Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data?



Abstract
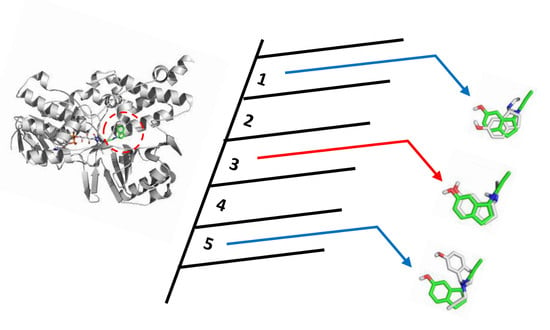
:
1. Introduction
2. Results
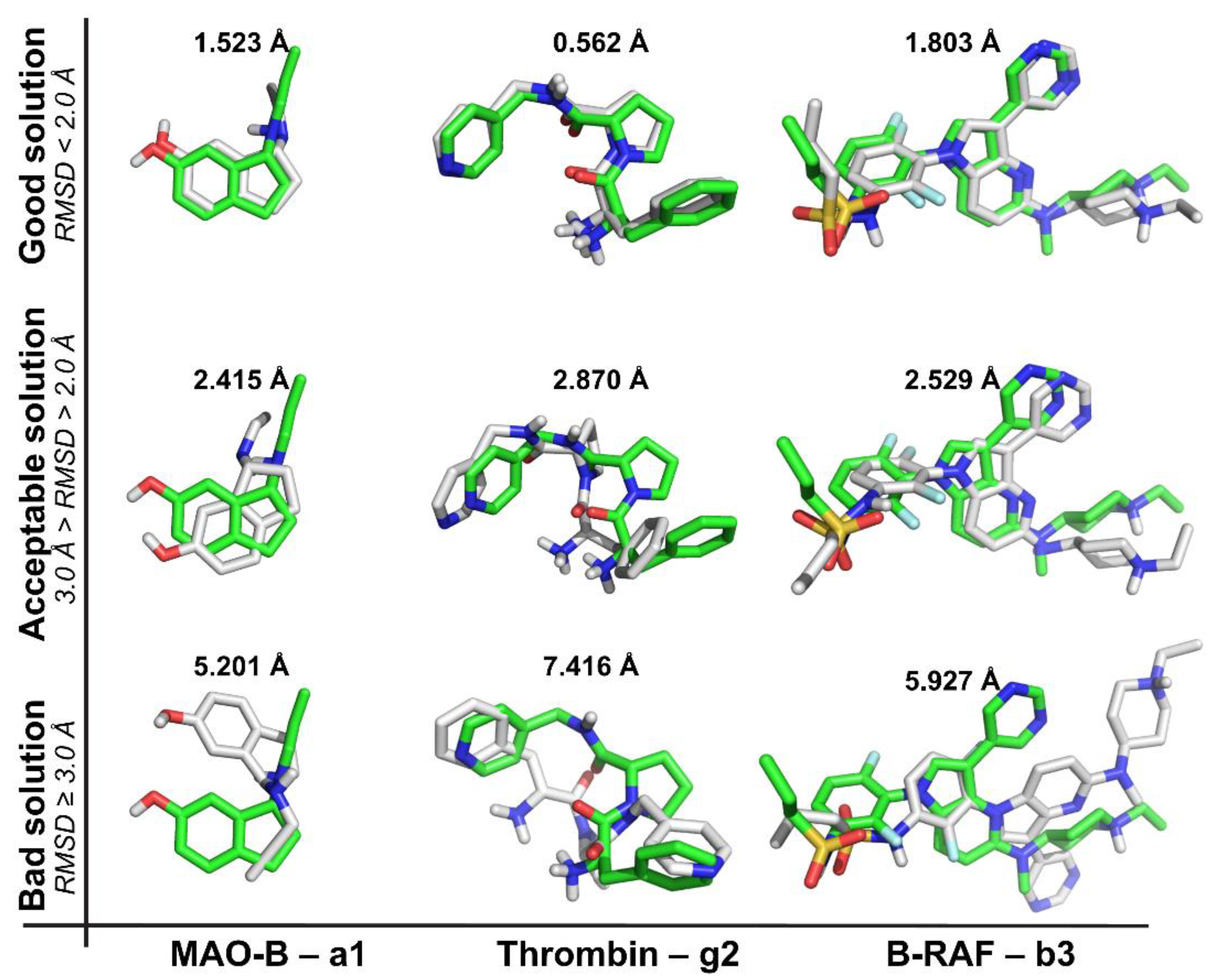
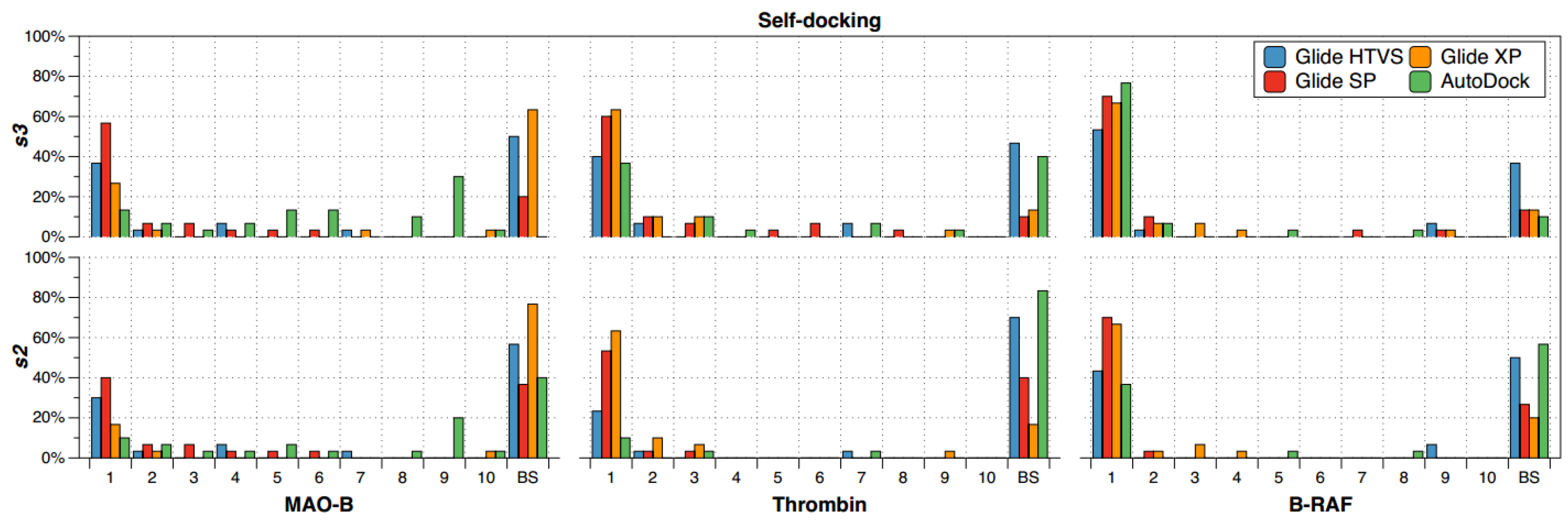
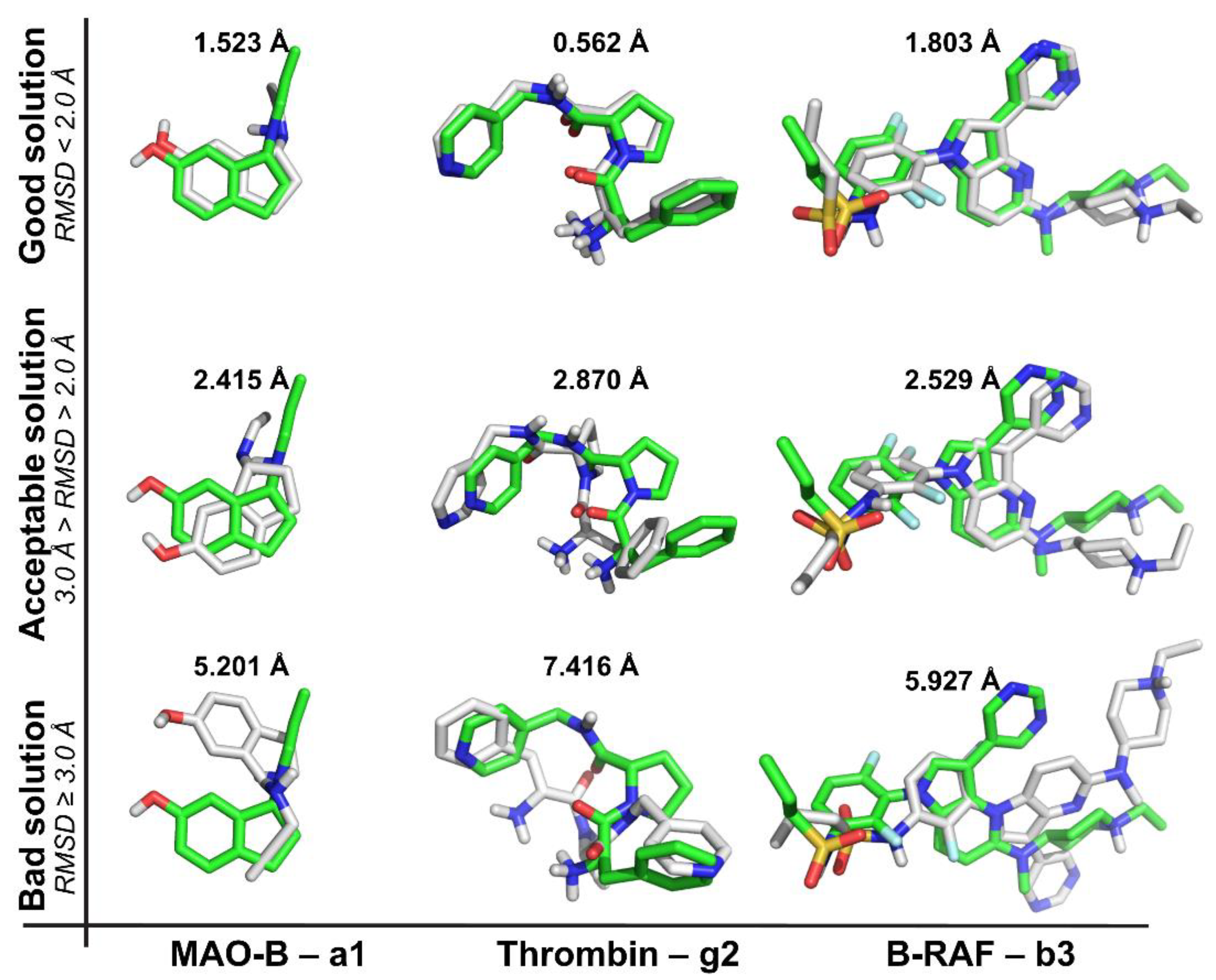
