1. Introduction
In 2018, liver cancer was the sixth most common cancer and the fourth leading cause of cancer deaths worldwide [
1]. The highest incidence of this cancer can be seen in East Asia, Southeast Asia, and North and Southern Africa [
2]. Based on the database of the International Agency for Research on Cancer (IARC), there were more than 69,000 new cancer cases in Myanmar in 2018 and liver cancers were in the top 5 in terms of incidence, mortality, and prevalence by cancer site [
1]. Currently, the Ministry of Health and Sports from Myanmar supports the implementation of the National Cancer Control Plan, focusing on priority activities and maximizing efforts in line with the respective mandates, priorities, and areas of expertise of the partner and to achieve better results for cancer prevention, care, and control.
Testing, annual screenings, and early intervention for cancers are currently inadequate on many accounts, which include the rise in population, an inadequate supply of drugs, the cost of treatments, the side effects of several synthetic medicines, and increasing resistance to the drugs used. In most rural areas, herbal medicine has been used for decades by traditional practitioners to treat cancer problems. Medicinal plants have long been used in the treatment of liver diseases or the maintenance of a healthy liver. Yacon, or
Smallanthus sonchifolius ((Poepp. & Endl.) H. Rob.), is a plant belonging to the Asteraceae family, native to the Andean regions of South America [
3]. The plant contents include phenolic acids, flavonoids, and sesquiterpene lactones [
4,
5]. Yacon has been used as a functional food with multiple beneficial effects on the body, including as an antimicrobial, as an antioxidant, hypolipidemic effects, and probiotic substances [
3,
6]. The plant was cultivated in Myanmar in the 2000s. It has become increasingly popular as medicated green tea for diabetes patients and its use is wide-spread.
In recent years, Yacon has emerged as a potential anti-cancer agent. Previous in vitro studies indicated that the crude extract of Yacon and the phytochemicals derived from the plants exerted the cytotoxicity against breast cancer [
7], colon cancer [
7,
8], and cervical cancer [
9,
10]. The anticancer property was attributed to sesquiterpene lactones in Yacon [
9,
10,
11]. In addition, Yacon has been well-known to have antioxidant effects because of an abundant amount of polyphenols, which are found at high quantities in leaves or stems of the plant [
6]. Recent studies have indicated that antioxidants might possess anti-tumor and hepatoprotective effects, although the mechanism needs further investigation [
12].
This research aimed to evaluate the effects of Yacon leaf extract (YLE) on liver cancer in vitro using hepatocellular carcinoma HepG2 cell line, which is the most commonly used in drug metabolism and hepatotoxicity studies. HepG2 cells are nontumorigenic with high proliferation rates and an epithelial-like morphology that performs many differentiated hepatic functions [
13]. The medicinal plant is of high pharmacological importance, but it is still not reported for its chemotherapeutic potential as an alternative medicine for liver cancer disease. Our results may provide scientific evidence for the therapeutic potential of this plant, as a functional food, on liver cancer.
3. Discussion
Yacon (
S. sonchifolius), a common edible plant grown throughout the world, is well known for its anti-diabetic properties [
14]. It is also demonstrated to have several other pharmacological properties, including anti-inflammatory, anti-oxidant, anti-allergic, and anti-cancer effects [
15]. The cytotoxicity potential of hexane, methanol, and dichloromethane extracts of Yacon leaves was assessed against MCF-7 and HT-29 cell lines by using the AlamarBlue
® assay [
7]. Sesquiterpene lactones from Yacon leaves, such as enhydrin, uvedalin, and their derivatives, also exhibited cytotoxic activity against MGC80-3 [
16], HeLa, HL-60, and B16-F10 cell lines [
9]. However, there has been no report yet to evaluate the anti-liver cancer activity of
S. sonchifolius on liver cancer cells. Therefore, this study aimed to investigate the anti-cancer effect of YLE on HepG2 cells in terms of inhibiting cell proliferation and migration. In addition, we also examined the YLE effect on cell cycle and ROS generation in this cell line.
