Influence of Extraction Conditions on Ultrasound-Assisted Recovery of Bioactive Phenolics from Blueberry Pomace and Their Antioxidant Activity

 , and
, and 
Abstract
1. Introduction
2. Results and Discussion
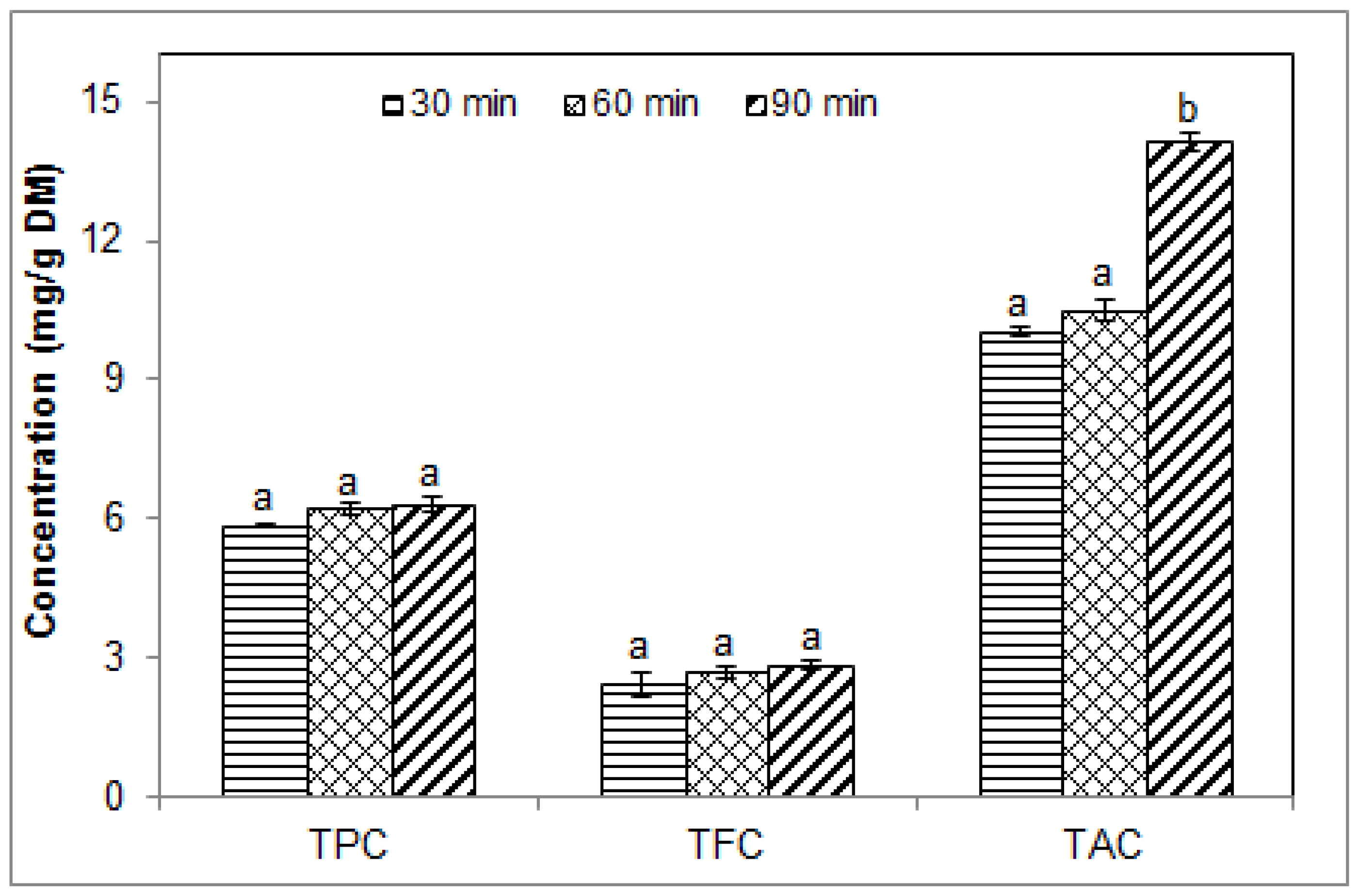
2.1. Effect of Sonication Time on the Phenolic Contents of Blueberry Pomace Water Extracts
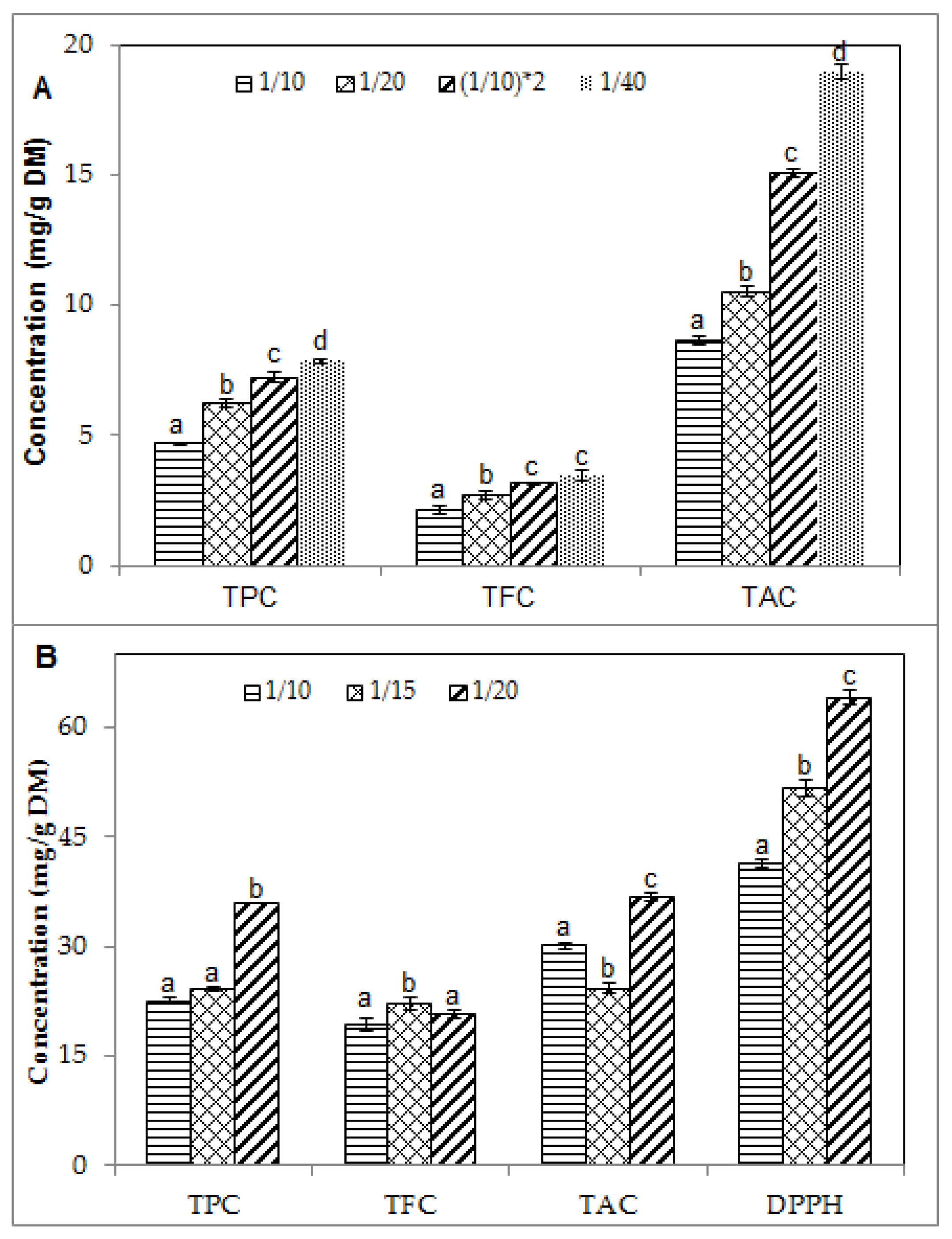
2.2. Effect of Solid/Liquid Ratio on the Phenolic Contents and Antioxidant Activity of Blueberry Pomace Extracts
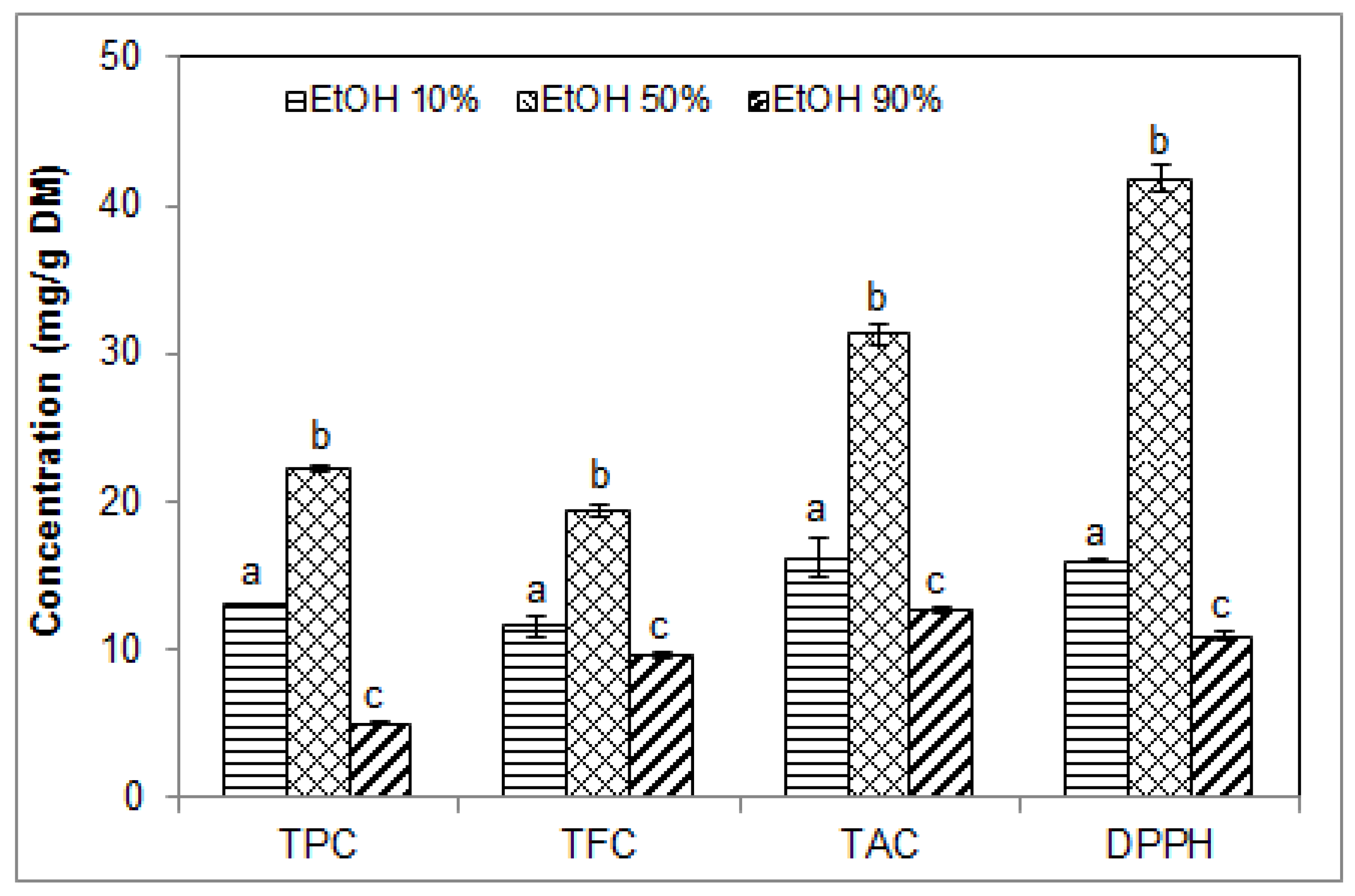
2.3. Effect of Ethanol Concentration on the Phenolic Contents and Antioxidant Activity of Blueberry Pomace Extracts
