Identification of a 3-Alkylpyridinium Compound from the Red Sea Sponge Amphimedon chloros with In Vitro Inhibitory Activity against the West Nile Virus NS3 Protease

 , , , and
, , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
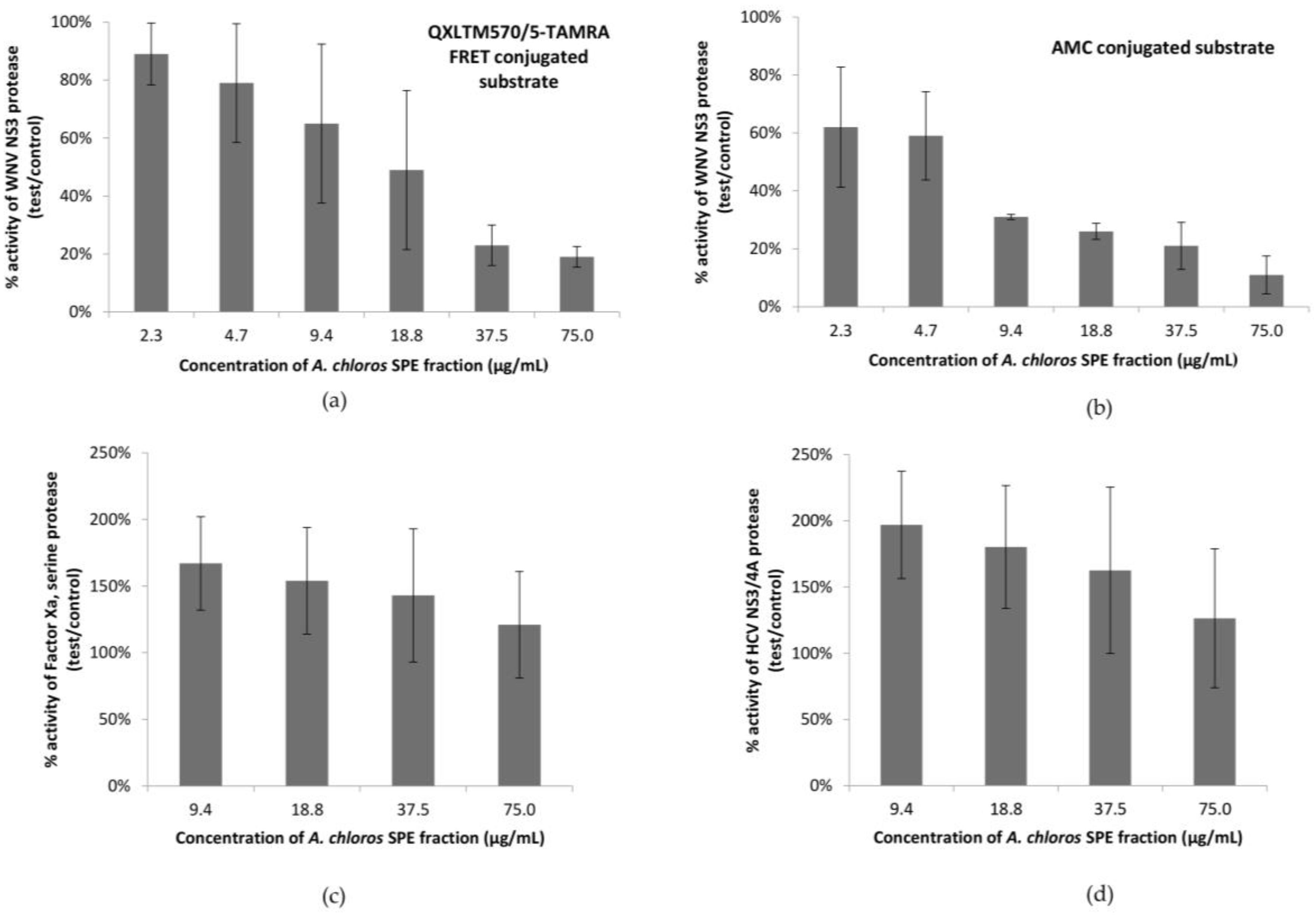
