Abstract
Ugi four component reaction (Ugi-4CR) isocyanide-based multicomponent reactions were used to synthesize diN-substituted glycyl-phenylalanine (diNsGF) derivatives. All of the synthesized compounds were characterized by spectroscopic and spectrometric techniques. In order to evaluate potential biological applications, the synthesized compounds were tested in computational models that predict the bioactivity of organic molecules by using only bi-dimensional molecular information. The diNsGF derivatives were predicted as cholinesterase inhibitors. Experimentally, all of the synthesized diNsGF derivatives showed moderate inhibitory activities against acetylcholinesterase (AChE) and poor activities against butyrylcholinesterase (BuChE). Compound 7a has significant activity and selectivity against AChE, which reveals that the diNsGF scaffold could be improved to reach novel candidates by combining other chemical components of the Ugi-4CR in a high-throughput combinatorial screening experiment. Molecular docking experiments of diNsGF derivatives inside AChE suggest that these compounds placed the phenylalanine group at the peripheral site of AChE. The orientations and chemical interactions of diNsGF derivatives were analyzed, and the changeable groups were identified for future exploration of novel candidates that could lead to the improvement of diNsGF derivative inhibitory activities.
1. Introduction
Multicomponent reactions (MCRs) have clear advantages with respect to conventional multistep reactions due to their potential to synthesize complicated organic molecules with low costs and quick reaction times [1,2]. It is common to use MCRs to obtain biologically relevant scaffold structures, mainly isocyanide-based MCRs, where the Ugi four component reaction (Ugi-4CR) has been particularly important for these endeavors [3,4,5,6,7,8]. Ugi-4CRs are easy and straightforward to perform; isocyanides derived from enantiopure α-amino acids (isocyanoacetates) could serve as substrates for this reaction. There are many successful reports that have obtained relevant biological scaffolds by using isocyanoacetate-based Ugi-4CRs [9,10].
The use of Ugi-4CRs by using isocyanoacetates as substrates has been documented in some diverse examples in the last few decades. For instance, Bonne et al. reported the development of a three-component synthesis of 5-methoxyoxazoles and a four-component synthesis of furopyrrolones on the basis of the unique reactivity of methyl α-(p-nitrophenyl)-α-isocyanoacetate [11]. On the other hand, Nenajdenko et al. reported the use of a 4-methyl-2,6,7-trioxabicyclo(2.2.2)octyl derivative (OBO-ester) of isocyanoacetic acid instead of an ethyl ester of R-isocyanoacids compounds in Ugi-4CR with subsequent deprotection that gives access to dipeptides with preserved configuration at the C-terminal amino acid [12]. The same authors synthesized fully protected natural and unnatural N-acetylcysteine, dipeptide Cys–Gly, glutathione, and homoglutathione derivatives by the Ugi-4CR using various benzylthio aldehydes and ketones as carbonyl building blocks. They used the ethyl isocyanoacetate as a peptide synthesis precursor, but they replaced it by the OBO-ester of the isocyanoacetic acid in cases where higher reactivity of an isocyanide was required [13]. In another report, Zhou et al. developed a protocol in which cyclohexylisocyanide and methyl 2-isocyanoacetate were used as convertible isocyanides for the Ugi/de-Boc/cyclization/Suzuki synthesis of biaryl-substituted 1,4-benzodiazepine-2,5-diones [14]. Finally, it is relevant to mention the approach described by Kotha [15] involving solid–liquid phase-transfer-catalyzed dialkylation of ethyl isocyanoacetate to synthesize symmetrically α,α-disubstituted α-amino acids.
The use of isocyanoacetates as Ugi-4CR substrates is relevant at present for the synthesis of novel bioactive compounds. In this work, diN-substituted glycyl-phenylalanine (diNsGF) derivatives were synthesized by using isocyanoacetate-based Ugi-4CRs. To identify possible biological applications of the novel compounds, they were tested in computational predictive models previously developed by our group. In these models, their possible role as cholinesterase inhibitors was detected. Our compounds were evaluated against the enzymes acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). Experimental biological activities and molecular modeling experiments demonstrate that the diNsGF derivatives have potential for the design of future potent AChE inhibitors, where the presence of the phenylalanine group guarantees the selectivity of these compounds for AChE versus BuChE.
2. Results
2.1. Synthesis of diN-Substituted Glycyl-Phenylalanine (diNsGF) Derivatives
An isocyanide MCR approach based on the well-known Ugi-4CR protocol [16] was implemented for the development of new diNsGF derivatives. Two different elements of diversity were varied for the preparation of these compounds, i.e., the carboxylic acid and the amino components, while the DL-Phenylalanine ethyl ester isocyanide and paraformaldehyde were unchanged as shown in Scheme 1. Five novel diNsGF derivatives were synthesized in moderate to good yields by a two-step procedure, followed by either N-terminus or C-terminus deprotection (Scheme 1). The pure compounds were obtained after flash chromatographic purification and their purity was confirmed by spectroscopy characterization.
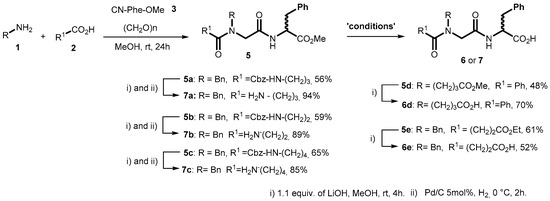
Scheme 1.
The Ugi four component reaction (Ugi-4CR) for the synthesis of diNsGF derivatives.
In an initial approach, a solution-phase Ugi-4CR protocol was employed for the synthesis of diNsGF derivatives, which proved to be suitable for β, γ, and δ amino acids (compounds 7a–c) but gave poor results when all reagents were added in one step. To solve this problem, we preformed the imine for 1 h and then the acid and isocyanide component was added, which provided good efficiency in the multicomponent preparation of the previously designed diNsGF compounds as compared with the literature [17]. Then, Cbz-protected compounds 7a–c were selected for the ester deprotection, employing a basic hydrolysis with LiOH in methanolic solution. Followed by catalytic hydrogenation using palladium in carbon, the zwitterion pseudo-peptide amino acids 7a–c were succesfully obtained. By using the previously described methodology, succinic acid monomethyl ester and benzoic acid were employed to the synthesis of compounds 6d and 6e, respectively. It was possible to obtain two novel dicarboxylic acid derivatives in good overall yield after both ester protected groups in the structure skeleton were deprotected, employing a basic hydrolysis with LiOH in methanolic solution (Scheme 1).
The novel diNsGF derivatives were fully characterized by spectroscopy (FT-IR, 1H, 13C-NMR, and two-dimensional NMR) and mass spectrometry. The FT-IR spectra of 7a–c and 6d–e were compared to each other, clearly showing the incorporation of the peptidic moiety for all Ugi adducts (see the Supplementary Material). For example, the new bands appearing at 2930 cm−1 and 1700 cm−1 correspond to the Csp3-H and the carbonyl stretching, respectively. Similarly, there are other new bands, such as the amine stretching and the band around 1420 cm−1, which were also attributed to aliphatic segments for 7a–c.