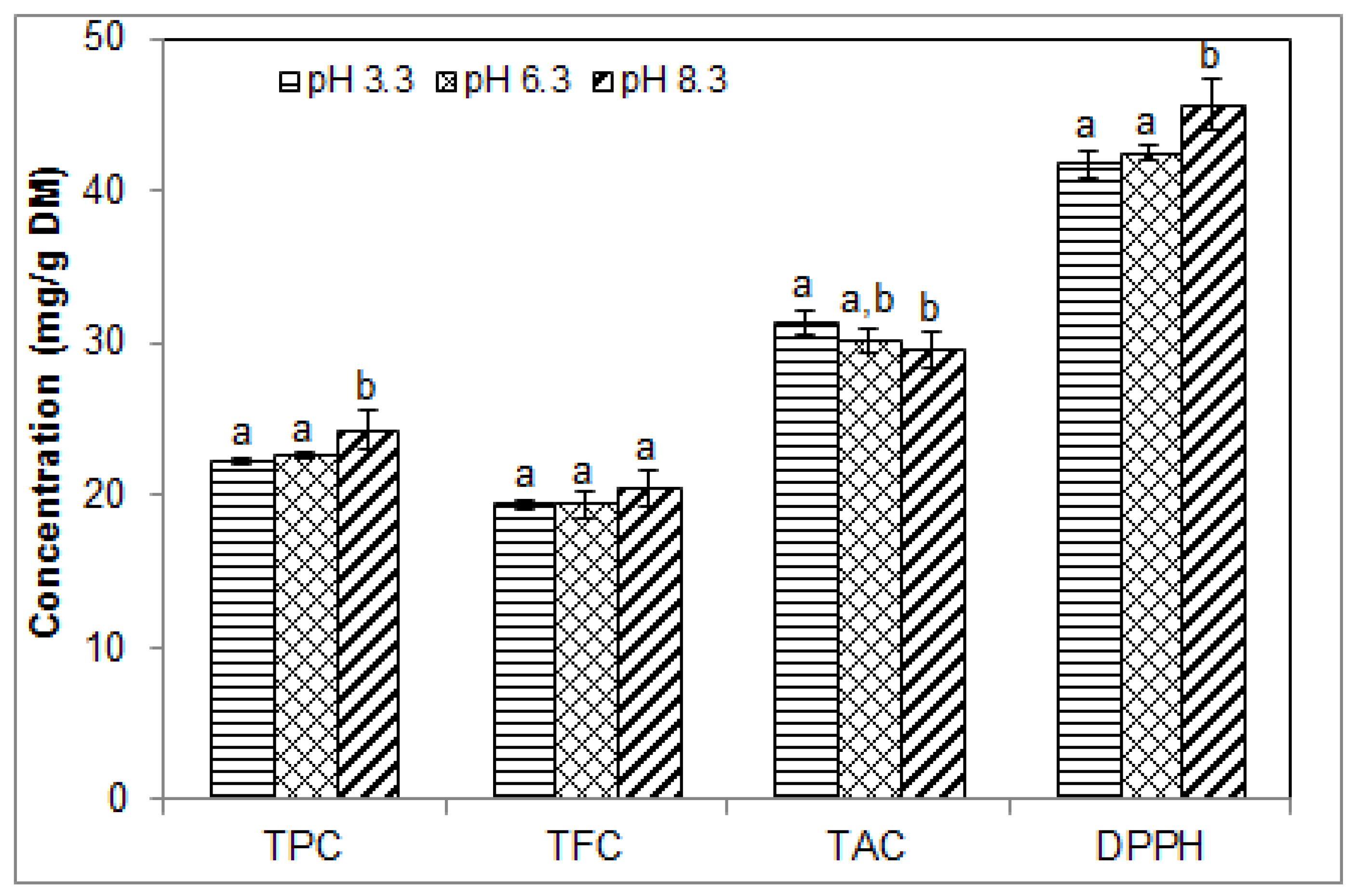
2.4. Effect of pH on the Phenolic Contents and Antioxidant Activity of Blueberry Pomace Extracts
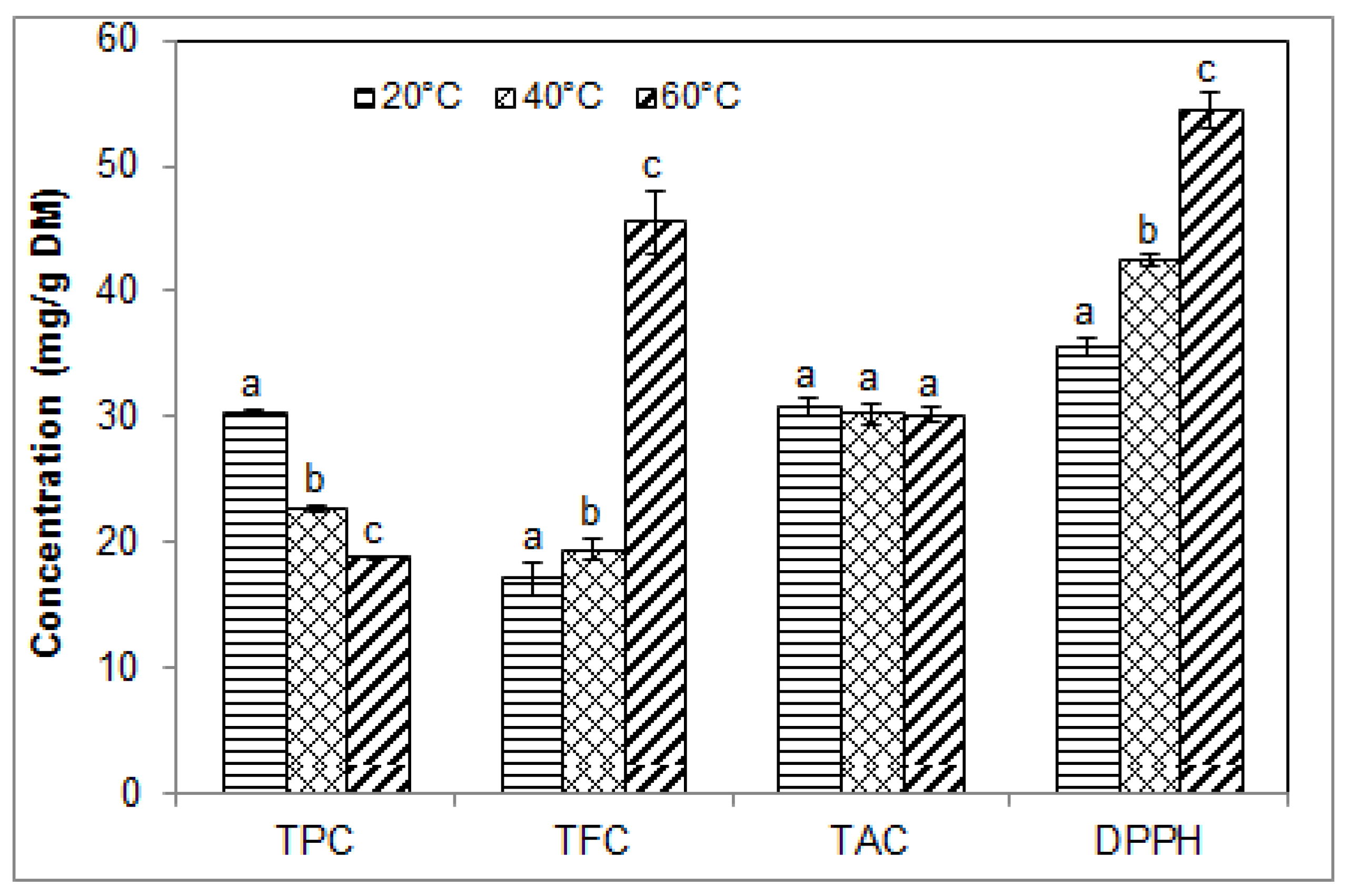
2.5. Effect of Temperature on the Phenolic Contents and Antioxidant Activity of Blueberry Pomace Extracts
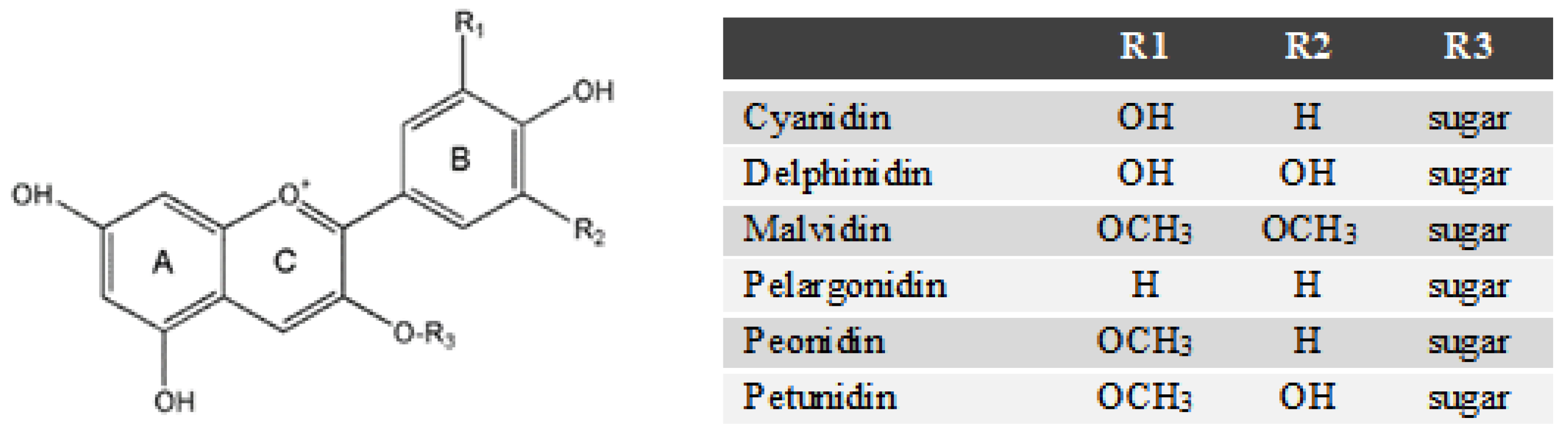
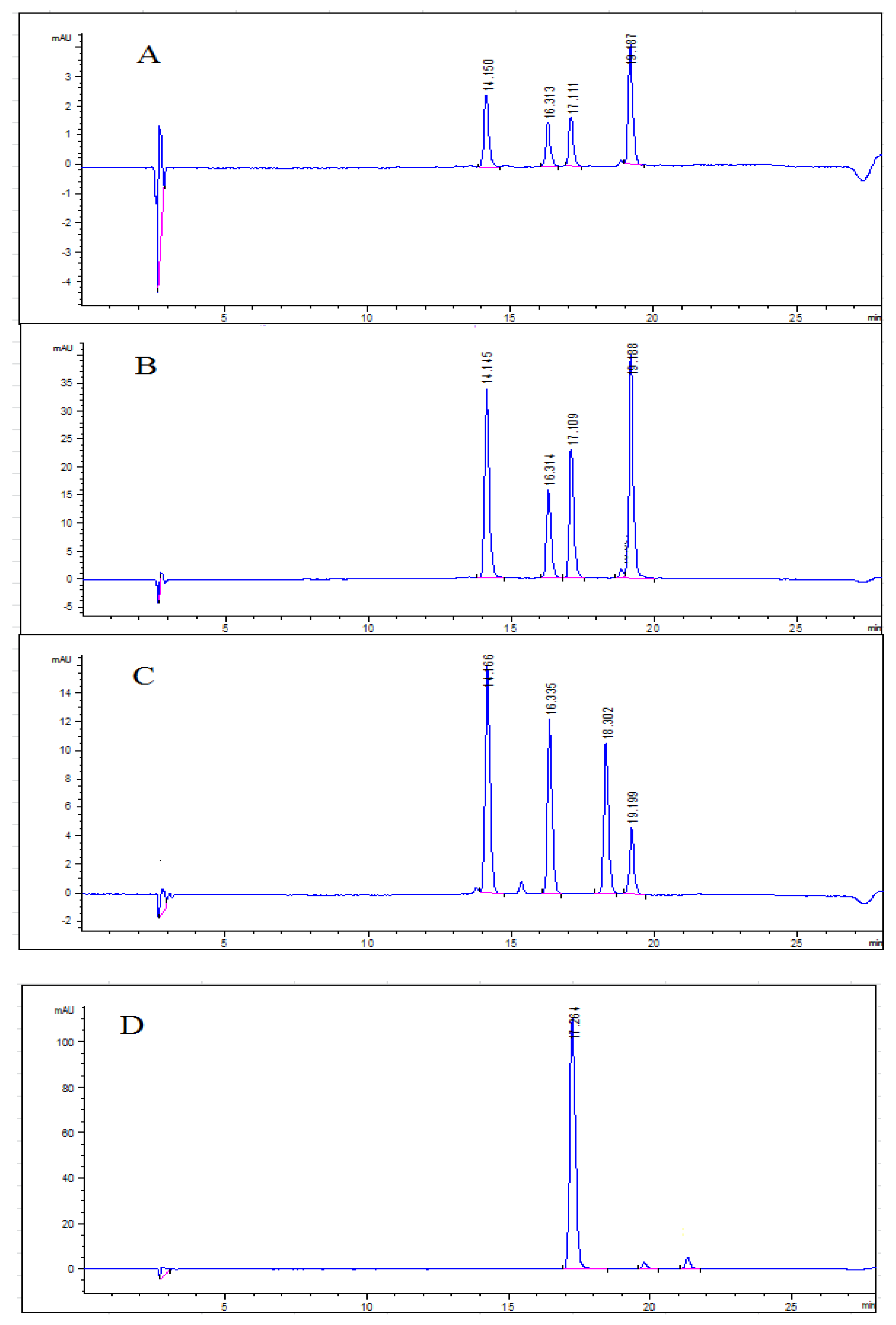
2.6. Anthocyanin Profiles in Water Extracts and Ethanolic Extracts from Blueberry Pomace
3. Materials and Methods
3.1. Plant Material and Chemicals
3.2. Ultrasound-Assisted Extraction (USAE)
3.3. Chemicals Analyses of Extracts