2.1. A. chloros Demonstrates Inhibition of West Nile Virus NS3 Protease
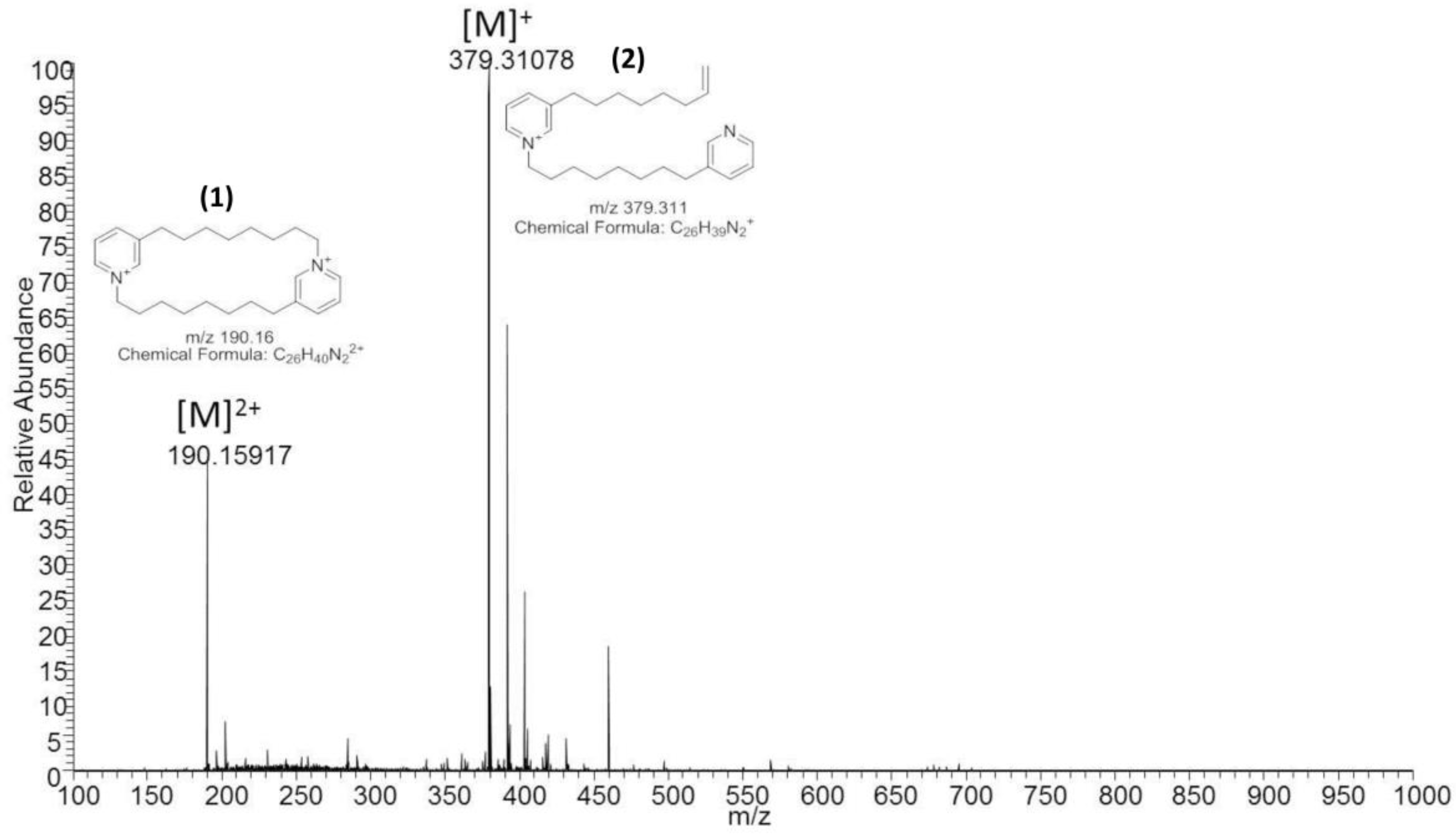
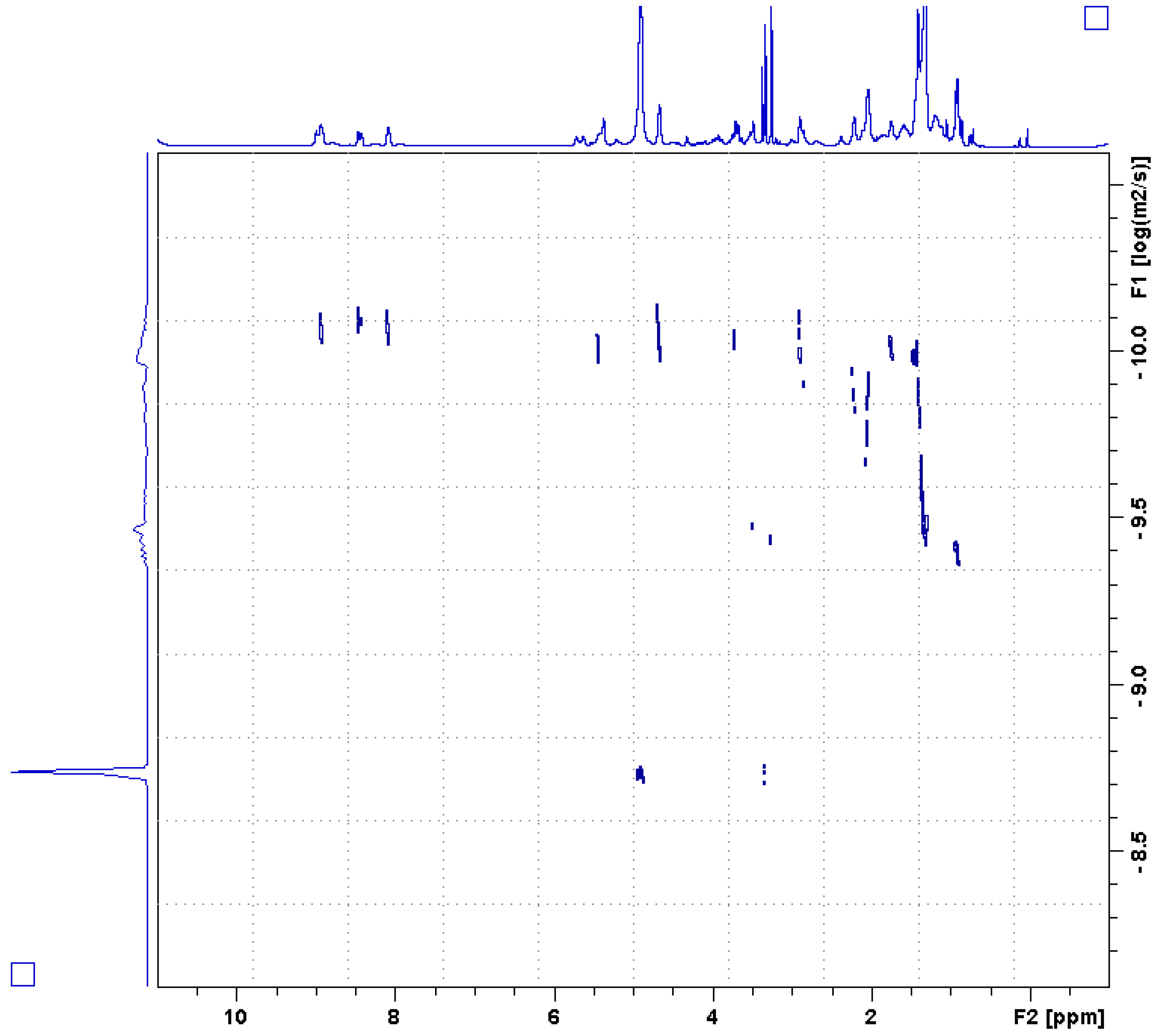
2.2. Analytical Chemistry Reveals 3-Alkylpyridinium as the Bioactive Compound
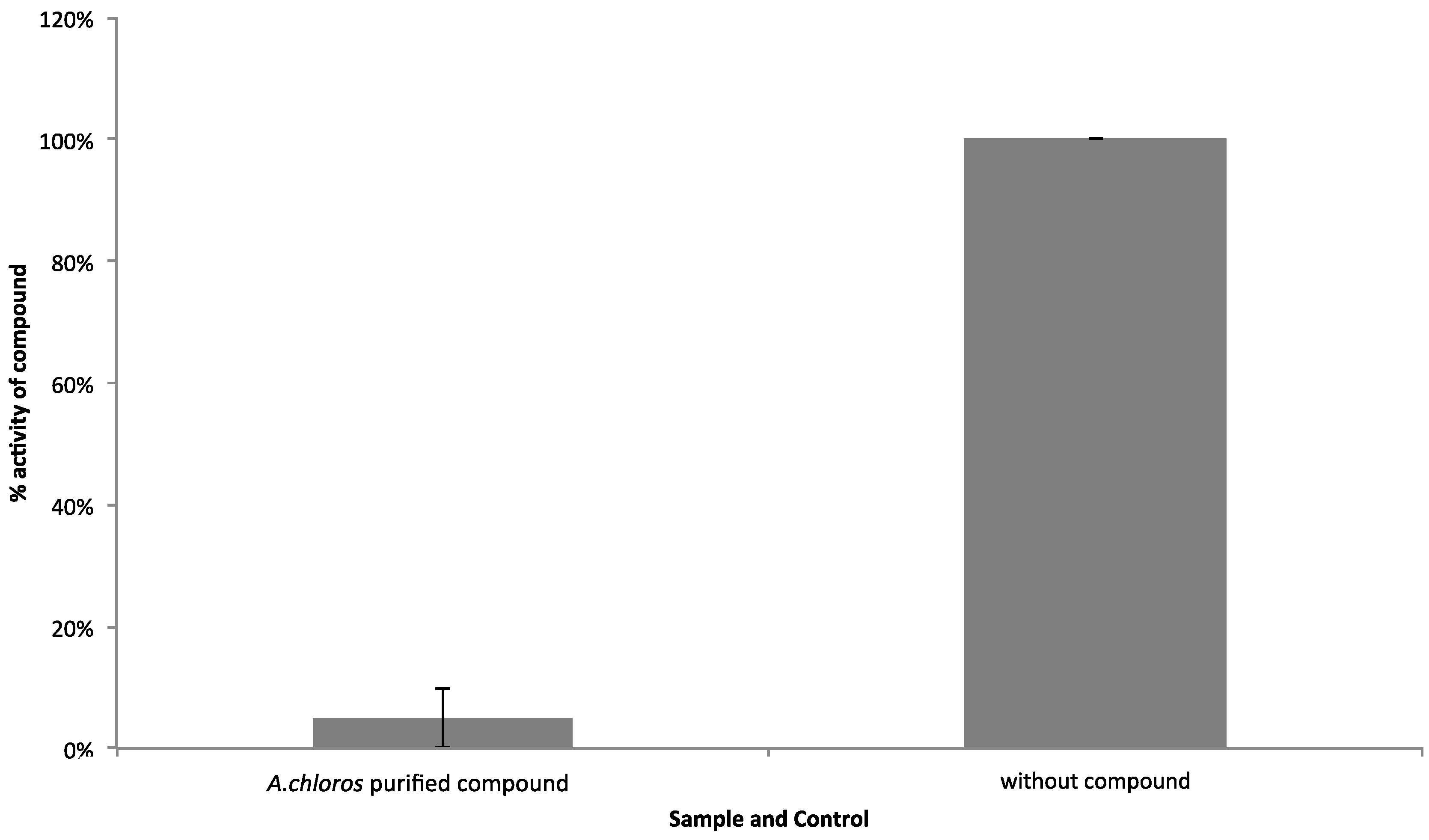
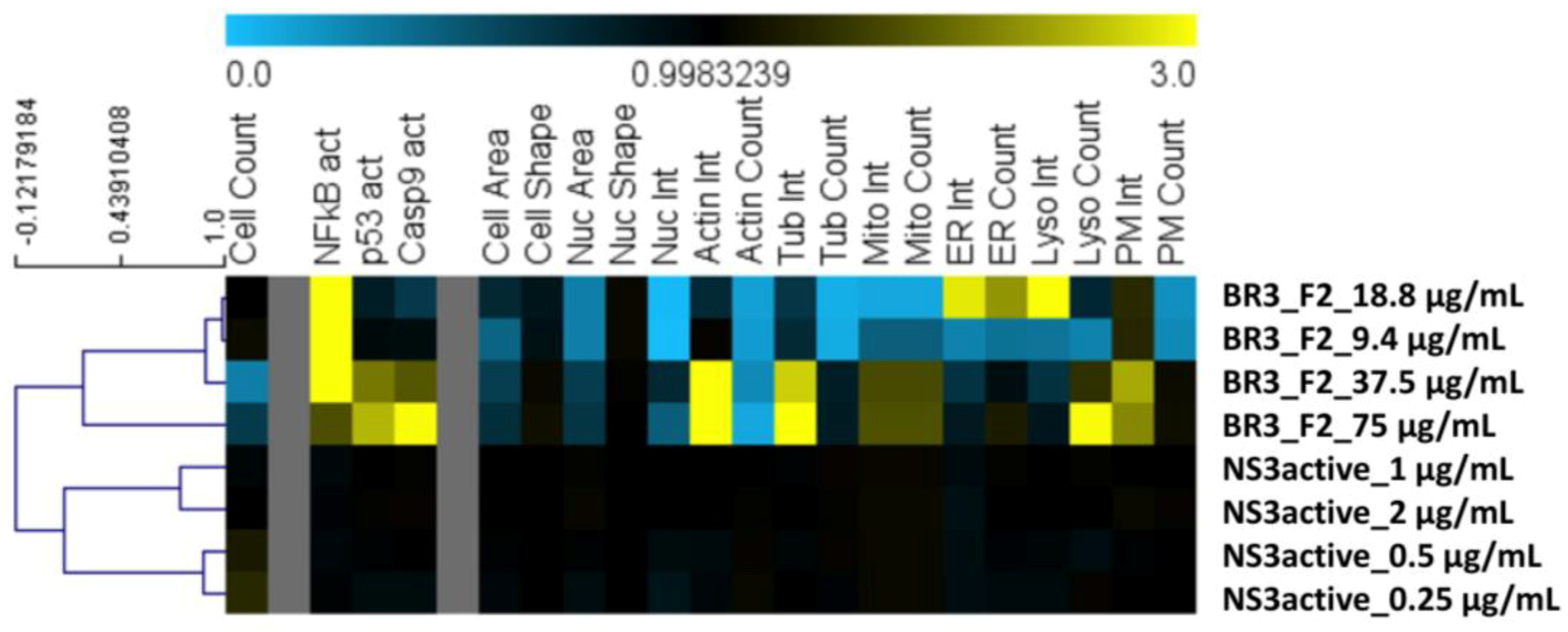
2.3. Cytological Profiling Reveals the Bioactive 3-Alkylpyridinium Salt as Negligibly Cytotoxic
3. Discussion
4. Materials and Methods
4.1. A. chloros Sponge Collection
4.2. A. chloros Sponge Extraction
4.3. Liquid Chromatography-Mass Spectrometry (LC-MS) of A. chloros SPE Fraction
4.4. Nuclear Magnetic Resonance (NMR) of A. chloros 3-Alkyl Pyridinium
4.5. West Nile Virus (WNV) NS3 Protease Inhibition Assay
4.6. HCV NS3/4A Protease Inhibition Assay
4.7. Thrombin Serine Protease Inhibition Assay
4.8. Cytological Profiling by High-Content Screening (HCS)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FDA Office of the Commissioner. Reports-Targeted Drug Development: Why Are Many Diseases Lagging Behind? Office of the Commissioner: Silver Spring, MD, USA, 2015. Available online: https://www.fdanews.com/ext/resources/files/07-15/7-15-FDA-Report.pdf?1518595108 (accessed on 23 June 2016).