The NMR spectra were similar for all compounds. The signal of chiral carbon that appears at 56 ppm from the Attached Proton Test (APT) experiment was the key to the elucidation of the structure, especially in the Heteronuclear Single Quantum Coherence (HSQC) experiment. This signal allowed us to begin the assigment of carbons and protons, and, therefore, to define the structures with the help of another experiment (mono and two-dimensional). The mass of each compound was obtained with a deviation less than 3 ppm, and spectra did not show any fragmentation. This illustrates the stability of this class of compounds against the strong conditions in which these experiments were performed (see the Supplementary Material Figures S1–S40).
2.2. Predictions of the AchE Inhibitory Activities by Using Two-Dimensional (2D) Autocorrelation Models
The idea was to develop novel compounds with a potential biological activity designed by two possible approaches. Firstly, compounds were designed with both amino and carboxylic groups to improve the interactions with the target by acting like a zwitterion pseudo-peptide amino acid (compounds 7a–c). Secondly, similar compounds were designed with two carboxylic groups (compounds 6d and 6e). Following our protocol, we planned to identify potential activities for the synthesized compounds.
With this in mind, we used the 2D autocorrelation models recently reported in reference [18] to predict the potential AChE inhibitory activities of the synthesized diNsGF derivatives. These models were trained with diverse AChE inhibitors with activities between pIC50 2 and 10, where IC50 was measured in M. According to these models, compounds 7a–c and 6d–e were predicted to have activities between pIC50 6 and 7.5, which indicates that they could inhibit the AChE enzyme.
A question arose about the capacities of our 2D autocorrelation models to predict diNsGF derivatives. For this, we tested whether the applicability domain (AD) of these models included the novel synthesized structures. The AD was previously estimated for the 2D autocorrelation models to evaluate their ability to tolerate new molecules by using William’s plots [19]. We could confirm that the diNsGF derivatives were inside the AD if the leverages of their independent 2D autocorrelation variables were within the range of the leverages of the molecules used for training the models. It means that calculated leverages should be below the warning leverage h*, which had values around 0.34 and 0.38 for the previously developed models.
The calculated leverages for compounds 7a–c and 6d–e were below 0.34 for the tested models. An exception was found only for compound 6e, which had a leverage value of 0.44 (higher than the warning leverage h*) for one of the 2D autocorrelation models. However, it was included within the AD range of the remaining models.
After these computational experiments, we concluded that our 2D autocorrelation models predicted that the diNsGF derivatives are potential AChE inhibitors. We consider these predictions to be reliable since the studied compounds are included in the AD of the predictive models.
2.3. Results of the AChE and BuChE Inhibitory Activities
The previously synthesized compounds were tested against AChE and BuChE. As shown in Table 1, the new diNsGF compounds had AChE inhibitory activities in a micromolar range. AChE inhibitory potency was expressed as the half of maximal inhibitory concentration, IC50. The inhibitory activity was determined by the modified Ellman’s method. The alkaloid galantamine was used as the reference drug. Table 1 shows that most of the studied compounds were able to inhibit in vitro both enzymes AChE and BuChE but in a very broad range of concentrations. At the same time, it proves that all new synthesized diNsGF compounds are significantly more active towards AChE than BuChE, with the exception of compound 6e. This result confirms that the previously reported 2D autocorrelation models had the capacity to identify novel active compounds against AChE. With respect to the moderate activities found for the synthesized compounds, we consider that the newly synthesized diNsGF derivatives can serve as base molecules for the design of novel selective AChE inhibitors.

Table 1.
Inhibitory activity of diNsGF derivatives from AChE and BuChE enzymes. 1
Compounds 6d, 7a, 7b, and 7c showed good AChE inhibitory activities, with IC50 values of 10.64, 3.67, 41.31, and 12.56 µM, respectively. Meanwhile, compound 6e showed the lowest IC50 value against BuChE (135 µM). Compounds 7a, 7b, and 7c have free amine groups attached to carbon chains of three, two, and four methylene groups, respectively. The differences in IC50 values for these compounds reflect possible effects of the chain length for improving the interactions that their amino groups may have with residues of the AChE binding site. The distance achieved by the amino group through three methylene groups (compound 7a) was adequate to achieve stable interactions between the inhibitor and the enzyme, enhancing its inhibitory capacity and its selectivity for AChE.
An in silico analysis (ADME/Tox) was done to predict some properties of the novel diNsGF derivatives (Table 1). The properties that have values within the acceptable ranges are: (i) logPo/w, which estimates how the behavior of each molecule will be in terms of lipophilicity [20] and permeability though the membrane [21]; (ii) logS, which indicates the drug transport, uptake, and bioavailability [22]; (iii) human oral absorption, and (iv) logKp (skin permeability), which indicates possibilities of administration and effects on bioavailability [23], a risk assessment of hazardous chemicals, and measurements of transdermal drug release rates [24,25]. The relevance of in silico ADME/Tox assays is that these theoretical features help to delimit the space within which there is a greater probability of finding compounds with a tendency to achieve a successful clinical development [26].
2.4. Results of the Kinetic Assays
Based on the obtained results in the cholinesterase inhibition, and with the aim to assess the kinetic mode of AChE inhibition of the target compounds, the most active compound 7a was subjected to a kinetic study. For this purpose, the rate of enzyme activity was measured at eight different concentrations of substrate ATC. The obtained Michaelis–Menten parameters along with the Lineweaver–Burk plots (Figure 1) allowed us to conclude that compound 7a would display a mixed-type inhibition against AChE with variation in Km and Vmax values. The compound 7a showed Km values of 0.045, 0.125, and 1.049 μM and Vmax of 2.95 × 10−3, 2.56 × 10−4, and 6.72 × 10−4 mol/min, respectively.
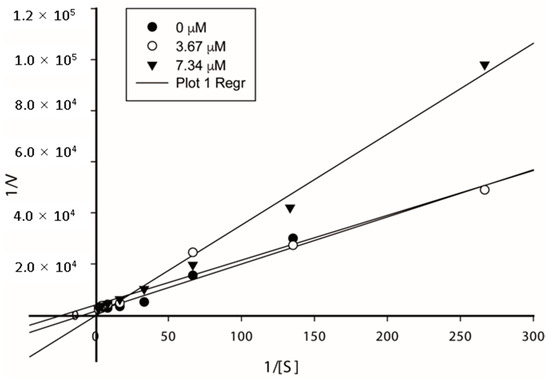
Figure 1.
The Lineweaver–Burk plot of AChE (0.02 U) with an acetylthiocholine substrate in the absence and presence of inhibitor 7a.