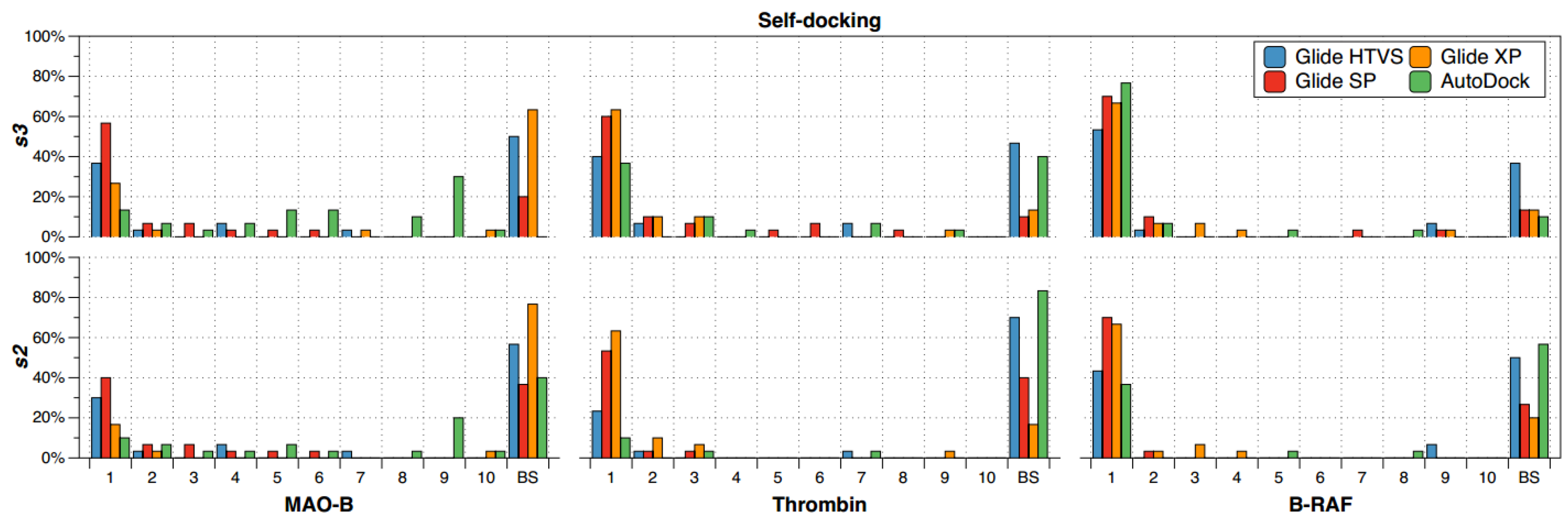
2.1. Self-Docking
- (a)
- The recurrence of the top first scoring position was higher for B-RAF inhibitors by using all the docking methods.
- (b)
- The recurrence of the top first scoring position was higher when the docking methods Glide SP and XP were used.
