
Asymmetric Primaquine and Halogenaniline Fumardiamides as Novel Biologically Active Michael Acceptors



 ,
,  , and
, and
Abstract
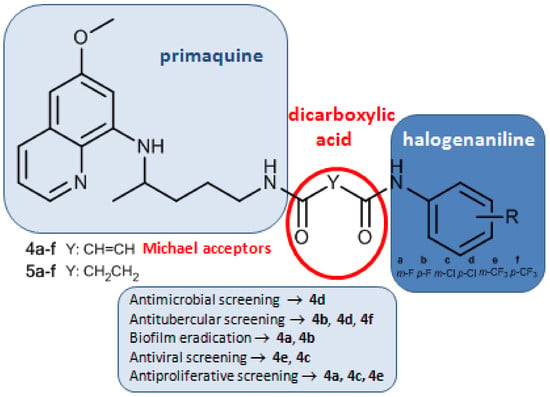
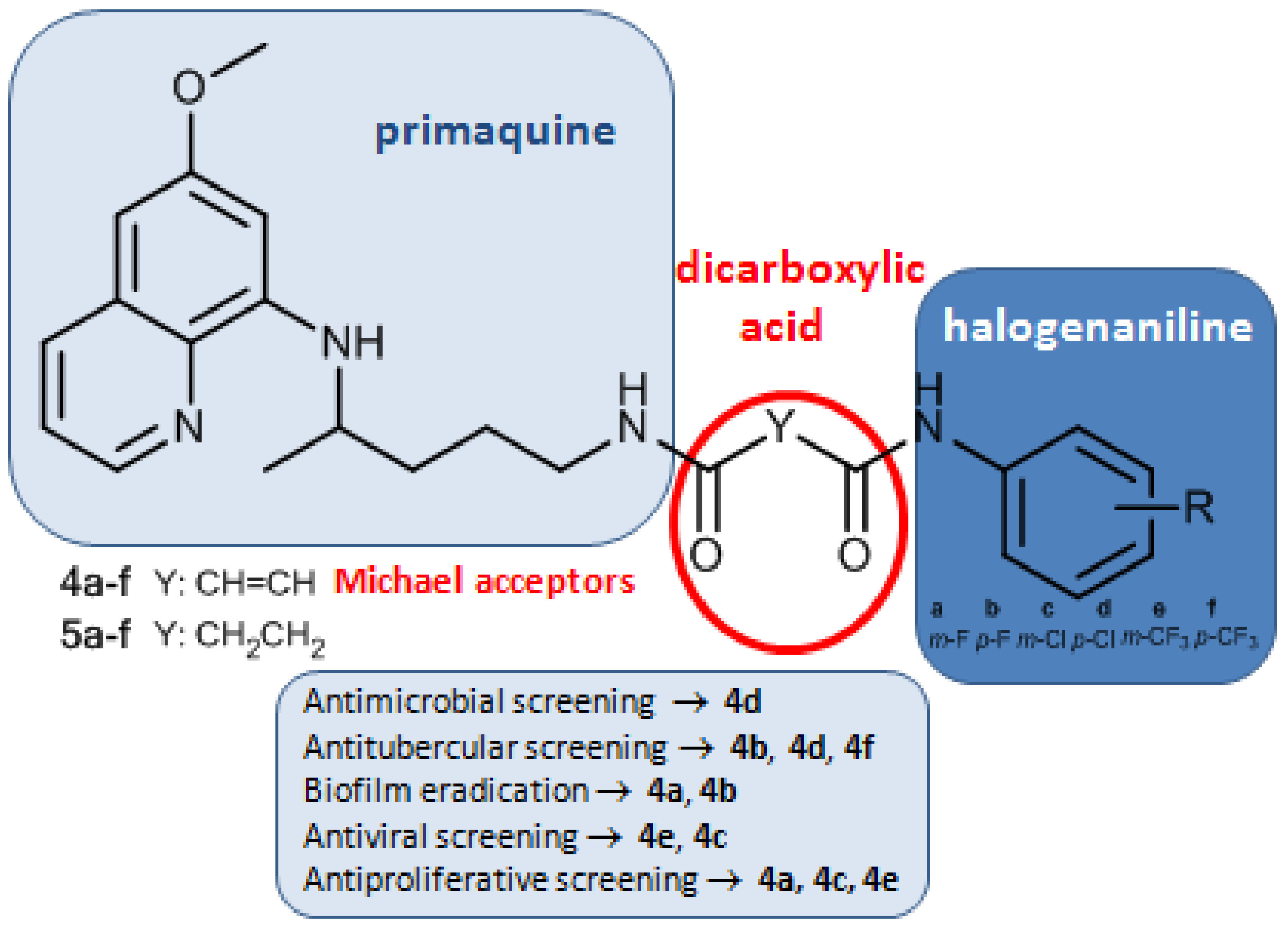
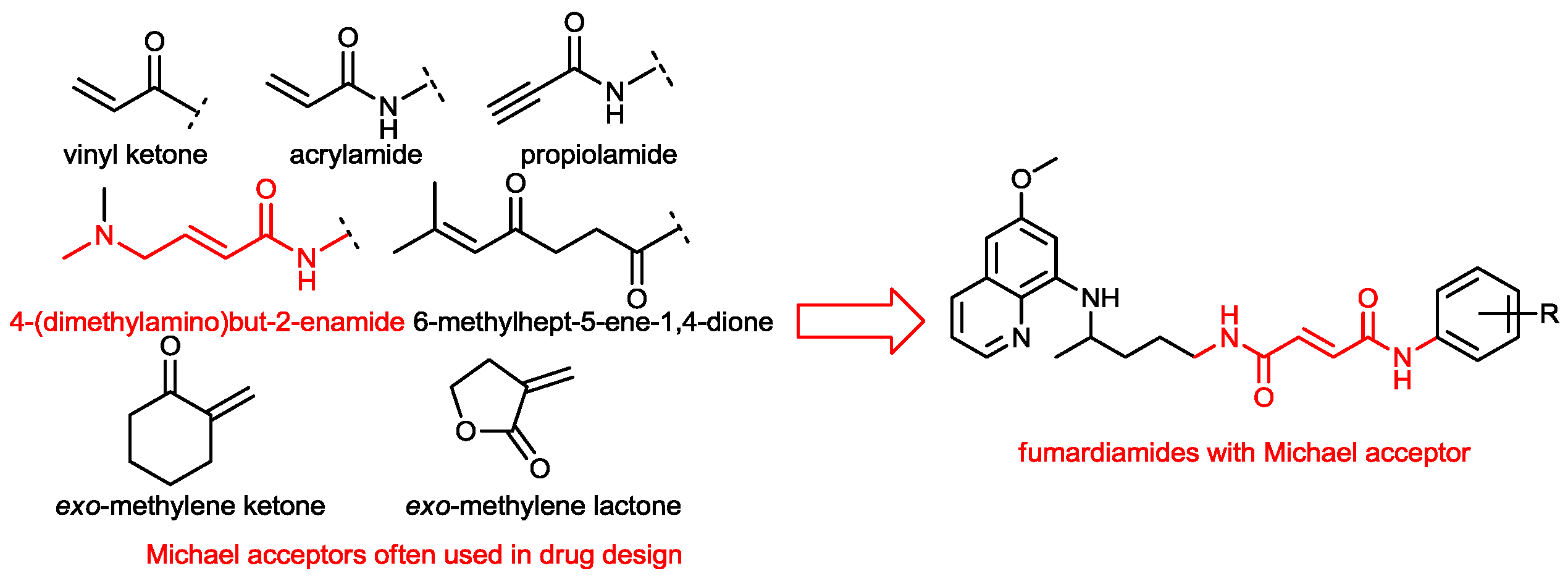
1. Introduction
2. Results and Discussion
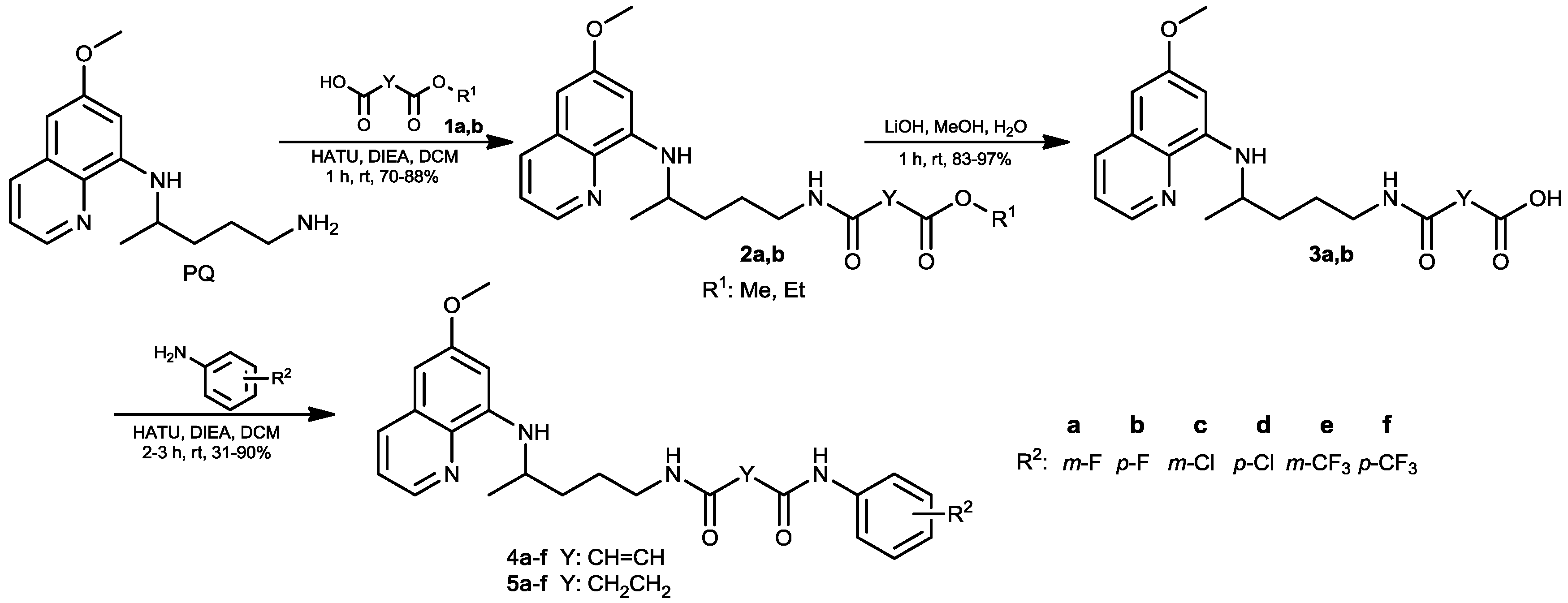
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Antibacterial and Antifungal Activity
2.2.2. Biofilm Eradication Assay
2.2.3. Antiviral Evaluation
2.2.4. Cytostatic Activity
2.2.5. Interaction with Glutathione (GSH)
3. Materials and Methods
3.1. Chemistry
3.1.1. Materials and General Methods
3.1.2. General Procedure for the Synthesis of Esters 2a,b
3.1.3. General Procedure for the Synthesis of Carboxylic Acids 3a,b
3.1.4. General Procedure for the Synthesis of Fumardiamides 4a–f and Succindiamides 5a–f