2.5. Docking Results
The synthesized diNsGF derivatives were predicted as AChE inhibitors by using 2D autocorrelation models, and this result was experimentally confirmed. Their activities were in the micromolar range; therefore, the synthesized compounds could be classified as moderate inhibitors. After the identification of the diNsGF derivatives as AChE inhibitors, the next step was the rational modification of substituents to improve the activities. With this in mind, it had become pertinent to study the putative interactions between our synthesized compounds and the binding site of AChE.
Therefore, the synthesized diNsGF derivatives (R and S enantiomers) were docked inside the AChE binding site to understand their ligand–protein interactions at the atomic level. The docking results are depicted in Figure 2. As can be seen in Figure 2A, all compounds (considering one or both enantiomers) placed the phenylalanine group at the peripheral site of the AChE pocket, forming a hydrogen bond (HB) between the carboxylate of the ligands and the backbone NH group of the residue F288, and π–π stacking interactions between the phenyl group of the ligands and the indole of the residue W279. This π–π stacking interaction with the residue W279 has been previously described for the AChE–donepezil complex [27] and other AChE inhibitors reported in the literature [28,29]. As in other AChE inhibitors, this interaction explains why the majority of diNsGF derivatives exhibited selectivity for AChE over BuChE. In BuChE, the peripheral site contains alanine instead of tryptophane (A277 instead of W279); therefore, the π–π stacking interaction between diNsGF derivatives and the peripheral site of AChE cannot be established between these compounds and BuChE.
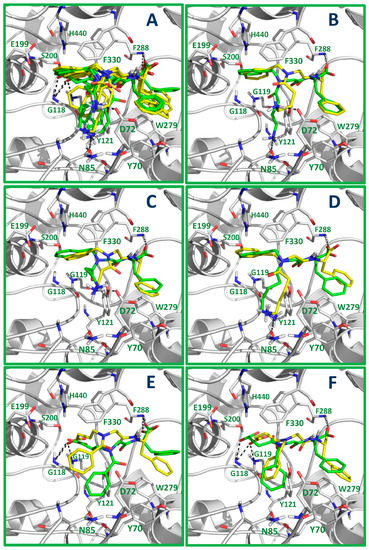
Figure 2.
Molecular docking models for diNsGF derivatives inside the AChE binding site. Docking poses of all the compounds (A); Docking poses of compounds 7a (B), 7b (C), 7c (D), 6d (E), and 6e (F), where R and S enantiomers are represented in green and yellow, respectively. Protein residues that form the main interactions with the inhibitors are in stick representation.
The compounds that contain an NH3+ group (7a, 7b, and 7c) can form HBs with the side chain CO of the residue N85 (Figure 2B,D) or a salt bridge with the side chain carboxylate of D72 (Figure 2B,C). At the same time, these compounds form a cation–π interaction between the phenyl of the benzylamine of the ligand and the positively charged imidazole of the residue H440 (Figure 2B–D). An HB interaction was formed with the side chain CO of the residue N85 when the NH3+ group was a large linear chain containing four methylene groups (compound 3, Figure 2D), while a salt bridge was formed with D72 when the NH3+ group was a short linear chain containing two methylene groups (compound 2, Figure 2C). When the NH3+ group was a linear chain containing three methylene groups, both possibilities were obtained (one for each enantiomer) according to docking experiments (compound 1, Figure 2B). The compounds that contain carboxyalkyl groups at the N of the benzoylamide (6d) or benzylamine (6e) can form HBs with the side chain CO of the residues G118 and G119 (Figure 2E,F), while the phenyl groups are oriented to the pocket near the residues Y121, D72, and W84.
3. Materials and Methods
3.1. Preparation of Compounds
The starting material was purchased from either Merck or Sigma-Aldrich. Monitoring of the reactions was through thin layer chromatography (TLC) using silica gel 60 (Merck, Darmstadt, Germany). Melting points (uncorrected) were determined on an Electrothermal IA9100 melting point apparatus (Stone, Staffs, UK). All products were characterized by spectral data (IR, 1H and 13C NMR, MS). 1H and 13C NMR spectra (400 MHz for 1H and 100 MHz for 13C) were recorded on an AM-400 spectrometer (Bruker, Rheinstetten, Germany) using DMSO-d6 as solvents. The chemical Shift (δ) of the solvent is reported at 2.5 for 1H-NMR and 40.09 for 13C-NMR, δ and J values are reported in ppm and Hz, respectively. The signals are represented as follows: Singlet (s), doublet (d), triplet (t), multiplet (m). IR spectra (KBr pellets, 500–4000 cm−1) were obtained from a NEXUS 670 FT-IR spectrometer (Thermo Nicolet, Madison, WI, USA). High-resolution mass spectra (HRMS-ESI) were obtained from Thermo Fisher Scientific Exactive Plus mass spectrometer. The analysis was performed at heater temperature, 50 °C; sheath gas flow, 5; sweep gas flow rate, 0, and spray voltage, 3.0 kV at negative mode. The accurate mass measurements were performed at a resolution of 140,000. The absorbance of biological assay was measured at 405 nm using a microtiter plate reader (Multiskan EX, Thermo, Vantaa, Finland). ADME7Tox prediction was made with QuikProp module of the Schrodinger suite (Schrödinger, LLC: New York, NY, USA).
(S)-Ethyl-2-Amino-3-Phenylpropionate: This compound was prepared following the methodology reported by Guo et al. [30]. SOCl2 (5.88 mL) was added dropwise at 0 °C to a suspension of L-phenylalanine 1 (4.46 g, 27.0 mmol) in 50 mL ethanol. The resulting mixture was then refluxed for 4 h. The volatiles were concentrated under reduced pressure and the resulting crude was treated with saturated aqueous Na2CO3 until pH 8–9. The mixture was extracted with Ethyl Acetate (2 × 50 mL). The combined organic phase was dried over Na2SO4 and evaporated under reduced pressure, obtaining the product as a white solid (5.6 g, 97%).
N-Formyl-l-Phenylalanine Ethyl ester: This compound was prepared following the methodology reported by Monge et al. [31]. Acetic anhydride (14 mL, 21 mmol, 7 equiv.) was added dropwise to a solution of (S)-Ethyl-2-Amino-3-Phenylpropionate (4.0 g, 21 mmol, 1 equiv.) in Formic acid (39 mL, 21 mmol, 50 equiv.) at 0 °C. The reaction was stirred at room temperature (rt) for 2 h, quenched with H2O (40 mL), and then extracted with CH2Cl2 (5 × 70 mL). The combined organic layers were washed with NaHCO3 (sat.) (5 × 70 mL) brine, dried (Na2SO4), and evaporated under reduced pressure to give the product as a colorless oil (4.4 g, 95%).
