Cycloartane and Oleanane Glycosides from the Tubers of Eranthis cilicica

Abstract
:
1. Introduction
2. Results and Discussion
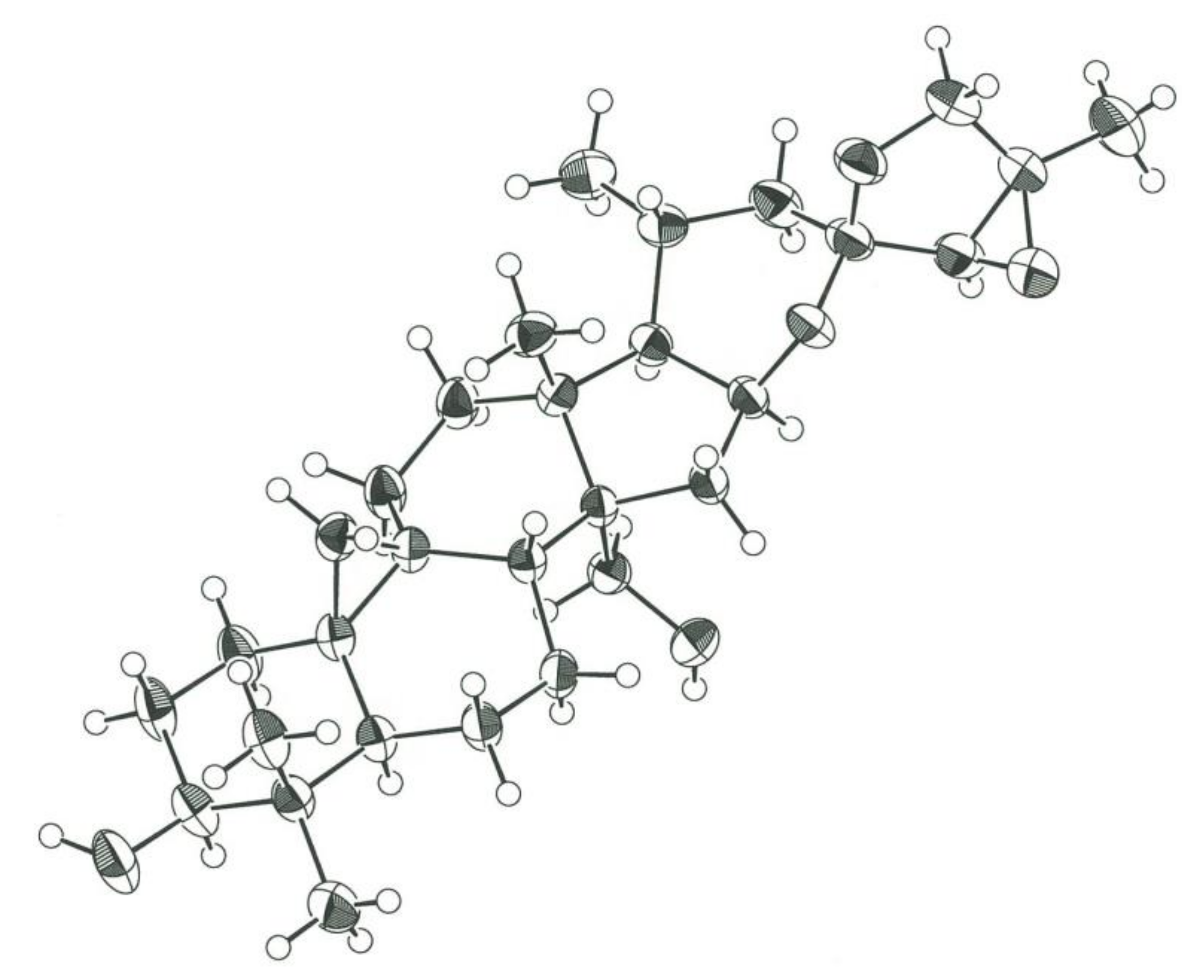
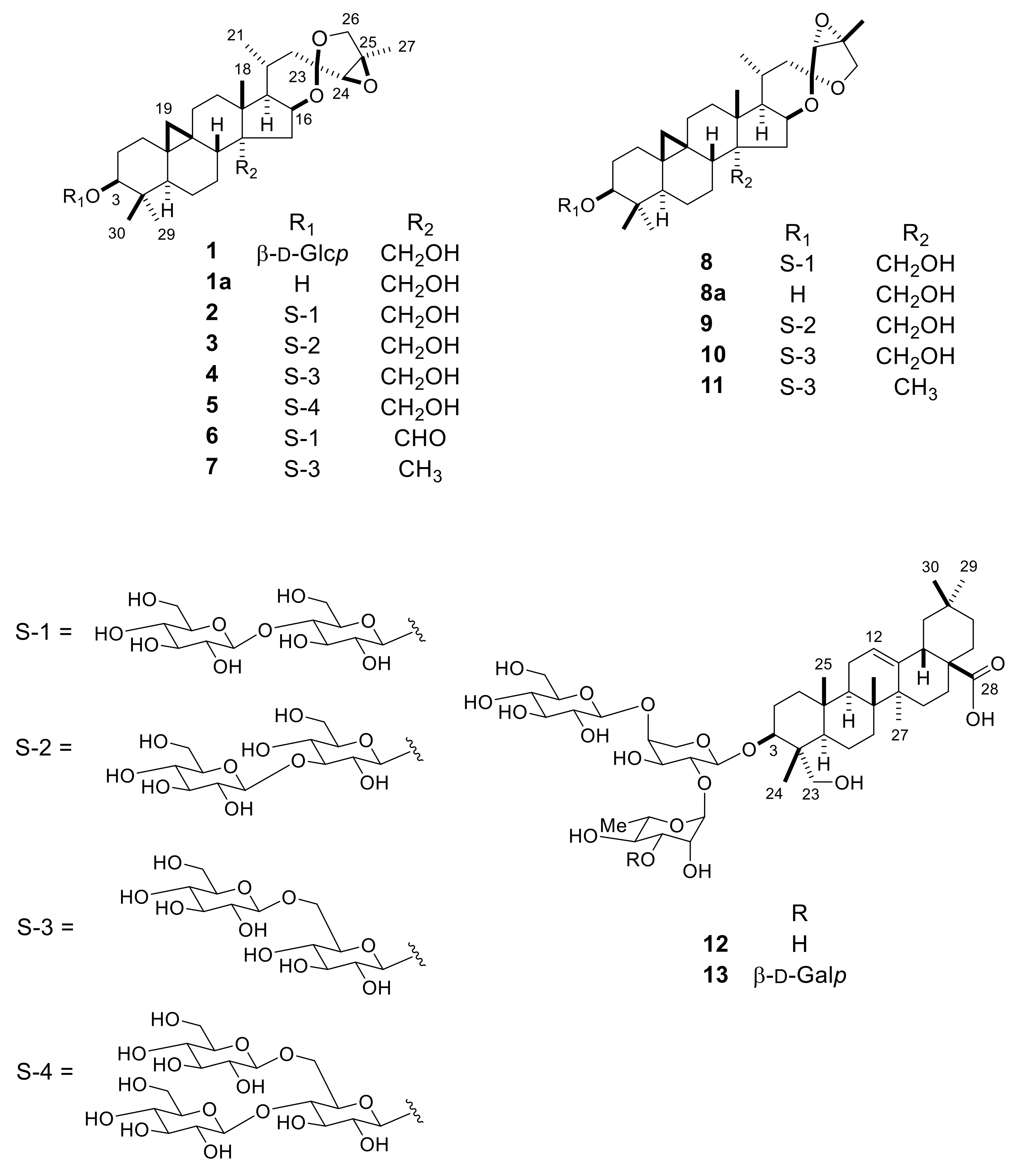
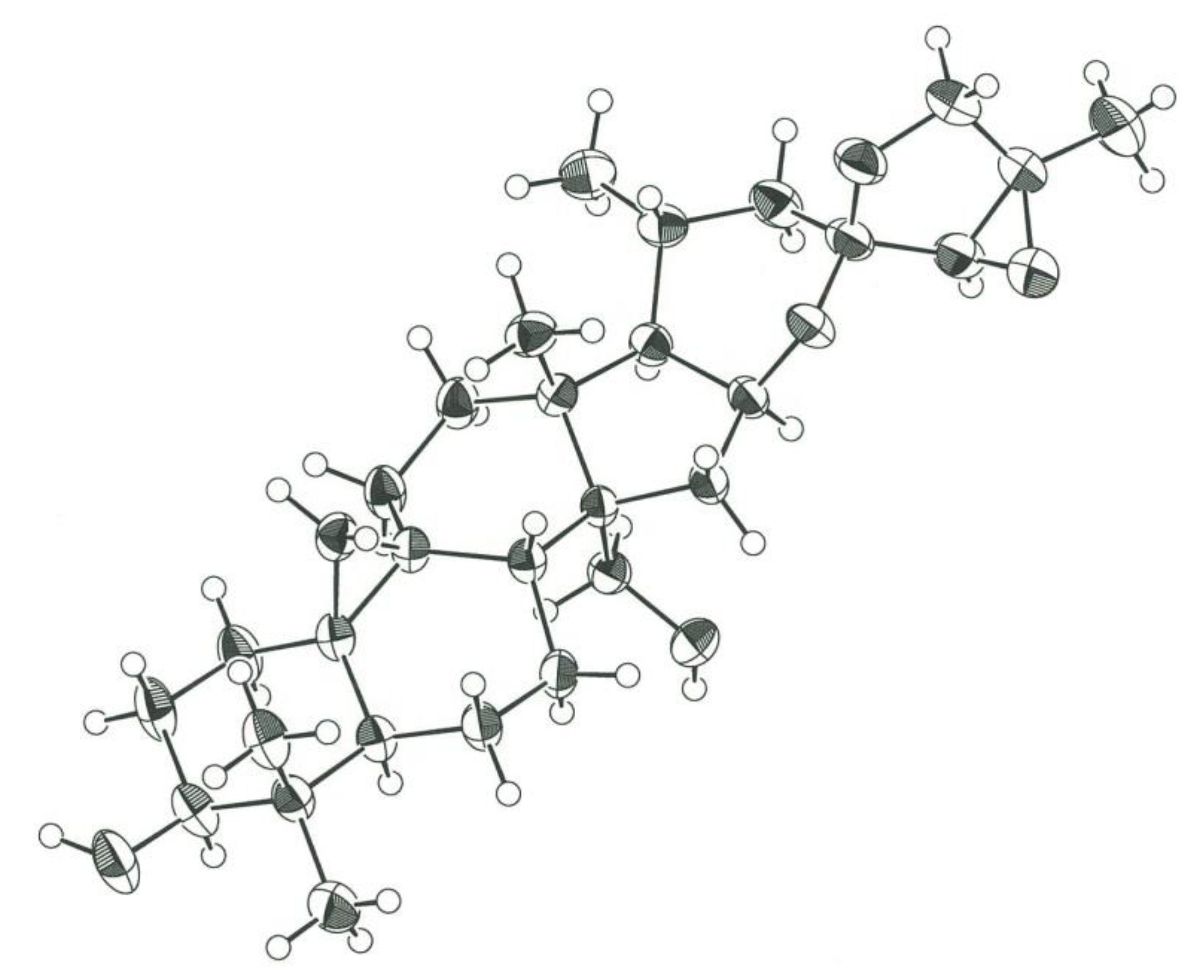
2.1. Isolation and Structure Elucidation of 1–13
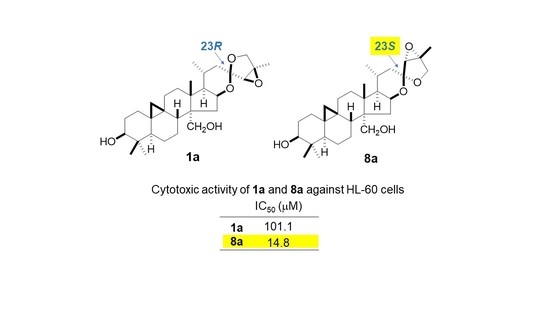
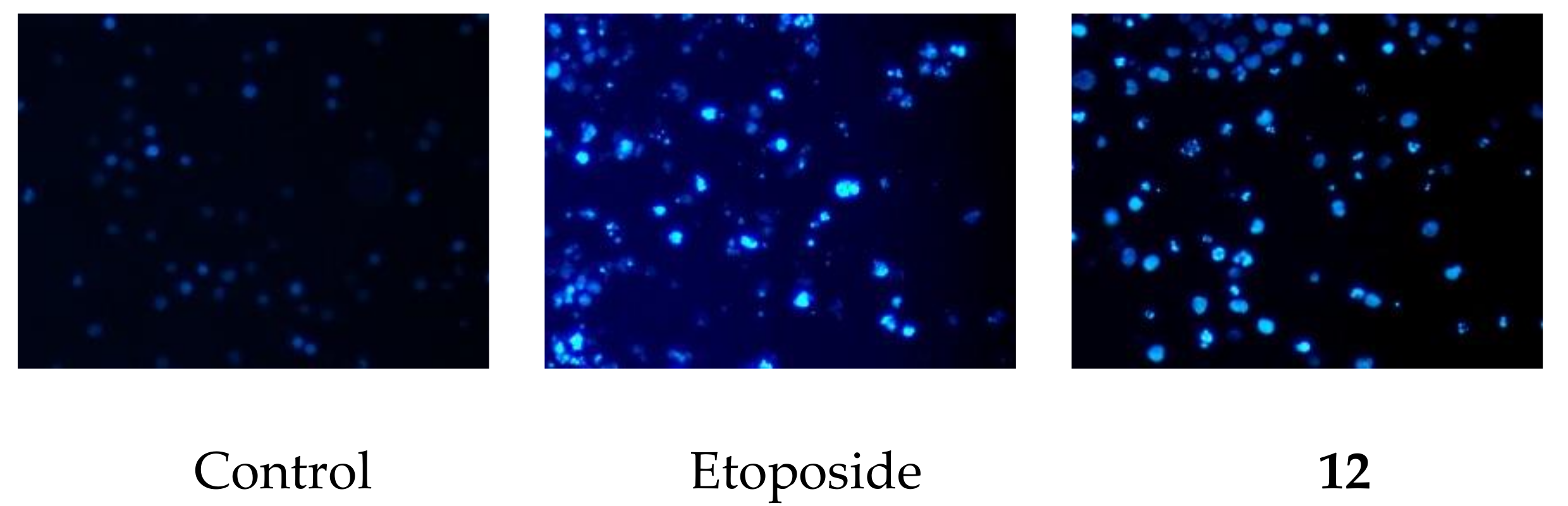
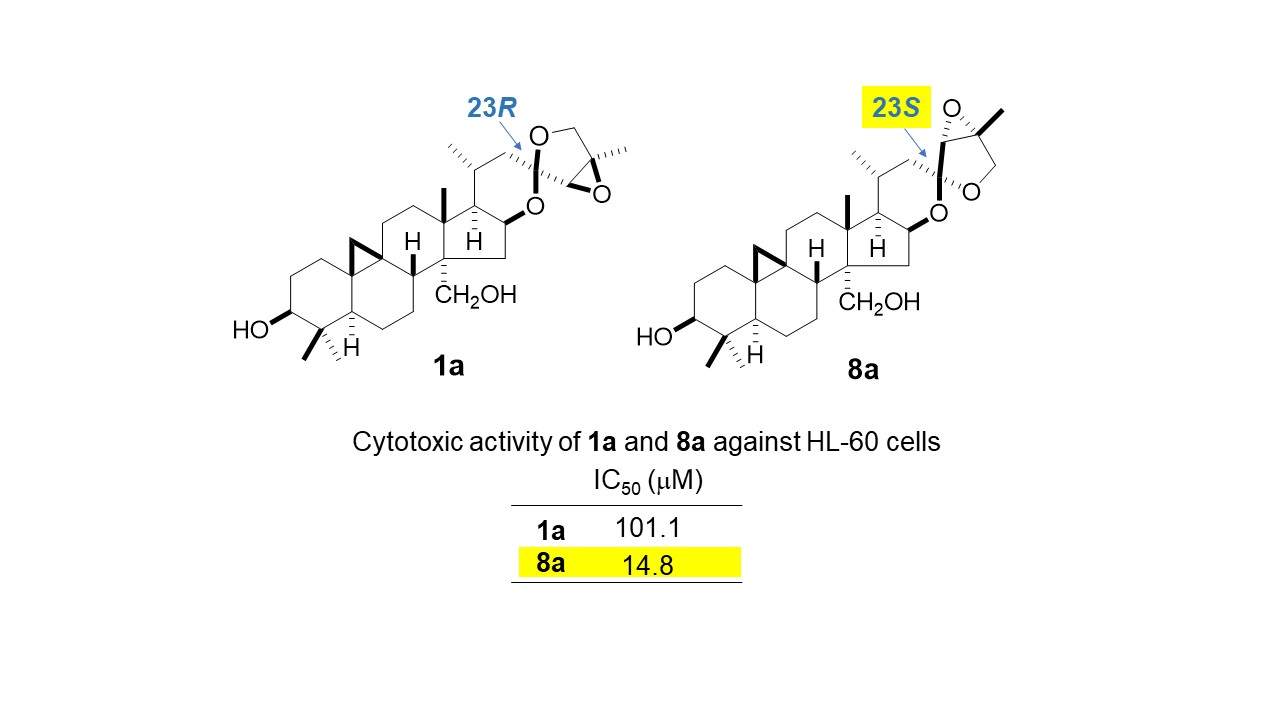
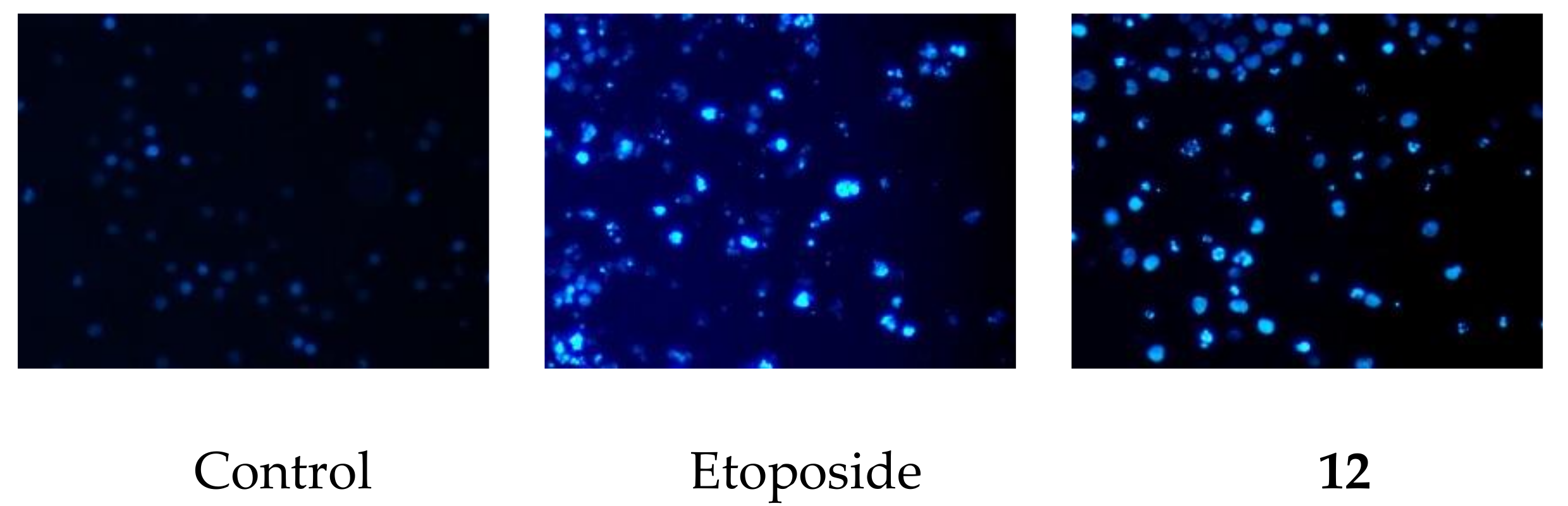
2.2. Cytotoxic Activity of 1a, 2, 8, 8a, 12, and 13
3. Materials and Methods
3.1. General Experimental Procedures and Plant Material
3.2. Extraction and Isolation
3.3. Structural Characterization
3.4. Cytotoxic Activity
3.5. DAPI Staininig
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kuroda, M.; Kubo, S.; Masatani, D.; Matsuo, Y.; Sakagami, H.; Mimaki, Y. Aestivalosides A–L, twelve pregnane glycosides from the seeds of Adonis aestivalis. Phytochemistry 2018, 150, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Kuroda, M.; Yokosuka, A.; Sakagami, H.; Mimaki, Y. Amurensiosides L–P, five new cardenolide glycosides from the roots of Adonis amurensis. Nat. Prod. Commun. 2015, 10, 27–32. [Google Scholar] [PubMed]