- (c)
- In the self-docking study, Autodock was the worst method to get the best solution at the top first scoring position.
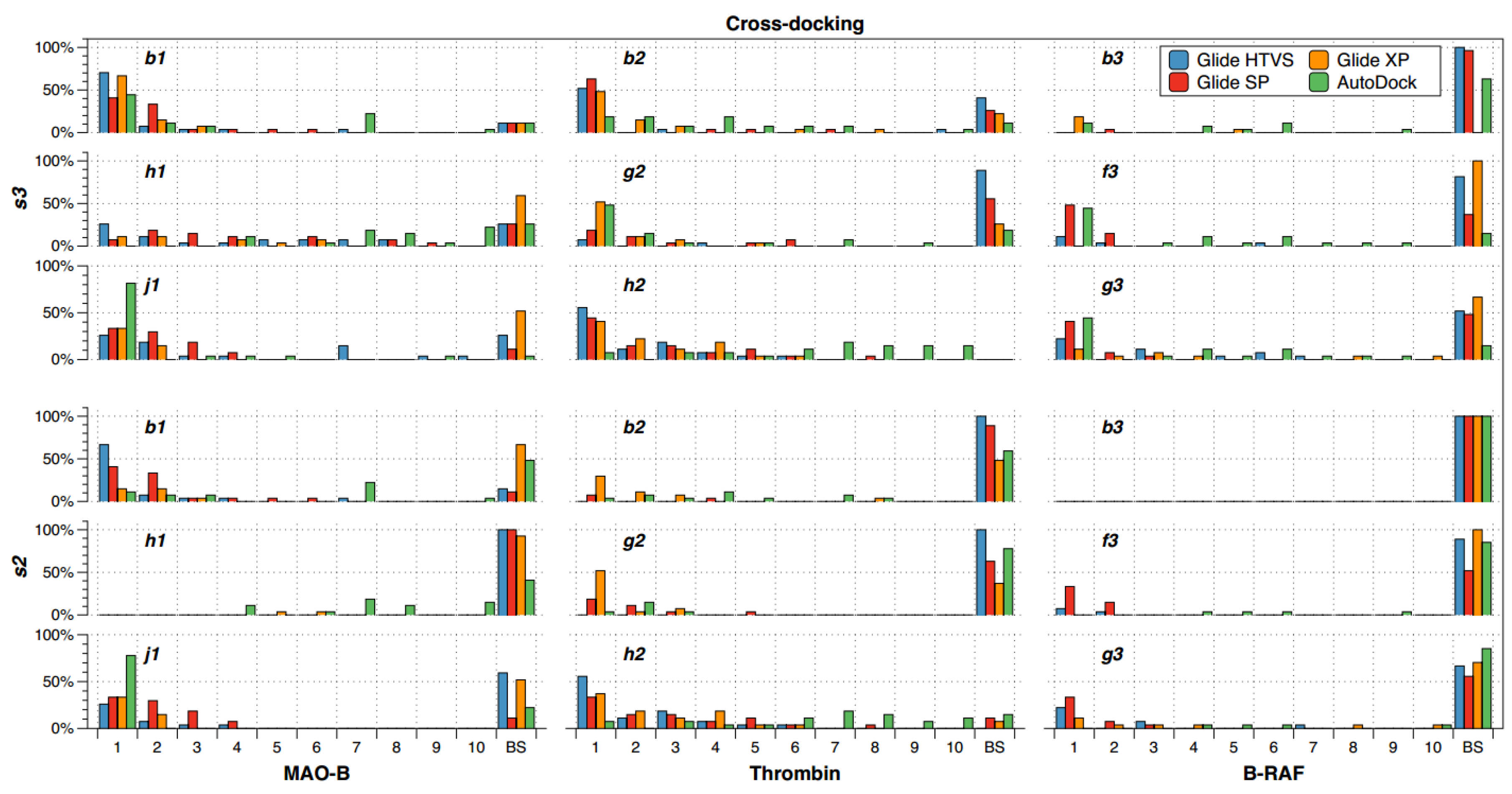
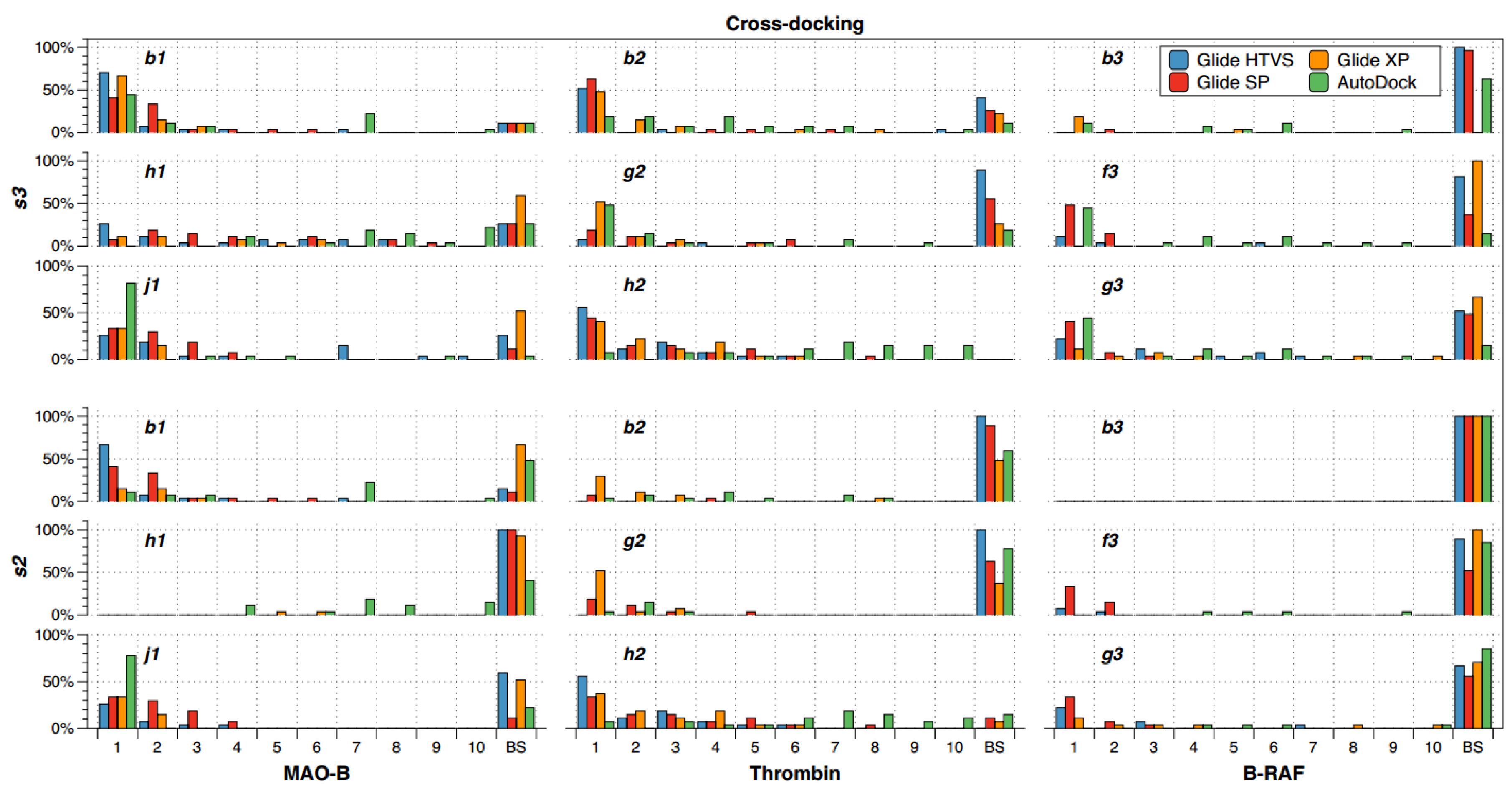
2.2. Cross-Docking
- (a)
- There are compounds that are well oriented in most of the crystal structures, but there are others that are conflictive. For instance, compound h2 had good orientations in almost all the crystallographic structures of thrombin, but b2 and g2 cross-docking experiments failed in most of the instances.
