Asymmetric Electrophilic Difluoromethylthiolation of Indanone-Based β-Keto Esters Using Difluoromethanesulfonyl Hypervalent Iodonium Ylides

 and
and 
Abstract
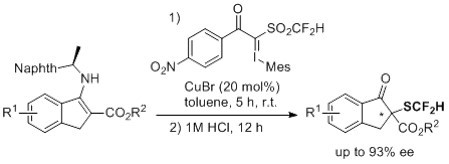
:
1. Introduction
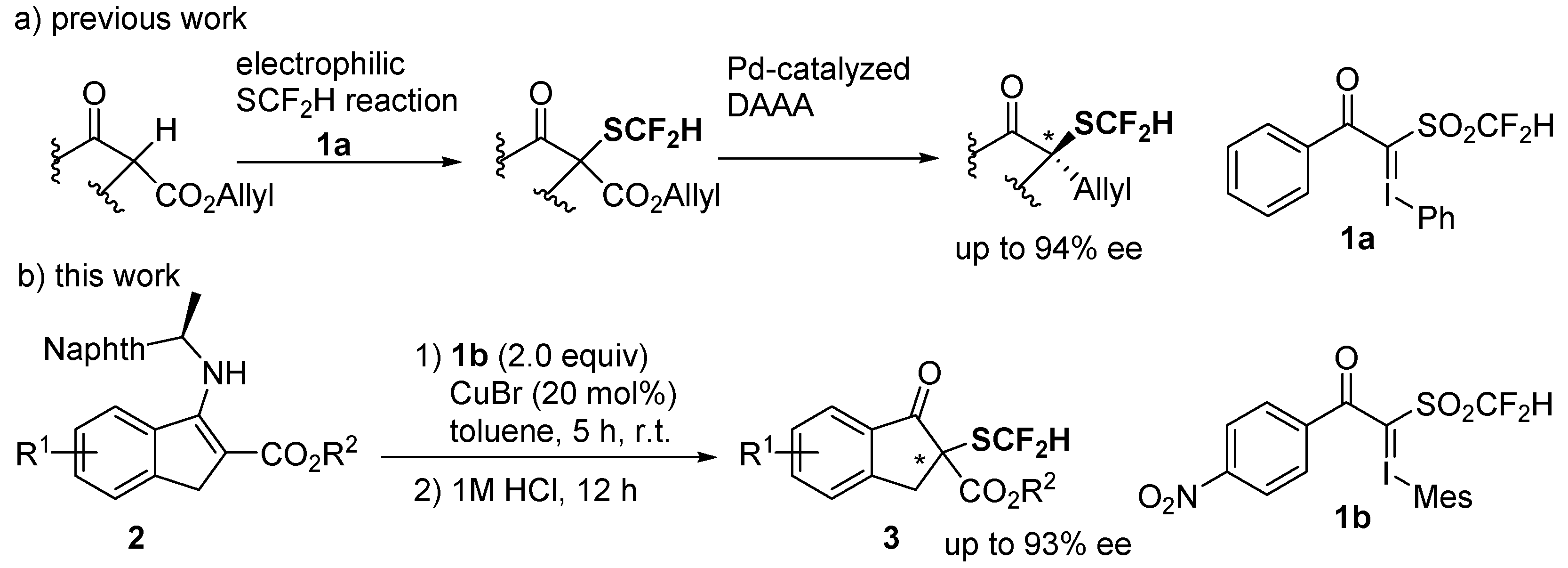
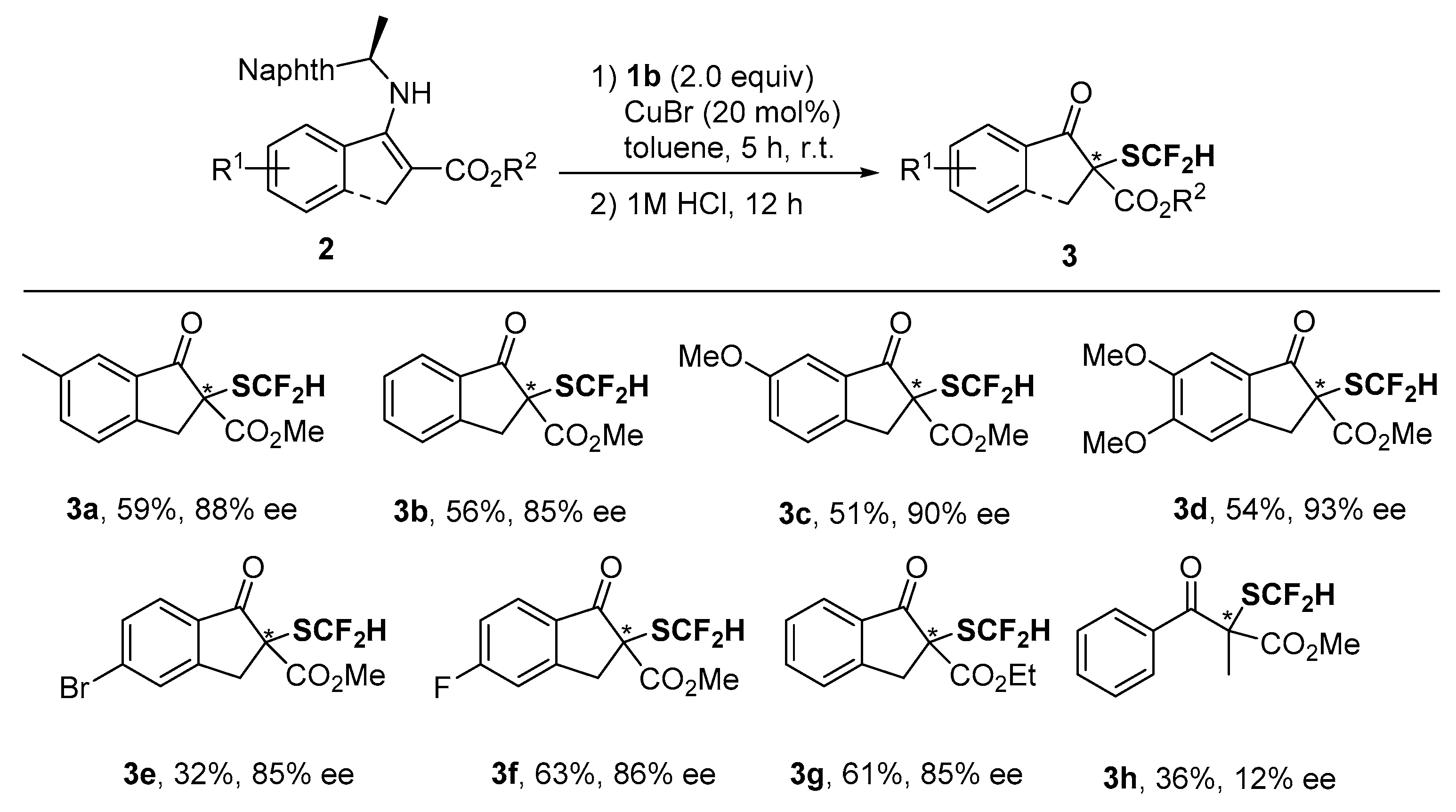
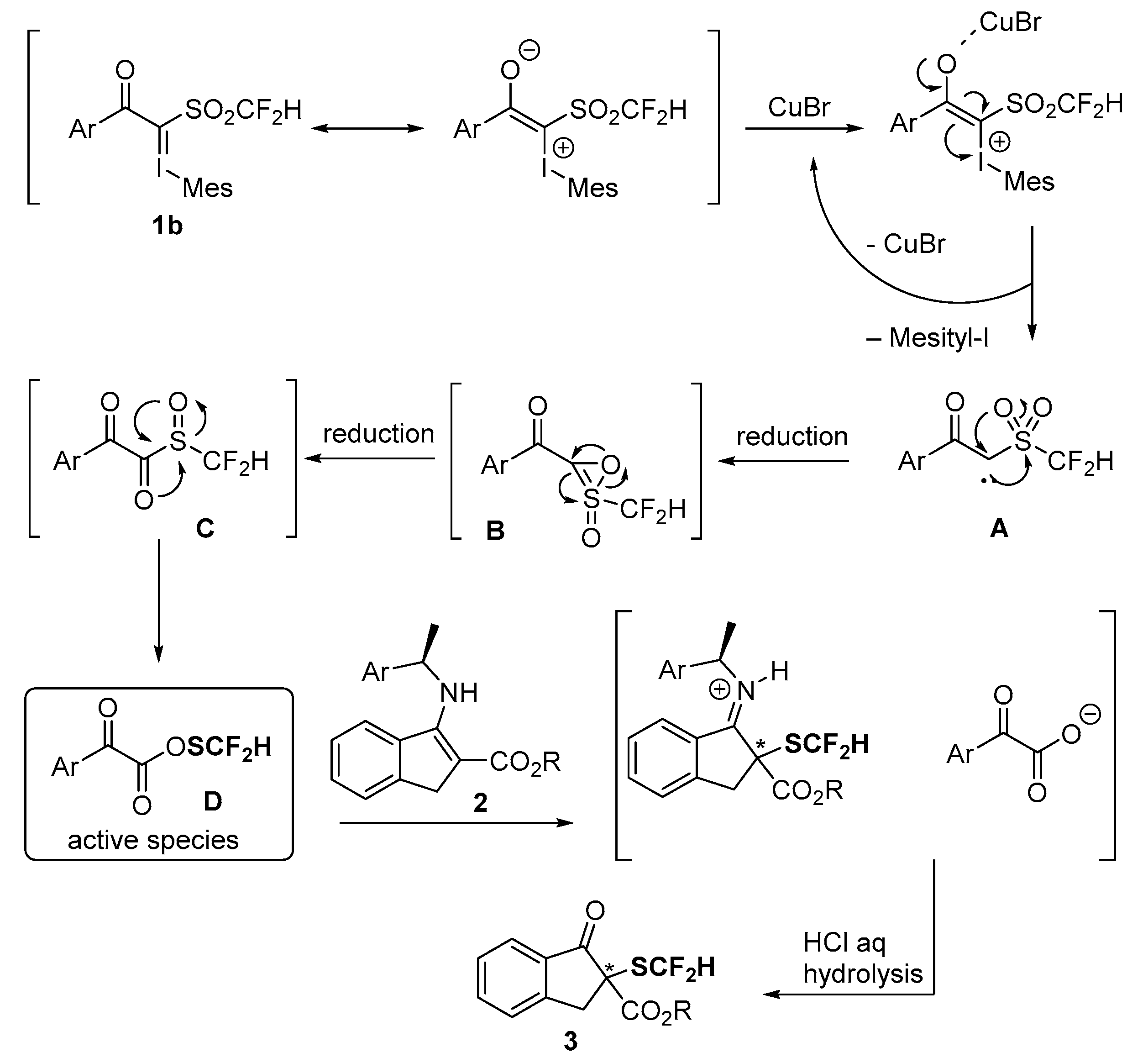
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Chiral Enamine (General Procedure)
3.3. Representative Procedure for the Diastereoselective Difluoromethylthiolation
General Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Toulgoat, F.; Alazet, S.; Billard, T. Direct trifluoromethylthiolation reactions: The “renaissance” of an old concept. Eur. J. Org. Chem. 2014, 2014, 2415–2428. [Google Scholar] [CrossRef]
- Xu, X.-H.; Matsuzaki, K.; Shibata, N. Synthetic methods for compounds having CF3–S units on carbon by trifluoromethylation, trifluoromethylthiolation, triflylation, and related reactions. Chem. Rev. 2015, 115, 731–764. [Google Scholar] [CrossRef]
- Xiong, H.-Y.; Pannecoucke, X.; Besset, T. Recent advances in the synthesis of SCF2H- and SCF2FG-containing molecules. Chem. Eur. J. 2016, 22, 16734–16749. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Bonesi, S.; Postigo, A. Late stage trifluoromethylthiolation strategies for organic compounds. Org. Biomol. Chem. 2016, 14, 7150–7182. [Google Scholar] [CrossRef]
- Erickson, J.A.; McLoughlin, J.I. Hydrogen bond donor properties of the difluoromethyl group. J. Org. Chem. 1995, 60, 1626–1631. [Google Scholar] [CrossRef]
- Chachignon, H.; Kondrashov, E.V.; Cahard, D. Asymmetric construction of chiral carbon centers featuring the SCF3 motif: Direct trifluoromethylthiolation versus building block approach. Fluorine Notes 2017, 113, 5. [Google Scholar] [CrossRef]
