
Synthesis, Docking and Biological Evaluation of a Novel Class of Imidazothiazoles as IDO1 Inhibitors

 , ,
, ,
Abstract
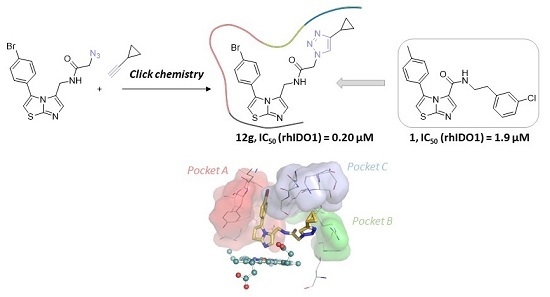
1. Introduction
2. Results
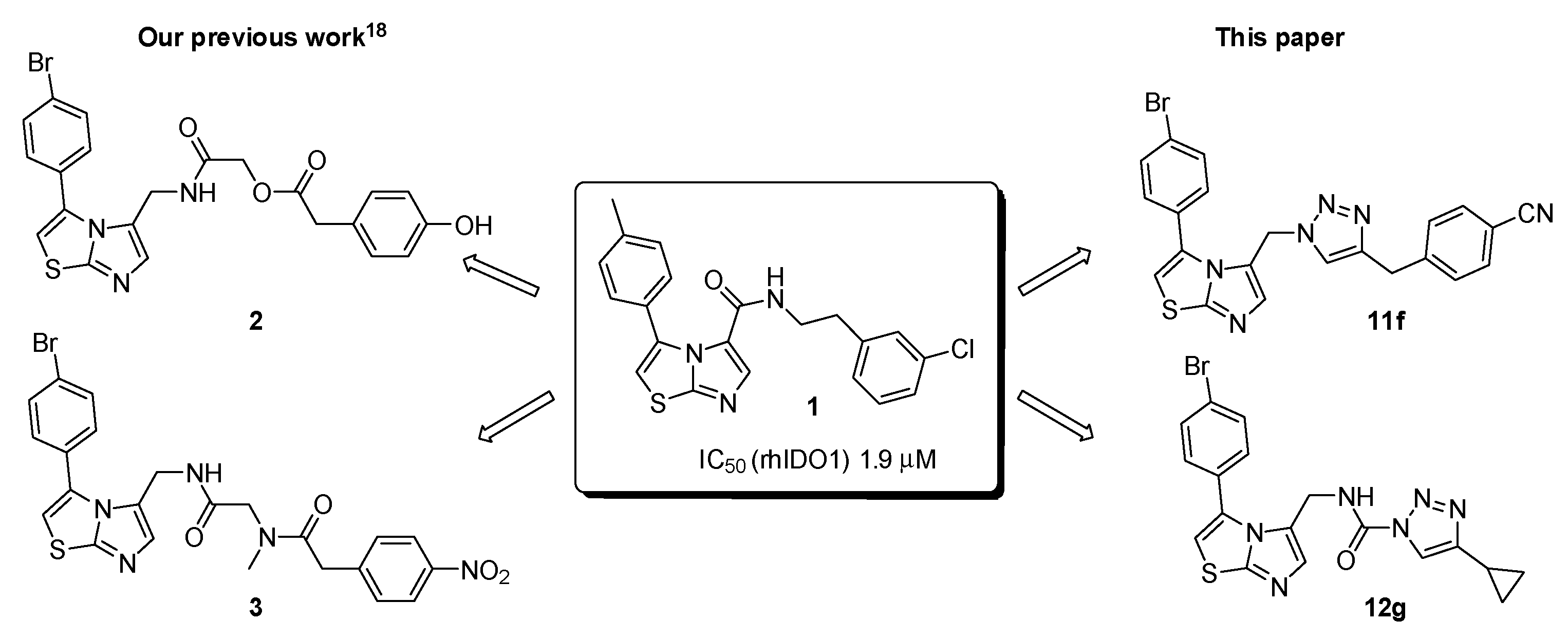
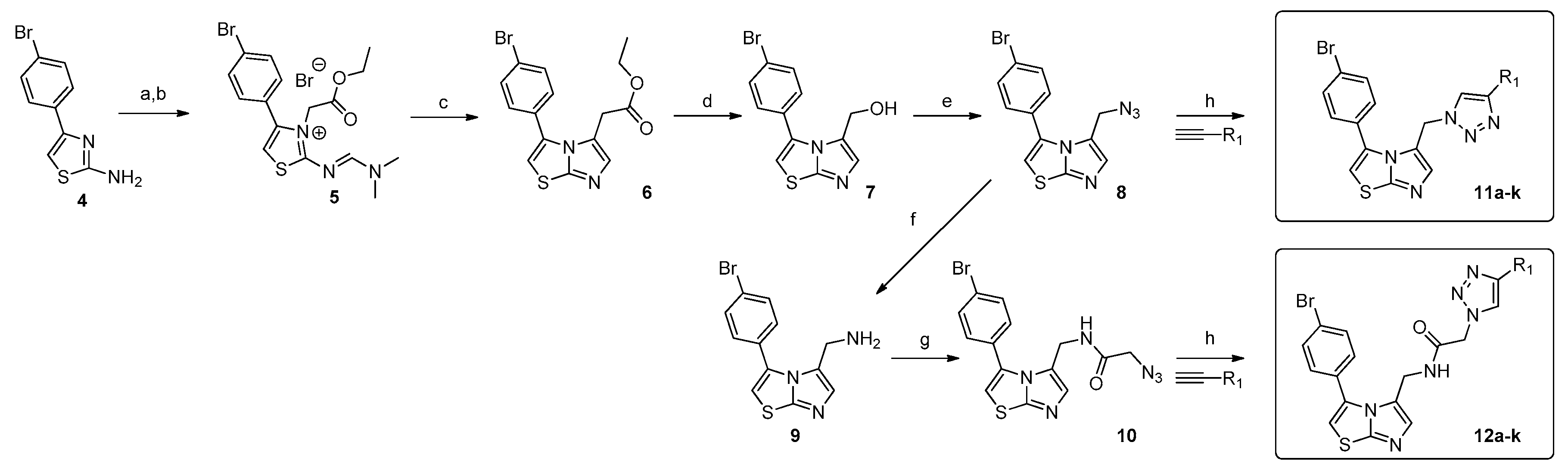
2.1. Chemistry
2.2. Biological Evaluation
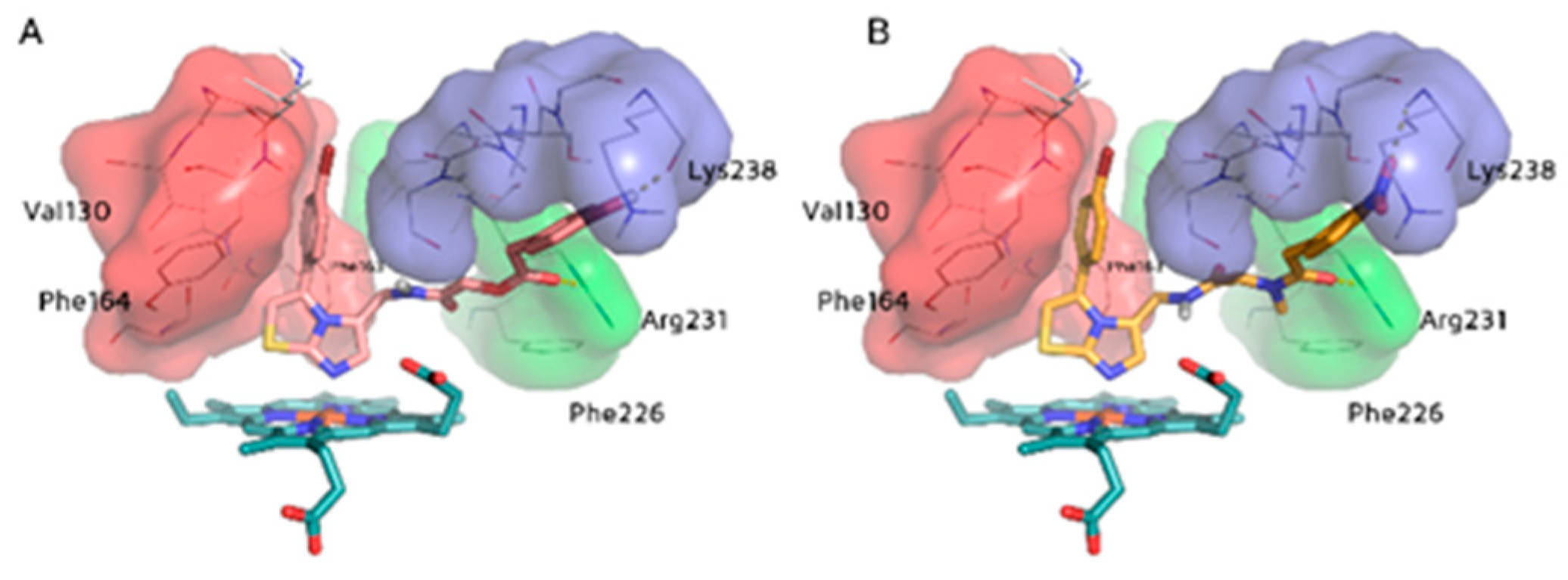
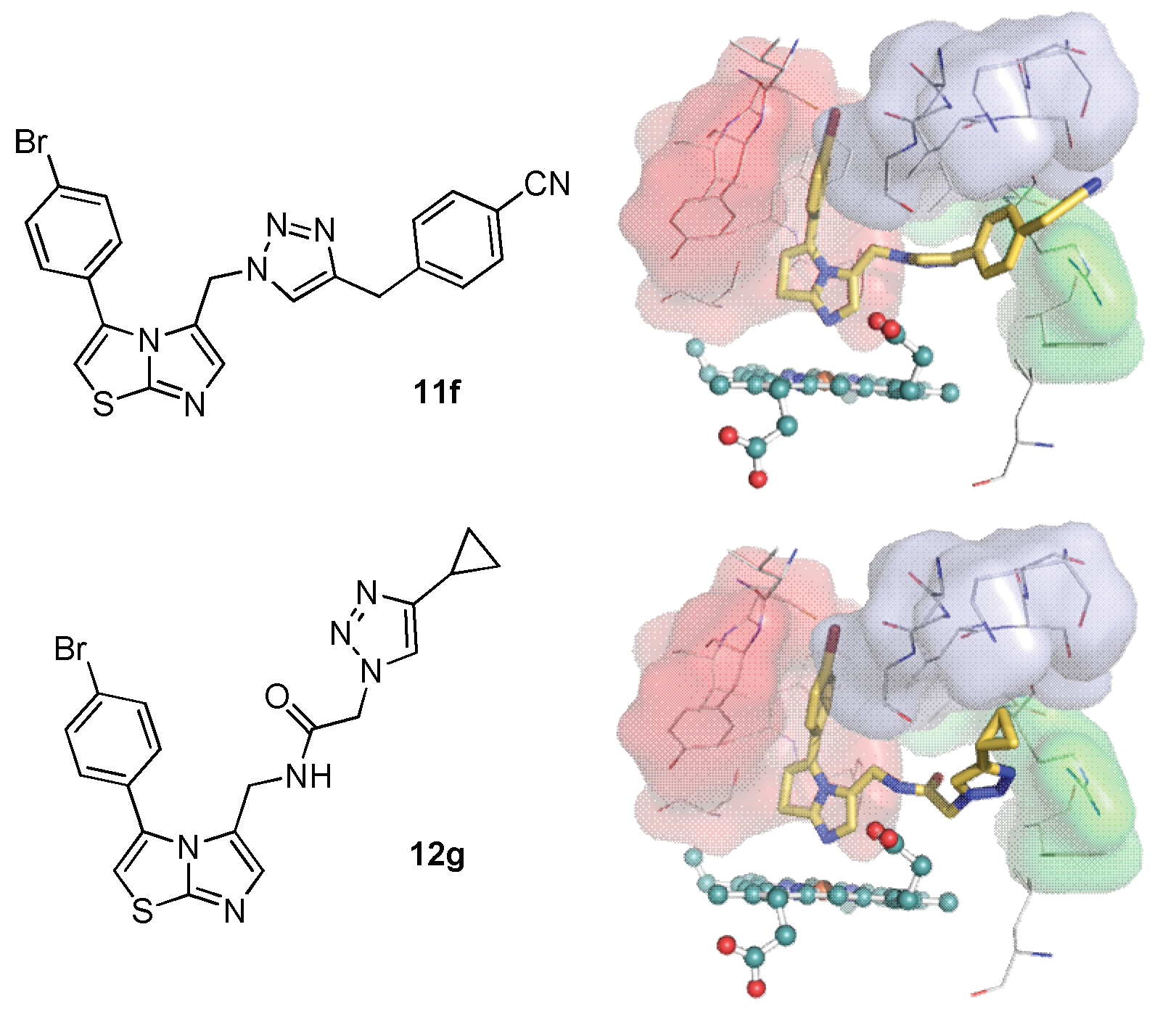
2.3. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. General Chemistry
3.1.2. Synthesis of 2-azido-N-((3-(4-bromophenyl)imidazo[2,1-b]thiazol-5-yl)methyl)acetamide, (10)
3.1.3. General Procedure for the Synthesis of Compounds 11a–11k and 12a–12k
3.1.4. Characterization of Compounds 11a–11k and 12a–12k
3.2. Biology: rhIDO1 Enzymatic Assay
3.3. Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase Pathways of Pathogenic Inflammation and Immune Escape in Cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef]
- Gostner, J.M.; Becker, K.; Uberall, F.; Fuchs, D. The Potential of Targeting Indoleamine 2,3-dioxygenase for Cancer Treatment. Expert Opin. Ther. Targets 2015, 19, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Mondal, A.; Dey, S.; Laury-Kleintop, L.D.; Muller, A.J. Inflammatory Reprogramming with IDO1 Inhibitors: Turning Immunologically Unresponsive ‘Cold’ Tumors ‘Hot’. Trends Cancer 2018, 4, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Smith, C.; DuHadaway, J.B.; Sutanto-Ward, E.; Prendergast, G.C.; Bravo-Nuevo, A.; Muller, A.J. IDO1 is an Integral Mediator of Inflammatory Neovascularization. EBioMedicine 2016, 14, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: Results of the phase 3 ECHO-301/KEYNOTE-252 study. J. Clin. Oncol. 2018, 36, 108. [Google Scholar] [CrossRef]
- Garber, K. A New Cancer Immunotherapy Suffers a Setback. Science 2018, 360, 588. [Google Scholar] [CrossRef]
- Tojo, S.; Kohno, T.; Tanaka, T.; Kamioka, S.; Ota, Y.; Ishii, T.; Kamimoto, K.; Asano, S.; Isobe, Y. Crystal Structures and Structure-Activity Relationships of Imidazothiazole Derivatives as IDO1 Inhibitors. ACS Med. Chem. Lett. 2014, 5, 1119–1123. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and Biology Of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef]
- Zarganes-Tzizikas, T.; Dömling, A. Modern Multicomponent Reactions for Better Drug Syntheses. Org. Chem. Front. 2014, 1, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click Chemistry Reactions in Medicinal Chemistry: Applications Of the 1,3-Dipolar Cycloaddition Between Azides and Alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef]
