Supersaturation Potential of Amorphous Active Pharmaceutical Ingredients after Long-Term Storage

 ,
,
Abstract

1. Introduction
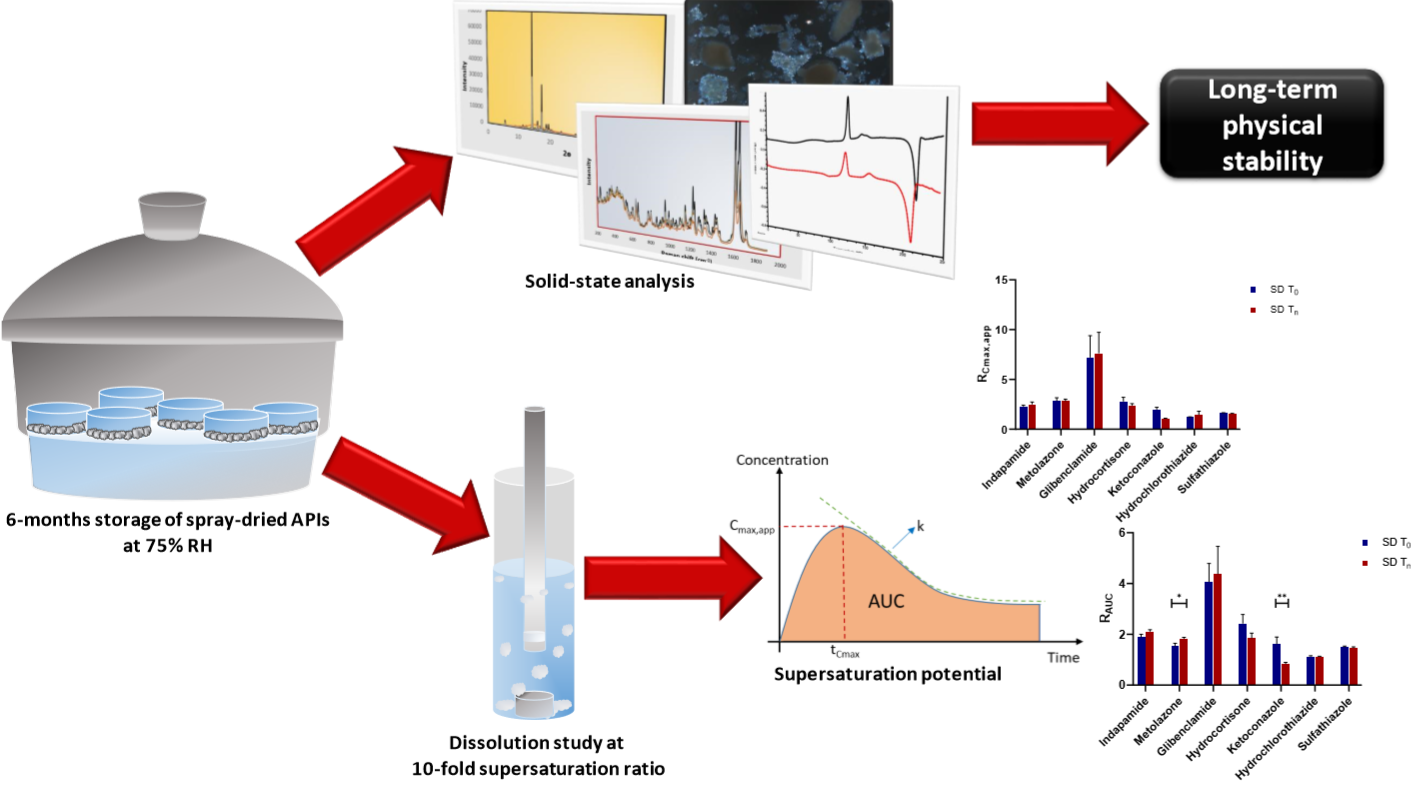
2. Results and Discussion
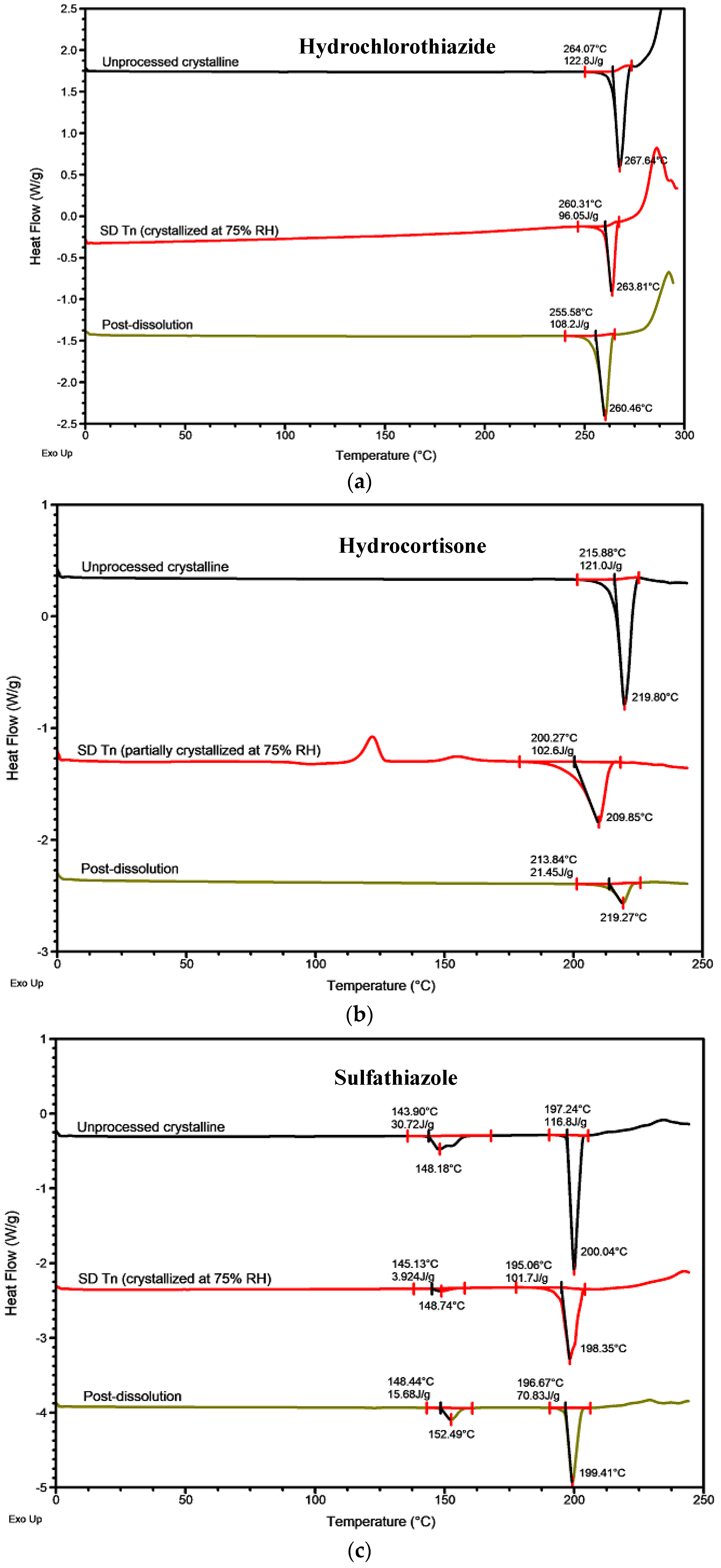
2.1. Physical Stability and Crystallization Behavior of Spray-Dried Amorphous APIs
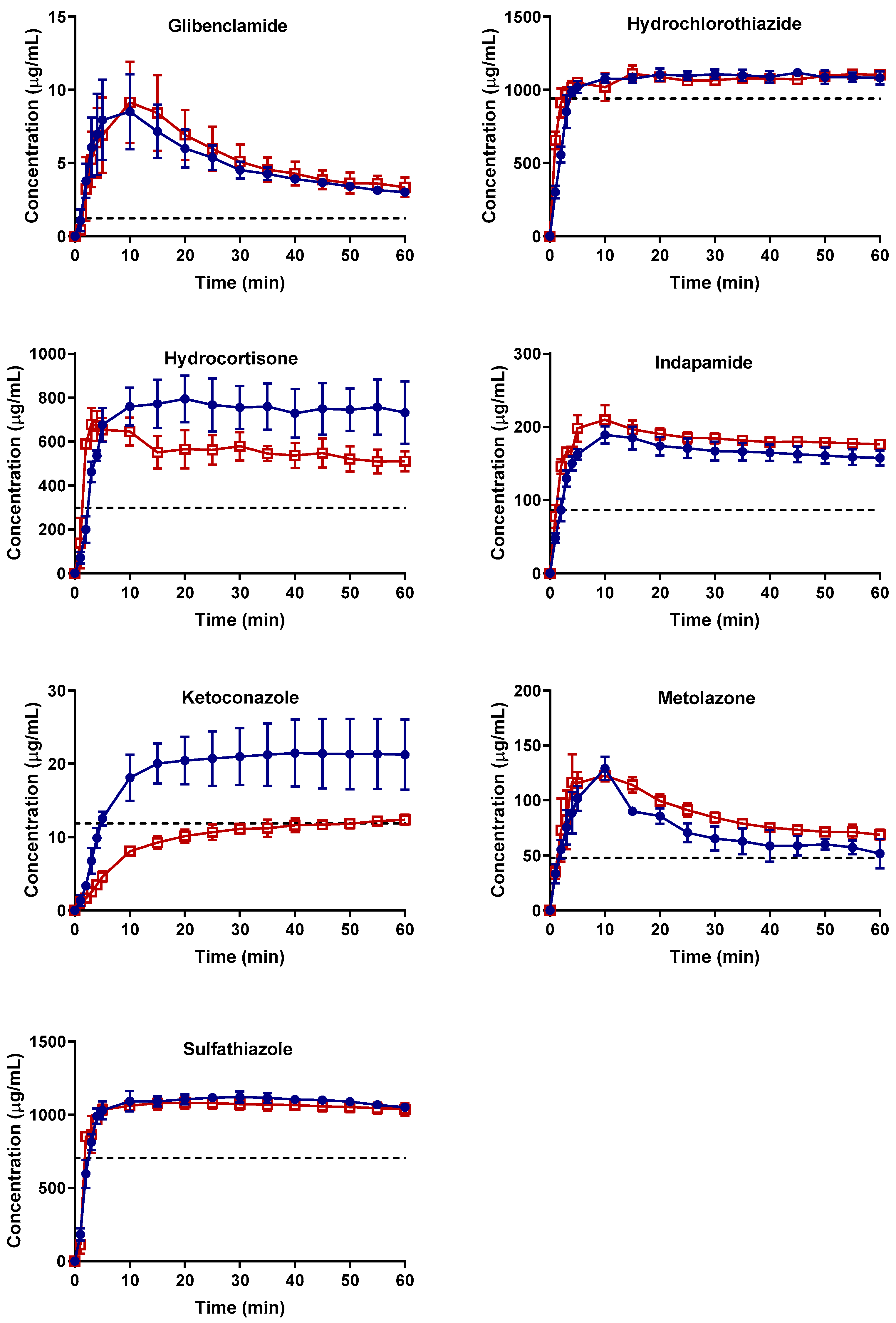
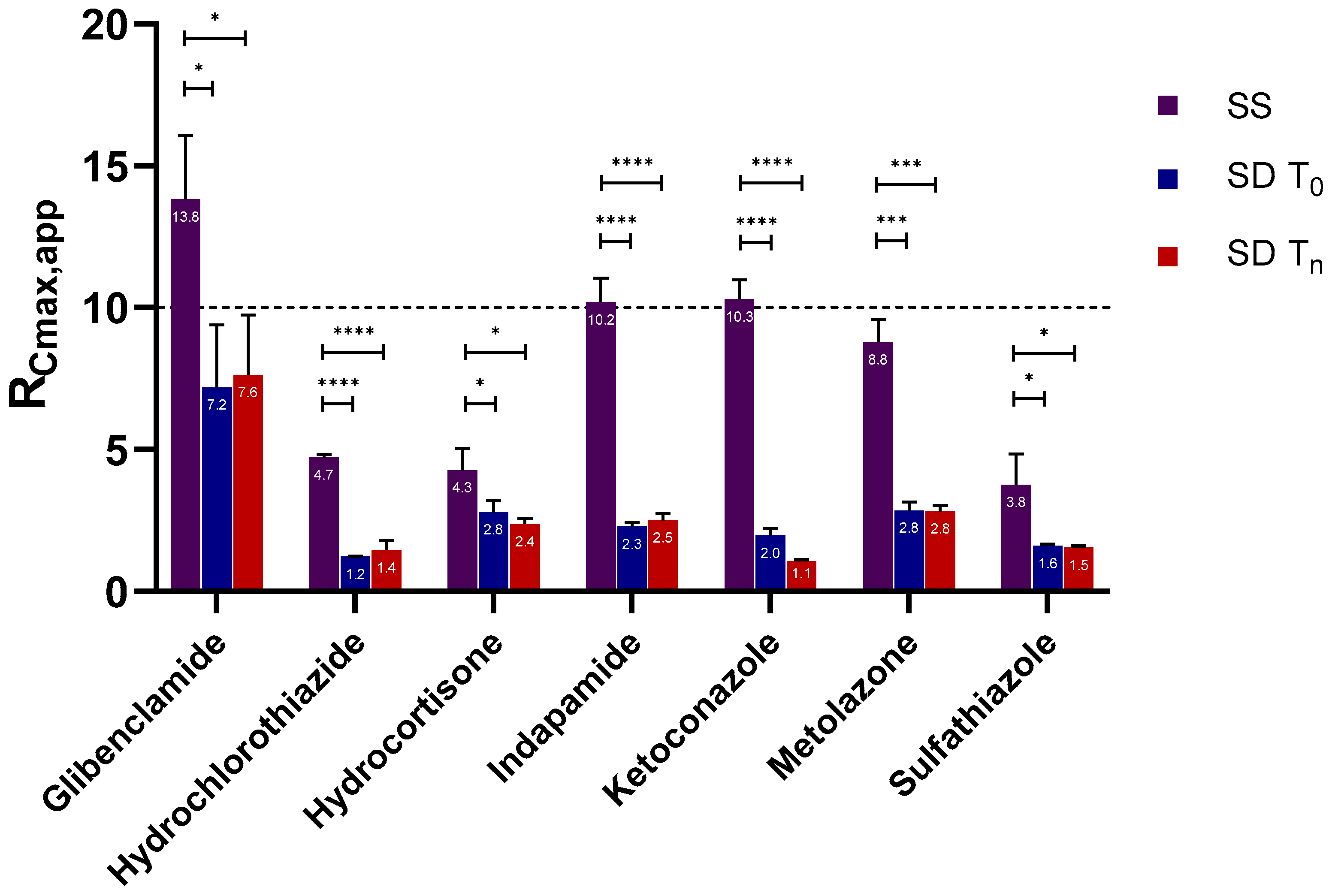
2.2. Supersaturation Potential and Crystallization Kinetics as a Function of Physical Stability
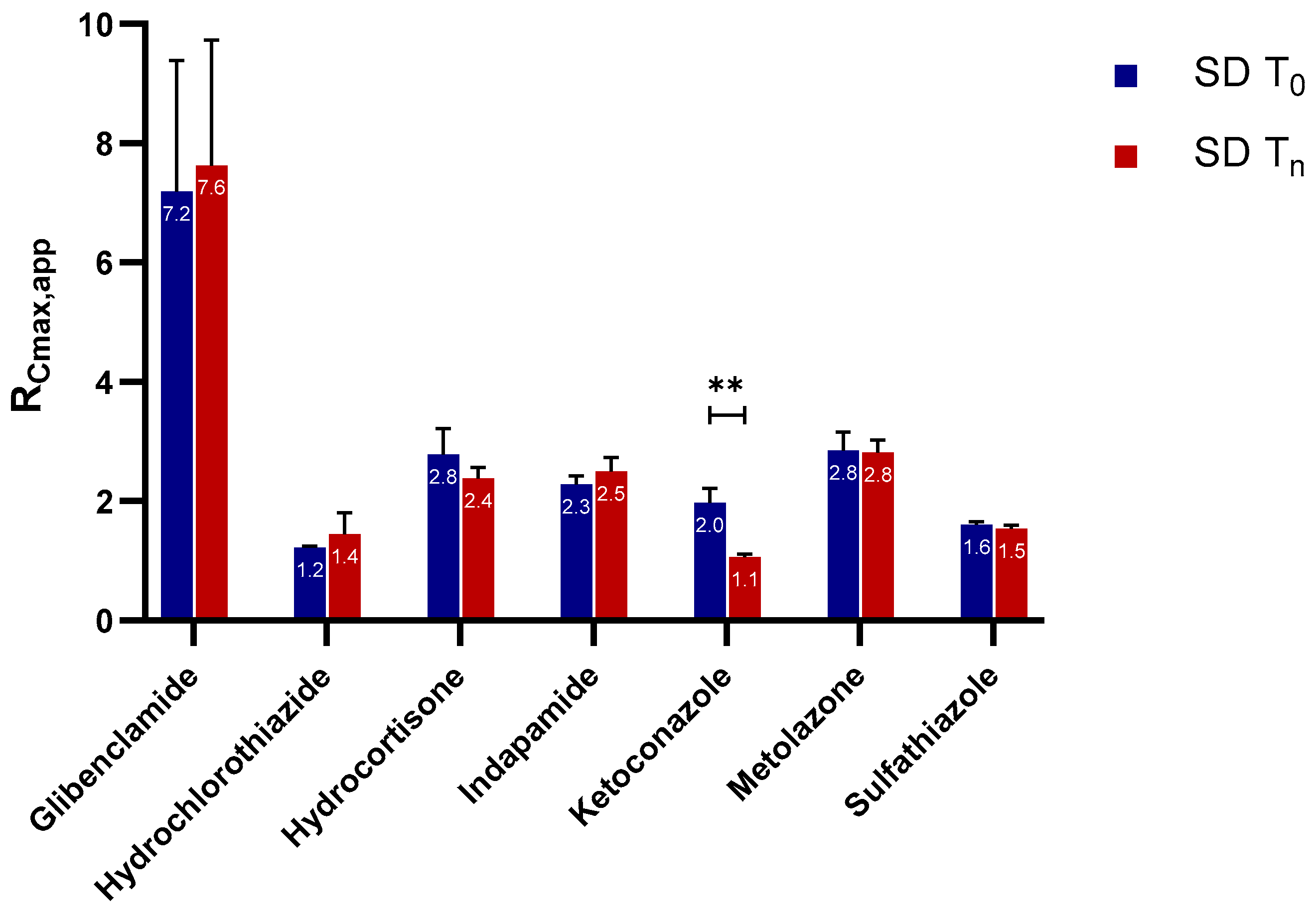
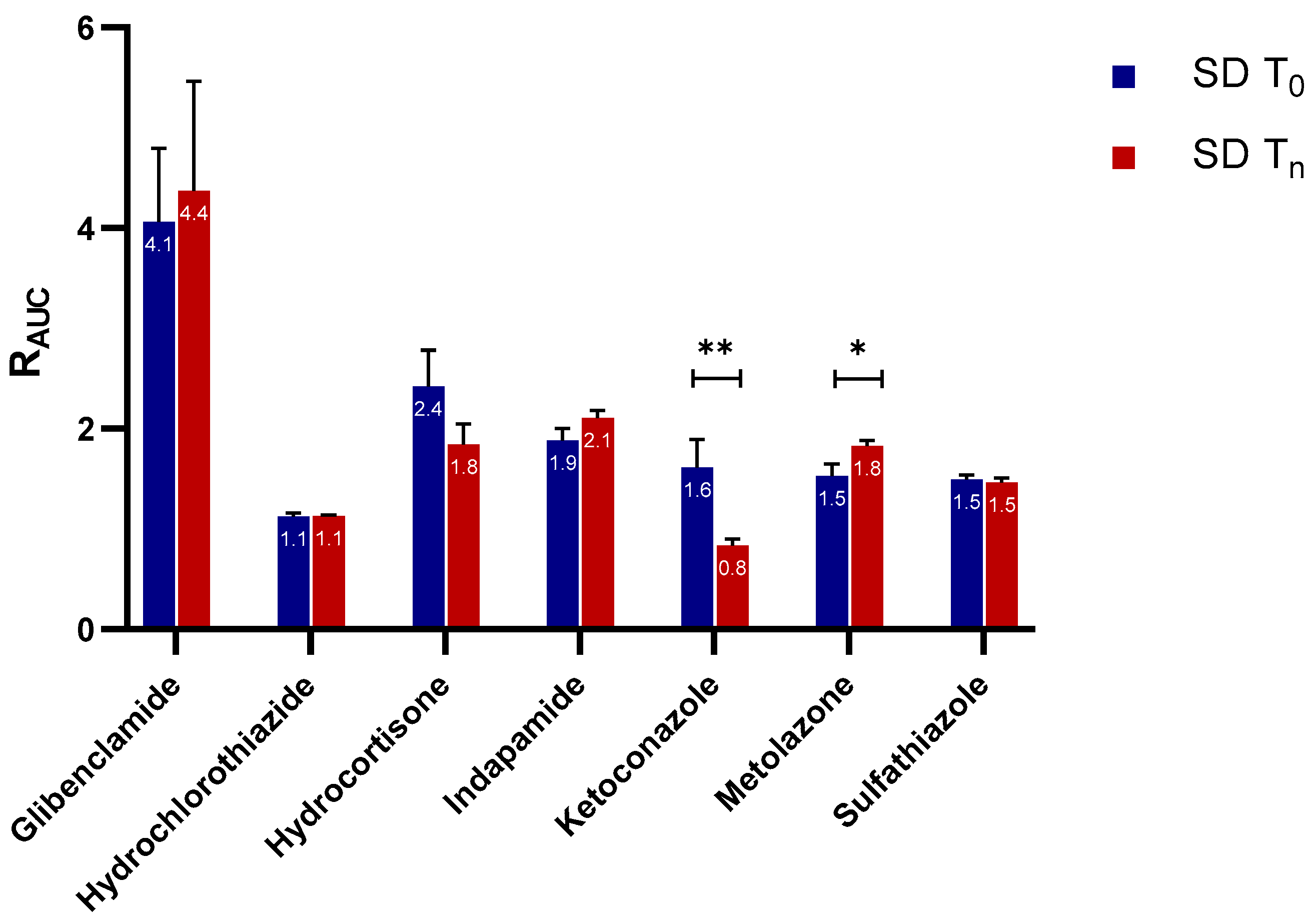
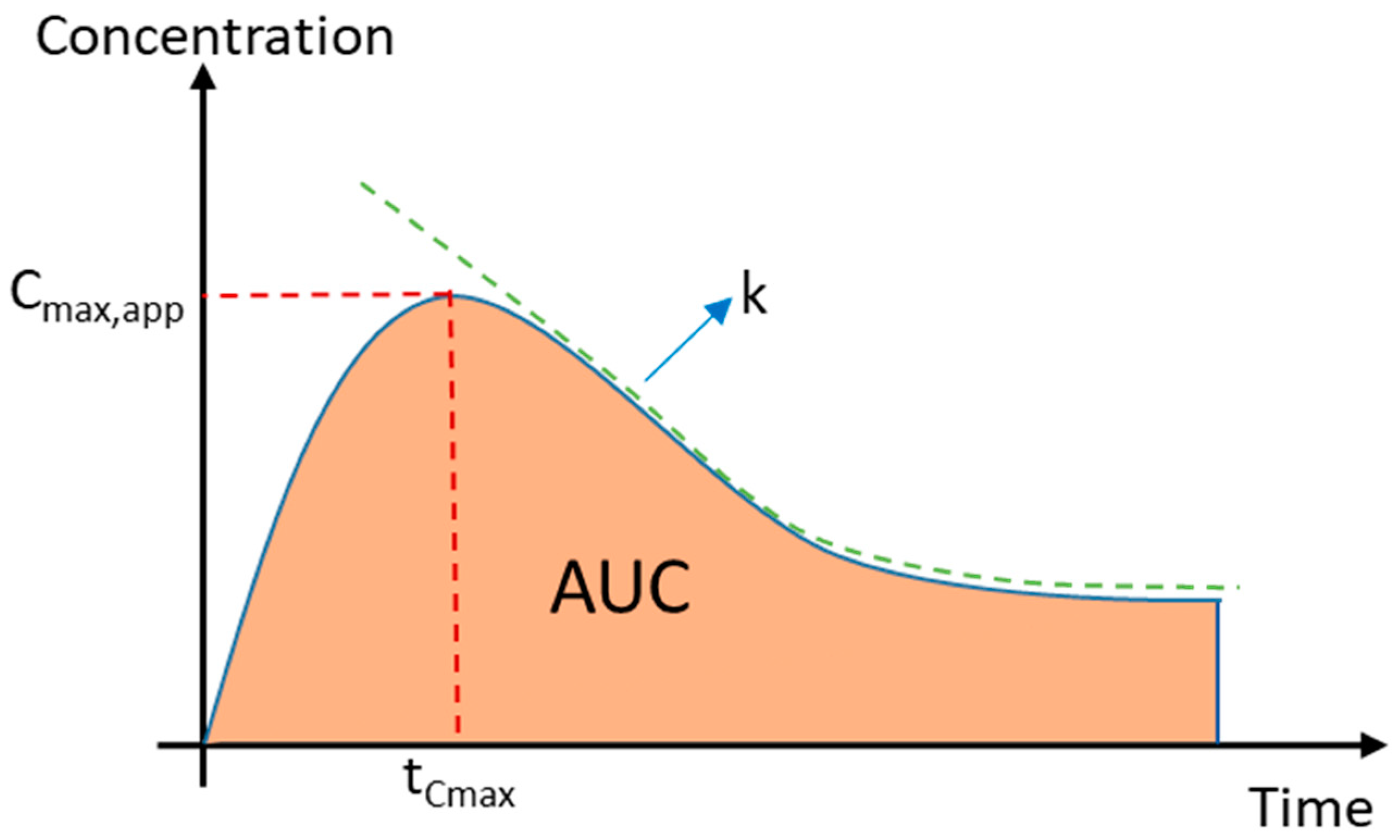
2.2.1. Apparent Cmax and AUC Ratio of Fresh and Aged and/or Crystallized Spray-Dried Solids
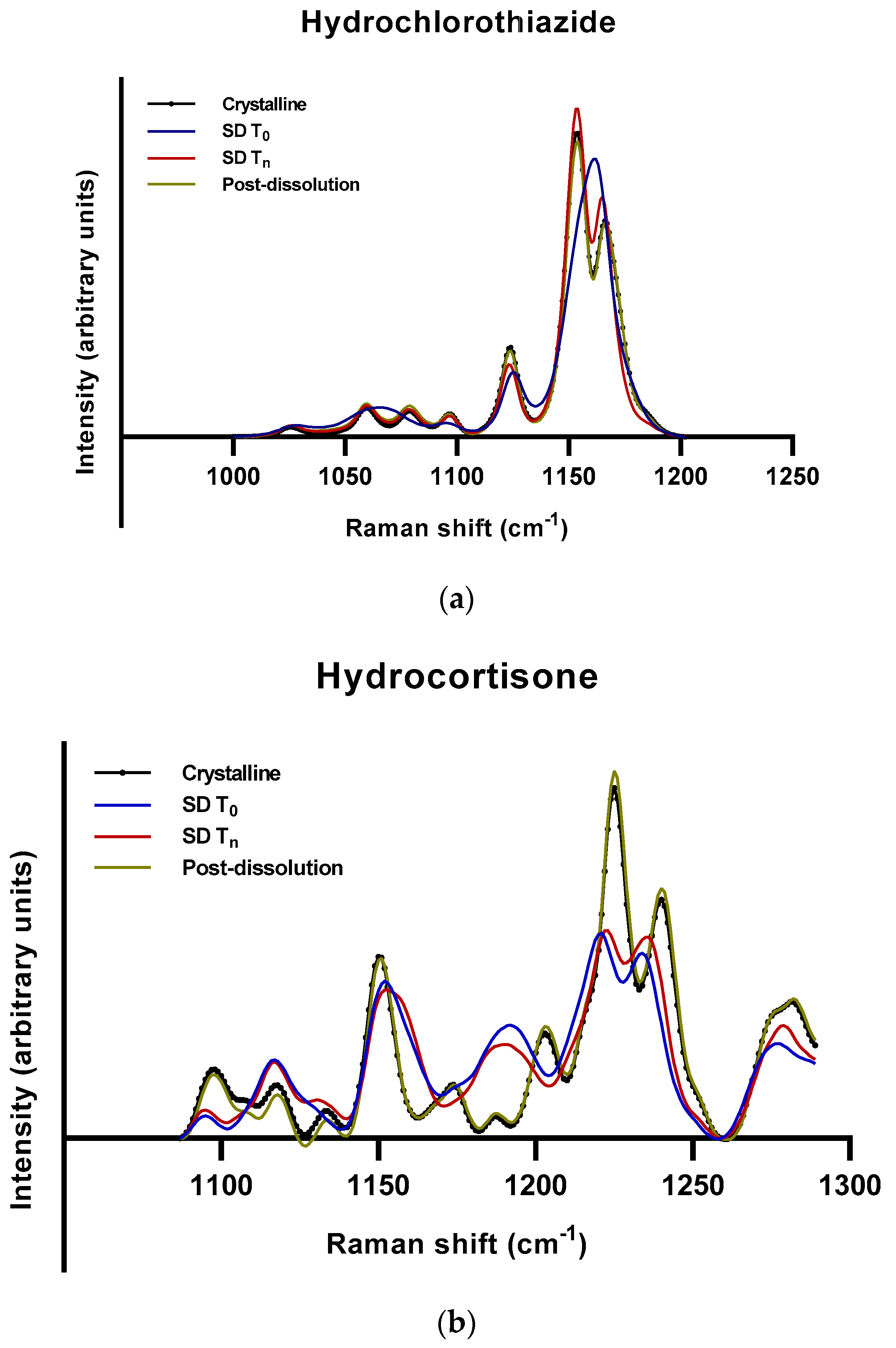
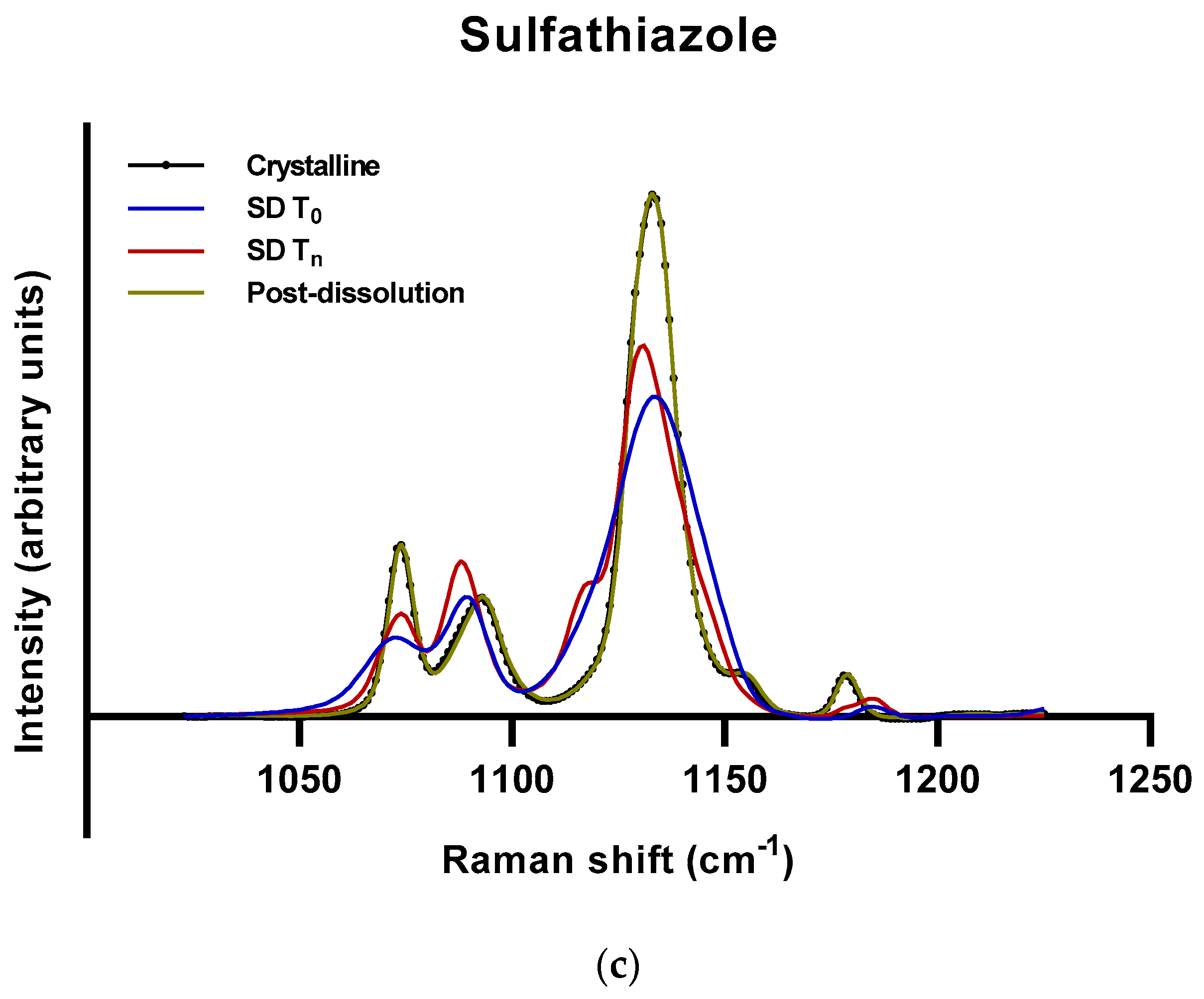
2.2.2. Roles of Crystallization Pathways, Polymorphs, and Impurities on the Supersaturation and/or Kinetic Solubility of Spray-Dried Solids
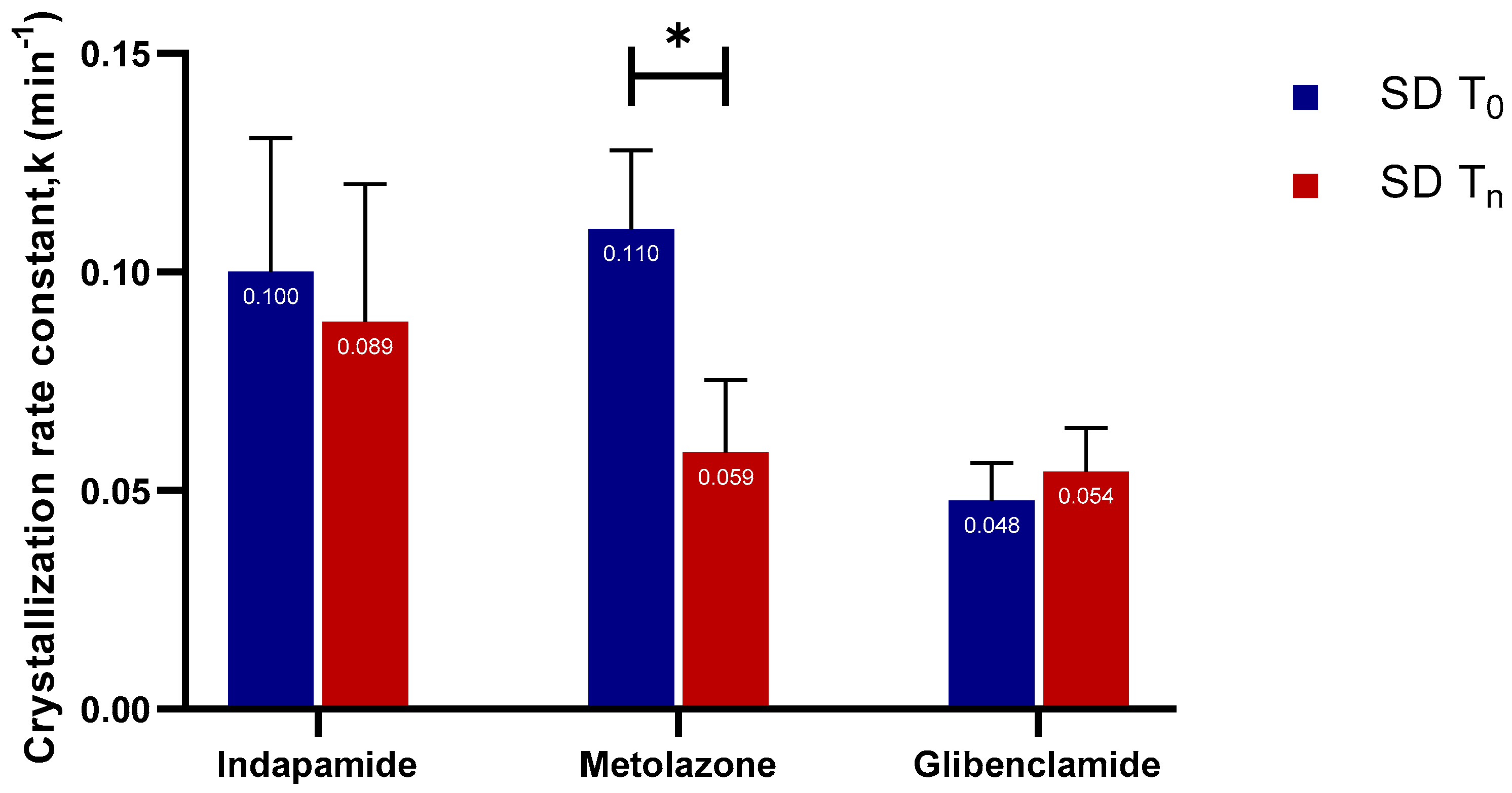
2.2.3. Crystallization Rate Constant (k) and Crystallization Kinetics
3. Materials and Methods
3.1. Materials
3.2. Preparation of Amorphous APIs and Physical Stability Study
3.3. Solid-State Analysis
3.3.1. Powder X-ray Diffraction (PXRD)
3.3.2. Modulated Differential Scanning Calorimetry (MDSC)
3.3.3. Raman Spectroscopy
3.4. Calculation of Amorphous Content Using Raman Spectra
3.5. In Vitro Supersaturation under Non-sink Conditions
3.6. In Vitro Supersaturation Determined by a Solvent Shift Method