3.3.1. Determination of Total Phenolic Content (TPC)
3.3.2. Determination of Total Flavonoid Content (TFC)
3.3.3. Determination of Total Anthocyanin Content (TAC) and Identification of Anthocyanins
3.3.4. Determination of Antioxidant Activity
3.4. Statistical Analyses
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Mudgal, V.; Madaan, N.; Mudgal, A.; Mishra, S. Dietary polyphenols and human health. Asian J. Biochem. 2010, 5, 154–162. [Google Scholar] [CrossRef]
- Seeram, N.P. Berry fruits for cancer prevention, current status and future prospects. J. Agric. Food Chem. 2008, 56, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Reque, M.P.; Steffens, S.R.; da Silva, M.A.; Jablonski, A.; Flôres, H.S.; de Rios, O.A.; de Jong, V.E. Characterization of blueberry fruits (Vaccinium spp.) and derived products. Food Sci. Technol. Campinas. 2014, 34, 773–779. [Google Scholar] [CrossRef]
- He, O.; Zhang, L.L.; Yue, X.Y.; Liang, J.; Jiang, J.; Gao, X.L.; Yue, P.X. Optimization of ultrasound-assisted extraction of phenolic compounds and anthocyanins from blueberry (Vaccinium ashei) wine pomace. Food Chem. 2016, 204, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.P.; Massarioli, P.A.; Denny, C.; dos Santos, F.L.; Franchin, M.; Pereira, E.G.; Vieira, T.M.; Rosalen, L.P.; de