- (b)
- The selection of the docking method was crucial in some cases; for instance, h1 had good orientations in almost all the crystallographic structures of MAO-B when AutoDock was used, but almost all cross-docking experiments for this compound failed when Glide HTVS, SP and XP were used.
- (c)
- B-RAF was a conflictive target for cross-docking. All its inhibitors were bad oriented in almost all crystallographic structures by using all docking methods. Particularly, compound b3 failed in all the instances.
3. Discussion and Recommendations
4. Materials and Methods
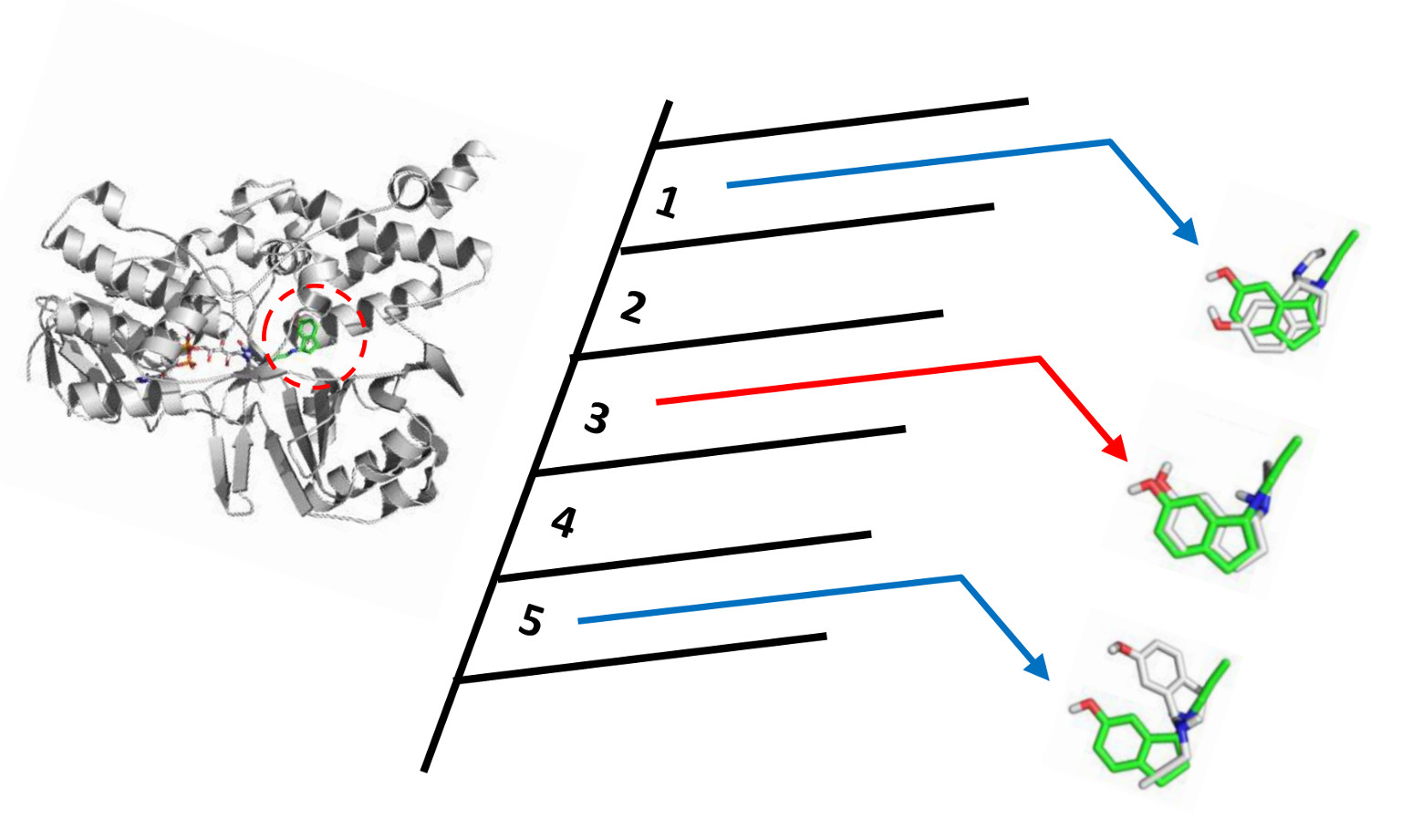
4.1. Self-Docking and Cross-Docking
4.2. Protein Targets under Study
4.3. Docking Experiments
- (a)
- Self-docking of each ligand inside its own protein structural conformation.