- Kubo, S.; Kuroda, M.; Matsuo, Y.; Masatani, D.; Sakagami, H.; Mimaki, Y. New cardenolides from the seeds of Adonis aestivalis. Chem. Pharm. Bull. 2012, 60, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Kubo, S.; Uchida, S.; Sakagami, H.; Mimaki, Y. Amurensiosides A–K, 11 new pregnane glycosides from the roots of Adonis amurensis. Steroids 2010, 75, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, A.; Sano, T.; Hashimoto, K.; Sakagami, H.; Mimaki, Y. Triterpene glycosides from the whole plant of Anemone hupehensis var. japonica and their cytotoxic activity. Chem. Pharm. Bull. 2009, 57, 1425–1430. [Google Scholar] [CrossRef]
- Mimaki, Y.; Watanabe, K.; Matsuo, Y.; Sakagami, H. Triterpene glycosides from the tubers of Anemone coronaria. Chem. Pharm. Bull. 2009, 57, 724–729. [Google Scholar] [CrossRef]
- Mimaki, Y.; Nadaoka, I.; Yasue, M.; Ohtake, Y.; Ikeda, M.; Watanabe, K.; Sashida, Y. Neocimicigenosides A and B, cycloartane glycosides from the rhizomes of Cimicifuga racemosa and their effects on CRF-stimulated ACTH secretion from AtT-20 cells. J. Nat. Prod. 2006, 69, 829–832. [Google Scholar] [CrossRef]
- Watanabe, K.; Mimaki, Y.; Sakagami, H.; Sashida, Y. Cycloartane glycosides from the rhizomes of Cimicifuga racemosa and their cytotoxic activities. Chem. Pharm. Bull. 2002, 50, 121–125. [Google Scholar] [CrossRef]
- Mimaki, Y.; Yokosuka, A.; Hamanaka, M.; Sakuma, C.; Yamori, T.; Sashida, Y. Triterpene saponins from the roots of Clematis chinensis. J. Nat. Prod. 2004, 67, 1511–1516. [Google Scholar] [CrossRef]