Ethyl 2-isocyano-3-phenylpropionate: This compound was prepared following the methodology reported by Monge et al. [31]. Et3N (14 mL, 20 mmol, 5 equiv.) was added to a stirred solution of N-Formamide ester (4.4 g, 20 mmol, I equiv.) in dry THF (50 mL) at −20 °C under a nitrogen atmosphere. A solution of POCl3 (2.3 mL, 20 mmol, 1.3 equiv.) in dry THF (10 mL) was added dropwise and the reaction mixture was allowed to warm to 0 °C. After stirring for 2 h, ice-cold water (65 mL) was added and the mixture was extracted with Et2O (4 × 50 mL). The combined organic layers were washed with brine, dried (Na2SO4), and evaporated under reduced pressure, and the resulting crude product was purified by flash column chromatography on silica gel to give the product as a yellow oil (3.2 g, 79%).
General Ugi-4CR-Based Procedure: A suspension of the amine (0.5 mmol, 1 equiv.) and the aldehyde (0.5 mmol, 1 equiv.) in MeOH (1 mL) was stirred for 1 h at rt. Et3N (0.5 mmol, 1 equiv.) was added when α-amino acid methyl ester hydrochlorides were employed. The carboxylic acid (0.5 mmol, 1 equiv.) and the isocyanide (0.5 mmol, 1 equiv.) were then added, and the reaction mixture was stirred at rt for 24 h. The volatiles were concentrated under reduced pressure and the resulting crude product was dissolved in 50 mL of CH2Cl2. The organic phase was washed sequentially with a saturated aqueous solution of citric acid (25 mL), aqueous 10% NaHCO3 (25 mL), and brine (25 mL), and then dried over anhydrous Na2SO4 and concentrated under reduced pressure. The resulting amorphous solid was used in the deprotection step without further purification [17].
General procedure for ethyl ester deprotection: A solution of lithium hydroxide (0.25 mmol, 2 equiv.) in water (1 mL) was added to a solution of the appropriate ester (0.25 mmol, 1 equiv.) in THF (1 mL). The mixture was homogenized with methanol (1 mL) and stirred at rt for 2 h. The solvent was evaporated to dryness and the residue was dissolved in water. The pH was set acidic with 1 M HCl and the desired product was extracted with EtOAc. The combined organic phases were washed with brine, dried with anhydrous Na2SO4, and the solvent removed in vacuo. The crudes were purified employing column chromatography. The desired products were confirmed for thin-layer chromatography (TLC) revelated with bromocresol green indicator [32].
General procedure for Cbz deprotection [33]: Pd/C (5 wt%, 5 mol%) was added to a solution of 5a–c (0.96 mmol) in THF:MeOH (1:1, 10 mL) and the mixture was stirred under a hydrogen atmosphere at rt for 1 h. After that, the mixture was filtered through celite (Merck, Darmstadt, Germany), the filtrate was evaporated, and the resultant residue was purified by column chromatography on silica gel (CHCl3:MeOH 9:1) to produce compounds 7a–c.
10-Amino-2,6-dibenzyl-4,7-dioxo-3,6-diazadecanoic acid (compound 7a): a suspension of the benzylamine (54.6 µL, 0.5 mmol) and the p-formaldehyde (15 mg, 0.5 mmol) in MeOH (1 mL) was stirred for 1 h at rt. After, 4-(((benzyloxy)carbonyl) amino)butanoic acid (118.55 mg, 0.5 mmol) and the ethyl 2-isocyano-3-phenylpropionate (101.5 mg, 0.5 mmol) were then added, and the reaction mixture was stirred at rt for 24 h according to the Ugi-4CR-based procedure. The resulting Cbz and ethyl protected compound was unprotected through the general deprotection procedure. Flash column chromatography purification (DCM:MeOH 5:1) allowed us to obtain 7a as a mixture of conformers. Yield: 93% (52 mg); white solid; m.p.: 212.5–214.5 °C; IR-FT (KBr, cm−1): 3440, 3025, 2921, 2853, 1638, 1625, 1580, 1400; 1H NMR (400 MHz, DMSO-d6) δ 7.86 (d, J = 7.6 Hz, 1H), 7.29 (d, J = 7.1 Hz, 2H), 7.25–7.12 (m, 8H), 4.61 (d, J = 15.2 Hz, 1H), 4.25 (d, J = 15.0 Hz, 1H), 4.20 (s, 1H), 3.90 (d, J = 17.6 Hz, 1H), 3.70 (d, J = 17.4 Hz, 1H), 3.13 (d, J = 10.7 Hz, 1H), 2.83 (d, J = 10.7 Hz, 1H), 2.79 (s, 3H), 2.51 (s, 2.11H), 1.81 (s, 2H); 13C NMR (100 MHz, DMSO) δ 174.80, 172.83, 168.10, 139.99, 138.23, 129.92, 129.62, 129.19, 128.21, 128.14, 127.47, 127.31, 126.26, 56.37, 50.27, 50.20, 38.73, 38.08, 29.81, 23.27. HRMS (ESI, m/z): calculated for C22H26N3O4− [M − H]− 396.1929 found 396.1926.
9-Amino-2,6-dibenzyl-4,7-dioxo-3,6-diazanonanoic acid (compound 7b); a suspension of the benzylamine (54.6 µL, 0.5 mmol) and the p-formaldehyde (15 mg, 0.5 mmol) in MeOH (1 mL) was stirred for 1 h at rt. After, 3-(((benzyloxy)carbonyl) amino)propanoic acid (118.6 mg, 0.5 mmol) and the ethyl 2-isocyano-3-phenylpropionate (101.5 mg, 0.5 mmol) were then added, and the reaction mixture was stirred at rt for 24 h according to the Ugi-4CR-based procedure. The resulting Cbz and ethyl protected compound was unprotected through the general deprotection procedure. Flash column chromatography purification (DCM:MeOH 5:1) allowed us to obtain 7b as a mixture of conformers. Yield: 88% (51 mg); white solid; m.p.: 213.4–215.1 °C; IR-FT (KBr, cm−1): 3482, 3054, 3017, 2921, 1653, 1596, 1387; 1H NMR (400 MHz, DMSO-d6) δ 7.88 (d, J = 8.08 Hz, 1Ha), 7.30 (d, J = 7.40 Hz, 2H), 7.23–7.17 (m, 8H), 4.55 (d, J = 15.11 Hz, 1H), 4.22 (m, 1H), 4.20 (d, J = 15.48 Hz, 1H), 3.91 (d, J = 17.43 Hz, 1H), 3.77 (d, 1H), 3.12 (d, J1 = 4.13 Hz, J1 = 13.62 Hz, 1H), 2.99 (m, 2H), 2.82 (d, J1 = 8.88 Hz, J1 = 13.64 Hz, 1H), 2.69 (m, 2H). 13C NMR (100 MHz, DMSO) δ 174.45, 171.48, 167.50, 139.81, 137.82, 129.70, 129.19, 128.85, 128.33, 128.25, 127.58, 127.42, 126.26, 56.26, 50.21, 49.79, 38.15, 35.88, 30.89. HRMS (ESI, m/z): calculated for C21H24N3O4− [M − H]−, 382.1772 found 382.1771.