- (b)
- Cross-docking of three selected ligands inside the remaining nine protein structures.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yuriev, E.; Ramsland, P.A. Latest developments in molecular docking: 2010–2011 in review. J. Mol. Recognit. 2013, 26, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 2004, 57, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Elokely, K.M.; Doerksen, R.J. Docking challenge: Protein sampling and molecular docking performance. J. Chem. Inf. Model. 2013, 53, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.A.; Jalaie, M.; Robertson, D.H.; Lewis, R.A.; Vieth, M. Lessons in molecular recognition: The effects of ligand and protein flexibility on molecular docking accuracy. J. Med. Chem. 2004, 47, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.J.; Nandigam, R.K.; Erickson, J.A.; Vieth, M. Lessons in molecular recognition. 2. Assessing and improving cross-docking accuracy. J. Chem. Inf. Model. 2007, 47, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Ballante, F.; Marshall, G.R. An Automated Strategy for Binding-Pose Selection and Docking Assessment in Structure-Based Drug Design. J. Chem. Inf. Model. 2016, 56, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the Differential Activity of Thrombin Inhibitors Using Docking, QSAR, Molecular Dynamics, and MM-GBSA. PLoS ONE 2015, 10, e0142774. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Romero, L.; Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Insights into the interactions between maleimide derivates and GSK3β combining molecular docking and QSAR. PLoS ONE 2014, 9, e102212. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.; Zilocchi, S.; Tiznado, W.; Collina, S. Docking and quantitative structure–activity relationship studies for imidazo[1,2−a]pyrazines as inhibitors of checkpoint kinase-1. Med. Chem. Res. 2012, 21, 1912–1920. [Google Scholar] [CrossRef]
- Caballero, J.; Vergara-Jaque, A.; Fernández, M.; Coll, D. Docking and quantitative structure–activity relationship studies for sulfonyl hydrazides as inhibitors of cytosolic human branched-chain amino acid aminotransferase. Mol. Divers. 2009, 13, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.; Quiliano, M.; Alzate-Morales, J.H.; Zimic, M.; Deharo, E. Docking and quantitative structure–activity relationship studies for 3-fluoro-4-(pyrrolo[2,1-f][1,2,4]triazin-4-yloxy)aniline, 3-fluoro-4-(1H-pyrrolo[2,3-b]pyridin-4-yloxy)aniline, and 4-(4-amino-2-fluorophenoxy)-2-pyridinylamine derivatives as c-Met kinase inhibitors. J. Comput. Aided Mol. Des. 2011, 25, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Alzate-Morales, J.H.; Caballero, J.; Vergara-Jaque, A.; González-Nilo, F.D. Insights into the structural basis of N2 and O6 substituted guanine derivatives as cyclin-dependent kinase 2 (CDK2) inhibitors: Prediction of the binding modes and potency of the inhibitors by docking and ONIOM calculations. J. Chem. Inf. Model. 2009, 49, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Alzate-Morales, J.H.; Vergara-Jaque, A.; Caballero, J. Computational Study on the Interaction of N1 Substituted Pyrazole Derivatives with B-Raf Kinase: An Unusual Water Wire Hydrogen-Bond Network and Novel Interactions at the Entrance of the Active Site. J. Chem. Inf. Model. 2010, 50, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is It Reliable to Use Common Molecular Docking Methods for Comparing the Binding Affinities of Enantiomer Pairs for Their Protein Target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, R.T. Structure-based drug design: Docking and scoring. Curr. Prot. Peptide Sci. 2007, 8, 312–328. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Hubálek, F.; Li, M.; Herzig, Y.; Sterling, J.; Edmondson, D.E.; Mattevi, A. Crystal structures of monoamine oxidase B in complex with four inhibitors of the N-propargylaminoindan class. J. Med. Chem. 2004, 47, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of Human Monoamine Oxidase B Complexes with Selective Noncovalent Inhibitors: Safinamide and Coumarin Analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Li, M.; Hubalek, F.; Restelli, N.; Edmondson, D.E.; Mattevi, A. Insights into the Mode of Inhibition of Human Mitochondrial Monoamine Oxidase B from High-Resolution Crystal Structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Hubalek, F.; Li, M.; Herzig, Y.; Sterling, J.; Edmondson, D.E.; Mattevi, A. Binding of Rasagiline-Related Inhibitors to Human Monoamine Oxidases: A Kinetic and Crystallographic Analysis. J. Med. Chem. 2005, 48, 8148–8154. [Google Scholar] [CrossRef] [PubMed]
- Milczek, E.M.; Bonivento, D.; Binda, C.; Mattevi, A.; Mcdonald, I.A.; Edmondson, D.E. Structural and Mechanistic Studies of Mofegiline Inhibition of Human Recombinant Monoamine Oxidase B. J. Med. Chem. 2008, 51, 8019–8026. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Aldeco, M.; Geldenhuys, W.J.; Tortorici, M.; Mattevi, A.; Edmondson, D.E. Molecular Insights into Human Monoamine Oxidase B Inhibition by the Glitazone Anti-Diabetes Drugs. ACS Med. Chem. Lett. 2012, 3, 39–42. [Google Scholar] [CrossRef] [PubMed]
