Involvement of CYP4F2 in the Metabolism of a Novel Monophosphate Ester Prodrug of Gemcitabine and Its Interaction Potential In Vitro

Abstract
1. Introduction
2. Results
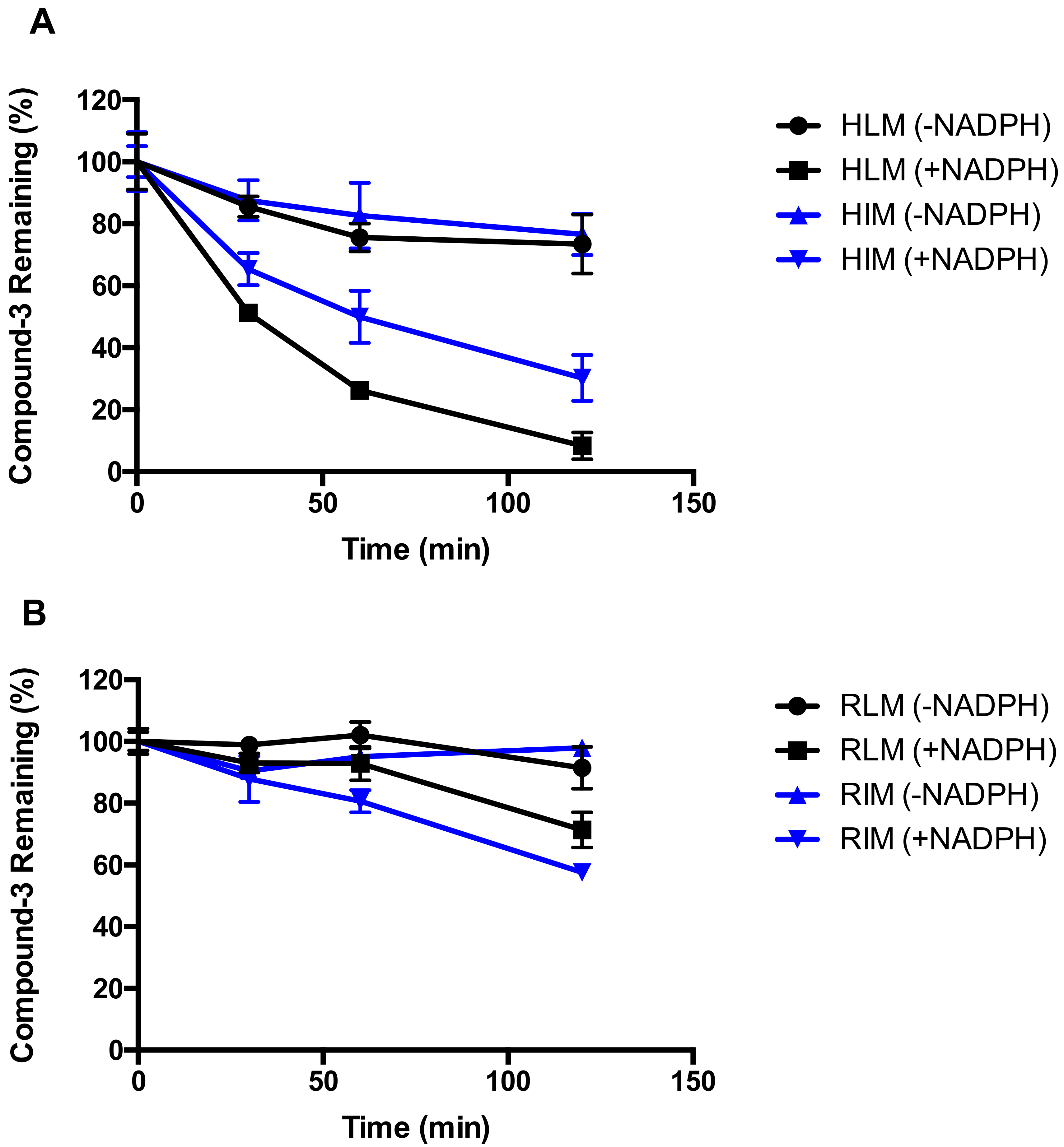
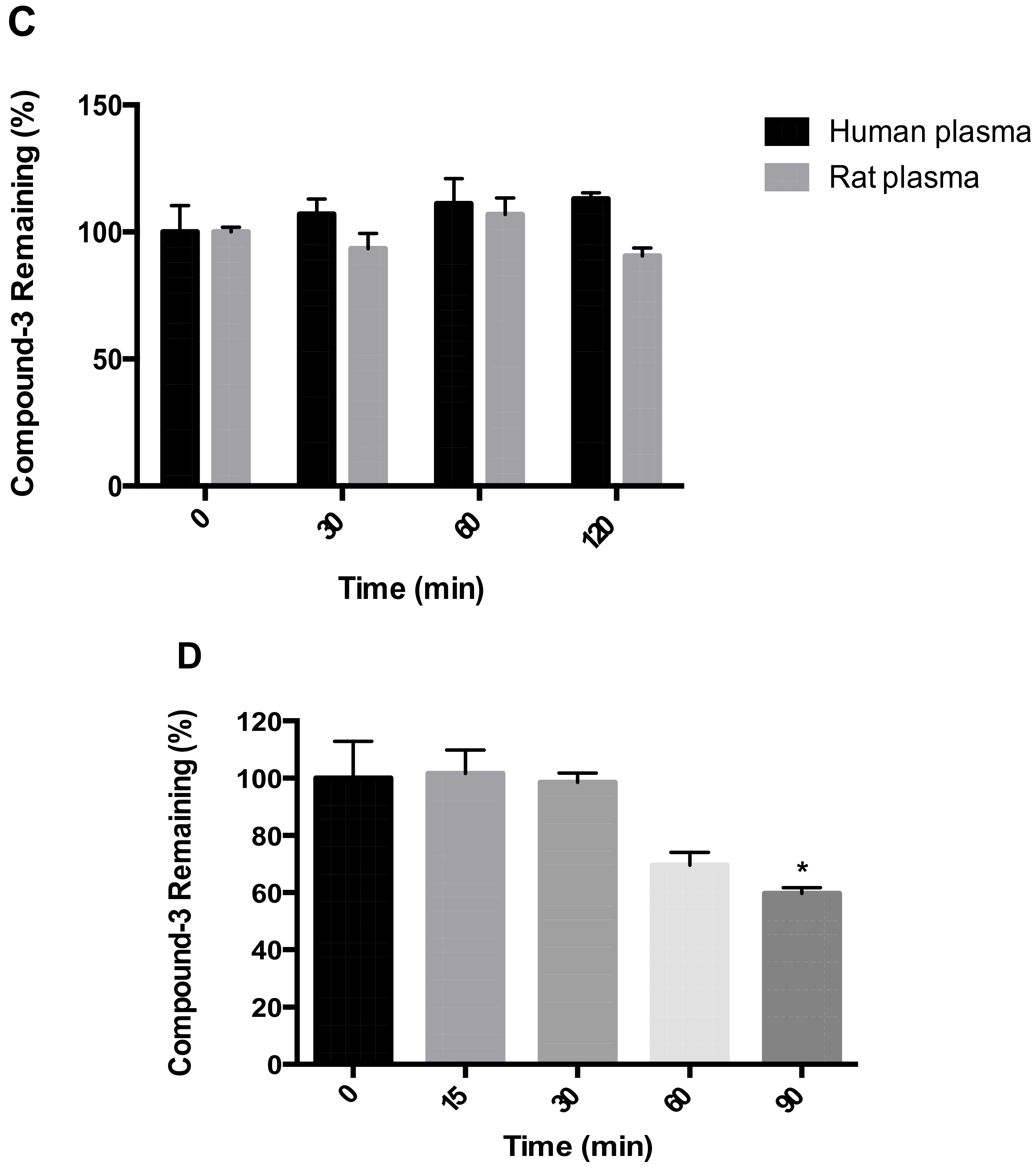
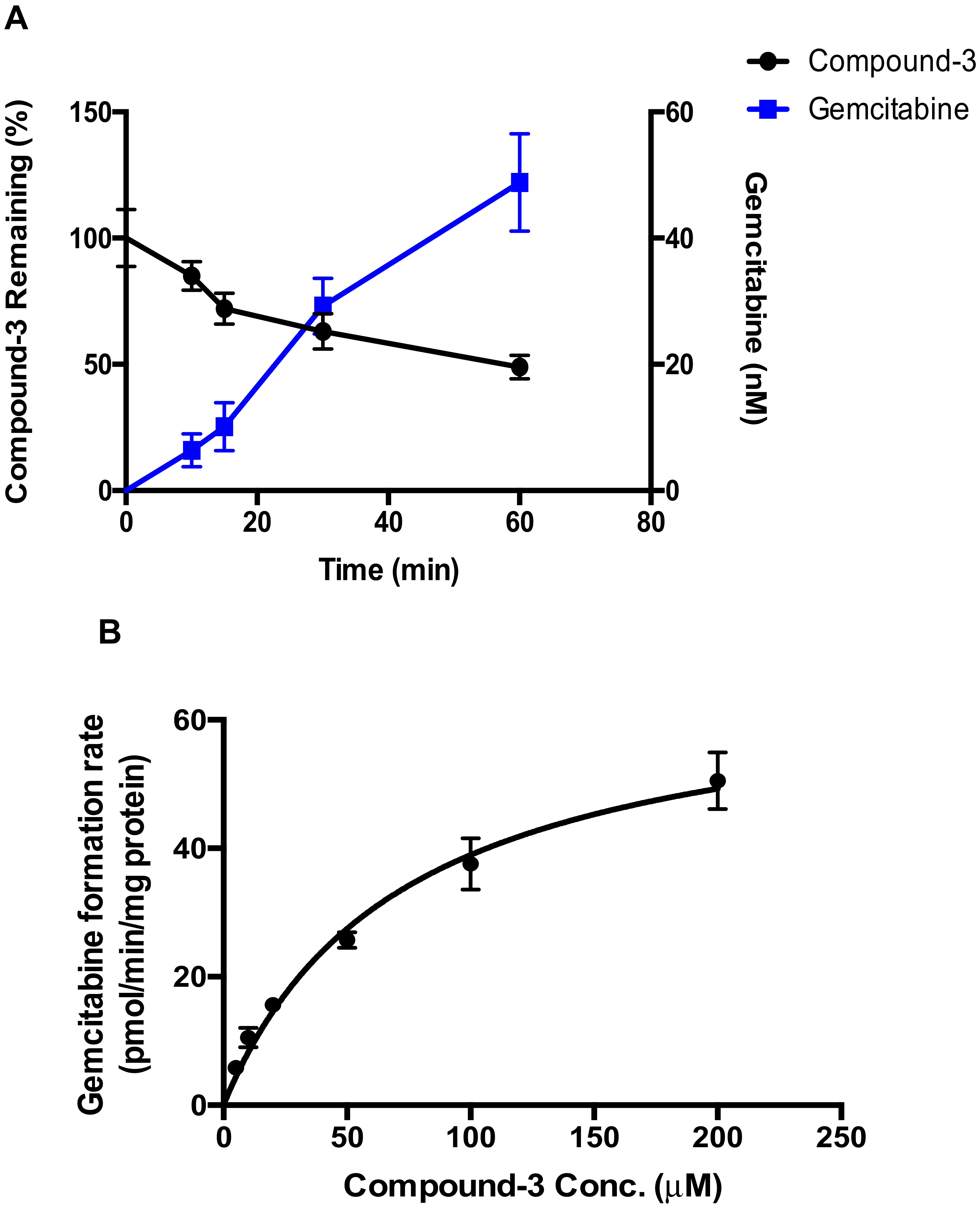
2.1. Metabolic Stability of Compound-3
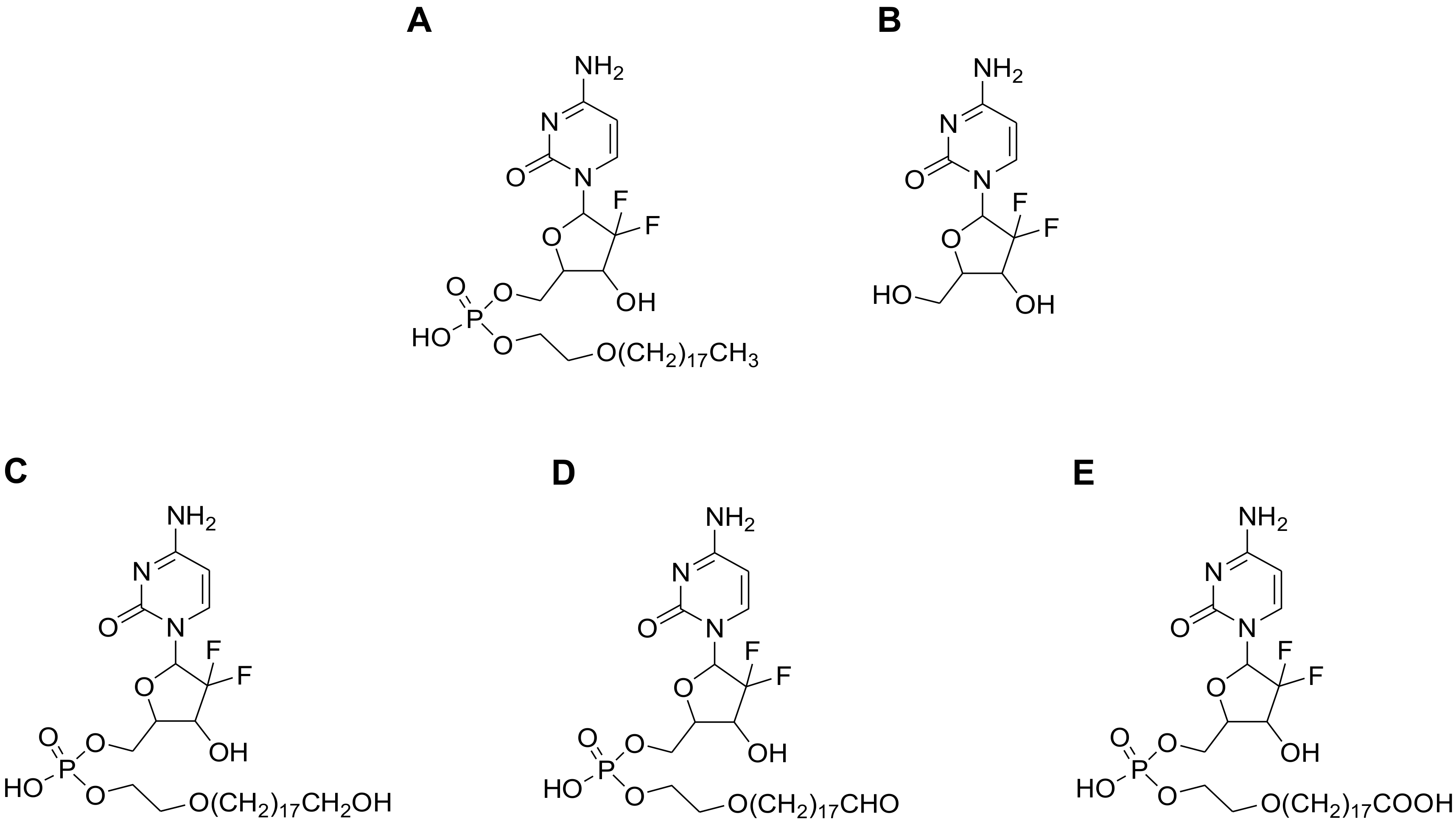
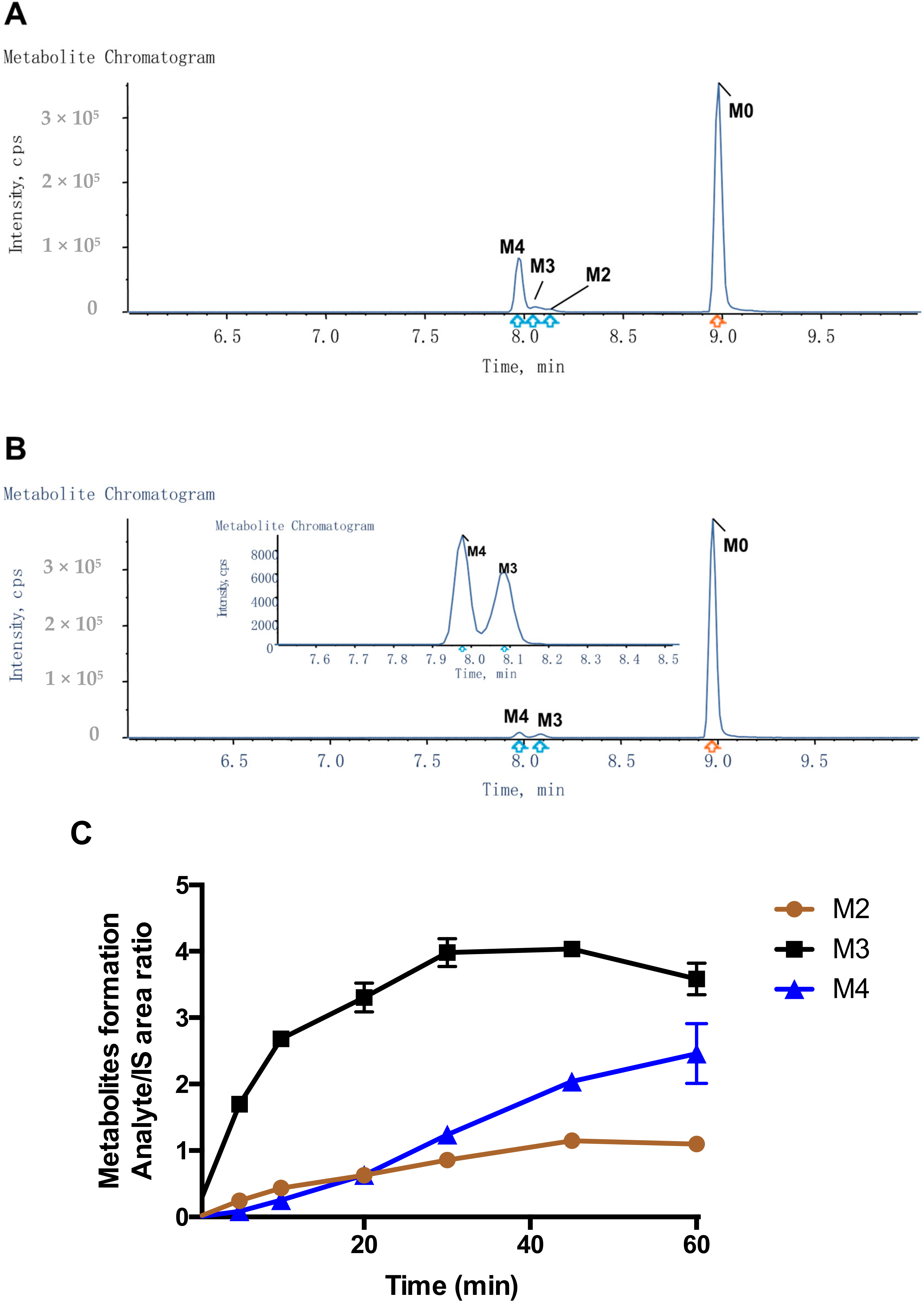
2.2. Detection of Compound-3 Metabolites
2.3. Hydrolysis of Compound-3 by Alkaline Phosphatase
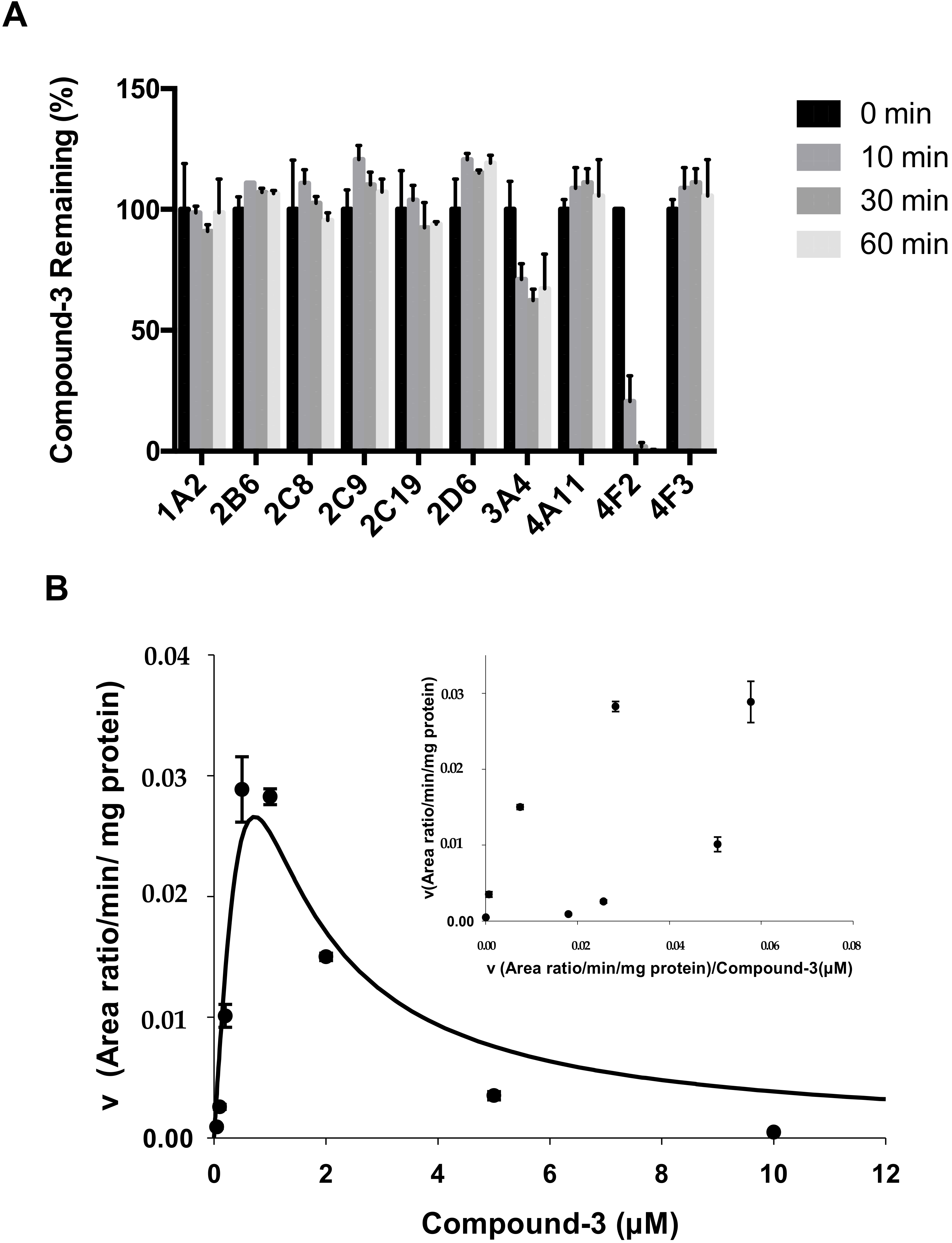
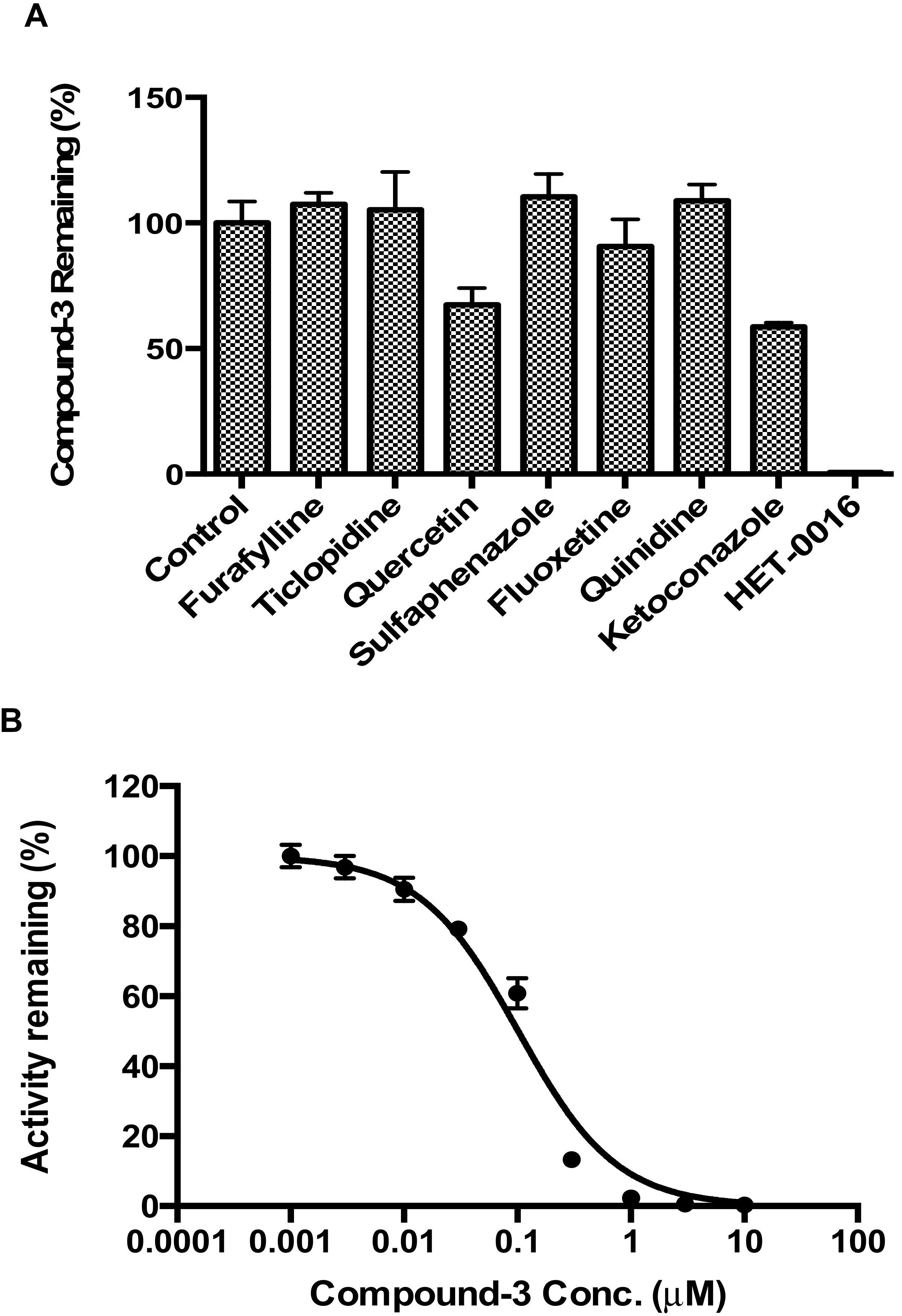
2.4. Identification of CYP Isozymes Involved in Compound-3 Metabolism
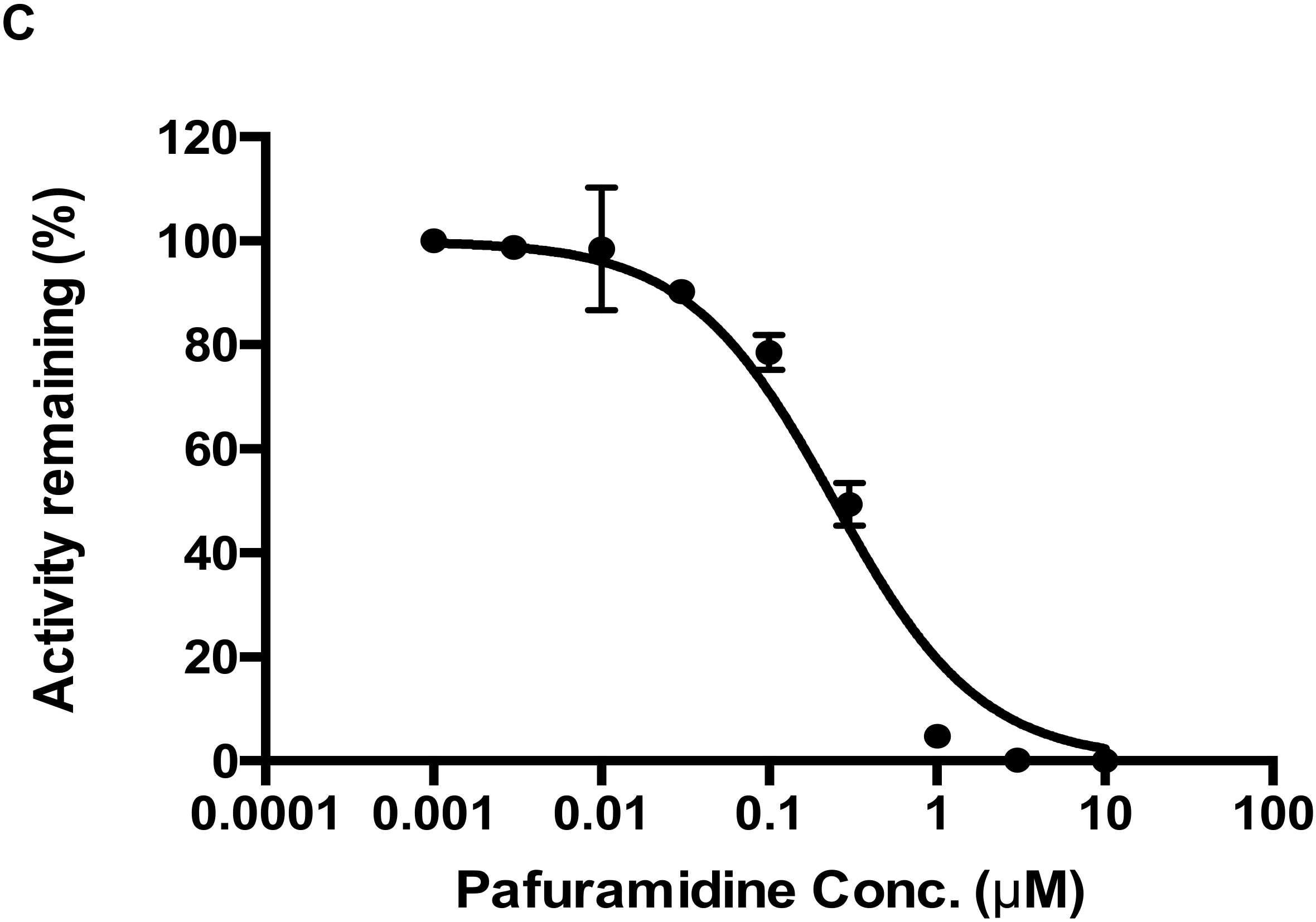
2.5. CYP4F2 Mediated Interaction
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Metabolic Stability of Compound-3 in Various In Vitro Matrices
4.3. Hydrolysis of Compound-3 by Alkaline Phosphatase
4.4. Identification of CYP Isozymes Involved in Compound-3 Metabolism
4.5. Evaluation of CYP4F2-Mediated Drug Interactions
4.6. LC-MS/MS Analysis
4.7. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CYP | cytochrome P450 |
| HRMS | high resolution mass spectrometry |