11-Amino-2,6-dibenzyl-4,7-dioxo-3,6-diazaundecanoic acid (compound 7c); a suspension of the benzylamine (54.6 µL, 0.5 mmol) and the p-formaldehyde (15 mg, 0.5 mmol) in MeOH (1 mL) was stirred for 1 h at rt. After, 5-(((benzyloxy)carbonyl) amino)pentanoic acid (125.6 mg, 0.5 mmol) and the ethyl 2-isocyano-3-phenylpropionate (101.5 mg, 0.5 mmol) were then added, and the reaction mixture was stirred at rt for 24 h according to the Ugi-4CR-based procedure. The resulting Cbz and ethyl protected compound was unprotected through the general deprotection procedure. Flash column chromatography purification (DCM:MeOH 5:1) allowed us to obtain 7c as a mixture of conformers. Yield: 85% (56 mg); white solid; m.p.: 218.9–220.5 °C; IR-FT (KBr, cm−1): 3448, 3062, 3028, 2926, 1659, 1640, 1627, 1387, 1209, 1026; 1H NMR (400 MHz, DMSO-d6) δ 7.75 (d, J = 7.93 Hz, 1H), 7.32–7.16 (m 10H), 4.45 (d, J = 15.18 Hz, 1H), 4.28 (d, J = 15.08 Hz, 1H), 4.21 (d, J1 = 8.01 Hz, J2 = 12.81 Hz, 1H), 3.85 (d, J = 17.24 Hz, 1H), 3.75 (d, J = 17.79 Hz, 1H), 3.09–3.11 (d, 1H), 2.85 (dd, J1 = 8.15 Hz, J2 = 13.49 Hz, 1H), 2.72 (m, 2H), 2.26 (m, 2H), 1.55 (m, 4H). 13C NMR (101 MHz, DMSO) δ 174.36, 173.14, 167.53, 139.59, 138.18, 129.70, 129.21, 128.85, 128.30, 128.20, 128.12, 127.20, 126.29, 56.02, 50.13, 49.60, 38.70, 38.12, 31.90, 27.22, 21.81. HRMS (ESI, m/z): calculated for C23H28N3O4− [M − H]−, 410.2085 found 410.2083.
6-Benzamido-2-benzyl-4-oxo-3,6-diazadecanedioic acid (compound 6d); triethylamine (69.7 µL, 0.5 mmol) was added to a suspension of methyl 4-aminobutanoate (76.8 mg, 0.5 mmol) in MeOH (1 mL), stirring for 10 min. Then, p-formaldehyde (15 mg, 0.5 mmol) was added and the mixture was stirred for 1 h at rt. After, benzoic acid (61 mg, 0.5 mmol) and the ethyl 2-isocyano-3-phenylpropionate (101.5 mg, 0.5 mmol) were added, and the reaction mixture was stirred at rt for 24 h according to the Ugi-4CR-based procedure. The resulting ethyl and methyl protected compound was unprotected through the general deprotection procedure. Flash column chromatography purification (DCM:MeOH 3:1) allowed us to obtain compound 6d as a mixture of conformers. Yield: 70% (61 mg); white solid; m.p.: 144.3–146 °C; IR-FT (KBr, cm−1): 3443, 2921, 2850, 1727, 1667, 1638, 1612; 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H,), 8.34 (d, J = 8.18 Hz, 1H), 7.46–7.12 (m, 10H), 4.50 (m, 1H, H), 3.77 (d, J = 5.13 Hz, 2H), 3.20 (m, 2H), 3.06 (m, 1H), 2.88 (m, 1H), 2.23 (t, 2H), 1.73 (t, 2H). 13C NMR (101 MHz, DMSO) δ 174.56, 173.12, 171.58, 168.58, 137.93, 136.86, 129.70, 129.53, 128.70, 128.62, 127.03, 126.88, 53.88, 51.66, 45.49, 37.02, 31.68, 22.35, HRMS (ESI, m/z): calculated for C22H23N2O6− [M − H]−, 411.1562 found 411.1571.
2,6-Dibenzyl-4,7-dioxo-3,6-diazadecanedioic acid (compound 6e); a suspension of the benzylamine (54.6 µL, 0.5 mmol) and the p-formaldehyde (15 mg, 0.5 mmol) in MeOH (1 mL) was stirred for 1 h at rt. After, 4-ethoxy-4-oxobutanoic acid (64 µL, 0.5 mmol) and the ethyl 2-isocyano-3-phenylpropionate (101.5 mg, 0.5 mmol) were added, and the reaction mixture was stirred at rt for 24 h according to the Ugi-4CR-based procedure. The resulting ethyl protected compound was unprotected through the general deprotection procedure. Flash column chromatography purification (DCM:MeOH 3:1) allowed us to obtain compound 6e as a mixture of conformers. Yield: 52% (60 mg); white solid; m.p.: 163.4–164.8 °C; IR-FT (KBr, cm−1): 3461, 2923, 2858, 1745, 1724, 1667, 1638; 1H NMR (400 MHz, DMSO-d6) δ 12.42 (s, 1H), 8.37 (d, J = 8.31 Hz, 1H), 7.31–7.13 (m, 10H), 4.54 (dd, J1 = 4.04 Hz, J2 = 9.00 Hz, 1H), 4.38 (d, J = 15.22 Hz, 1H), 4.28 (d, J = 15.23 Hz, 1H), 3.90 (d, J = 17.51 Hz, 1H), 3.82 (d, 1H), 3.10 (dd, J1 = 4.61 Hz, J2 = 13.77 Hz, 1H), 2.87 (dd, J1 = 4.76 Hz, J2 = 9.09 Hz, 1H), 2.62 (t, J = 6.50 Hz, 1H), 2.47 (t, J = 6.53 Hz, 1H), 2.42 (m, 2H). 13C NMR (101 MHz, DMSO) δ 174.39, 173.16, 172.52, 168.38, 137.98, 137.94, 129.61, 129.13, 128.82, 128.65, 128.06, 127.48, 127.31, 126.90, 53.83, 49.56, 49.40, 37.24, 29.67, 27.79, HRMS (ESI, m/z): calculated for C22H23N2O6− [M − H]−, 411.1562 found 411.1570.
3.2. Computational Predictions of Bioactivities
The molecular structures of the diNsGF derivatives (compounds 7a, 7b, 7c, 6d, and 6e) were sketched in MarvinSketch (ChemAxon, Budapest, Hungary). Two-dimensional (2D) autocorrelation descriptors with lags between 1 and 8 were calculated for these structures by using the Dragon software v3.0 (DRAGON, Milano Chemometrics, Milano, Italy). Two-dimensional (2D) autocorrelation descriptors capture the interdependence between atomic properties (masses, van der Waals volumes, electronegativities, and polarizabilities) in a molecular graph [34,35]. The activities of the diNsGF derivatives were predicted by using computational 2D autocorrelation models recently reported by our group, trained for the identification of potential AChE inhibitors [18]. These models were trained with a broad dataset of around 300 AChE inhibitors, and they were validated by using different statistical tests. We previously demonstrated the ability of these models to predict diverse AChE inhibitors; therefore, they were used in the current work for predicting the potential AChE inhibitory activities of the synthesized diNsGF derivatives.