- De Colibus, L.; Li, M.; Binda, C.; Lustig, A.; Edmondson, D.E.; Mattevi, A. Three-Dimensional Structure of Human Monoamine Oxidase a (Mao A): Relation to the Structures of Rat Mao a and Human Mao B. Proc. Natl. Acad. Sci. USA 2005, 102, 12864–12869. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Markotan, T.; Coppo, F.; Tomczuk, B.; Crysler, C.; Eisennagel, S.; Spurlino, J.; Gremminger, L.; Soll, R.M.; Giardino, E.C.; et al. Oxyguanidines. Part 2: Discovery of a novel orally active thrombin inhibitor through structure-based drug design and parallel synthesis. Bioorg. Med. Chem. Lett. 2004, 14, 3727–3731. [Google Scholar] [CrossRef] [PubMed]
- Ruehmann, E.; Betz, M.; Heine, A.; Klebe, G. Fragments Can Bind Either More Enthalpy or Entropy-Driven: Crystal Structures and Residual Hydration Pattern Suggest Why. J. Med. Chem. 2015, 58, 6960–6971. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Kreutter, K.D.; Pan, W.; Crysler, C.; Spurlino, J.; Player, M.P.; Tomczuk, B.; Lu, T. 2-(2-Chloro-6-fluorophenyl)acetamides as Potent Thrombin Inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6266–6269. [Google Scholar] [CrossRef] [PubMed]
- Biela, A.; Khayat, M.; Tan, H.; Kong, J.; Heine, A.; Hangauer, D.; Klebe, G. Impact of ligand and protein desolvation on ligand binding to the S1 pocket of thrombin. J. Mol. Biol. 2012, 418, 350–366. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Mulichak, A.M.; Lewis, S.D.; Shafer, J.A. Crystal structure of human alpha-thrombin complexed with hirugen and p-amidinophenylpyruvate at 1.6 A resolution. Arch. Biochem. Biophys. 1995, 322, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Meneyrol, J.; Follmann, M.; Lassalle, G.; Wehner, V.; Barre, G.; Rousseaux, T.; Altenburger, J.M.; Petit, F.; Bocskei, Z.; Schreuder, H.; et al. 5-Chlorothiophene-2-carboxylic acid [(S)-2-[2-methyl-3-(2-oxopyrrolidin-1-yl)benzenesulfonylamino]-3-(4-methylpiperazin-1-yl)-3-oxopropyl]amide (SAR107375), a selective and potent orally active dual thrombin and factor Xa inhibitor. J. Med. Chem. 2013, 56, 9441–9456. [Google Scholar] [CrossRef] [PubMed]
- Biela, A.; Sielaff, F.; Terwesten, F.; Heine, A.; Steinmetzer, T.; Klebe, G. Ligand binding stepwise disrupts water network in thrombin: Enthalpic and entropic changes reveal classical hydrophobic effect. J. Med. Chem. 2012, 55, 6094–6110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Waizenegger, I.C.; Baum, A.; Steurer, S.; Stadtmuller, H.; Bader, G.; Schaaf, O.; Garin-Chesa, P.; Schlattl, A.; Schweifer, N.; Haslinger, C.; et al. A Novel RAF Kinase Inhibitor with DFG-Out-Binding Mode: High Efficacy in BRAF-Mutant Tumor Xenograft Models in the Absence of Normal Tissue Hyperproliferation. Mol. Cancer Ther. 2016, 15, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Xie, P.; Ventocilla, C.; Zhou, G.; Vultur, A.; Chen, Q.; Liu, Q.; Herlyn, M.; Winkler, J.; Marmorstein, R. Identification of a Novel Family of BRAF(V600E) Inhibitors. J. Med. Chem. 2012, 55, 5220–5230. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Lawhorn, B.G.; Philp, J.; Graves, A.P.; Shewchuk, L.; Holt, D.A.; Gatto, G.J.; Kallander, L.S. GSK114: A selective inhibitor for elucidating the biological role of TNNI3K. Bioorg. Med. Chem. Lett. 2016, 26, 3355–3358. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.D.; Grina, J.; Newhouse, B.; Welch, M.; Topalov, G.; Littman, N.; Callejo, M.; Gloor, S.; Martinson, M.; Laird, E.; et al. Potent and selective pyrazole-based inhibitors of B-Raf kinase. Bioorg. Med. Chem. Lett. 2008, 18, 4692–4695. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Patrick, D.R.; Batorsky, R.S.; Ho, M.L.; Do, H.T.; Zhang, S.Y.; Kumar, R.; Rusnak, D.W; Takle, A.K.; Wilson, D.M.; et al. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res. 2006, 66, 11100–11105. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Wenglowsky, S.; Miknis, G.; Rast, B.; Buckmelter, A.J.; Ely, R.J.; Schlachter, S.; Laird, E.R.; Randolph, N.; Callejo, M.; et al. Non-oxime inhibitors of B-Raf(V600E) kinase. Bioorg. Med. Chem. Lett. 2011, 21, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Okaniwa, M.; Hirose, M.; Arita, T.; Yabuki, M.; Nakamura, A.; Takagi, T.; Kawamoto, T.; Uchiyama, N.; Sumita, A.; Tsutsumi, S.; et al. Discovery of a Selective Kinase Inhibitor (TAK-632) Targeting Pan-RAF Inhibition: Design, Synthesis, and Biological Evaluation of C-7-Substituted 1,3-Benzothiazole Derivatives. J. Med. Chem. 2013, 56, 6478–6494. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.; Alzate-Morales, J.H.; Vergara-Jaque, A. Investigation of the differences in activity between hydroxycycloalkyl N1 substituted pyrazole derivatives as inhibitors of B-Raf kinase by using docking, molecular dynamics, QM/MM, and fragment-based de novo design: Study of binding mode of diastereomer compounds. J. Chem. Inf. Model. 2011, 51, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, Y.; Tanaka, I.; Yao, M. Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2010, 26, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Bouzida, D.; Gehlhaar, D.K.; Rejto, P.A.; Arthurs, S.; Colson, A.B.; Freer, S.T.; Larson, V.; Luty, B.A.; Marrone, T.; et al. Deciphering common failures in molecular docking of ligand-protein complexes. J. Comput. Aided Mol. Des. 2000, 14, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Gutierrez, C.; Adasme-Carreño, F.; Fuentes, E.; Palomo, I.; Caballero, J. Computational study of the binding orientation and affinity of PPARγ agonists: Inclusion of ligand-induced fit by cross-docking. RSC Adv. 2016, 6, 64756–64768. [Google Scholar] [CrossRef]