Alencar, M.S. Winery by-products, extraction optimization, phenolic composition and cytotoxic evaluation to act as a new source of scavenging of reactive oxygen species. Food Chem. 2015, 181, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Sójka, M.; Kołodziejczyk, K.; Milala, J. Polyphenolic and basic chemical composition of black chokeberry industrial by-products. Ind. Crop. Prod. 2013, 51, 77–78. [Google Scholar] [CrossRef]
- Su, M.S.; Silva, L.J. Antioxidant activity, anthocyanins, and phenolics of rabbiteye blueberry (Vaccinium ashei) by-products as affected by fermentation. Food Chem. 2006, 97, 447–451. [Google Scholar] [CrossRef]
- Klavins, L.; Kviesisa, J.; Nakurteb, I.; Klavins, M. Berry press residues as a valuable source of polyphenolics, extraction optimisation and analysis. Food Sci. Technol. 2018, 93, 583–591. [Google Scholar] [CrossRef]
- Struck, S.; Plaza, M.; Turner, C.; Rohm, H. Berry pomace—A review of processing and chemical analysis of its polyphenols. Int. J. Food Sci. Technol. 2016, 51, 1305–1318. [Google Scholar] [CrossRef]
- Statistic Canada. Estimates, Production and Farm Gate Value of Fresh and Processed Fruits. Available online: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=3210036401 (accessed on 5 June 2018).