3.3. AChE and BuChE Assays
AChE hydrolyzes acetylcholine, which mediates transmission at some synapses in the brain, including synapses involved in the acquisition of short-term memory. Therefore, AChE inhibitors lead to the restoration of the levels of acetylcholine, which is needed for patients with a failing memory, such as Alzheimer’s disease (AD) patients. AChE inhibitors, such as donepezil [27], galantamine [36], and rivastigmine [37], have been approved for AD treatment during the last few decades, but the presence of several side effects has been a challenge for the prolonged use of these drugs [38,39,40]. In the clinical treatment of AD, selective AChE inhibitors have shown better therapeutic effects when compared with nonselective inhibitors. Therefore, the development of novel selective AChE inhibitors is an important approach for AD treatment.
Inhibitory activities were evaluated using the methods by [41]. The samples were dissolved in 96-well plates (50 μL) in phosphate buffer (K2HPO4 8 mM, NaH2PO4 2.3 mM, NaCl 150 mM, and 0.5% Tween 20, pH 7.6). A solution of acetylcholinesterase (AChE)/butyrylcholinesterase (BuChE) (50 μL, 0.20 unit/mL) from Electrophorus electricus and equine serum was added to the same buffer, respectively. The assay solutions, except for the substrate, were pre-incubated with the enzymes for 30 min at room temperature (rt). Then, the substrate solution, composed of Na2HPO4 (40 mM), acetylthiocholine (ATC)/butyrylthiocholine (BTC) (0.24 mM), and 5,5′-dithio-bis(2-nitrobenzoic acid) (0.2 mM, DTNB, Ellman’s reagent), was added. The absorbance of the reaction was measured at 405 nm for 5 min. AChE/BuChE inhibition was determined for each compound. The enzymatic activity was calculated as a percentage and then compared with the control, which was composed of buffer and an enzyme solution. The compounds were tested in dilution intervals ranging from 500 to 0.98 µg/mL. Each assay was run in triplicate and each reaction was repeated three times. The IC50 values were determined using a regression analysis. The alkaloid galantamine was used as the reference compound. The galantamine IC50 was developed in three courses ranging from 250 to 0.0.031 μg/mL with an average of 0.57 μM for AchE and 7.96 μM for BuChE. DMSO was diluted to a concentration in excess of 1 in 10,000, and no inhibitory action in neither AChE nor BuChE was detected in separate prior experiments.
3.4. Kinetic Assays
The kinetic studies of the most active compound were performed according to Ellman’s method with some modifications to the 96-well plate. The assays were carried out in a total volume of 200 μL per well containing AChE from Electrophorus electricus (0.20 unit/mL) in PBS, the synthesized compound in DMSO, PBS at IC50, 2× IC50 concentration, and substrate solution. The general procedure was as follows: AChE and the synthesized compounds 7a to IC50 and twice concentrations in PBS were mixed and incubated at rt for 30 min. After incubation, substrate from 3.8 × 10−3–0.48 mM was added into the mixture. The reaction was recorded for 10 min measuring the absorbance at 405 nm using a Microtiter plate reader (Multiskan EX, Thermo). Data were plotted using GraphPad Prism v4.03 software (GraphPad Software Inc., San Diego, CA, USA) to obtain the Michaelis–Menten constant (Km) and maximal velocity (Vmax) values. Km values were expressed in μM and Vmax as mol/min/mg protein and correspond to the mean ± SD of three independent assays in triplicate.
3.5. Molecular Docking
Here, we report the docking experiments that were performed to understand how diNsGF derivatives interact with AChE, which is a necessary task for future rational design for improving AChE inhibitory activities.
Docking was used to obtain the putative binding modes of the diNsGF derivatives inside the AChE binding site. Glide SP and XP methods, contained in Maestro software v9.0 (Schrödinger, LLC: New York, NY, USA), were employed to perform the docking experiments [42,43]. Glide docking searches ligand orientations by using hierarchical filters in a defined receptor grid space. Filters include a sampling of the positional, conformational, and orientational space of the ligands with a final evaluation of their energy interactions with the protein residues inside the grid.
The protein structural coordinates were extracted from the X-ray crystal structure of the AChE–donepezil complex (Protein Data Bank (PDB) code 1EVE). This structure was modified with the Protein Preparation Wizard in Maestro (Schrödinger, LLC: New York, NY, USA): hydrogen atoms were added, followed by the adjustment of bond orders, protonation states for protonatable residues were adjusted to match pH = 7.4, and water molecules were deleted. Finally, it was subjected to geometry optimization by using an OPLS2005 force field [44].
The sketched structures of diNsGF derivatives were converted into three-dimensional structures with the Maestro software facilities. A grid box of 18 Å × 18 Å × 18 Å was centered on the center of mass of the donepezil in the modified protein structure. Ligand minimizations in the receptor field were carried out with a distance-dependent dielectric of 2.0. Afterwards, the lowest energy poses were subjected to a Monte Carlo procedure that sampled the nearby torsional minima, and the best poses were determined by using the GlideScore scoring function, where terms for buried polar groups and steric clashes were considered [45].
4. Conclusions
In this work, we report the synthesis of novel diNsGF derivatives through Ugi-4CR in mild conditions, a simple starting material, and moderate yields. The synthesized compounds 7a–7c and 6d–6e were purified and characterized. Their potential use as bioactive compounds was explored by using predictive models. Previously constructed 2D autocorrelation models identified possible AChE inhibitory activities in our compounds; therefore, experiments were prepared to confirm this hypothesis.
The obtained compounds were tested against AChE and BuChE enzymes, showing moderate inhibition values and good selectivity. The binding modes of the compounds were studied by using molecular docking and the essential interactions between the compounds and the AChE binding site were characterized. These models suggest that rational modifications of the substituents at the N of the glycine in the diNsGF derivatives (R and R1 groups) based on the described AChE-ligand models provide a basis for the development of novel AChE inhibitors through the use of the Ugi-4CR in a few steps. This facilitates the possibility of exploring many different substituents to improve the interactions between the diNsGF derivatives and the AChE binding site. For the design of novel diNsGF derivatives in the future, researchers could benefit from the main advantages of Ugi-4CRs, such as quick reaction times, diminished costs, and the prevention of time-consuming product purification.
Other dipeptidic derivatives could be explored in the future with an aromatic amino acid instead of the phenylalanine to preserve the π–π stacking interactions with the residue W279 (an interaction that was previously described for the AChE–donepezil complex). The potential of the diNsGF derivatives as AChE inhibitors was demonstrated in this work. Their orientations inside the AChE binding site indicate that the aromatic group present in the phenylalanine is important for the AChE inhibitory activity. In the future, we could explore the use of different aromatic groups in the design of diN-substituted glycyl-tyrosine and glycyl-tryptophan derivatives as AChE inhibitors.