- Shao, X.; Wang, X.; Yang, T.; Lu, L.; Shen, Q. An electrophilic hypervalent iodine reagent for trifluoromethylthiolation. Angew. Chem. Int. Ed. 2013, 52, 3457–3460. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, T.; Cheng, X.; Shen, Q. Enantioselective electrophilic trifluoromethylthiolation of β-ketoesters: A case of reactivity and selectivity bias for organocatalysis. Angew. Chem. Int. Ed. 2013, 52, 12860–12864. [Google Scholar] [CrossRef]
- Bootwicha, T.; Liu, X.; Pluta, R.; Atodiresei, I.; Rueping, M. N-trifluoromethylthiophthalimide: A stable electrophilic SCF3-reagent and its application in the catalytic asymmetric trifluoromethylsulfenylation. Angew. Chem. Int. Ed. 2013, 52, 12856–12859. [Google Scholar] [CrossRef]
- Deng, Q.-H.; Rettenmeier, C.; Wadepohl, H.; Gade, L.H. Copper–boxmi complexes as highly enantioselective catalysts for electrophilic trifluoromethylthiolations. Chem. Eur. J. 2014, 20, 93–97. [Google Scholar] [CrossRef]
- Zhu, X.L.; Xu, J.H.; Cheng, D.J.; Zhao, L.J.; Liu, X.Y.; Tan, B. In situ generation of electrophilic trifluoromethylthio reagents for enantioselective trifluoromethylthiolation of oxindoles. Org. Lett. 2014, 16, 2192–2195. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Shen, Q.; Lu, L. Cinchona alkaloid-catalyzed enantioselective trifluoromethylthiolation of oxindoles. Chin. J. Chem. 2014, 32, 678–680. [Google Scholar] [CrossRef]
- Rueping, M.; Liu, X.; Bootwicha, T.; Pluta, R.; Merkens, C. Catalytic enantioselective trifluoromethylthiolation of oxindoles using shelf-stable N-(trifluoromethylthio)phthalimide and a cinchona alkaloid catalyst. Chem. Commun. 2014, 50, 2508–2511. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; An, R.; Zhang, X.; Luo, J.; Zhao, X. Enantioselective trifluoromethylthiolating lactonization catalyzed by an indane-based chiral sulfide. Angew. Chem. Int. Ed. 2016, 55, 5846–5850. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wu, M.; Wan, H.; Wang, J.; Wang, G.; Guoa, H.; Sun, S. Efficient catalytic α-trifluoromethylthiolation of aldehydes. New. J. Chem. 2016, 40, 6550–6553. [Google Scholar] [CrossRef]
- Li, M.; Xue, X.-S.; Cheng, J.-P. Mechanism and origins of stereoinduction in natural cinchona alkaloid catalyzed asymmetric electrophilic trifluoromethylthiolation of β-keto esters with N-trifluoromethylthiophthalimide as electrophilic SCF3 source. ACS Catal. 2017, 7, 7977–7986. [Google Scholar] [CrossRef]
- Zeng, J.L.; Chachignon, H.; Ma, J.A.; Cahard, D. Nucleophilic trifluoromethylthiolation of cyclic sulfamidates: Access to chiral β- and γ-SCF3 amines and α-amino esters. Org. Lett. 2017, 19, 1974–1977. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liang, Y.; Ji, J.; Luo, J.; Zhao, X. Chiral selenide-catalyzed enantioselective allylic reaction and intermolecular difunctionalization of alkenes: Efficient construction of C-SCF3 stereogenic molecules. J. Am. Chem. Soc. 2018, 140, 4782–4786. [Google Scholar] [CrossRef]
- Jin, M.Y.; Li, J.; Huang, R.; Zhou, Y.; Chung, L.W.; Wang, J. Catalytic asymmetric trifluoromethylthiolation of carbonyl compounds via a diastereo and enantioselective Cu-catalyzed tandem reaction. Chem. Commun. 2018, 54, 4581–4584. [Google Scholar] [CrossRef]
- Gelat, F.; Poisson, T.; Biju, A.T.; Pannecoucke, X.; Besset, T. Trifluoromethylthiolation of α-chloroaldehydes: Access to quaternary SCF3-containing centers. Eur. J. Org. Chem. 2018, 2018, 3693–3696. [Google Scholar] [CrossRef]
- Chachignon, H.; Kondrashov, E.V.; Cahard, D. Diastereoselective electrophilic trifluoromethylthiolation of chiral oxazolidinones: Access to enantiopure α-SCF3 alcohols. Adv. Synth. Catal. 2018, 360, 965–971. [Google Scholar] [CrossRef]