- Yokosuka, A.; Iguchi, T.; Kawahata, R.; Mimaki, Y. Cytotoxic bufadienolides from the whole plants of Helleborus foetidus. Phytochem. Lett. 2018, 23, 94–99. [Google Scholar] [CrossRef]
- Mimaki, Y.; Matsuo, Y.; Watanabe, K.; Sakagami, H. Furostanol glycosides from the rhizomes of Helleborus orientalis. J. Nat. Med. 2010, 64, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Sakagami, H.; Mimaki, Y. Four new steroidal saponins from the rhizomes of Helleborus orientalis. Heterocycles 2005, 65, 775–785. [Google Scholar]
- Mimaki, Y.; Watanabe, K.; Sakuma, C.; Sakagami, H.; Sashida, Y. Novel polyoxygenated spirostanol glycosides from the rhizomes of Helleborus orientalis. Helv. Chim. Acta 2003, 86, 398–407. [Google Scholar] [CrossRef]
- Watanabe, K.; Mimaki, Y.; Sakagami, H.; Sashida, Y. Bufadienolide and spirostanol glycosides from the rhizomes of Helleborus orientalis. J. Nat. Prod. 2003, 66, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Mimaki, Y.; Yokosuka, A.; Kuroda, M.; Hamanaka, M.; Sakuma, C.; Sashida, Y. New bisdesmosidic triterpene saponins from the roots of Pulsatilla chinensis. J. Nat. Prod. 2001, 64, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Mimaki, Y.; Kuroda, M.; Asano, T.; Sashida, Y. Triterpene saponins and lignans from the roots of Pulsatilla chinensis and their cytotoxic activity against HL-60 cells. J. Nat. Prod. 1999, 62, 1279–1283. [Google Scholar] [CrossRef]