Supplementary Materials
The following are available online. Figure S1: FT-IR spectrum in KBr for 7a. Figure S2: 400 MHz 1H-NMR spectrum in DMSO-d6 for 7a. Figure S3: 400 MHz 13C-NMR spectrum in DMSO-d6 for 7a. Figure S4: 400 MHz APT spectrum in DMSO-d6 for 7a. Figure S5: 400 MHz HSQC spectrum in DMSO-d6 for 7a. Figure S6: 400 MHz HMBC spectrum in DMSO-d6 for 7a. Figure S7: 400 MHz COSY spectrum in DMSO-d6 for 7a. Figure S8: High-resolution mass spectrometry-ESI spectrum of 7a. Figure S9: FT-IR spectrum in KBr for 7b. Figure S10: 400 MHz 1H-NMR spectrum in DMSO-d6 for 7b. Figure S11: 400 MHz 13C-NMR spectrum in DMSO-d6 for 7b. Figure S12: 400 MHz APT spectrum in DMSO-d6 for 7b. Figure S13: 400 MHz HSQC spectrum in DMSO-d6 for 7b. Figure S14: 400 MHz HMQC spectrum in DMSO-d6 for 7b. Figure S15: 400 MHz COSY spectrum in DMSO-d6 for 7b. Figure S16: High-resolution mass spectrometry-ESI spectrum of 7b. Figure S17: FT-IR spectrum in KBr for 7c. Figure S18: 400 MHz 1H-NMR spectrum in DMSO-d6 for 7c. Figure S19: 400 MHz 13C-NMR spectrum in DMSO-d6 for 7c. Figure S20: 400 MHz APT spectrum in DMSO-d6 for 7c. Figure S21: 400 MHz HSQC spectrum in DMSO-d6 for 7c. Figure S22: 400 MHz HMBC spectrum in DMSO-d6 for 7c. Figure S23: 400 MHz COSY spectrum in DMSO-d6 for 7c. Figure S24: High-resolution mass spectrometry-ESI spectrum of 7c. Figure S25: FT-IR spectrum in KBr for 6d. Figure S26: 400 MHz 1H-NMR spectrum in DMSO-d6 for 6d. Figure S27: 400 MHz 13C-NMR spectrum in DMSO-d6 for 6d. Figure S28: 400 MHz APT spectrum in DMSO-d6 for 6d. Figure S29: 400 MHz HSQC spectrum in DMSO-d6 for 6d. Figure S30: 400 MHz HMQC spectrum in DMSO-d6 for 6d. Figure S31: 400 MHz COSY spectrum in DMSO-d6 for 6d. Figure S32: High-resolution mass spectrometry-ESI spectrum of 6d. Figure S33: FT-IR spectrum in KBr for 6e. Figure S34: 400 MHz 1H-NMR spectrum in DMSO-d6 for 6e. Figure S35: 400 MHz 13C-NMR spectrum in DMSO-d6 for 6e. Figure S36: 400 MHz APT spectrum in d6-DMSO for 6e. Figure S37: 400 MHz, HSQC spectrum in DMSO-d6 for 6e. Figure S38: 400 MHz, HMQC spectrum in DMSO-d6 for 6e. Figure S39: 400 MHz, COSY spectrum in DMSO-d6 for 6e. Figure S40: High-resolution mass spectrometry-ESI spectrum of 6e.
Author Contributions
Conceptualization, J.C.; Formal analysis, A.F.d.l.T. and M.G.; Funding acquisition, J.C.; Methodology, L.P.-P., A.F.d.l.T., and J.L.V.-L.; Project administration, J.C.; Resources, M.G. and J.C.; Software, J.L.V.-L.; Supervision, J.C.; Writing–original draft, L.P.-P., A.F.d.l.T., and J.C.; Writing–review & editing, A.F.d.l.T. and J.C.
Funding
This research was funded by Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT), grant number 1170718. Luis Prent-Peñaloza is grateful for the CONICYT scholarship funds associated with the folio No. 63130155.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Slobbe, P.; Ruijter, E.; Orru, R.V.A. Recent applications of multicomponent reactions in medicinal chemistry. Med. Chem. Commun. 2012, 3, 1189–1218. [Google Scholar] [CrossRef]
- Ahmadi, T.; Mohammadi Ziarani, G.; Gholamzadeh, P.; Mollabagher, H. Recent advances in asymmetric multicomponent reactions (AMCRs). Tetrahedron Asymmetry 2017, 28, 708–724. [Google Scholar] [CrossRef]
- Ghabraie, E.; Balalaie, S.; Mehrparvar, S.; Rominger, F. Synthesis of functionalized β-lactams and pyrrolidine-2,5-diones through a metal-free sequential Ugi-4CR/cyclization reaction. J. Org. Chem. 2014, 79, 7926–7934. [Google Scholar] [CrossRef] [PubMed]
- Brauer, M.C.N.; Neves Filho, R.A.W.; Westermann, B.; Heinke, R.; Wessjohann, L.A. Synthesis of antibacterial 1,3-diyne-linked peptoids from an Ugi-4CR/Glaser coupling approach. Beilstein J. Org. Chem. 2015, 11, 25–30. [Google Scholar] [CrossRef]
- Borah, P.; Borah, J.M.; Chowdhury, P. Microwave (MW) irradiated Ugi four-component reaction (Ugi-4CR): Expedited synthesis of steroid-amino acid conjugatesA novel class of hybrid compounds. Steroids 2015, 98, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Polindara-García, L.A.; Montesinos-Miguel, D.; Vazquez, A. An efficient microwave-assisted synthesis of cotinine and iso-cotinine analogs from an Ugi-4CR approach. Org. Biomol. Chem. 2015, 13, 9065–9071. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Zarezadeh, N.; Abbasi, A. One-pot tandem Ugi-4CR/S(N)Ar approach to highly functionalized quino [2,3-b][1,5]benzoxazepines. Mol. Divers. 2016, 20, 483–495. [Google Scholar] [CrossRef]
- Van der Poorten, O.; Van Den Hauwe, R.; Hollanders, K.; Maes, B.U.W.; Tourwé, D.; Jida, M.; Ballet, S. Rapid construction of substituted 3-amino-1,5-benzothiazepin-4(5H)-one dipeptide scaffolds through an Ugi-4CR-Ullmann cross-coupling sequence. Org. Biomol. Chem. 2018, 16, 1242–1246. [Google Scholar] [CrossRef]
- Bauer, S.M.; Armstrong, R.W. Total Synthesis of Motuporin (Nodularin-V). J. Am. Chem. Soc. 1999, 121, 6355–6366. [Google Scholar] [CrossRef]
- Bayer, T.; Riemer, C.; Kessler, H. A new strategy for the synthesis of cyclopeptides containing diaminoglutaric acid. J. Pept. Sci. 2001, 7, 250–261. [Google Scholar] [CrossRef]
- Bonne, D.; Dekhane, M.; Zhu, J. Modulating the reactivity of α-Isocyanoacetates: Multicomponent synthesis of 5-Methoxyoxazoles and Furopyrrolones. Angew. Chem. Int. Ed. 2007, 46, 2485–2488. [Google Scholar] [CrossRef] [PubMed]