3.2. Biological Evaluation
3.2.1. In Vitro Antibacterial Susceptibility Assay (MIC Determination)
3.2.2. In Vitro Antifungal Susceptibility Testing
3.2.3. Minimum Biofilm Eradication Assay
3.2.4. Antiviral Evaluation
3.2.5. Cytostatic Activity
3.2.6. Interaction with Glutathione (GSH)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry, 2nd ed.; Oxford University Press: New York, NY, USA, 2012. [Google Scholar]
- Smaill, J.B.; Rewcastle, G.W.; Loo, J.A.; Greis, K.D.; Chan, O.H.; Reyner, E.L.; Lipka, E.; Showalter, H.D.; Vincent, P.W.; Elliott, W.L.; et al. Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions. J. Med. Chem. 2000, 43, 1380–1397. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Shimamura, T.; Shah, K.; LaFramboise, T.; Glatt, K.A.; Liniker, E.; Borgman, C.L.; Haringsma, H.J.; Feng, W.; Weir, B.A.; et al. The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272. Oncogene 2007, 26, 5023–5027. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Coleman, R.E.; Cortés, J.; Janni, W. Advances in the management of HER2-positive early breast cancer. Crit. Rev. Oncol. Hematol. 2017, 119, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent modifiers: A chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J. Med. Chem. 2017, 60, 839–885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; You, Q.; Zhang, X. Prodrug strategy for cancer cell-specific targeting: A recent overview. Eur. J. Med. Chem. 2017, 139, 542–563. [Google Scholar] [CrossRef] [PubMed]
- Compound Summary for CID 5281081. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/entacapone#section=Top (accessed on 25 May 2018).