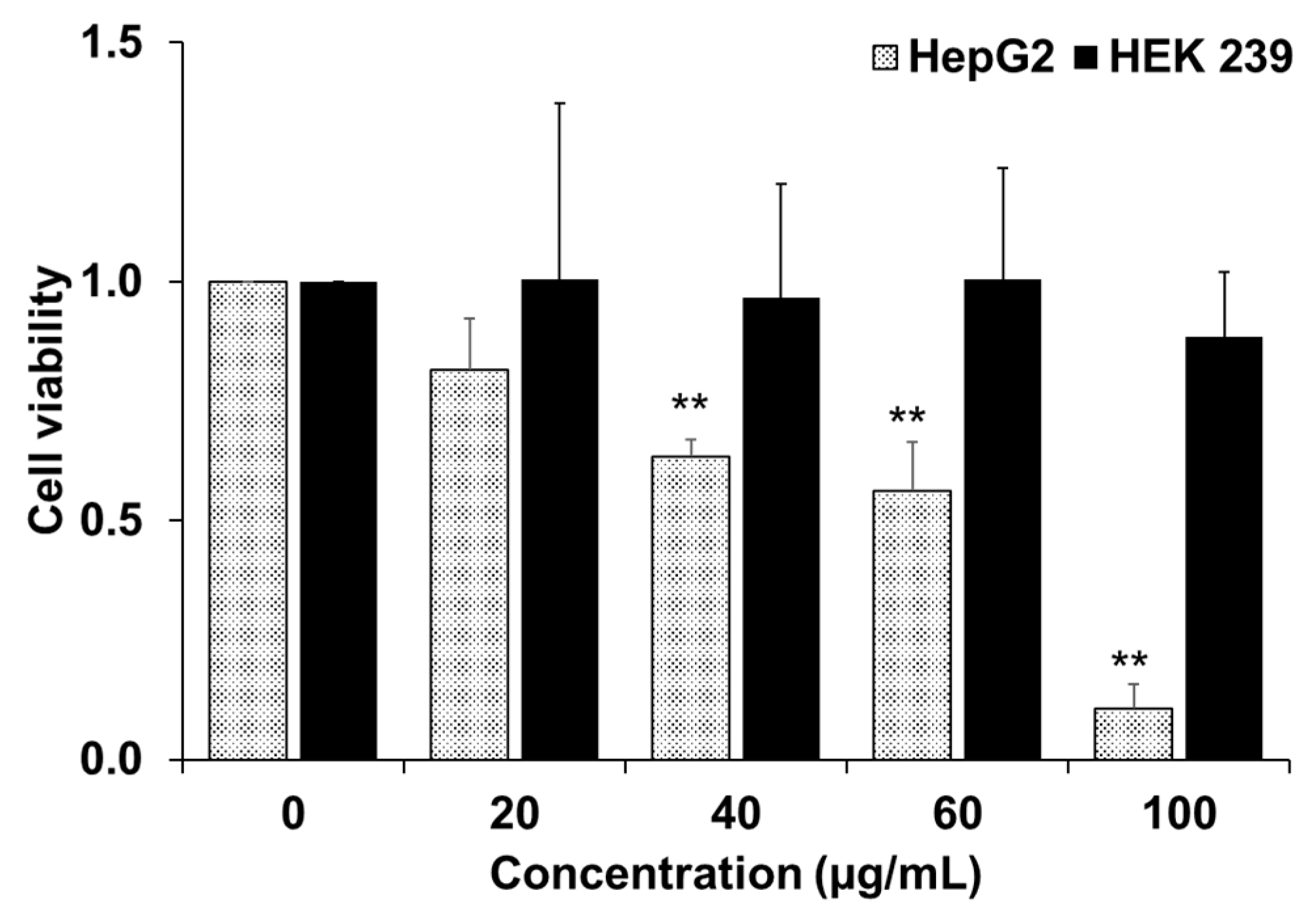
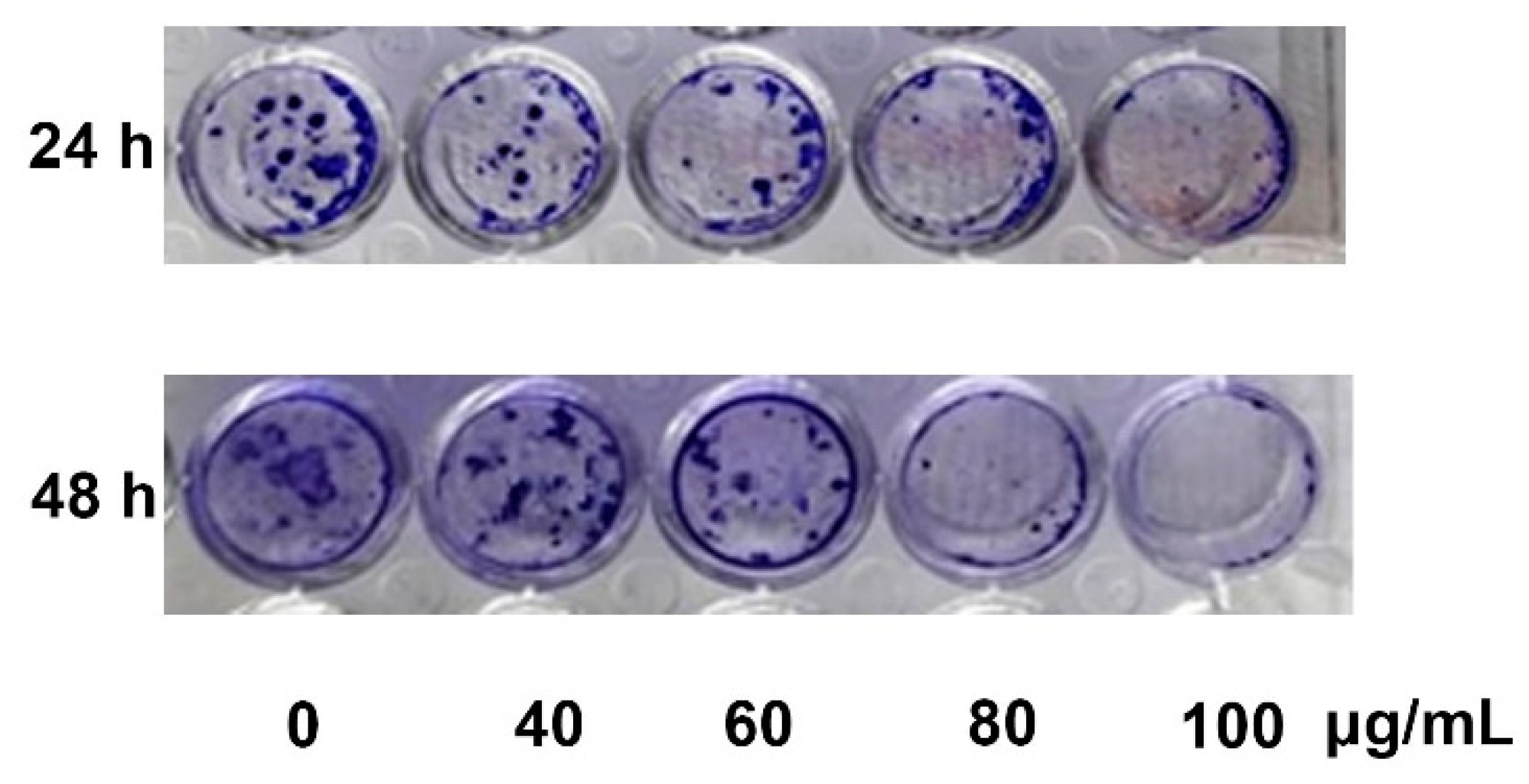
In the current study, YLE was found to show a potent inhibitory effect on HepG2 cell survival. Firstly, we examined the toxicity of YLE on liver cancer HepG2 cells. The calculated IC50 (58.2 ± 1.9 µg/mL) implied the promising inhibitory effect against these cancer cells. Indeed, the colony formation of HepG2 cells was significantly suppressed with increasing concentration of YLE up to more than 90% at 100 µg/mL, in comparison with the control, after 