- Campbell, G.L.; Marfin, A.A.; Lanciottia, R.S.; Gublera, D.J. West Nile virus. Lancet Infect. Dis. 2002, 2, 519–529. [Google Scholar] [CrossRef]
- Leyssen, P.; De Clercq, E.; Neyts, J. Perspectives for the treatment of infections with Flaviviridae. Clin. Microbiol. Rev. 2000, 13, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.P.; Shi, P.-Y. West Nile virus drug discovery. Viruses 2013, 5, 2977–3006. [Google Scholar] [CrossRef] [PubMed]
- Brinton, M.A. Replication cycle and molecular biology of the West Nile virus. Viruses 2013, 6, 13–53. [Google Scholar] [CrossRef] [PubMed]
- Cregar-Hernandez, L.; Jiao, G.-S.; Johnson, A.T.; Lehrer, A.T.; Wong, T.A.S.; Margosiak, S.A. Small molecule pan-dengue and West Nile virus NS3 protease inhibitors. Antivir. Chem. Chemother. 2011, 21, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV/AIDS 2015, 7, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Salam, K.A.; Akimitsu, N. Hepatitis C virus NS3 inhibitors: current and future perspectives. Biomed. Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Patick, A.K.; Potts, K.E. Protease inhibitors as antiviral agents. Clin. Microbiol. Rev. 1998, 11, 614–627. [Google Scholar] [PubMed]
- Schmitz, J.F.; Hollenbeak, K.H.; Campbell, D.C. Marine natural products: halitoxin, toxic complex of several marine sponges of the genus Haliclona. J. Org. Chem. 1978, 43, 3916–3922. [Google Scholar] [CrossRef]
- Davies-Coleman, M.T.; Faulkner, D.J.; Dubowchik, G.M.; Roth, G.P.; Polson, C.; Fairchild, C. A new EGF-active polymeric pyridinium alkaloid from the sponge Callyspongia fibrosa. J. Org. Chem. 1993, 58, 5925–5930. [Google Scholar] [CrossRef]
- Oku, N.; Nagai, K.; Shindoh, N.; Terada, Y.; van Soest, R.W.M.; Matsunaga, S.; Fusetani, N. Three new cyclostellettamines, which inhibit histone deacetylase, from a marine sponge of the genus Xestospongia. Bioorganic. Med. Chem. Lett. 2004, 14, 2617–2620. [Google Scholar] [CrossRef] [PubMed]
- Zovko, A.; Viktorsson, K.; Lewensohn, R.; Kološa, K.; Filipič, M.; Xing, H.; Kem, W.R.; Paleari, L.; Turk, T. APS8, a polymeric alkylpyridinium salt blocks α7 nAChR and induces apoptosis in non-small cell lung carcinoma. Mar. Drugs 2013, 11, 2574–2594. [Google Scholar] [CrossRef] [PubMed]
- Grandič, M.; Zovko, A.; Frangež, R.; Turk, T.; Sepčić, K. Binding and permeabilization of lipid bilayers by natural and synthetic 3-alkylpyridinium polymers. Bioorganic. Med. Chem. 2012, 20, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.H.; Whyment, A.D.; Foster, A.; Gordon, K.H.; Milne, B.F.; Jaspars, M. Analysis of the structure and electrophysiological actions of halitoxins: 1,3 alkyl-pyridinium salts from Callyspongia ridleyi. J. Membr. Biol. 2000, 176, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Shinoda, K.; Fusetani, N. Cribrochalinamine oxides A and B, antifungal Beta-substituted pyridines with an azomethine N-oxide from a marine sponge Cribrochalina sp. Tetrahedron Lett. 1993, 34, 5953–5954. [Google Scholar] [CrossRef]
- De Oliveira, J.H.H.L.; Seleghim, M.H.R.; Timm, C.; Grube, A.; Köck, M.; Nascimento, G.G.F.; Martins, A.C.T.; Silva, E.G.O.; de Souza, A.O.; Minarini, P.R.R.; et al. Antimicrobial and antimycobacterial activity of cyclostellettamine alkaloids from sponge Pachychalina sp. Mar. Drugs 2006, 4, 1–8. [Google Scholar] [CrossRef]
- Dasari, V.R.R.K.; Muthyala, M.K.K.; Nikku, M.Y.; Donthireddy, S.R.R. Novel Pyridinium compound from marine actinomycete, Amycolatopsis alba var. nov. DVR D4 showing antimicrobial and cytotoxic activities in vitro. Microbiol. Res. 2012, 167, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sun, D. Macrocyclic drugs and synthetic methodologies toward macrocycles. Molecules 2013, 18, 6230–6268. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; Deng, Z.; Rogerson, A.K.; McLachlan, A.S.; Richards, J.J; Nilsson, M.; Morris, G.A. Quantitative interpretation of diffusion-ordered NMR spectra: Can we rationalize small molecule diffusion coefficients? Angew. Chem. Int. Ed. 2013, 52, 3199–3202. [Google Scholar] [CrossRef] [PubMed]