- Da, S.R.; Rocha-Santos, T.A.P.; Duarte, A.C. Supercritical fluid extraction of bioactive compounds. TrAC Trends Anal. Chem. 2016, 76, 40–51. [Google Scholar]
- Pandey, A.; Tripathi, S. Concept of standardization, extraction and pre phytochemical screening strategies for herbal drug. J. Pharmacogn. Phytochem. 2014, 2, 115–119. [Google Scholar]
- Chemat, F.; Abert-Vian, M.; Cravotto, G. Green extraction of natural products, concept and principles. Int. J. Mol. Sci. 2012, 13, 8615–8627. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Weller, C.L. Recent advances in extraction of nutraceuticals from plants. Trends Food Sci. Technol. 2006, 17, 300–312. [Google Scholar] [CrossRef]
- Castro-López, C.; Ventura-Sobrevilla, J.M.; González-Hernández, M.D.; Rojas, R.; Ascacio-Valdés, J.A.; Aguilar, C.N.; Martínez-Ávila, G.C.G. Impact of extraction techniques on antioxidant capacities and phytochemical composition of polyphenol-rich extracts. Food Chem. 2017, 237, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Bendicho, C.; De La Calle, I.; Pena, F.; Costas, M.; Cabaleiro, N.; Lavilla, I. Ultrasound-assisted pretreatment of solid samples in the context of green analytical chemistry. Trends Anal. Chem. 2012, 31, 50–60. [Google Scholar] [CrossRef]
- Armenta, S.; Garrigues, S.; De Guardia, M. The role of green extraction techniques in Green Analytical Chemistry. Trends Anal. Chem. 2015, 71, 2–8. [Google Scholar] [CrossRef]
- Esclapez, M.D.; Garcĭa-Pérez, J.V.; Mulet, A.; Cárcel, J.A. Ultrasound-assisted extraction of natural products. Food Eng. Rev. 2011, 3, 108–120. [Google Scholar] [CrossRef]
- Chemat, F.; Rombaut, N.; Sicaire, A.G.; Meullemiestre, A.; Fabiano-Tixier, A.S.; Abert-Vian, M. Ultrasound assisted extraction of food and natural products. Mechanisms, techniques, combinations, protocols and applications. A review. Ultrason. Sonochem. 2017, 34, 540–560. [Google Scholar] [CrossRef] [PubMed]
- Abad, R.B.; Bou-Maroun, E.; Reparet, J.M.; Blanquet, J.; Cayot, N. Impact of lipid extraction on the dearomatisation of an Eisenia foetida protein powder. Food Chem. 2010, 119, 459–466. [Google Scholar]
- Galvan d’Alessandro Kriaa, K.; Nikov, I.; Dimitrov, K. Ultrasound assisted extraction of polyphenols from black chokeberry. Sep. Purif. Technol. 2012, 93, 42–47. [Google Scholar] [CrossRef]
- Do, Q.D.; Angkawijaya, A.E.; Tran-Nguyen, P.L.; Huynh, L.H.; Soetaredjo, F.E.; Ismadji, S.; Ju, Y.H. Effect of extraction solvent on total phenol content, total flavonoid content, and antioxidant activity of Limnophila aromatica. J. Food Drug Anal. 2014, 22, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, N.; Wang, S.X.; Kou, P.; Zhao, C.J.; Fu, Y.J. Ultrasound-negative pressure cavitation extraction of phenolic compounds from blueberry leaves and evaluation of its DPPH radical scavenging activity. Food Bioprod. Process. 2018, 108, 69–80. [Google Scholar] [CrossRef]
- Ćujić, N.; Šavikin, K.; Jankovic, T.; Pljevljakušić, D.; Zdunić, G.; Ibric, S. Optimization of polyphenols extraction from dried chokeberry using maceration as traditional technique. Food Chem. 2016, 194, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Lapornik, B.; Prošek, M.; Wondra, G.A. Comparison of extracts prepared from plant by-products using different solvents and extraction time. J. Food Eng. 2005, 71, 214–222. [Google Scholar] [CrossRef]
- Xu, D.P.; Zheng, J.; Zhou, Y.; Li, Y.; Li, S.; Li, H.B. Ultrasound-assisted extraction of natural antioxidants from the flower of Limonium sinuatum: Optimization and comparison with conventional methods. Food Chem. 2017, 217, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cao, Y.L.; Jiang, J.G.; Lin, Q.S.; Chen, J.; Zhu, L. Response surface optimization of ultrasound-assisted flavonoids extraction from the flower of Citrus aurantium L. var. amara Engl. J. Sep. Sci. 2010, 33, 1349–1355. [Google Scholar] [PubMed]
- Safdar, M.N.; Kausar, T.; Jabbar, S.; Mumtaz, A.; Ahad, K.; Saddozai, A.A. Extraction and quantification of polyphenols from kinnow (Citrus reticulate L.) peel using ultrasound and maceration techniques. J. Food Drug Anal. 2017, 25, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Złotek, U.; Mikulska, S.; Nagajek, M.; Swieca, M. The effect of different solvents and number of extraction steps on the polyphenol content and antioxidant capacity of basil leaves (Ocimum basilicum L.) extracts. Saudi J. Biol. Sci. 2016, 23, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Kaderides, K.; Goula, A.M.; Adamopoulos, K.G. A process for turning pomegranate peels into a valuable food ingredient using ultrasound-assisted extraction and encapsulation. Innov. Food Sci. Emerg. Technol. 2015, 31, 204–215. [Google Scholar] [CrossRef]