3.6.1. Stock Solution Preparation
3.6.2. Solvent Shift Experiment
3.7. Calculation of the Apparent Maximum Concentration, Area under the Curve, and Crystallization Rate Constant
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Andrews, G.P. Advances in solid dosage form manufacturing technology. Philos. Trans. R. Soc. A 2007, 365, 2935–2949. [Google Scholar] [CrossRef] [PubMed]
- Byrn, S.R.; Pfeiffer, R.R.; Stowell, J.G. Solid-State Chemistry of Drugs, 2nd ed.; SSCI, Inc.: West Lafayette, IN, USA, 1999; ISBN1 0967067103. ISBN2 9780967067100. [Google Scholar]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley–VCH: Weinheim, Germany, 2004. [Google Scholar]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today 2012, 9, e71–e174. [Google Scholar] [CrossRef] [PubMed]
- Horter, D.; Dressman, J. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv. Drug Deliv. Rev. 1997, 25, 3–14. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.S.; Dulin, W. A biopharmaceutical classification-based Right-First-Time formulation approach to reduce human pharmacokinetic variability and project cycle time from First-In-Human to clinical Proof-Of-Concept. Pharm. Dev. Technol. 2012, 17, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef]
- Edueng, K.; Mahlin, D.; Bergström, C.A.S. The Need for Restructuring the Disordered Science of Amorphous Drug Formulations. Pharm. Res. 2017, 34, 1754–1772. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility advantage of amorphous pharmaceuticals, part 3: Is maximum solubility advantage experimentally attainable and sustainable? J. Pharm. Sci. 2011, 100, 4349–4356. [Google Scholar] [CrossRef]
- Chawla, G.; Bansal, A.K. A comparative assessment of solubility advantage from glassy and crystalline forms of a water-insoluble drug. Eur. J. Pharm. Sci. 2007, 32, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility advantage of amorphous pharmaceuticals: I. A thermodynamic analysis. J. Pharm. Sci. 2010, 99, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Vyazovkin, S.; Dranca, I. Physical Stability and Relaxation of Amorphous Indomethacin. J. Phys. Chem. B 2005, 109, 18637–18644. [Google Scholar] [CrossRef] [PubMed]
- Vyazovkin, S.; Dranca, I. Effect of Physical Aging on Nucleation of Amorphous Indomethacin. J. Phys. Chem. B 2007, 111, 7283–7287. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Zografi, G. The Relationship Between the Glass Transition Temperature and the Water Content of Amorphous Pharmaceutical Solids. Pharm. Res. 1994, 11, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Andronis, V.; Yoshioka, M.; Zografi, G. Effects of Sorbed Water on the Crystallization of Indomethacin from the Amorphous State. J. Pharm. Sci. 1997, 86, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Katkov, I.I.; Levine, F. Prediction of the glass transition temperature of water solutions: Comparison of different models. Cryobiology 2004, 49, 62–82. [Google Scholar] [CrossRef]