24 h or 48 h treatment. Taken together, these results suggested that YLE demonstrates a long-term suppressive effect on cell proliferation of HepG2 in a concentration-dependent manner.
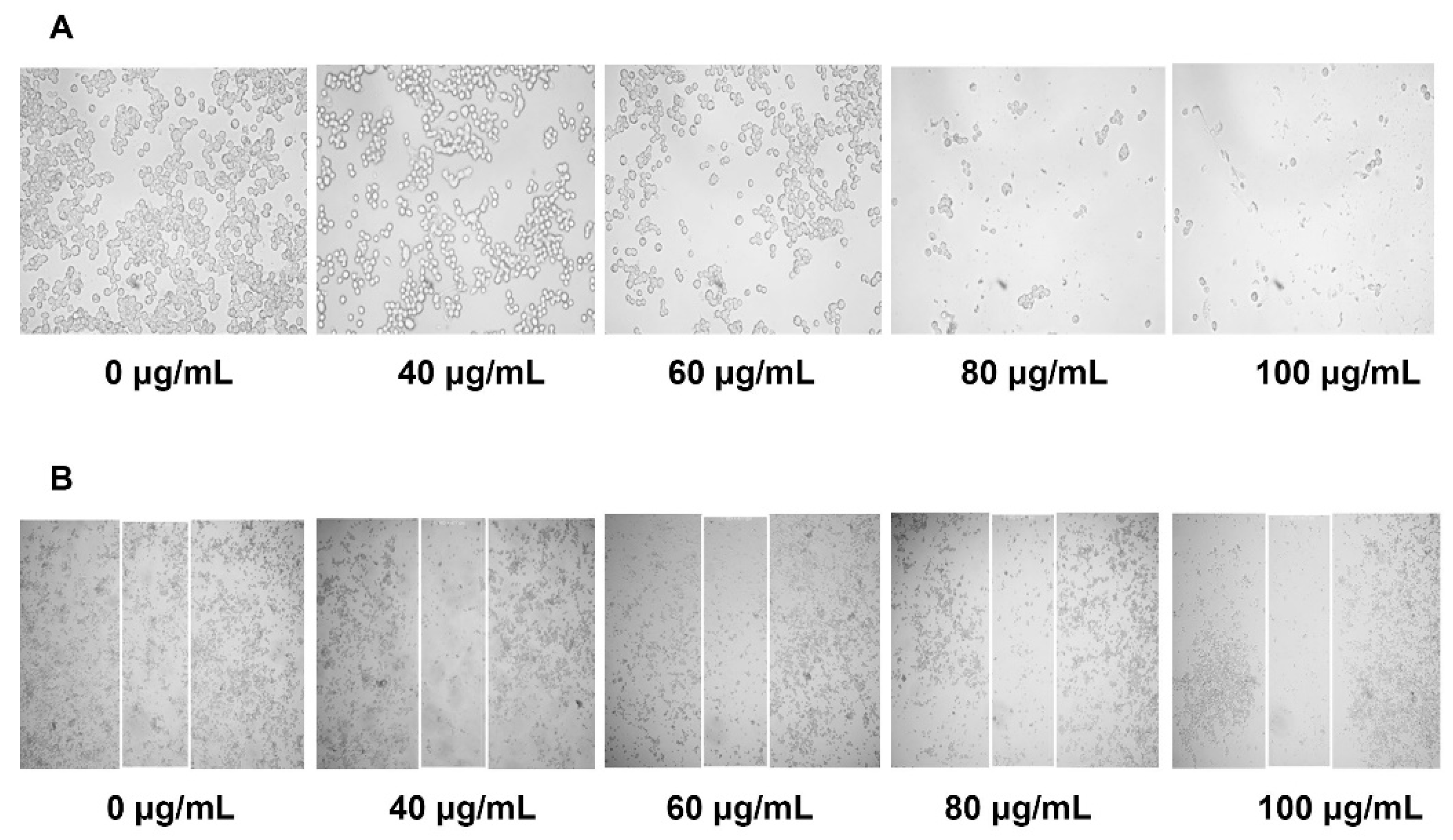
It is known that metastasis is one of the leading causes of death from cancer and it is complicated to examine [
17]. During tumor metastasis, malignant cells migrate into neighboring healthy tissues, contributing to tumor development. In this study, YLE could effectively prevent the migration of HepG2 in a