- Luo, J.; Cao, Q.; Cao, X.; Zhao, X. Selenide-catalyzed enantioselective synthesis of trifluorome- thylthiolated tetrahydronaphthalenes by merging desymmetrization and trifluoromethylthiolation. Nat. Commun. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Maeno, M.; Sasaki, K.; Guo, M.; Hashimoto, M.; Shiro, M.; Shibata, N. Synthesis of chiral nonracemic α-difluoromethylthio compounds with tetrasubstituted stereogenic centers via a palladium-catalyzed decarboxylative asymmetric allylic alkylation. Org. Lett. 2018, 20, 7044–7048. [Google Scholar] [CrossRef]
- Arimori, S.; Matsubara, O.; Takada, M.; Shiro, M.; Shibata, N. Difluoromethanesulfonyl hypervalent iodonium ylides for electrophilic difluoromethylthiolation reactions under copper catalysis. R. Soc. Open Sci. 2016, 3, 160102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Matsubara, O.; Jia, S.; Tokunaga, E.; Shibata, N. Difluoromethylthiolation of phenols and related compounds with a HF2CSO2Na/Ph2PCl/Me3SiCl system. Org. Lett. 2017, 19, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Gu, Y.; Lu, L.; Shen, Q. N-difluoromethylthiophthalimide: A shelf-stable, electrophilic reagent for difluoromethylthiolation. J. Am. Chem. Soc. 2015, 137, 10547. [Google Scholar] [CrossRef] [PubMed]
- Christoffers, J.; Baro, A.; Werner, T. α-Hydroxylation of β-dicarbonyl compounds. Adv. Synth. Catal. 2004, 346, 143–151. [Google Scholar] [CrossRef]
- Toullec, P.Y.; Bonaccorsi, C.; Mezzetti, A.; Togni, A. Expanding the scope of asymmetric electrophilic atom-transfer reactions: Titanium- and ruthenium-catalyzed hydroxylation of β-ketoesters. Proc. Natl. Acad. Sci. USA 2004, 101, 5810–5814. [Google Scholar] [CrossRef]