- Knopp, M.M.; Wendelboe, J.; Holm, R.; Rades, T. Effect of amorphous phase separation and crystallization on the in vitro and in vivo performance of an amorphous solid dispersion. Eur. J. Pharm. Biopharm. 2018, 130, 290–295. [Google Scholar] [CrossRef]
- Novakovic, D.; Isomaki, A.; Pleunis, B.; Fraser-Miller, S.J.; Peltonen, L.; Laaksonen, T.; Strachan, C.J. Understanding Dissolution and Crystallization with Imaging: A Surface Point of View. Mol. Pharm. 2018, 15, 5361–5373. [Google Scholar] [CrossRef]
- Bravo-Osuna, I.; Ferrero, C.; Jiménez-Castellanos, M.R. Influence of moisture content on the mechanical properties of methyl methacrylate–starch copolymers. Eur. J. Pharm. Biopharm. 2007, 66, 63–72. [Google Scholar] [CrossRef]
- Dawoodbhai, S.; Rhodes, C.T. The effect of moisture on powder flow and on compaction and physical stability of tablets. Drug Dev. Ind. Pharm. 1989, 15, 1577–1600. [Google Scholar] [CrossRef]
- Boistelle, R.; Astier, J.P. Crystallization mechanisms in solution. J. Cryst. Growth 1988, 90, 14–30. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Zhang, G.G.Z.; Zhou, D.; Gao, Y.; Taylor, L.S. Understanding the Behavior of Amorphous Pharmaceutical Systems during Dissolution. Pharm. Res. 2010, 27, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Greco, K.; Bogner, R. Crystallization of Amorphous Indomethacin during Dissolution: Effect of Processing and Annealing. Mol. Pharm. 