- Tsukamoto, Y. The Grand Dictionary of Horticulture, Exec. ed.; Shogakukan: Tokyo, Japan, 1988; Vol. 2, pp. 89–92. ISBN 4093051119. [Google Scholar]
- Watanabe, K.; Mimaki, Y.; Sakuma, C.; Sashida, Y. Eranthisaponins A and B, two new bisdesmosidic triterpene saponins from the tubers of Eranthis cilicica. J. Nat. Prod. 2003, 66, 879–882. [Google Scholar] [CrossRef]
- Kuroda, M.; Uchida, S.; Watanabe, K.; Mimaki, Y. Chromones from the tubers of Eranthis cilicica and their antioxidant activity. Phytochemistry 2009, 70, 288–293. [Google Scholar] [CrossRef]
- Malinow, M.R.; Gardner, J.O.; Nelson, J.T.; McLaughlin, P.; Upson, B.; Rosemarie, A.H. Effects of α-and β-tigogenin cellobiosides on cholesterol absorption. Steroids 1986, 48, 197–211. [Google Scholar] [CrossRef]
- Sautour, M.; Miyamoto, T.; Lacaille-Dubois, M.A. Steroidal saponins from Smilax medica and their antifungal activity. J. Nat. Prod. 2005, 68, 1489–1493. [Google Scholar] [CrossRef]
- Matsuo, Y.; Watanabe, K.; Mimaki, Y. Triterpene glycosides from the underground parts of Caulophyllum thalictroides. J. Nat. Prod. 2009, 72, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Mimaki, Y. Lignans from Santalum album and their cytotoxic activities. Chem. Pharm. Bull. 2010, 58, 587–590. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | 1 | 1a | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 8a | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 32.1 | 32.5 | 32.1 | 32.0 | 32.1 | 32.0 | 31.3 | 32.1 | 32.1 | 32.4 | 32.1 | 32.1 | 32.0 |
| 2 | 29.8 | 31.2 | 29.7 | 29.6 | 29.9 | 29.7 | 29.5 | 30.2 | 29.8 | 31.1 | 29.8 | 30.0 | 29.9 |
| 3 | 88.6 | 77.9 | 88.7 | 88.6 | 88.4 | 88.7 | 88.3 | 88.5 | 88.7 | 77.8 | 88.7 | 88.4 | 88.5 |
| 4 | 41.2 | 41.0 | 41.1 | 41.0 | 41.1 | 41.1 | 41.0 | 41.2 | 41.2 | 41.0 | 41.2 | 41.2 | 41.1 |
| 5 | 47.5 | 47.5 | 47.5 | 47.4 | 47.3 | 47.4 | 46.4 | 47.3 | 47.5 | 47.4 | 47.5 | 47.4 | 47.2 |
| 6 | 21.1 | 21.5 | 21.1 | 21.0 | 21.1 | 21.0 | 20.0 | 20.8 | 21.2 | 21.5 | 21.2 | 21.2 | 20.8 |
| 7 | 27.4 | 27.6 | 27.4 | 27.3 | 27.4 | 27.4 | 26.7 | 26.2 | 27.5 | 27.6 | 27.5 | 27.5 | 26.2 |
| 8 | 48.3 | 48.5 | 48.3 | 48.3 | 48.4 | 48.3 | 41.9 | 47.5 | 48.5 | 48.7 | 48.5 | 48.5 | 47.6 |
| 9 | 19.7 | 19.8 | 19.8 | 19.7 | 19.7 | 19.7 | 19.4 | 19.6 | 19.8 | 19.8 | 19.8 | 19.8 | 19.5 |
| 10 | 26.6 | 27.0 | 26.6 | 26.5 | 26.5 | 26.5 | 27.6 | 26.5 | 26.7 | 26.9 | 26.6 | 26.7 | 26.5 |
| 11 | 27.3 | 27.4 | 27.2 | 27.2 | 27.1 | 27.1 | 27.6 | 26.3 | 27.2 | 27.2 | 27.2 | 27.1 | 26.1 |
| 12 | 32.6 | 32.7 | 32.6 | 32.5 | 32.5 | 32.5 | 33.6 | 33.2 | 32.6 | 32.6 | 32.6 | 32.6 | 33.1 |