- Zhdanko, A.G.; Nenajdenko, V.G. Nonracemizable isocyanoacetates for multicomponent reactions. J. Org. Chem. 2009, 74, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Zhdanko, A.G.; Gulevich, A.V.; Nenajdenko, V.G. One-step synthesis of N-acetylcysteine and glutathione derivatives using the Ugi reaction. Tetrahedron 2009, 65, 4692–4702. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, W.; Yan, B. Use of cyclohexylisocyanide and methyl 2-isocyanoacetate as convertible isocyanides for microwave-assisted fluorous synthesis of 1,4-benzodiazepine-2,5-dione library. J. Comb. Chem. 2010, 12, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Kotha, S. The building block approach to unusual α-amino acid derivatives and peptides. Acc. Chem. Res. 2003, 36, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Marcaccini, S.; Torroba, T. The use of the Ugi four-component condensation. Nat. Protoc. 2007, 2, 632–639. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, A.F.; Rivera, D.G.; Ferreira, M.A.B.; Corrêa, A.G.; Paixão, M.W. Multicomponent combinatorial development and conformational analysis of prolyl peptide–peptoid hybrid catalysts: Application in the direct asymmetric michael addition. J. Org. Chem. 2013, 78, 10221–10232. [Google Scholar] [CrossRef]
- Velázquez-Libera, J.L.; Caballero, J.; Toropova, A.P.; Toropov, A.A. Estimation of 2D autocorrelation descriptors and 2D Monte Carlo descriptors as a tool to build up predictive models for acetylcholinesterase (AChE) inhibitory activity. Chemom. Intell. Lab. Syst. 2019, 184, 14–21. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.D.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef]
- Sakuratani, Y.; Kasai, K.; Noguchi, Y.; Yamada, J. Comparison of Predictivities of Log P Calculation Models Based on Experimental Data for 134 Simple Organic Compounds. QSAR Comb. Sci. 2007, 26, 109–116. [Google Scholar] [CrossRef]
- Souza, E.S.; Zaramello, L.; Kuhnen, C.A.; Junkes, B.D.S.; Yunes, R.A.; Heinzen, V.E.F. Estimating the octanol/water partition coefficient for aliphatic organic compounds using semi-empirical electrotopological index. Int. J. Mol. Sci. 2011, 12, 7250–7264. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.J.; Xia, K.; Zhang, W.; Xu, X.J. ADME evaluation in drug discovery. 4. Prediction of aqueous solubility based on atom contribution approach. J. Chem. Inf. Comput. Sci. 2004, 44, 266–275. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Zhang, W.; Xu, X. ADME evaluation in drug discovery. 7. Prediction of oral absorption by correlation and classification. J. Chem. Inf. Model. 2007, 47, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Potts, R.O.; Guy, R.H. Predicting skin permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lian, G.; Han, L. Prediction of human skin permeability using artificial neural network (ANN) modeling. Acta Pharmacol. Sin. 2007, 28, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov. Today 2001, 6, 357–366. [Google Scholar] [CrossRef]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef]
- De la Torre, P.; Astudillo-Saavedra, L.; Caballero, J.; Quiroga, J.; Alzate-Morales, J.; Cabrera, M.; Trilleras, J. A novel class of selective acetylcholinesterase inhibitors: Synthesis and evaluation of (E)-2-(Benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles. Molecules 2012, 17, 12072–12085. [Google Scholar] [CrossRef]
- Ambure, P.; Kar, S.; Roy, K. Pharmacophore mapping-based virtual screening followed by molecular docking studies in search of potential acetylcholinesterase inhibitors as anti-Alzheimer’s agents. BioSystems 2014, 116, 10–20. [Google Scholar] [CrossRef]
- Guo, F.; Chang, D.; Lai, G.; Zhu, T.; Xiong, S.; Wang, S.; Wang, Z. Enantioselective and regioselective friedel–crafts alkylation of pyrroles with nitroalkenes catalyzed by a tridentate schiff base–copper complex. Chem. A Euro. J. 2011, 17, 11127–11130. [Google Scholar] [CrossRef]
- Monge, D.; Jensen, K.L.; Marín, I.; Jørgensen, K.A. Synthesis of 1,2,4-triazolines: Base-catalyzed hydrazination/cyclization cascade of α-isocyano esters and amides. Org. Lett. 2011, 13, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Bland, N.D.; Campbell, R.K.; Pollastri, M.P. Evaluation of pyrrolidine and pyrazolone derivatives as inhibitors of trypanosomal phosphodiesterase B1 (TbrPDEB1). Tetrahedron Lett. 2015, 56, 2832–2835. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Tsuritani, T.; Yasukata, T. A Mild method for the synthesis of carbamate-protected guanidines using the burgess reagent. Org. Lett. 2014, 16, 1868–1871. [Google Scholar] [CrossRef] [PubMed]
- González, M.; Caballero, J.; Helguera, A.; Garriga, M.; González, G.; Fernández, M. 2D Autocorrelation modelling of the inhibitory activity of cytokinin-derived cyclin-dependent kinase inhibitors. Bull. Math. Biol. 2006, 68, 735–751. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Tundidor-Camba, A.; Caballero, J.M. 2D Autocorrelation modeling of the activity of trihalobenzocycloheptapyridine analogues as farnesyl protein transferase inhibitors. Mol. Simul. 2005, 31, 575–584. [Google Scholar] [CrossRef]
- Greenblatt, H.M.; Guillou, C.; Guénard, D.; Argaman, A.; Botti, S.; Badet, B.; Thal, C.; Silman, I.; Sussman, J.L. The complex of a bivalent derivative of galanthamine with torpedo acetylcholinesterase displays drastic deformation of the active-site gorge: Implications for structure-based drug design. J. Am. Chem. Soc. 2004, 126, 15405–15411. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, P.; Millard, C.B.; Harel, M.; Dvir, H.; Enz, A.; Sussman, J.L.; Silman, I. Kinetic and structural studies on the interaction of cholinesterases with the anti-Alzheimer drug rivastigmine. Biochemistry 2002, 41, 3555–3564. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, CD001190. [Google Scholar] [CrossRef]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, CD005593. [Google Scholar] [CrossRef]
- Jackson, S.; Ham, R.J.; Wilkinson, D. The safety and tolerability of donepezil in patients with Alzheimer’s disease. Br. J. Clin. Pharmacol. 2004, 58, 1–8. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom Force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 7a, 7b, 7c, 6d, and 6e are available from the authors. |



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).