dose-dependent manner. Therefore, YLE could contribute to hinder metastasis progression in hepatocellular carcinoma.
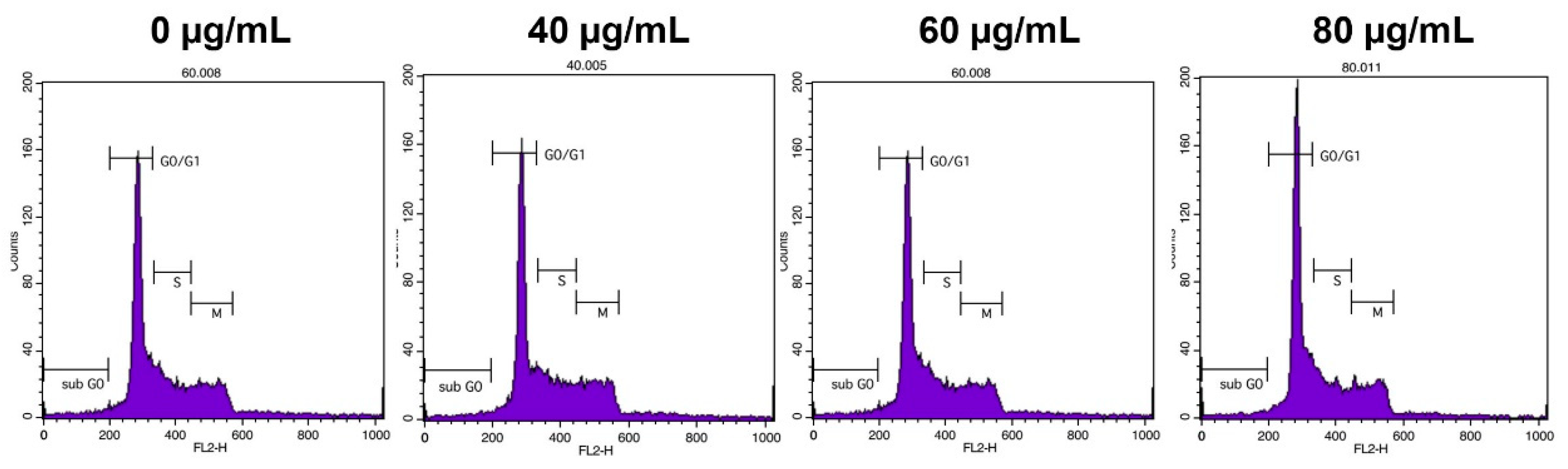
To control cancer growth, inhibition of the progression of the cell cycle is one of the essential strategies [
18]. In this study, flow cytometry analysis demonstrated that the extract dose-dependently increased the percentage of cells of the G0/G1 phase of the HepG2 cell cycle. This observation reveals that YLE induces cell cycle arrest at the G0/G1 phase of the cell cycle.