- Matthews, D.A.; Dragovich, P.S.; Webber, S.E.; Fuhrman, S.A.; Patick, A.K.; Zalman, L.S.; Hendrickson, T.F.; Love, R.A.; Prins, T.J.; Marakovits, J.T.; et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA 1999, 96, 11000–11007. [Google Scholar] [CrossRef] [PubMed]
- Buzdar, A.U.; Robertson, J.F.; Eiermann, W.; Nabholtz, J.M. An overview of the pharmacology and pharmacokinetics of the newer generation aromatase inhibitors anastrozole, letrozole, and exemestane. Cancer 2002, 95, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Somberg, J.C.; Molnar, J. The pleiotropic effects of ethacrynic acid. Am. J. Ther. 2009, 16, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.M. The toxicology of microcystins. Toxicon 1998, 36, 953–962. [Google Scholar] [CrossRef]
- Eusugi, S.; Fujisawa, N.; Yoshida, J.; Watanabe, M.; Dan, S.; Yamori, T.; Shiono, Y.; Kimura, K.; Pyrrocidine, A. A metabolite of endophytic fungi, has a potent apoptosis-inducing activity against HL60 cells through caspase activation via the Michael addition. J. Antibiot. 2016, 69, 133–140. [Google Scholar] [CrossRef]
- Ramsay, J.R.; Suhrbier, A.; Aylward, J.H.; Ogbourne, S.; Cozzi, S.J.; Poulsen, M.G.; Baumann, K.C.; Welburn, P.; Redlich, G.L.; Parsons, P.G. The sap from Euphorbia peplus is effective against human nonmelanoma skin cancers. Br. J. Dermatol. 2011, 164, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Swanson, N.; Anderson, L.L.; Melgaard, A.; Xu, Z.; Berman, B. Ingenol mebutate gel for actinic keratosis. N. Engl. J. Med. 2012, 366, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Picato® gel–FDA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202833lbl.pdf. (accessed on 9 June 2018).
- Sebök, B.; Bonnekoh, B.; Geisel, J.; Mahrle, G. Antiproliferative and cytotoxic profiles of antipsoriatic fumaric acid derivatives in keratinocyte cultures. Eur. J. Pharmacol. 1994, 270, 79–87. [Google Scholar] [CrossRef]
- Smith, D. Fumaric acid esters for psoriasis: A systematic review. Ir. J. Med. Sci. 2017, 186, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Kocaadam, B.; Şanlier, N. Curcumin, an active component of turmeric (Curcuma longa), and its effects on health. Crit. Ver. Food Sci. Nutr. 2017, 57, 2889–2895. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Pinto, D.C.G.A. Plant secondary metabolites as anticancer agents: Successes in clinical trials and therapeutic application. Int. J. Mol. Sci. 2018, 19, 263. [Google Scholar] [CrossRef] [PubMed]
- Pavić, K.; Perković, I.; Cindrić, M.; Pranjić, M.; Martin-Kleiner, I.; Kralj, M.; Schols, D.; Hadjipavlou-Litina, D.; Katsori, A.-M.; Zorc, B. Novel semicarbazides and ureas of primaquine with bulky aryl or hydroxyalkyl substituents: Synthesis, cytostatic and antioxidative activity. Eur. J. Med. Chem. 2014, 86, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Perković, I.; Antunović, M.; Marijanović, I.; Pavić, K.; Ester, K.; Kralj, M.; Vlainić, J.; Kosalec, I.; Schols, D.; Hadjipavlou-Litina, D.; et al. Novel urea and bis-urea primaquine derivatives with hydroxyphenyl and halogenphenyl substituents: Synthesis and biological evaluation. Eur. J. Med. Chem. 2016, 124, 622–636. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.D. Natural cinnamic acids, synthetic derivatives and hybrids with antimicrobial activity. Molecules 2014, 19, 292–349. [Google Scholar] [CrossRef] [PubMed]