- Riva, B.; Griglio, A.; Serafini, M.; Cordero-Sanchez, C.; Aprile, S.; Di Paola, R.; Gugliandolo, E.; Alansary, D.; Biocotino, I.; Lim, D.; et al. Pyrtriazoles, a Novel Class of Store-Operated Calcium Entry Modulators: Discovery, Biological Profiling, and in Vivo Proof-of-Concept Efficacy in Acute Pancreatitis. J. Med. Chem. 2018, 61, 9756–9783. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.; Griglio, A.; Aprile, S.; Seiti, F.; Travelli, C.; Pattarino, F.; Grosa, G.; Sorba, G.; Genazzani, A.A.; Gonzalez-Rodriguez, S.; et al. Targeting Transient Receptor Potential Vanilloid 1 (TRPV1) Channel Softly: The Discovery of Passerini Adducts as a Topical Treatment for Inflammatory Skin Disorders. J. Med. Chem. 2018, 61, 4436–4455. [Google Scholar] [CrossRef] [PubMed]
- Pirali, T.; Ciraolo, E.; Aprile, S.; Massarotti, A.; Berndt, A.; Griglio, A.; Serafini, M.; Mercalli, V.; Landoni, C.; Campa, C.C.; et al. Identification of a Potent Phosphoinositide 3-Kinase Pan Inhibitor Displaying a Strategic Carboxylic Acid Group and Development of Its Prodrugs. ChemMedChem 2017, 12, 1542–1554. [Google Scholar] [CrossRef]
- Fallarini, S.; Massarotti, A.; Gesù, A.; Giovarruscio, S.; Coda Zabetta, G.; Bergo, R.; Giannelli, B.; Brunco, A.; Lombardi, G.; Sorba, G.; et al. In Silico-Driven Multicomponent Synthesis Of 4,5- and 1,5-Disubstituted Imidazoles as Indoleamine 2,3-Dioxygenase Inhibitors. Med. Chem. Commun. 2016, 7, 409–419. [Google Scholar] [CrossRef]