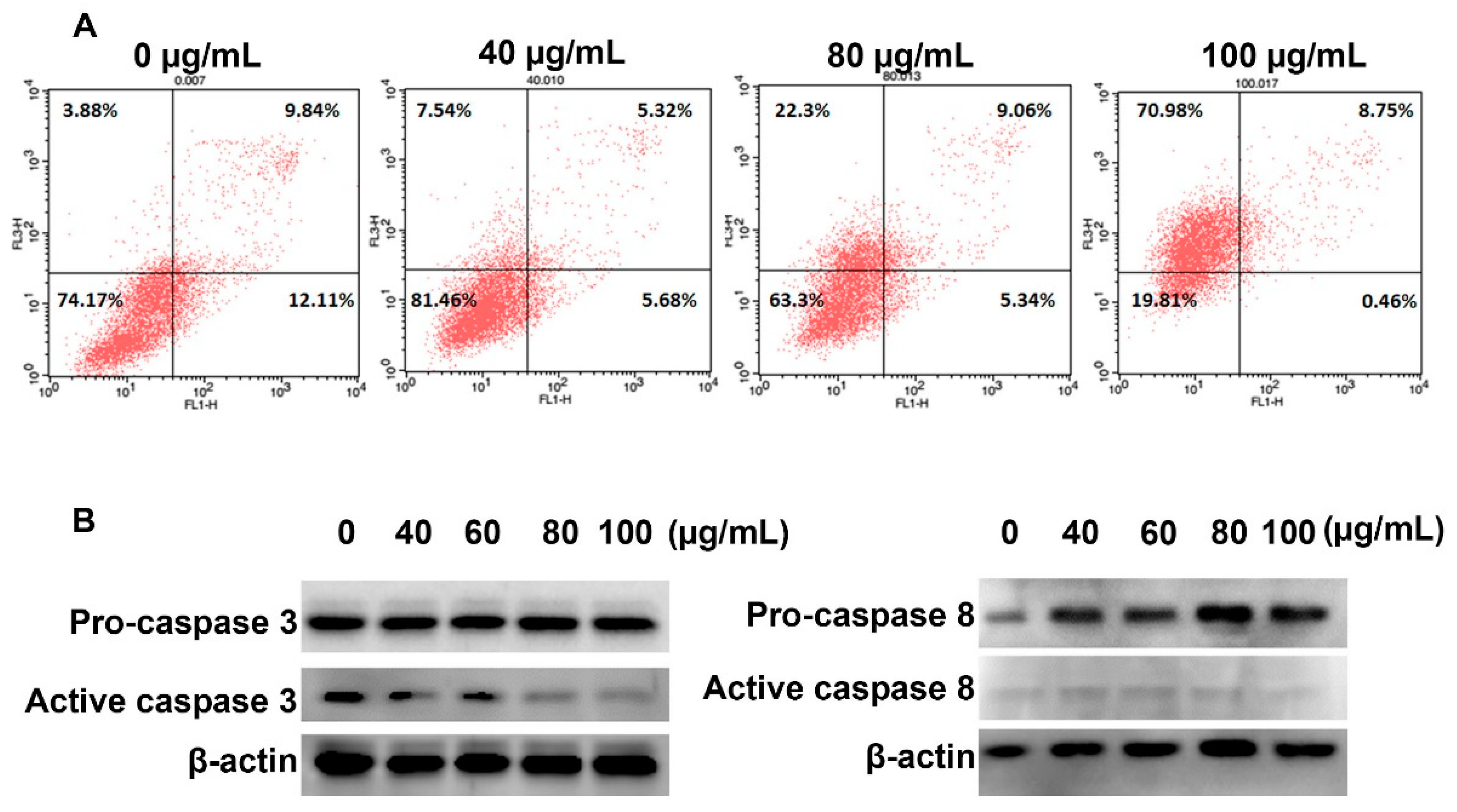
In term of inducing cancer cell death, we found that YLS induced necrosis in HepG2 cells. The number of cells in the necrosis subpopulation significantly increased from 3.88% (control group) to 70.98% (100 μg/mL YLE-treatment group), whereas YLE did not activate caspase 3 or caspase 8, which regulate apoptosis in cells.