| HLM or HIM | human liver or intestinal microsomes |
| IS | internal standard |
| RLM or RIM | rat liver or intestinal microsomes |
References
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.-R.; et al. Discovery of a β-d-2′-deoxy-2′-α-fluoro-2′-β-c-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis c virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, M.; Zolopa, A.; Squires, K.; Ruane, P.; Coakley, D.; Kearney, B.; Zhong, L.J.; Wulfsohn, M.; Miller, M.D.; Lee, W.A. Phase I/II study of the pharmacokinetics, safety and antiretroviral activity of tenofovir alafenamide, a new prodrug of the HIV reverse transcriptase inhibitor tenofovir, in HIV-infected adults. J. Antimicrob. Chemother. 2014, 69, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Murakami, E.; Tolstykh, T.; Bao, H.Y.; Niu, C.R.; Steuer, H.M.M.; Bao, D.H.; Chang, W.; Espiritu, C.; Bansal, S.; Lam, A.M.; et al. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285, 34337–34347. [Google Scholar] [CrossRef] [PubMed]
- Sinokrot, H.; Smerat, T.; Najjar, A.; Karaman, R. Advanced prodrug strategies in nucleoside and non-nucleoside antiviral agents: A review of the recent five years. Molecules 2017, 22, 1736. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y.; Rattan, H.S.; Balzarini, J. The protide prodrug technology: From the concept to the clinic. J. Med. Chem. 2018, 61, 2211–2226. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.D.; Saneja, A.; Gupta, P.K.; Gupta, P.N. Recent advances in drug delivery strategies for improved therapeutic efficacy of gemcitabine. Eur. J. Pharm. Sci. 2016, 93, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Moysan, E.; Bastiat, G.; Benoit, J.P. Gemcitabine versus modified gemcitabine: A review of several promising chemical modifications. Mol. Pharm. 2013, 10, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Wickremsinhe, E.; Bao, J.; Smith, R.; Burton, R.; Dow, S.; Perkins, E. Preclinical absorption, distribution, metabolism, and excretion of an oral amide prodrug of gemcitabine designed to deliver prolonged systemic exposure. Pharmaceutics 2013, 5, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of protide technology to gemcitabine: A successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.X.; Lu, J.; Li, J.J.; Wang, M.Y.; Xu, Y.T.; Wang, Y.D.; Zhang, H.J. Enhanced antitumor activity of monophosphate ester prodrugs of gemcitabine: In vitro and in vivo evaluation. J. Pharm. Sci. 2016, 105, 2966–2973. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Aho, U.; Nilsson, B.I.; Peters, G.J.; Pastor-Anglada, M.; Rasch, W.; Sandvold, M.L. Gemcitabine chemoresistance in pancreatic cancer: Molecular mechanisms and potential solutions. Scand. J. Gastroenterol. 2009, 44, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Michaels, S.; Wang, M.Z. The revised human liver cytochrome p450 “pie”: Absolute protein quantification of CYP4F and CYP3A enzymes using targeted quantitative proteomics. Drug Metab. Dispos. 2014, 42, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.L.; Edson, K.Z.; Totah, R.A.; Rettie, A.E. Chapter Eight-Cytochrome p450 ω-Hydroxylases in Inflammation and Cancer; Hardwick, J.P., Ed.; Advances Pharmacology Academic Press: Cambridge, MA, USA, 2015; Volume 74, pp. 223–262. [Google Scholar]
- Andrews, L.D.; Zalatan, J.G.; Herschlag, D. Probing the origins of catalytic discrimination between phosphate and sulfate monoester hydrolysis: Comparative analysis of alkaline phosphatase and protein tyrosine phosphatases. Biochemistry 2014, 53, 6811–6819. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Guengerich, F.P. Survey of human oxidoreductases and cytochrome p450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 2015, 28, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Kalsotra, A.; Strobel, H.W. Cytochrome P4504F subfamily: At the crossroads of eicosanoid and drug metabolism. Pharm. Ther. 2006, 112, 589–611. [Google Scholar] [CrossRef] [PubMed]
- Edson, K.Z.; Prasad, B.; Unadkat, J.D.; Suhara, Y.; Okano, T.; Guengerich, F.P.; Rettie, A.E. Cytochrome p450-dependent catabolism of vitamin K: Ω-Hydroxylation catalyzed by human CYP4F2 and CYP4F11. Biochemistry 2013, 52, 8276–8285. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Z.; Saulter, J.Y.; Usuki, E.; Cheung, Y.-L.; Hall, M.; Bridges, A.S.; Loewen, G.; Parkinson, O.T.; Stephens, C.E.; Allen, J.L.; et al. CYP4F enzymes are the major enzymes in human liver microsomes that catalyze the o-demethylation of the antiparasitic prodrug db289 [2,5-bis(4-amidinophenyl)furan-bis-o-methylamidoxime]. Drug Metab. Dispos. 2006, 34, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Z.; Wu, J.Q.; Bridges, A.S.; Zeldin, D.C.; Kornbluth, S.; Tidwell, R.R.; Hall, J.E.; Paine, M.F. Human enteric microsomal CYP4F enzymes o-demethylate the antiparasitic prodrug pafuramidine. Drug Metab. Dispos. 