. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Heo, Y.; Lee, S. Metal-Free Decarboxylative Three-Component Coupling Reaction for the Synthesis of Propargylamines. Org. Lett. 2013, 15, 3322–3325. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Huang, S. A Microwave-Assisted Three-Component Synthesis of Arylaminomethyl-Acetylenes: A Facile Access to Terminal Alkynes. Synlett 2014, 25, 407–410. [Google Scholar] [CrossRef]
- Das, B.G.; Nallagonda, R.; Ghorai, P. Direct Substitution of Hydroxy Group of π-Activated Alcohols with Electron-Deficient Amines Using Re2O7 Catalyst. J. Org. Chem. 2012, 77, 5577–5583. [Google Scholar] [CrossRef] [PubMed]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef] [PubMed]
- Dallinger, D.; Kappe, C.O. Why flow means green—Evaluating the merits of continuous processing in the context of sustainability. Curr. Opin. Green Sustain. Chem. 2017, 7, 6–12. [Google Scholar] [CrossRef]
- Jensen, K.F. Flow chemistry—Microreaction technology comes of age. AIChE J. 2017, 63, 858–869. [Google Scholar] [CrossRef]
- Fitzpatrick, D.E.; Ley, S.V. Engineering chemistry for the future of chemical synthesis. Tetrahedron 2018, 74, 3087–3100. [Google Scholar] [CrossRef]
- Baumann, M.; Baxendale, I.R. The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry. Beilstein J. Org. Chem. 2015, 11, 1194–1219. [Google Scholar] [CrossRef]
- Lummiss, J.A.M.; Morse, P.D.; Beingessner, R.L.; Jamison, T.F. Towards More Efficient, Greener Syntheses through Flow Chemistry. Chem. Rec. 2017, 17, 667–680. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Dombrowski, A.W. Emerging Trends in Flow Chemistry and Applications to the Pharmaceutical Industry. J. Med. Chem. 2019, 62, 6422–6468. [Google Scholar] [CrossRef] [PubMed]
- McQuade, D.T.; Seeberger, P.H. Applying Flow Chemistry: Methods, Materials, and Multistep Synthesis. J. Org. Chem. 2013, 78, 6384–6389. [Google Scholar] [CrossRef] [PubMed]
- Movsisyan, M.; Delbeke, E.I.P.; Berton, J.K.E.T.; Battilocchio, C.; Ley, S.V.; Stevens, C.V. Taming hazardous chemistry by continuous flow technology. Chem. Soc. Rev. 2016, 45, 4892–4928. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Baxendale, I.R.; Ley, S.V.; Nikbin, N.; Smith, C.D.; Tierney, J.P. A modular flow reactor for performing Curtius rearrangements as a continuous flow process. Org. Biomol. Chem. 2008, 6, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.; Hopes, P. Use of a Curtius rearrangement as part of the multikilogram manufacture of a pyrazine building block. Org. Process Res. Dev. 2018, 22, 77–81. [Google Scholar] [CrossRef]
- Born, S.C.; Edwards, C.E.R.; Martin, B.; Jensen, K.F. Continuous, on-demand generation and separation of diphenylphosphoryl azide. Tetrahedron 2018, 74, 3137–3142. [Google Scholar] [CrossRef]
- Smith, C.J.; Smith, C.D.; Nikbin, N.; Ley, S.V.; Baxendale, I.R. Flow synthesis of organic azides and the multistep synthesis of imines and amines using a new monolithic triphenylphosphine reagent. Org. Biomol. Chem. 2011, 9, 1927–1937. [Google Scholar] [CrossRef]
- For larger reaction scales a series of scavenger columns were used. An automated switching mechanism as described in the literature can be used to streamline this process; please see: Kitching, M.O.; Dixon, O.E.; Baumann, M.; Baxendale, I.R. Flow-Assisted Synthesis: A Key Fragment of SR 142948A. Eur. J. Org. Chem. 2017, 6540–6553. [Google Scholar] [CrossRef]