| 13 | 45.5 | 45.5 | 45.4 | 45.4 | 45.4 | 45.4 | 46.5 | 46.3 | 45.3 | 45.3 | 45.3 | 45.3 | 46.3 |
| 14 | 51.5 | 51.6 | 51.5 | 51.5 | 51.5 | 51.5 | 64.0 | 44.7 | 51.6 | 51.6 | 51.6 | 51.6 | 44.5 |
| 15 | 38.2 | 38.3 | 38.2 | 38.1 | 38.1 | 38.1 | 36.1 | 44.4 | 37.6 | 37.6 | 37.6 | 37.6 | 43.8 |
| 16 | 75.3 | 75.4 | 75.3 | 75.2 | 75.3 | 75.3 | 74.2 | 75.2 | 73.7 | 73.7 | 73.7 | 73.7 | 73.1 |
| 17 | 56.9 | 57.0 | 56.9 | 56.7 | 56.8 | 56.8 | 57.1 | 56.6 | 57.1 | 57.1 | 57.1 | 57.1 | 56.7 |
| 18 | 22.0 | 22.1 | 22.0 | 21.9 | 22.0 | 22.0 | 20.5 | 20.6 | 22.0 | 22.0 | 22.0 | 22.0 | 20.5 |
| 19 | 30.6 | 30.9 | 30.6 | 30.5 | 30.6 | 30.5 | 28.1 | 30.0 | 30.6 | 30.8 | 30.6 | 30.7 | 30.1 |
| 20 | 23.9 | 23.9 | 23.9 | 23.8 | 23.8 | 23.8 | 23.1 | 23.7 | 26.5 | 26.4 | 26.5 | 26.4 | 26.2 |
| 21 | 20.7 | 20.7 | 20.7 | 20.6 | 20.6 | 20.6 | 20.9 | 20.7 | 20.5 | 20.4 | 20.5 | 20.4 | 20.3 |
| 22 | 37.7 | 37.8 | 37.7 | 37.6 | 37.6 | 37.6 | 36.8 | 37.6 | 36.8 | 36.7 | 36.8 | 36.7 | 36.5 |
| 23 | 106.1 | 106.1 | 106.1 | 106.0 | 106.1 | 106.1 | 106.2 | 106.1 | 106.1 | 106.1 | 106.1 | 106.1 | 106.0 |
| 24 | 62.0 | 62.1 | 62.0 | 62.0 | 62.0 | 62.0 | 62.1 | 62.0 | 63.4 | 63.4 | 63.4 | 63.4 | 63.3 |
| 25 | 62.4 | 62.4 | 62.4 | 62.4 | 62.4 | 62.4 | 62.4 | 62.5 | 63.1 | 63.1 | 63.1 | 63.1 | 63.1 |