2007, 35, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zollinger, M.; Borell, H.; Zimmerlin, A.; Patten, C.J. CYP4F enzymes are responsible for the elimination of fingolimod (FTY720), a novel treatment of relapsing multiple sclerosis. Drug Metab. Dispos. 2011, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Tim, T.; Jenni, C.; Laurie, K.; Etsuko, U.; Brain, O.; Kathy, V.S.; Herve, M.-M.; Irma, M.G.; Lawrence, C.T. CYP4F2 is the major cytochrome p450 enzyme involved in CMX001 metabolism. Drug Metab. Rev. 2013, 45, 84–85. [Google Scholar]
- Davit, B.; Reynolds, K.; Yuan, R.; Ajayi, F.; Conner, D.; Fadiran, E.; Gillespie, B.; Sahajwalla, C.; Huang, S.M.; Lesko, L.J. Fda evaluations using in vitro metabolism to predict and interpret in vivo metabolic drug-drug interactions: Impact on labeling. J. Clin. Pharmacol. 1999, 39, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Tilson, H.; Hines, L.E.; McEvoy, G.; Weinstein, D.M.; Hansten, P.D.; Matuszewski, K.; Le Comte, M.; Higby-Baker, S.; Hanlon, J.T.; Pezzullo, L.; et al. Recommendations for selecting drug-drug interactions for clinical decision support. Am. J. Health-Syst. Pharm. 