- Dent, M.; Dragović-Uzelac, V.; Penić, M.; Brnčić, M.; Bosiljkov, T.; Levaj, B. The effect of extraction solvents, temperature and time on the composition and mass fraction of polyphenols in Dalmatian wild sage (Salvia officinalis L.) extracts. Food Technol. Biotechnol. 2013, 51, 84–91. [Google Scholar]
- Librán, C.M.; Mayor, L.; Garcia-Castello, E.M.; Vidal-Brotons, D. Polyphenol extraction from grape wastes, solvent and pH effect. Agric. Sci. 2013, 4, 56–62. [Google Scholar]
- Cheng, A.; Chen, X.; Wang, W.; Gong, Z.; Liu, L. Contents of extractable and non-extractable polyphenols in the leaves of blueberry. Czech J. Food Sci. 2013, 31, 275–282. [Google Scholar] [CrossRef]
- Kalt, W.; McDonald, J.E.; Donner, H. Anthocyanins, phenolics, and antioxidant capacity of processed lowbush blueberry products. J. Food Sci. 2000, 65, 390–393. [Google Scholar] [CrossRef]
- Ruenroengklin, N.; Zhong, J.; Duan, X.; Yang, B.; Li, J.; Jiang, Y. Effects of various temperatures and pH values on the extraction yield of phenolics from litchi fruit pericarp tissue and the antioxidant activity of the extracted anthocyanins. Int. J. Mol. Sci. 2008, 9, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Amendola, D.; De Faveri, D.M.; Spigno, G. Grape marc phenolics, extraction kinetics, quality and stability of extracts. J. Food Eng. 2010, 97, 384–392. [Google Scholar] [CrossRef]
- Li, D.; Meng, X.; Li, B. Profiling of anthocyanins from blueberries produced in China using HPLC-DAD-MS and exploratory analysis by principal component analysis. J. Food Compos. Anal. 2016, 47, 1–7. [Google Scholar] [CrossRef]
- You, Q.; Wang, B.; Chen, F.; Huang, Z.; Wang, X.; Luo, P.G. Comparison of anthocyanins and phenolics in organically and conventionally grown blueberries in selected cultivars. Food Chem. 2011, 125, 201–208. [Google Scholar] [CrossRef]
- Barnes, J.S.; Nguyen, H.P.; Shen, S.; Schug, K.A. General method for extraction of blueberry anthocyanins and identification using high performance liquid chromatography–electrospray ionization-ion trap-time of flight-mass spectrometry. J. Chromatogr. A 2009, 1216, 4728–4735. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, X.; Hu, X.; Li, T.; Fu, X.; Liu, R.H. Comparison of phytochemical profiles, antioxidant and cellular antioxidant activities of different varieties of blueberry (Vaccinium spp.). Food Chem. 2017, 217, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Tournour, H.H.; Marcela, S.A.; Magalhaes, L.M.; Barreiros, L.; Queiroz, J.; Cunha, L.M. Valorization of grape pomace, extraction of bioactive phenolics with antioxidant properties. Ind. Crop. Prod. 2015, 74, 397–406. [Google Scholar] [CrossRef]
- Herald, T.J.; Gadgil, P.; Tilley, M. High-throughput microplate assays for screening flavonoid content and DPPH-scavenging activity in sorghum bran and flour. J. Sci. Food Agric. 2012, 92, 2326–2331. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Standards of catechin, cyanidin, delphinidin, gallic acid, malvidin, pelargonidin, peonidin and petunidin are commercially available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solid/Liquid Ratio | Temperature (°C) | Time (min) | pH | Ethanol (% v/v) in Water | |
|---|---|---|---|---|---|
| Runs 1–3 (Figure 1) | 1/20 | 40 | 30–60–90 | 5.0 | 0 |
| Runs 4–8 (Figure 2A) | 1/10–(1/10) × 2 *–1/20–1/40 | 40 | 60 | 3.3 | 0 |
| Runs 9–11 (Figure 2B) | 1/10–1/15–1/20 | 40 | 60 | 3.3 | 50 |
| Runs 12–14 (Figure 3) | 1/15 | 40 | 40 | 3.3 | 10–50–90 |
| Runs 15–17 (Figure 4) | 1/15 | 40 | 40 | 3.3–6.3–8.3 | 50 |
| Runs 18–20 (Figure 5) | 1/15 | 20–40–60 | 40 | 3.3 | 50 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bamba, B.S.B.; Shi, J.; Tranchant, C.C.; Xue, S.J.; Forney, C.F.; Lim, L.-T. Influence of Extraction Conditions on Ultrasound-Assisted Recovery of Bioactive Phenolics from Blueberry Pomace and Their Antioxidant Activity. Molecules 2018, 23, 1685. https://doi.org/10.3390/molecules23071685
Bamba BSB, Shi J, Tranchant CC, Xue SJ, Forney CF, Lim L-T. Influence of Extraction Conditions on Ultrasound-Assisted Recovery of Bioactive Phenolics from Blueberry Pomace and Their Antioxidant Activity. Molecules. 2018; 23(7):1685. https://doi.org/10.3390/molecules23071685
Chicago/Turabian StyleBamba, Bio Sigui Bruno, John Shi, Carole C. Tranchant, Sophia Jun Xue, Charles F. Forney, and Loong-Tak Lim. 2018. "Influence of Extraction Conditions on Ultrasound-Assisted Recovery of Bioactive Phenolics from Blueberry Pomace and Their Antioxidant Activity" Molecules 23, no. 7: 1685. https://doi.org/10.3390/molecules23071685
APA StyleBamba, B. S. B., Shi, J., Tranchant, C. C., Xue, S. J., Forney, C. F., & Lim, L.-T. (2018). Influence of Extraction Conditions on Ultrasound-Assisted Recovery of Bioactive Phenolics from Blueberry Pomace and Their Antioxidant Activity. Molecules, 23(7), 1685. https://doi.org/10.3390/molecules23071685