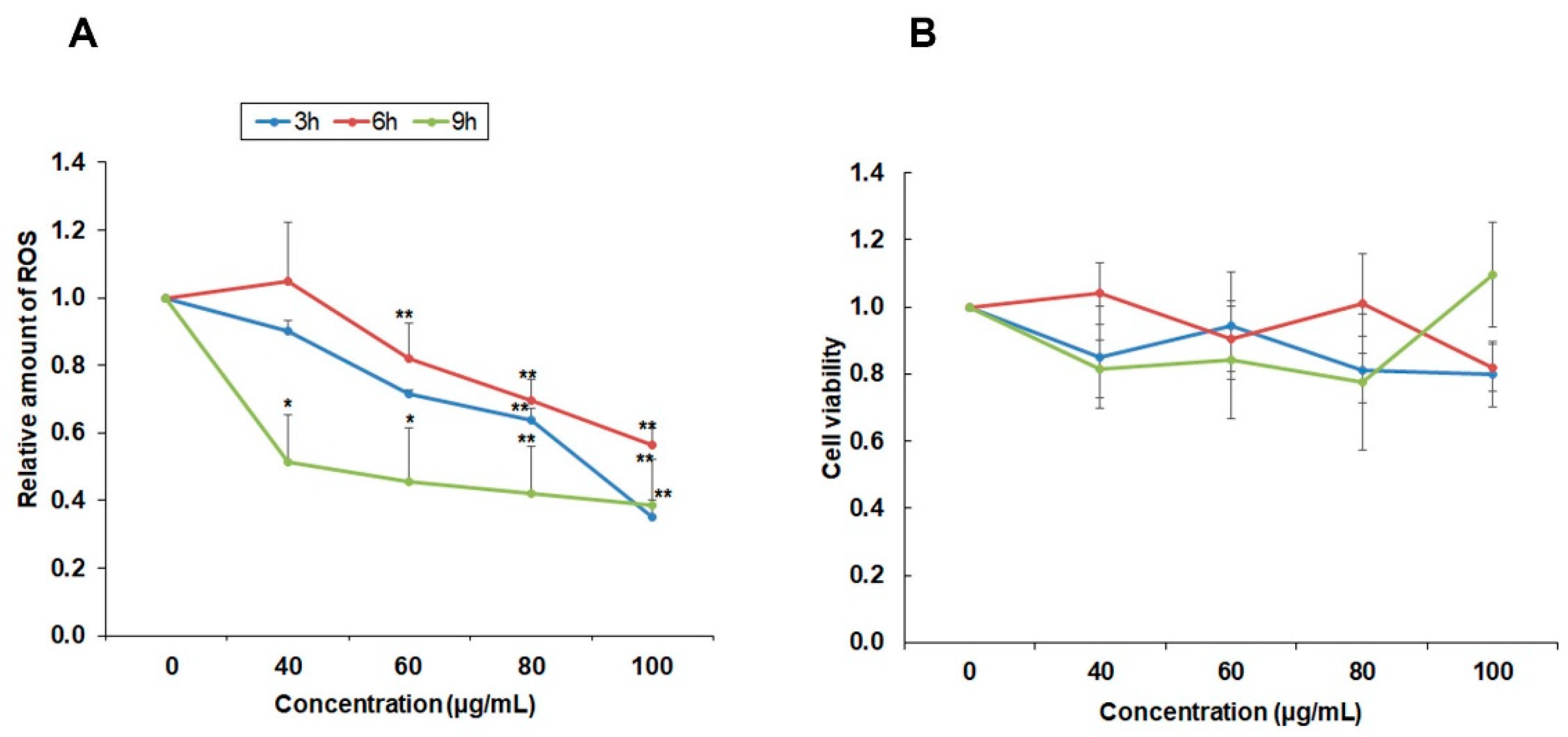
High levels of ROS in cancer cells have been found in almost all cancer cells due to high metabolomic activity and they have specific functions in cancer cell development [
19]. ROS have been reported to be involved in cell proliferation, cell survival, cell cycle progression, and angiogenesis. Therefore, suppressing ROS may be a useful strategy in cancer treatment [
20]. Since YLE has been reported to contain a large number of polyphenols [
4], we sought to evaluate the antioxidant effect of YLE on HepG2 cells. YLE could notably reduce ROS in HepG2 in both dose and time-dependent manners. These observations may support the inhibitory effect of YLE on HepG2 cell proliferation.
The presence of sesquiterpene lactones, such as polymatin B, enhydrin, and uvedalin, was also confirmed by HPLC coupled with high-resolution mass tandem analysis [
5]. These compounds have been demonstrated to have cytotoxicity as well as to induce apoptosis or necrosis in several cancer cell lines [
11]. In the current study, we partly confirmed the presence of these sesquiterpenoids sharing the common fragment characteristic of the uvedalin moiety. These compounds may be significant as chemical defenses for human hepatocellular carcinoma HepG2 cells. Although the effect of individual constituents of this plant extract on HepG2 cells needs to be further investigated, the findings in the current study suggest that