2016, 73, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Clinical Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications: Guidance for Industry. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf (accessed on 18 April 2018).
Sample Availability: Compound-3 was supplied by PharmaResources, Ltd. and pafuramidine was purchased from MedChemexpress. Please refer to Materials and Methods for the sources of all other reagents. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Metabolic Reaction | Retention Time (min) | Observed Mass (m/z) | Calculated Mass (m/z) | Elemental Composition | Mass Error (ppm) | Matrix | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALP | HLM | RLM | HIM | RIM | Human Hepatocytes | |||||||
| M0 | Compound-3 | 8.98 | 640.3538 | 640.3538 | C29H52F2N3O8P | −8.8 | √ | √ | √ | √ | √ | √ |
| M1 | Gemcitabine | 2.62 | 264.0782 | 264.0796 | C9H11F2N3O4 | 0.2 | √ | |||||
| M2 | Oxidation to aldehyde | 8.14 | 654.3331 | 654.3331 | C29H50F2N3O9P | 0.8 | √ | √ | √ | √ | ||
| M3 | Oxidation to hydroxyl | 8.05 | 656.3489 | 656.3487 | C29H52F2N3O9P | −9.6 | √ | √ | √ | √ | √ | |
| M4 | Oxidation to carboxylic acid | 7.97 | 670.3282 | 670.3280 | C29H50F2N3O10P | −7.6 | √ | √ | √ | √ | √ | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, Y.; Lu, J.; Qi, H.; Cheng, I.; Zhang, H. Involvement of CYP4F2 in the Metabolism of a Novel Monophosphate Ester Prodrug of Gemcitabine and Its Interaction Potential In Vitro. Molecules 2018, 23, 1195. https://doi.org/10.3390/molecules23051195
Wang Y, Li Y, Lu J, Qi H, Cheng I, Zhang H. Involvement of CYP4F2 in the Metabolism of a Novel Monophosphate Ester Prodrug of Gemcitabine and Its Interaction Potential In Vitro. Molecules. 2018; 23(5):1195. https://doi.org/10.3390/molecules23051195
Chicago/Turabian StyleWang, Yedong, Yuan Li, Jia Lu, Huixin Qi, Isabel Cheng, and Hongjian Zhang. 2018. "Involvement of CYP4F2 in the Metabolism of a Novel Monophosphate Ester Prodrug of Gemcitabine and Its Interaction Potential In Vitro" Molecules 23, no. 5: 1195. https://doi.org/10.3390/molecules23051195
APA StyleWang, Y., Li, Y., Lu, J., Qi, H., Cheng, I., & Zhang, H. (2018). Involvement of CYP4F2 in the Metabolism of a Novel Monophosphate Ester Prodrug of Gemcitabine and Its Interaction Potential In Vitro. Molecules, 23(5), 1195. https://doi.org/10.3390/molecules23051195