- Kakwani, M.D.; Suryavanshi, P.; Ray, M.; Rajan, M.G.R.; Majee, S.; Samad, A.; Devarajan, P.; Degani, M.S. Design, synthesis and antimycobacterial activity of cinnamide derivatives: A molecular hybridization approach. Bioorg. Med. Chem. Lett. 2011, 21, 1997–1999. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Baltas, M.; Bedos-Belval, F. Cinnamic acid derivatives as anticancer agents-a review. Curr. Med. Chem. 2011, 18, 1672–1703. [Google Scholar] [CrossRef] [PubMed]
- Pavić, K.; Perković, I.; Gilja, P.; Kozlina, F.; Ester, K.; Kralj, M.; Schols, D.; Hadjipavlou-Litina, D.; Pontiki, E.; Zorc, B. Design, synthesis and biological evaluation of novel primaquine-cinnamic acid conjugates of amide and acylsemicarbazide type. Molecules 2016, 21, 1629. [Google Scholar] [CrossRef] [PubMed]
- Pavić, K.; Perković, I.; Pospíšilová, Š.; Machado, M.; Fontinha, D.; Prudêncio, M.; Jampilek, J.; Coffey, A.; Endersen, L.; Rimac, H.; et al. Primaquine hybrids as promising antimycobacterial and antimalarial agents. Eur. J. Med. Chem. 2018, 143, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Vlainić, J.; Kosalec, I.; Pavić, K.; Hadjipavlou-Litina, D.; Pontiki, E.; Zorc, B. Insights into biological activity of ureidoamides with primaquine and amino acid moieties. J. Enzyme Inhib. Med. Chem. 2018, 33, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Levatić, J.; Pavić, K.; Perković, I.; Uzelac, L.; Ester, K.; Kralj, M.; Kaiser, M.; Rottmann, M.; Supek, F.; Zorc, B. Machine learning prioritizes synthesis of primaquine ureidoamides with high antimalarial activity and attenuated cytotoxicity. Eur. J. Med. Chem. 2018, 146, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Beus, M.; Rajić, Z.; Maysinger, D.; Mlinarić, Z.; Antunović, M.; Marijanović, I.; Fontinha, D.; Prudêncio, M.; Held, J. SAHA-primaquine hybrids (sahaquines) as potential anticancer and antimalarial compounds. Chem. Open. submitted.
- Chemicalize, 2017, ChemAxon Ltd. Available online: http://www.chemicalize.org (accessed on 5 April 2018).
- Hufnagel, D.A.; Price, J.E.; Stephenson, R.E.; Kelley, J.; Benoit, M.F.; Chapman, M.R. Thiol starvation induces redox-mediated dysregulation of Escherichia coli biofilm components. J. Bacteriol. 2018, 200, e00389-17. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.F.; Davey, L. Disulfide bonds: A key modification in bacterial extracytoplasmic proteins. J. Dent. Res. 2017, 96, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, M.E.; Abramite, J.A.; Anderson, D.P.; Aulabaugh, A.; Dahal, U.P.; Gilbert, A.M.; Li, C.; Montgomery, J.; Oppenheimer, S.R.; Ryder, T.; et al. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem. 2014, 57, 10072–10079. [Google Scholar] [CrossRef] [PubMed]
- Oravcova, V.; Zurek, L.; Townsend, A.; Clark, A.B.; Ellis, J.C.; Cizek, A. American crows as carriers of vancomycin-resistant enterococci with vanA gene. Environ. Microbiol. 2014, 16, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; The 8th informational supplement document; CLSI: Wayne, PA, USA, 2012; M100-S22. [Google Scholar]
- EUCAST, European Committee on Antimicrobial Susceptibility Testing. Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth micro dilution. EUCAST Discussion Document. Clin. Microbiol. Infect. 2013, 9, 1–10. [Google Scholar]