S. sonchifolius leaf could be recommended as a potential source of a chemopreventative agent against liver cancer.
4. Materials and Methods
4.1. Preparation of the Samples
The Yacon (Smallanthus sonchifolius ((Poepp. & Endl.) H. Rob.) leaves were collected from Pindaya Township, Shan State, in the eastern part of Myanmar. The sample was identified at the Department of Botany, University of Yangon. The leaves were cleaned, carefully dried in shadow, and powdered. About 2 g of ground powder was soaked in 20 mL methanol and shaken for 8 h at 180 rpm and 37 °C. The soaked substance was filtered throughout 110 mm filter paper (Hyundai, Seoul, South Korea). The solvent was eliminated with a rotary evaporator (SB-1200, EYELA, Shanghai, China) at 60 °C. The residue was placed in a freeze drier (Operon, Korea) to dry. The crude extract was kept in a refrigerator at 4 °C and protected from light. The experiment was performed in triplicate. Four-hundred mg of the sample extract was dissolved in 1 mL of dimethyl sulfoxide (DMSO) and then serially diluted to 100, 60, 40, and 20 μg/mL for further biological experiments. All other chemical reagents were from Sigma-Aldrich Chemical Company (St. Louis, MO, USA) unless otherwise noted.
4.2. Cell Line and Culture Medium
HepG2 and HEK 239 cell lines were obtained from the American Type Culture Collection (ATCC). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) media supplemented with 10% (v/v) fetal bovine serum and 1% (v/v), 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were cultured in an incubator at 37 °C and 5% CO2 humidified atmosphere. Media were changed every 2 to 3 days. Reagents and media for cell culture were purchased from GenDePOT (Katy, TX, USA).
4.3. Cytotoxicity Assay (MTT Assay)
The cellular toxicity of the YLE on cultured cells was measured using 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT). Cells were grown in a 96-well plate at a density of 1 × 10
4 cells per well. The cells are allowed to grow overnight in a cell culture incubator. Then, we treated cells with different concentrations of samples (0, 20, 40, 60, and 100 μg/mL) for 24 h. Later, cells were washed twice with phosphate buffer saline (PBS). MTT solution was added to each well (final concentration of MTT was 0.5 mg/mL) and the plate was incubated for 3 h. Finally, the medium was replaced by DMSO to solubilize the formazan crystals. The optical density was measured by absorbance at 595 nm with a microplate spectrophotometer (SpectraMax 190, Molecular Devices, San Jose, CA, USA). We identified the IC
50 value using the ED50 Plus v1.0 online software (National Institute of Respiratory Diseases (INER), Mexico) as described elsewhere [
21].
4.4. Cell Colony Formation Assay
Cells were seeded at a density of 1 × 103 cells/well in 6-well plates and allowed to attach overnight. Then, the cells were treated with YLE (0, 40, 60, 80, and 100 μg/mL) for 24 h or 48 h. After treatment, cells were continuously grown for another 2 weeks. The media were changed every 3 days. After 14 days, cells were washed with PBS and fixed with 3.7% formaldehyde solution for 30 min before being stained with 0.5% crystal violet for another 30 min at room temperature (RT). Cells were washed with water to remove the dye and then photographed.
4.5. Cell Migration Assay (Wound–Healing Assay)
We seeded cells in a 24-well plate (5 × 104 cells/well) and cultured at 37 °C, 5% CO2 for 24 h. The 80–90% cell confluence was observed before the scratch assay was performed. We stimulated a wound using a sterile 200 μL pipette tip. After washing with PBS to remove loosened debris, cells were treated with YLE (0, 40, 60, 80, and 100 μg/mL) for 24 h at 37 °C, 5% CO2. After treatment, cells were washed twice with PBS. The images of cells were captured under a CKX41 microscope (Olympus, Japan) at 400× magnification. Images were processed with ProgRes Capture Pro software v.2.8.8 (JENOPTIK Optical Systems, Jena, Germany).
4.6. Reactive Oxygen Species (ROS) Production Assay
The level of intracellular ROS was determined by the change in fluorescence resulting from the oxidation of the fluorescent probe dichlorofluorescein diacetate (DCF-DA). We seeded cells at a density of 1 × 106 cells/well in 6 wells plate and then treated them with 0, 40, 60, 80, and 100 ug/mL of YLE for 3, 6, and 9 h. After indicated periods, cells were washed twice with DPBS and stained with dichlorofluorescein diacetate (DCF-DA, 10 μM) at 37 °C for 30 min in the dark. After washing twice with PBS, the fluorescence generation was measured in a microplate reader, (excitation wavelength (ext.): 485 nm; emission wavelength (emi.): 535 nm). Data were expressed as the percentage of ROS relative to untreated control groups.