2010, 7, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Predecki, P.; Suryanarayanan, R. Use of glancing angle X-ray powder diffractometry to depth-profile phase transformations during dissolution of indomethacin and theophylline tablets. Pharm. Res. 2004, 21, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, M.; Kogermann, K.; Heinz, A.; Aaltonen, J.; Peltonen, L.; Strachan, C.; Yliruusi, J. Better understanding of dissolution behaviour of amorphous drugs by in situ solid-state analysis using Raman spectroscopy. Eur. J. Pharm. Biopharm. 2009, 71, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Edueng, K.; Mahlin, D.; Larsson, P.; Bergström, C.A.S. Mechanism-based selection of stabilization strategy for amorphous formulations: Insights into crystallization pathways. J. Control. Release 2017, 256, 193–202. [Google Scholar] [CrossRef]
- El-Zhry El-Yafi, A.K.; El-Zein, H. Technical crystallization for application in pharmaceutical material engineering: Review article. Asian J. Pharm. Sci. 2015, 10, 283–291. [Google Scholar] [CrossRef]
- Murphy, D.; Rodríguez-Cintrón, F.; Langevin, B.; Kelly, R.C.; Rodríguez-Hornedo, N. Solution-mediated phase transformation of anhydrous to dihydrate carbamazepine and the effect of lattice disorder. Int. J. Pharm. 2002, 246, 121–134. [Google Scholar] [CrossRef]