| 26 | 67.9 | 68.0 | 67.9 | 67.9 | 67.9 | 67.9 | 68.1 | 68.0 | 68.7 | 68.6 | 68.7 | 68.6 | 68.6 |
| 27 | 14.2 | 14.2 | 14.2 | 14.1 | 14.2 | 14.1 | 14.2 | 14.2 | 13.7 | 13.7 | 13.7 | 13.7 | 13.7 |
| 28 | 63.5 | 63.6 | 63.4 | 63.3 | 63.4 | 63.3 | 210.2 | 19.7 | 63.2 | 63.2 | 63.2 | 63.2 | 19.6 |
| 29 | 25.7 | 26.1 | 25.6 | 25.5 | 25.5 | 25.5 | 25.6 | 25.7 | 25.7 | 26.0 | 25.6 | 25.6 | 25.6 |
| 30 | 15.4 | 14.8 | 15.4 | 15.3 | 15.3 | 15.3 | 15.1 | 15.4 | 15.4 | 14.8 | 15.4 | 15.4 | 15.3 |
| 1′ | 106.8 | 106.4 | 106.1 | 106.6 | 106.3 | 106.4 | 106.8 | 106.4 | 106.2 | 106.7 | 106.6 | ||
| 2′ | 75.8 | 75.2 | 74.4 | 75.5 | 75.0 | 75.3 | 75.6 | 75.3 | 74.4 | 75.6 | 75.5 | ||
| 3′ | 78.7 | 76.9 | 88.7 | 78.2 | 76.5 | 77.0 | 78.5 | 76.9 | 88.9 | 78.5 | 78.5 | ||
| 4′ | 71.8 | 81.6 | 69.7 | 71.6 | 81.1 | 81.6 | 71.7 | 81.6 | 69.8 | 71.6 | 71.6 | ||
| 5′ | 78.3 | 76.2 | 77.7 | 77.0 | 74.9 | 76.3 | 77.1 | 76.2 | 77.9 | 77.1 | 77.0 | ||
| 6′ | 63.0 | 62.3 | 62.4 | 70.2 | 68.7 | 62.4 | 70.3 | 62.4 | 62.5 | 70.3 | 70.1 | ||
| 1′′ | 104.9 | 105.7 | 105.2 | 104.7 | 105.0 | 105.4 | 105.0 | 105.9 | 105.3 | 105.2 | |||
| 2′′ | 74.8 | 75.4 | 75.1 | 75.1 | 74.8 | 74.7 | 74.8 | 75.5 | 75.2 | 75.1 | |||
| 3′′ | 78.2 | 78.1 | 78.4 | 78.2 | 78.2 | 78.4 | 78.2 | 78.2 | 78.3 | 78.2 | |||
| 4′′ | 71.5 | 71.4 | 71.6 | 71.6 | 71.5 | 71.7 | 71.5 | 71.6 | 71.7 | 71.5 | |||