- Griglio, A.; Torre, E.; Serafini, M.; Bianchi, A.; Schmid, R.; Coda Zabetta, G.; Massarotti, A.; Sorba, G.; Pirali, T.; Fallarini, S. A Multicomponent Approach in the Discovery of Indoleamine 2,3-Dioxygenase 1 Inhibitors: Synthesis, Biological Investigation and Docking Studies. Bioorg. Med. Chem. Lett. 2018, 28, 651–657. [Google Scholar] [CrossRef]
- Röhrig, U.F.; Majjigapu, S.R.; Grosdidier, A.; Bron, S.; Stroobant, V.; Pilotte, L.; Colau, D.; Vogel, P.; Van den Eynde, B.J.; Zoete, V.; et al. Rational Design of 4-Aryl-1,2,3-Triazoles for Indoleamine 2,3-Dioxygenase 1 Inhibition. J. Med. Chem. 2012, 55, 5270–5290. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, U.F.; Majiigapu, S.R.; Caldelari, D.; Dilek, N.; Reichenbach, P.; Ascencao, K.; Irving, M.; Coukos, G.; Vogel, P.; Zoete, V.; et al. 1,2,3-Triazoles as Inhibitors Of Indoleamine 2,3-Dioxygenase 2 (IDO2). Bioorg. Med. Chem. Lett. 2016, 26, 4330–4333. [Google Scholar] [CrossRef]
- Alexandre, J.A.; Swan, M.K.; Latchem, M.J.; Boyall, D.; Pollard, J.R.; Hughes, S.W.; Westcott, J. New 4-Amino-1,2,3-Triazole Inhibitors of Indoleamine 2,3-Dioxygenase Form a Long-Lived Complex with the Enzyme and Display Exquisite Cellular Potency. ChemBioChem 2018, 19, 552–561. [Google Scholar] [CrossRef]
- OMEGA, version 2.4.6, OpenEye Scientific Software, Santa Fe, N.M. Available online: http://www.eyesopen.com (accessed on 3 March 2018).