- Towler, C.S.; Davey, R.J.; Lancaster, R.W.; Price, C.J. Impact of molecular speciation on crystal nucleation in polymorphic systems: The conundrum of gamma glycine and molecular ‘self poisoning’. J. Am. Chem. Soc. 2004, 126, 13347–13353. [Google Scholar] [CrossRef]
- Blagden, N.; Davey, R.J.; Lieberman, H.F.; Williams, L.; Payne, R.; Roberts, R.; Rowe, R.; Docherty, R. Crystal chemistry and solvent effects in polymorphic systems Sulfathiazole. J. Chem. Soc. Faraday Trans. 1998, 94, 1035–1044. [Google Scholar] [CrossRef]
- Weissbuch, I.; Addadi, L.; Leiserowitz, L. Molecular recognition at crystal interfaces. Science 1991, 253, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, F.; Sui, Y.; She, Z.; Zhai, W.; Wang, C.; Deng, Y. Effect of particle size on solubility, dissolution rate, and oral bioavailability: Evaluation using coenzyme Q10 as naked nanocrystals. Int. J. Nanomed. 2012, 7, 5733. [Google Scholar]
- Abu Bakar, M.R.; Nagy, Z.K.; Rielly, C.D.; Dann, S.E. Investigation of the riddle of sulfathiazole polymorphism. Int. J. Pharm. 2011, 414, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Kim, J.K. Pharmaceutical studies on the polymorphism of hydrochlorothiazide. Arch. Pharm. Res. 1984, 7, 47–52. [Google Scholar] [CrossRef]
- Suitchmezian, V.; Jess, I.; Nather, C. Structural, thermodynamic, and kinetic aspects of the trimorphism of hydrocortisone. J. Pharm. Sci. 2008, 97, 4516–4527. [Google Scholar] [CrossRef] [PubMed]