- Bugni, T.S.; Richards, B.; Bhoite, L.; Cimbora, D.; Harper, M.K.; Ireland, C.M. Marine natural product libraries for high-throughput screening and rapid drug discovery. J. Nat. Prod. 2008, 71, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.G.; Kuntz, I.D. UCSF Sparky - an NMR Display, Annotation and Assignment Tool. J. Cell Biochem. 1993, 53, 254. [Google Scholar] [CrossRef]
- Kremb, S.; Voolstra, C.R. High-resolution phenotypic profiling of natural products-induced effects on the single-cell level. Sci. Rep. 2017, 7, 44472. [Google Scholar] [CrossRef] [PubMed]
- Madaan, P.; Tyagi, V.K. Quaternary pyridinium salts: a review. J. Oleo Sci. 2008, 57, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Lippert, K.; Galinski, E.A. Enzyme stabilization be ectoine-type compatible solutes: protection against heating, freezing and drying. Appl. Microbiol. Biotechnol. 1992, 37, 61–65. [Google Scholar] [CrossRef]
- Krupa, J.C.; Mort, J.S. Optimization of detergents for the assay of cathepsins B, L, S, and K. Anal. Biochem. 2000, 283, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, C.; Kayser, O.; Müller, R.H. Enzymatic Degradation of Dynasan 114 SLN - Effect of Surfactants and Particle Size. J. Nanoparticle Res. 2002, 4, 121–129. [Google Scholar] [CrossRef]
- Tucker, S.J.; McClelland, D.; Jaspars, M.; Sepčić, K.; MacEwan, D.J.; Scott, R.H. The influence of alkyl pyridinium sponge toxins on membrane properties, cytotoxicity, transfection and protein expression in mammalian cells. Biochim. Biophys. Acta Biomembr. 2003, 1614, 171–181. [Google Scholar] [CrossRef]
- Kremb, S.; Müller, C.; Schmitt-Kopplin, P.; Voolstra, C.R. Bioactive Potential of Marine Macroalgae from the Central Red Sea (Saudi Arabia) Assessed by High-Throughput Imaging-Based Phenotypic Profiling. Mar. Drugs 2017, 15, 80. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from authors. |





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Rourke, A.; Kremb, S.; Duggan, B.M.; Sioud, S.; Kharbatia, N.; Raji, M.; Emwas, A.-H.; Gerwick, W.H.; Voolstra, C.R. Identification of a 3-Alkylpyridinium Compound from the Red Sea Sponge Amphimedon chloros with In Vitro Inhibitory Activity against the West Nile Virus NS3 Protease. Molecules 2018, 23, 1472. https://doi.org/10.3390/molecules23061472
O’Rourke A, Kremb S, Duggan BM, Sioud S, Kharbatia N, Raji M, Emwas A-H, Gerwick WH, Voolstra CR. Identification of a 3-Alkylpyridinium Compound from the Red Sea Sponge Amphimedon chloros with In Vitro Inhibitory Activity against the West Nile Virus NS3 Protease. Molecules. 2018; 23(6):1472. https://doi.org/10.3390/molecules23061472
Chicago/Turabian StyleO’Rourke, Aubrie, Stephan Kremb, Brendan M. Duggan, Salim Sioud, Najeh Kharbatia, Misjudeen Raji, Abdul-Hamid Emwas, William H. Gerwick, and Christian R. Voolstra. 2018. "Identification of a 3-Alkylpyridinium Compound from the Red Sea Sponge Amphimedon chloros with In Vitro Inhibitory Activity against the West Nile Virus NS3 Protease" Molecules 23, no. 6: 1472. https://doi.org/10.3390/molecules23061472
APA StyleO’Rourke, A., Kremb, S., Duggan, B. M., Sioud, S., Kharbatia, N., Raji, M., Emwas, A.-H., Gerwick, W. H., & Voolstra, C. R. (2018). Identification of a 3-Alkylpyridinium Compound from the Red Sea Sponge Amphimedon chloros with In Vitro Inhibitory Activity against the West Nile Virus NS3 Protease. Molecules, 23(6), 1472. https://doi.org/10.3390/molecules23061472