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Nicholls, A. Conformer Generation with OMEGA: Learning from the Data Set and the Analysis of Failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- FRED, version 3.0.0, OpenEye Scientific Software, Santa Fe, N.M. Available online: http://www.eyesopen.com (accessed on 3 March 2018).
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B.; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-Atom Contacts and Structure Validation for Proteins and Nucleic Acids. Nucleic Acids Res. 2007, 35, 375–383. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, version 1.3, Schrödinger LLC. 2010. Available online: https://pymol.org/2/ (accessed on 3 March 2018).
Sample Availability: Samples of the compounds 11f and 12g are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  | ||||
|---|---|---|---|---|---|
 | Cpd, Yield (%) | Enzymatic Assay Inhibition (%) at 1 µM a | Cpd, Yield (%) | Enzymatic Assay Inhibition (%) at 1 µM a | |
 | a | 11a, 77% | 30 ± 6.7 | 12a, 81% | 63 ± 4.3 |
 | b | 11b, 51% | 35 ± 2.7 | 12b, 77% | 61 ± 5.5 |
 | c | 11c, 87% | 57 ± 13 | 12c, 46% | 5 ± 1.2 |
 | d | 11d, 43% | 17 ± 1.2 | 12d, 83% | 56 ± 4.7 |
 | e | 11e, 83% | 28 ± 1.5 | 12e, 76% | 50 ± 4.5 |
 | f | 11f, 76% | 74 ± 5.8 | 12f, 68% | 55 ± 7.2 |
 | g | 11g, 44% | 61 ± 7.1 | 12g, 46% | 70 ± 9.2 |
 | h | 11h, 68% | 35 ± 2.4 | 12h, 26% | 63 ± 4.1 |
 | i | 11i, 45% | 0 | 12i, 82% | 45 ± 2.8 |
 | j | 11j, 41% | 81 ± 7.9 | 12j, 42% | 33 ± 4.5 |
 | k | 11k, 48% | 62 ± 5.6 | 12k, 55% | 50 ± 11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serafini, M.; Torre, E.; Aprile, S.; Massarotti, A.; Fallarini, S.; Pirali, T. Synthesis, Docking and Biological Evaluation of a Novel Class of Imidazothiazoles as IDO1 Inhibitors. Molecules 2019, 24, 1874. https://doi.org/10.3390/molecules24101874
Serafini M, Torre E, Aprile S, Massarotti A, Fallarini S, Pirali T. Synthesis, Docking and Biological Evaluation of a Novel Class of Imidazothiazoles as IDO1 Inhibitors. Molecules. 2019; 24(10):1874. https://doi.org/10.3390/molecules24101874
Chicago/Turabian StyleSerafini, Marta, Enza Torre, Silvio Aprile, Alberto Massarotti, Silvia Fallarini, and Tracey Pirali. 2019. "Synthesis, Docking and Biological Evaluation of a Novel Class of Imidazothiazoles as IDO1 Inhibitors" Molecules 24, no. 10: 1874. https://doi.org/10.3390/molecules24101874
APA StyleSerafini, M., Torre, E., Aprile, S., Massarotti, A., Fallarini, S., & Pirali, T. (2019). Synthesis, Docking and Biological Evaluation of a Novel Class of Imidazothiazoles as IDO1 Inhibitors. Molecules, 24(10), 1874. https://doi.org/10.3390/molecules24101874