- Schwalbe, R.; Steele-Moore, L.; Goodwin, A.C. (Eds.) Antimicrobial Susceptibility Testing Protocols; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Sheehan, D.J.; Espinel-Ingroff, A.; Steele, M.; Webb, C.D. Antifungal susceptibility testing of yeasts: A brief overview. Clin. Infect. Dis. 1993, 17, 494–500. [Google Scholar] [CrossRef]
- Stepanović, S.; Vuković, D.; Hola, V.; Di Bonaventura, G.; Djukić, S.; Cirković, I.; Ruzicka, F. Quantification of biofilm in microtiter plates: Overview of testing conditions and practical recommendations for assessment of biofilm production by Staphylococci. APMIS 2007, 115, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Tzioumaki, N.; Manta, S.; Tsoukala, E.; Vande Voorde, J.; Liekens, S.; Komiotis, D.; Balzarini, J. Synthesis and biological evaluation of unsaturated keto and exomethylene D-arabinopyranonucleoside analogs: Novel 5-fluorouracil analogs that target thymidylate synthase. Eur. J. Med. Chem. 2011, 46, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lescrinier, E.; Groaz, E.; Persoons, L.; Daelemans, D.; Herdewijn, P.; De Jonghe, S. Synthesis and biological evaluation of pyrrolo[2,1-f][1,2,4]triazine C-nucleosides with a ribose, 2′-deoxyribose, and 2′,3′-dideoxyribose sugar moiety. ChemMedChem. 2018, 13, 97–104. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Compd. | MIC (µg/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Staphylococcus aureus ATCC 6538 | Streptococcus pneumoniae MFBF 10373 | Enterococcus faecalis ATCC 29212 | Bacillus cereus ATCC 11778 | Bacillus subtilis ATCC 6633 | Acinetobacter baumannii MFBF 10913 | Mycobacterium tuberculosis H37Ra | Mycobacterium marinum CAMP 5644 | Candida albicans ATCC 90028 | |
| 4a | >100 | >100 | 128 | >100 | 50 | >100 | 128 | >256 | >100 |
| 4b | 12.5 | 12.5 | 64 | 25 | 25 | 25 | 32 | 64 | >100 |
| 4c | 12.5 | 25 | 256 | 25 | 50 | 25 | 128 | 128 | >100 |
| 4d | 6.1 | 12.5 | 64 | >100 | >100 | 12.5 | 128 | 64 | 12.5 |
| 4e | 50 | 50 | 128 | >100 | >100 | 25 | 128 | 256 | >100 |
| 4f | 50 | 50 | 64 | >100 | >100 | 25 | 64 | 64 | >100 |
| PQ 1 | 50 | 50 | 128 | 70 | 80 | 50 | 256 | 256 | >100 |
| TC 2 | 0.3 | 0.3 | – | 0.3 | 0.3 | 3 | – | – | – |
| CIP 3 | – | – | – | – | – | – | 16.0 | 0.3 | – |
| INH 4 | – | – | – | – | – | – | 5.0 | 64.0 | – |
| RIF 5 | – | – | – | – | – | – | 8.7 | 2.2 | – |
| Amph 6 | – | – | – | – | – | – | – | – | 0.5 |
| Compd. | MBEC (µg/mL) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Staphylococcus aureus ATCC 6538 | Streptococcus pneumonia MFBF 10373 | Enterococcus faecalis ATCC 29212 | Bacillus cereus ATCC 11778 | Bacillus subtilis ATCC 6633 | Escherichia coli ATCC 10536 | Pseudomonas aeruginosa ATCC 27853 | Serratia marcescens ATCC 10905 | Proteus mirabilis MFBF 10430 | Salmonella enteritidis MFBF 11945 | Acinetobacter baumannii MFBF 10913 | |
| 4a | 12.5 | 12.5 | 25 | >100 | 50 | 100 | 25 | 25 | >100 | >100 | 6.3 |
| 4b | 12.5 | 25 | 25 | 25 | 25 | 50 | 12.5 | >100 | 25 | >100 | 25 |
| 4c | 12.5 | 100 | 25 | 25 | 50 | 50 | 50 | >100 | >100 | >100 | 100 |
| 4d | 25 | 50 | 50 | >100 | >100 | 50 | >100 | >100 | >100 | >100 | 100 |
| 4e | 50 | 50 | 50 | >100 | >100 | 50 | 50 | >100 | >100 | >100 | 100 |
| 4f | 25 | 50 | 50 | >100 | >100 | 50 | 25 | >100 | >100 | >100 | 100 |
| PQ 1 | 50 | 70 | >100 | 70 | 80 | 65 | 65 | >100 | >100 | 70 | 50 |
| Gen 2 | 12.5 | 25 | 50 | 50 | 50 | 25 | 50 | 50 | 50 | 50 | 25 |