4.7. Cell Cycle Analysis
HepG2 cells at a concentration of 1 × 106/well cells were cultured in 6-well plates and treated with different concentrations of YLE for 24h. After treatment, the cells were washed with PBS and harvested using a centrifuge at 1300 rpm for 3 min. Then, cells were re-suspended and fixed with 70% ethanol at −20 °C for 1 week. After fixing, we harvested cells by washing with PBS and centrifuging at 2000 rpm for 3 min. Cells were incubated with RNase (200 μg/mL) at 37 °C for 30 min to remove cellular RNA, then stained with propidium iodide (PI, 100 μg/mL) for another 10 min at RT in the dark. Finally, the cells were analyzed by flow cytometry (BD FACSCalibur, BD Biosciences, USA) according to detected signals in the FL2 channel (ext: 488 nm, emi: 564–606 nm) while data were analyzed with Cell Quest Pro software (BD Biosciences, San Jose, CA, USA).
4.8. Annexin V/PI Assay
HepG2 cells were seeded in the 6-well plates (1 × 106 cells/well). After overnight incubation, we treated cells with indicated concentrations of YLE for 24 h. Cells were washed with PBS and harvested using a centrifuge at 2000 rpm for 3 min for the following analysis. To distinguish apoptotic and necrotic cell death in HepG2 cells, we used the BD Annexin V: FITC Apoptosis Detection Kit I (BD Biosciences, San Diego, CA, USA), according to the manufacture’s direction. The cells were analyzed by flow cytometry (BD FACSCalibur, San Jose, CA). Signals were detected in the FL1 channel (for Annexin V) and FL3 channel (for PI), while data were analyzed with Cell Quest Pro software.
4.9. Western Blotting
The HepG2 cells were seeded in a 6-well plate at 1 × 106 cells/well and treated with various concentrations of YLE for 24 h. The whole cells lysates were prepared by homogenizing cells in a prepared lysis buffer (20 mM HEPES (pH 7.6), 350 mM NaCl, 20% glycerol, 0.5 mM EDTA, 0.1 mM EGTA, 1% NP-40, 50 mM NaF, 0.1 mM DTT, 0.1 mM PMSF, and a protease inhibitor cocktail) on ice for 30 min, followed by collecting the supernatant using a centrifuge at 15,000 rpm for 10 min. The concentration of protein was identified using a Bradford assay. The equal amounts of proteins were separated by electrophoresis on 10% SDS gels and transferred to nitrocellulose membranes. Blots were blocked with 5% BSA for 1 h prior to incubating with the following primary antibodies: β-actin conjugated with HRP (Santa Cruz, 47778) as1:3000, pro-caspase 3 (rabbit, Santa Cruz H-277: sc-7148) as 1:1000, cleaved caspase 3 (rabbit, Abcam ab2302) as 1:1000, pro-caspase 8 (rabbit, Santa Cruz, sc-6134) as 1:1000, and cleaved caspase 8 (rabbit, Cell Signaling Technology, 9496) as 1:1000, at 4 °C overnight. After washing the membrane and incubating with goat anti-rabbit IgG (H + L) secondary antibody conjugated with HRP (GeneTex, GTX213110, 1:3000) for 1 h at RT, the bands of interest were detected using an EZWestern ECL kit (Daeillab Service, Seoul, South Korea) and then photographed on the LAS-4000 imaging system (GE Healthcare, Chicago, IL, USA).
4.10. LC-Q-TOF-MS Analysis of YLE
Since the cytotoxicity of YLE was attributed to its active ingredients, which were known to be sesquiterpenoids, we confirmed the presence of these compounds using liquid chromatography analysis in an HPLC system (Agilent Technologies, Santa Clara, CA, USA) linked to a G6530A ESI-Q-TOF MS spectrometer (Agilent Technologies). Chromatographic separations were conducted using a C18 column (2.1 × 150 mm, 3.5 μm, Agilent Technologies). The mobile phases were water with 0.1% formic acid (A) and acetonitrile with 0.1% formic acid (B). The gradient program consisted of the following: 0–15 min, 5–50% B; 15–20 min, 50% B; 20–30 min, 50–95% B; followed by 95% B for 10 min washing. The flow rate was 0.3 mL/min and the temperature was kept at 40 °C. The injection volume was 3 μL. The instrument was operated with an ESI source in positive ion mode. The mass determination was performed using the following MS conditions: Ion spray voltage of 400 kV; desolvation temperature, 350 °C; desolvation gas flow rate, 10 L/min. The fragmentor was set at 150 V. The full-scan mass spectra were acquired within an m/z range of 100 to 1200 m/z in the MS mode followed by the target MS/MS mode. Data acquisition and proceeding were performed using Mass Hunter Qualitative Analysis software (Agilent Technologies). The compounds were putatively identified based on their mass tandems in comparison with published data.
4.11. Statistical Analysis
In this study, we performed all experiments in triplicate and analyzed results using one-way ANOVA followed by a Dunnett’s test (SPSS version 25.0; Chicago, IL, USA). Probabilities of p < 0.05 were considered significant.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