- Meenan, P.A.; Anderson, S.R.; Klug, D.L. The influence of impurities and solvents on crystallization. In Handbook of Industrial Crystallization, 2nd ed.; Butterworth-Heinemann: Boston, MA, USA, 2002; pp. 67–100. [Google Scholar]
- Boistelle, R. Impurity effects in crystal growth from solution. In Interfacial Aspects of Phase Transformations; Springer: Dordrecht, The Netherlands, 1982; Volume 87, pp. 621–638. [Google Scholar]
- Chadha, R.; Gupta, S.; Shukla, G. Crystal habit, characterization and pharmacological activity of various crystal forms of arteether. Acta Pharm. Sin. B 2011, 1, 129–135. [Google Scholar] [CrossRef][Green Version]
- Morantz, D.J.; Mathur, K.K. Inhomogeneous distribution of impurities in organic single crystals and the effect of molecular parameters. J. Cryst. Growth 1972, 16, 147–158. [Google Scholar] [CrossRef]
- Edueng, K.; Bergström, C.A.S.; Gråsjö, J.; Mahlin, D. Long-term physical (in)stability of spray-dried amorphous drugs: Relationship with glass-forming ability and physicochemical properties. 2019. submitted for publication. [Google Scholar]
Sample Availability: Not available. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| API | Time to Start Crystallization (Day) | Time to Complete Crystallization (Day) | Crystallization Initiation Propensity | Crystal Growth | Estimated Amorphous Content at Day 168 (%) | Stability for 168 Days at 75% RH |
|---|---|---|---|---|---|---|
| Indapamide | >168 | >168 | Low | n.d. | 100 | High |