| Compd. | Cytotoxicity | EC50 1 (µM) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CC50 2 | MCC 3 | Para-Influenza-3 Virus | Reovirus-1 | Sindbis Virus | Coxsackie Virus B4 | Punta Toro Virus | Yellow Fever Virus | |||||||
| MTS | Visual CPE Score | MTS | Visual CPE Score | MTS | Visual CPE Score | MTS | Visual CPE Score | MTS | Visual CPE Score | MTS | Visual CPE Score | |||
| 4a | 46.9 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – |
| 4b | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – |
| 4c | >50 | 10 | >50 | >50 | 3.8 | >50 | 3.0 | >50 | >50 | >50 | 5.5 | >50 | >50 | >50 |
| 4d | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – |
| 4e | >50 | 10 | >50 | >50 | 3.1 | >50 | 5.3 | 4.2 | 3.1 | 2.7 | 5.4 | >50 | >50 | >50 |
| 4f | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – |
| PQ | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – | >50 | – |
| DS-10.000 4 | >100 | >100 | >100 | >100 | >100 | >100 | 4.0 | 10 | >100 | 34 | 16 | 8.9 | 1.6 | >100 |
| RIB 5 | >250 | >250 | 73 | 111 | 107 | 126 | >250 | 11 | >250 | >250 | 85 | 111 | 119 | >250 |
| MPA 6 | >100 | >100 | 1.0 | 0.8 | 0.6 | 0.8 | 12 | 1.7 | >100 | >100 | 11 | 8.9 | 0.5 | 0.8 |
| Compd. | IC50 (µM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Capan-1 | Hap1 | HCT-116 | NCI-H460 | DND-41 | HL-60 | K-562 | MM.1S | Z-138 | |
| 4a | 16.9 | 19.3 | 22.3 | 15.5 | 6.7 | 5.7 | 31.2 | 13.0 | 8.4 |
| 4b | 50.5 | 66.4 | >100 | 29.1 | >100 | >100 | 70.5 | >100 | >100 |
| 4c | 69.9 | >100 | 73.0 | 43.7 | 8.4 | >100 | >100 | >100 | 46.5 |
| 4d | 91.8 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 4e | 56.5 | >100 | >100 | 34.2 | 8.9 | >100 | 71.2 | >100 | 68.4 |
| 4f | 79.4 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| PQ 1 | 18.7 | 42.7 | 30.9 | 52.6 | 11.4 | 2.2 | 35.2 | 28.3 | 7.1 |
| DXT 2 | 0.75 | 1.17 | 7.66 | 1.30 | 0.94 | 1.24 | 1.22 | 3.38 | 5.42 |
| EPEG 3 | 0.15 | 0.04 | 1.35 | 0.09 | 0.03 | 0.03 | 0.01 | 0.97 | 0.02 |
| STS 4 | 0.66 | 3.56 | 0.78 | 1.66 | 6.96 | 13.10 | 0.23 | 1.61 | 0.40 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajić, Z.; Beus, M.; Michnová, H.; Vlainić, J.; Persoons, L.; Kosalec, I.; Jampílek, J.; Schols, D.; Keser, T.; Zorc, B. Asymmetric Primaquine and Halogenaniline Fumardiamides as Novel Biologically Active Michael Acceptors. Molecules 2018, 23, 1724. https://doi.org/10.3390/molecules23071724
Rajić Z, Beus M, Michnová H, Vlainić J, Persoons L, Kosalec I, Jampílek J, Schols D, Keser T, Zorc B. Asymmetric Primaquine and Halogenaniline Fumardiamides as Novel Biologically Active Michael Acceptors. Molecules. 2018; 23(7):1724. https://doi.org/10.3390/molecules23071724
Chicago/Turabian StyleRajić, Zrinka, Maja Beus, Hana Michnová, Josipa Vlainić, Leentje Persoons, Ivan Kosalec, Josef Jampílek, Dominique Schols, Toma Keser, and Branka Zorc. 2018. "Asymmetric Primaquine and Halogenaniline Fumardiamides as Novel Biologically Active Michael Acceptors" Molecules 23, no. 7: 1724. https://doi.org/10.3390/molecules23071724
APA StyleRajić, Z., Beus, M., Michnová, H., Vlainić, J., Persoons, L., Kosalec, I., Jampílek, J., Schols, D., Keser, T., & Zorc, B. (2018). Asymmetric Primaquine and Halogenaniline Fumardiamides as Novel Biologically Active Michael Acceptors. Molecules, 23(7), 1724. https://doi.org/10.3390/molecules23071724