| 5′′ | 78.4 | 78.6 | 78.3 | 78.1 | 78.5 | 78.4 | 78.4 | 78.7 | 78.4 | 78.3 | |||
| 6′′ | 62.3 | 62.3 | 62.5 | 62.3 | 62.5 | 62.7 | 62.4 | 62.5 | 62.6 | 62.5 | |||
| 1′′′ | 105.0 | ||||||||||||
| 2′′′ | 75.1 | ||||||||||||
| 3′′′ | 78.2 | ||||||||||||
| 4′′′ | 71.5 | ||||||||||||
| 5′′′ | 78.1 | ||||||||||||
| 6′′′ | 62.5 |
| Compound | IC50 (µM) |
|---|---|
| 1a | 101.1 ± 0.44 |
| 2 | >200 |
| 8 | >200 |
| 8a | 14.8 ± 1.00 |
| 12 | 10.6 ± 0.40 |
| 13 | 10.8 ± 0.53 |
| etoposide | 0.32 ± 0.01 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, K.; Mimaki, Y.; Fukaya, H.; Matsuo, Y. Cycloartane and Oleanane Glycosides from the Tubers of Eranthis cilicica. Molecules 2019, 24, 69. https://doi.org/10.3390/molecules24010069
Watanabe K, Mimaki Y, Fukaya H, Matsuo Y. Cycloartane and Oleanane Glycosides from the Tubers of Eranthis cilicica. Molecules. 2019; 24(1):69. https://doi.org/10.3390/molecules24010069
Chicago/Turabian StyleWatanabe, Kazuki, Yoshihiro Mimaki, Haruhiko Fukaya, and Yukiko Matsuo. 2019. "Cycloartane and Oleanane Glycosides from the Tubers of Eranthis cilicica" Molecules 24, no. 1: 69. https://doi.org/10.3390/molecules24010069
APA StyleWatanabe, K., Mimaki, Y., Fukaya, H., & Matsuo, Y. (2019). Cycloartane and Oleanane Glycosides from the Tubers of Eranthis cilicica. Molecules, 24(1), 69. https://doi.org/10.3390/molecules24010069