| Metolazone | >168 | >168 | Low | n.d. | 100 | High |
| Glibenclamide | 1 | >168 | High | Slow | 94 | Moderate |
| Hydrocortisone | 1 | >168 | High | Slow | 89 | Moderate |
| Ketoconazole | 1 | 7 | High | Rapid | 0 | Low |
| Hydrochlorothiazide | 1 | 1 | High | Rapid | 0 | Low |
| Sulfathiazole | 1 | 1 | High | Rapid | 0 | Low |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edueng, K.; Mahlin, D.; Gråsjö, J.; Nylander, O.; Thakrani, M.; Bergström, C.A.S. Supersaturation Potential of Amorphous Active Pharmaceutical Ingredients after Long-Term Storage. Molecules 2019, 24, 2731. https://doi.org/10.3390/molecules24152731
Edueng K, Mahlin D, Gråsjö J, Nylander O, Thakrani M, Bergström CAS. Supersaturation Potential of Amorphous Active Pharmaceutical Ingredients after Long-Term Storage. Molecules. 2019; 24(15):2731. https://doi.org/10.3390/molecules24152731
Chicago/Turabian StyleEdueng, Khadijah, Denny Mahlin, Johan Gråsjö, Olivia Nylander, Manish Thakrani, and Christel A.S. Bergström. 2019. "Supersaturation Potential of Amorphous Active Pharmaceutical Ingredients after Long-Term Storage" Molecules 24, no. 15: 2731. https://doi.org/10.3390/molecules24152731
APA StyleEdueng, K., Mahlin, D., Gråsjö, J., Nylander, O., Thakrani, M., & Bergström, C. A. S. (2019). Supersaturation Potential of Amorphous Active Pharmaceutical Ingredients after Long-Term Storage. Molecules, 24(15), 2731. https://doi.org/10.3390/molecules24152731