- An alternative, yet more complex approach, would be to use in-line analysis by IR to control the dosing rate of the triphenylphosphine reagent. For a leading example, please see: Lange, H.; Carter, C.F.; Hopkin, M.D.; Burke, A.; Goode, J.G.; Baxendale, I.R.; Ley, S.V. A breakthrough method for the accurate addition of reagents in multi-step segmented flow processing. Chem. Sci. 2011, 2, 765–769. [Google Scholar] [CrossRef]
- Pace, V.; Hoyos, P.; Castoldi, L.; Dominguez de Maria, P.; Alcantara, A.R. 2-Methyltetrahydrofuran (2-MeTHF): A biomass-derived solvent with broad application in organic chemistry. ChemSusChem 2012, 5, 1369–1379. [Google Scholar] [CrossRef]
- Aycock, D.F. Solvent applications of 2-methyltetrahydrofuran in organometallic and biphasic reactions. Org. Process Res. Dev. 2007, 11, 156–159. [Google Scholar] [CrossRef]
- Monticelli, S.; Castoldi, L.; Murgia, I.; Senatore, R.; Mazzeo, E.; Wackerlig, J.; Urban, E.; Langer, T.; Pace, V. Recent advancements on the use of 2-methyltetrahydrofuran in organometallic chemistry. Mon. Chem. 2017, 148, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Sasmal, P.K.; Chandrasekhar, A.; Sridhar, S.; Iqbal, J. Novel one-step method for the conversion of isothiocyanates to 2-alkyl(aryl)aminothiazoles. Tetrahedron 2008, 64, 11074–11080. [Google Scholar] [CrossRef]
- Arshadi, S.; Vessally, E.; Edjlali, L.; Hosseinzadeh-Khanmiri, R.; Ghorbani-Kalhor, E. N-Propargylamines: Versatile building blocks in the construction of thiazole cores. Beilstein J. Org. Chem. 2017, 13, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Scalacci, N.; Pelloja, C.; Radi, M.; Castagnolo, D. Microwave-Assisted Domino Reactions of Propargylamines with Isothiocyanates: Selective Synthesis of 2-Aminothiazoles and 2-Amino-4-methylenethiazolines. Synlett 2016, 27, 1883–1887. [Google Scholar] [CrossRef]
- Singh, R.P.; Gout, D.; Lovely, C.J. Tandem thioacylation-intramolecular hydrosulfenylation of propargyl amines—rapid access to 2-aminothiazolidines. Eur. J. Org. Chem. 2019, 2019, 1726–1740. [Google Scholar] [CrossRef]
- Xie, J.; Guo, Z.; Huang, Y.; Qu, Y.; Song, H.; Song, H.; Liu, Y.; Wang, Q. One-pot copper-catalyzed cascade bicyclization strategy for synthesis of 2-(1H-indol-1-yl)-4,5-dihydrothiazoles and 2-(1H-indol-1-yl)thiazol-5-yl aryl ketones with molecular oxygen as an oxygen source. Adv. Synth. Catal. 2019, 361, 490–495. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds reported are available from the authors. |









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donnelly, K.; Zhang, H.; Baumann, M. Development of a Telescoped Flow Process for the Safe and Effective Generation of Propargylic Amines. Molecules 2019, 24, 3658. https://doi.org/10.3390/molecules24203658
Donnelly K, Zhang H, Baumann M. Development of a Telescoped Flow Process for the Safe and Effective Generation of Propargylic Amines. Molecules. 2019; 24(20):3658. https://doi.org/10.3390/molecules24203658
Chicago/Turabian StyleDonnelly, Kian, Huan Zhang, and Marcus Baumann. 2019. "Development of a Telescoped Flow Process for the Safe and Effective Generation of Propargylic Amines" Molecules 24, no. 20: 3658. https://doi.org/10.3390/molecules24203658
APA StyleDonnelly, K., Zhang, H., & Baumann, M. (2019). Development of a Telescoped Flow Process for the Safe and Effective Generation of Propargylic Amines. Molecules, 24(20), 3658. https://doi.org/10.3390/molecules24203658