- Christoffers, J.; Werner, T.; Frey, W.; Baro, A. Straightforward synthesis of (R)-(−)-kjellmanianone. Chem. Eur. J. 2004, 10, 1042–1045. [Google Scholar] [CrossRef]
- Ishimaru, T.; Shibata, N.; Nagai, J.; Nakamura, S.; Toru, T.; Kanemasa, S. Lewis acid-catalyzed enantioselective hydroxylation reactions of oxindoles and β-keto esters using DBFOX ligand. J. Am. Chem. Soc. 2006, 128, 16488–16489. [Google Scholar] [CrossRef]
- Lu, M.; Zhu, D.; Lu, Y.; Zeng, X.; Tan, B.; Xu, Z.; Zhong, G. Chiral brønsted acid-catalyzed enantioselective α-hydroxylation of β-dicarbonyl compounds. J. Am. Chem. Soc. 2009, 131, 4562–4563. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, G.; Wang, Z.; Zhang, R.; Zhang, X.; Ding, K. Spiro-2,2′-bichroman-based bisoxazoline (SPANbox) ligands for ZnII-catalyzed enantioselective hydroxylation of β-keto esters and 1,3-diester. Chem. Sci. 2011, 2, 1141–1144. [Google Scholar] [CrossRef]
- Lian, M.; Li, Z.; Cai, Y.; Meng, Q.; Gao, Z. Enantioselective photooxygenation of β-keto esters by chiral phase-transfer catalysis using molecular oxygen. Chem. Asian J. 2012, 7, 2019–2023. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Wang, B.; Mu, H.; Zhang, H.; Song, Y.; Qu, J. Development of tartaric acid derived chiral guanidines and their application to catalytic enantioselective α-hydroxylation of β-dicarbonyl compounds. Org. Lett. 2013, 15, 3106–3109. [Google Scholar] [CrossRef] [PubMed]
- Racemic products 3 were prepared according to ref 24 and used as reference for ee determination by HPLC analysis.
- Recovery of chiral amine auxiliary: In the synthesis of 3d (3.2), water phase of the extraction was basified with NaOH aq to pH 14, and it was extracted with CH2Cl2 three times. The combined organic phase was dried by Na2SO4 and concentrated in vacuo to give the chiral amine, (R)-1-(naphthalen-1-yl)ethan-1-amine in 25% yield.
- Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. Trifluoromethanesulfonyl hypervalent iodonium ylide for copper-catalyzed trifluoromethylthiolation of enamines, indoles, and β-keto esters. J. Am. Chem. Soc. 2013, 135, 8782–8785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-H.; Yin, L.; Wang, Y.-M. General and efficient method for the preparation of β-enamino ketones and esters catalyzed by indium tribromide. Adv. Synth. Catal. 2006, 348, 184–190. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, F.; Li, X. Cross-dehydrogenative coupling between enamino esters and ketones: Synthesis of tetrasubstituted pyrroles. Org. Lett. 2012, 14, 1412–1415. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1a, 1b, 3 are available from the authors. |

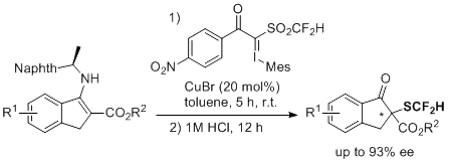{kind=link}
{kind=link}
{kind=link}
{kind=link}
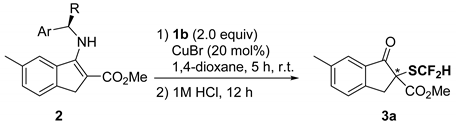
| Entry | Ar | R | 2 | Yield (%) 1 | Ee (%) 2 |
|---|---|---|---|---|---|
| 1 | Ph | Me | 2a | 79 | 57 |
| 2 | Ph | Et | 2b | 79 | 46 |
| 3 | 1-Naphthyl | Me | 2c | 57 | 62 |
| 4 | 4-MeC6H4 | Me | 2d | 80 | 57 |
| 5 | 4-MeOC6H4 | Me | 2e | 78 | 49 |
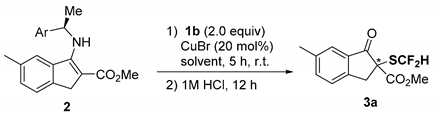
| Entry | Ar | 2 | Solvent | Yield (%) 1 | Ee (%) 2 |
|---|---|---|---|---|---|
| 1 | Ph | 2a | 1,4-dioxane | 79 | 57 |
| 2 | Ph | 2a | THF | 75 | 56 |
| 3 | Ph | 2a | CH2Cl2 | 74 | 67 |
| 4 | Ph | 2a | CHCl3 | 75 | 68 |
| 5 | Ph | 2a | Toluene | 76 | 69 |
| 6 | 1-Naphthyl | 2c | 1,4-dioxane | 57 | 62 |
| 7 | 1-Naphthyl | 2c | CH2Cl2 | 52 | 88 |
| 8 | 1-Naphthyl | 2c | CHCl3 | 65 | 87 |
| 9 | 1-Naphthyl | 2c | Toluene | 59 | 88 |
| 10 | 1-Naphthyl | 2c | CH3CN | 0 | – |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gondo, S.; Matsubara, O.; Chachignon, H.; Sumii, Y.; Cahard, D.; Shibata, N. Asymmetric Electrophilic Difluoromethylthiolation of Indanone-Based β-Keto Esters Using Difluoromethanesulfonyl Hypervalent Iodonium Ylides. Molecules 2019, 24, 221. https://doi.org/10.3390/molecules24020221
Gondo S, Matsubara O, Chachignon H, Sumii Y, Cahard D, Shibata N. Asymmetric Electrophilic Difluoromethylthiolation of Indanone-Based β-Keto Esters Using Difluoromethanesulfonyl Hypervalent Iodonium Ylides. Molecules. 2019; 24(2):221. https://doi.org/10.3390/molecules24020221
Chicago/Turabian StyleGondo, Satoshi, Okiya Matsubara, Hélène Chachignon, Yuji Sumii, Dominique Cahard, and Norio Shibata. 2019. "Asymmetric Electrophilic Difluoromethylthiolation of Indanone-Based β-Keto Esters Using Difluoromethanesulfonyl Hypervalent Iodonium Ylides" Molecules 24, no. 2: 221. https://doi.org/10.3390/molecules24020221
APA StyleGondo, S., Matsubara, O., Chachignon, H., Sumii, Y., Cahard, D., & Shibata, N. (2019). Asymmetric Electrophilic Difluoromethylthiolation of Indanone-Based β-Keto Esters Using Difluoromethanesulfonyl Hypervalent Iodonium Ylides. Molecules, 24(2), 221. https://doi.org/10.3390/molecules24020221