- Barreca, M.L.; Iraci, N.; De Luca, L.; Chimirri, A. Induced-fit docking approach provides insight into the binding mode and mechanism of action of HIV-1 integrase inhibitors. ChemMedChem 2009, 4, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Carpi, A.; Menabò, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Laux, G. MAO-inhibitors in Parkinson’s Disease. Exp. Neurobiol. 2011, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Szökő, É.; Tábi, T.; Riederer, P.; Vécsei, L.; Magyar, K. Pharmacological aspects of the neuroprotective effects of irreversible MAO-B inhibitors, selegiline and rasagiline, in Parkinson’s disease. J. Neural Transm. (Vienna) 2018. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.A.; Key, N.S.; Levy, J.H. Blood coagulation: Hemostasis and thrombin regulation. Anesth. Analg. 2009, 108, 1433–1446. [Google Scholar] [CrossRef] [PubMed]
- Stringer, K.A.; Lindenfeld, J. Hirudins: Antithrombin anticoagulants. Ann. Pharmacother. 1992, 26, 1535–1540. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Théroux, P. Platelet activation with unfractionated heparin at therapeutic concentrations and comparisons with a low-molecular-weight heparin and with a direct thrombin inhibitor. Circulation 1998, 97, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Alban, S. Pharmacological strategies for inhibition of thrombin activity. Curr. Pharm. Des. 2008, 14, 1152–1175. [Google Scholar] [CrossRef] [PubMed]
- Mercer, K.E.; Pritchard, C.A. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta 2003, 1653, 25–40. [Google Scholar] [CrossRef]
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Batt, D.; Warmuth, M. B-Raf kinase inhibitors for cancer treatment. Curr. Opin. Investig. Drugs 2007, 8, 452–456. [Google Scholar] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Caporuscio, F.; Rastelli, G.; Imbriano, C.; Del Rio, A. Structure-based design of potent aromatase inhibitors by high-throughput docking. J. Med. Chem. 2011, 54, 4006–4017. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hay, S.O.; Lane, A.L.; Caulfield, T.R.; Claussin, C.; Bertrand, J.; Masson, A.; Choudhry, S.; Fauq, A.H.; Maharvi, G.M.; Leissring, M.A. Optimization of peptide hydroxamate inhibitors of insulin-degrading enzyme reveals marked substrate-selectivity. J. Med. Chem. 2013, 56, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Osguthorpe, D.J.; Sherman, W.; Hagler, A.T. Generation of receptor structural ensembles for virtual screening using binding site shape analysis and clustering. Chem. Biol. Drug Des. 2012, 80, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Amaning, K.; Lowinski, M.; Vallee, F.; Steier, V.; Marcireau, C.; Ugolini, A.; Delorme, C.; Foucalt, F.; McCort, G.; Derimay, N.; et al. The use of virtual screening and differential scanning fluorimetry for the rapid identification of fragments active against MEK1. Bioorg. Med. Chem. Lett. 2013, 23, 3620–3626. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D. Computational methods applied to rational drug design. Open Med. Chem. J. 2016, 10, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MAO-B | |||
|---|---|---|---|
| PDB ID | Resolution (Å) | Ligand | Reference |
| 1S3E | 1.60 | a1: 5-hydroxy-N-propargyl-1(R)-aminoindan | [23] |
| 2V5Z | 1.60 | b1: Safinamide | [24] |
| 1S3B | 1.65 | c1:N-methyl-N-propargyl-1(R)-aminoindan | [23] |
| 1OJC | 2.40 | d1: 4-chloro-N-(2-hydrixyethyl)benzamide | [25] |
| 2C65 | 1.70 | e1: (1R)-4-({[ethyl(methyl)amino]carbonyl}oxy)-N-methyl-N-[(1E)-prop-2-en-1-ylidene]undan-1-aminium | [26] |
| 2VZ2 | 2.30 | f1: (1Z)-4-(4-fluorophenyl)-2-methylidenebutan-1-imine | [27] |
| 4A79 | 1.89 | g1: Pioglitazone | [28] |
| 1S2Y | 2.12 | h1: N-propargyl-1(S)-aminoindan | [23] |
| 2BYB | 2.20 | i1:N-methyl-N-[(1R)-1-methyl-2-phenylethyl]prop-2-en-1-amine | [29] |
| 1OJ9 | 2.30 | j1: 1,4-duphenyl-2-butene | [25] |
| Thrombin | |||
| PDB ID | Resolution (Å) | Ligand | Reference |
| 1T4U | 2.00 | a2: 2-methanesulfonyl-benzenesulfonic acid 3-methyl-5-((1-amidinoaminooxymethyl-cyclopropyl)methyloxy)-phenylester | [30] |
| 1T4V | 2.00 | b2: N-allyl-5-amidinoaminooxy-propyloxy-3-chloro-N-cyclopentylbenzamide | [30] |
| 3C27 | 2.18 | c2:N-[2-(carbamimidamidooxy)ethyl]-2-{6-cyano-3-[(2,2-difluoro-2-pyridin-2-ylethyl)amino]-2-fluorophenyl}acetamide | |
| 4UDW | 1.16 | d2:d-phenylalanyl-N-(2,5-dichlorobenzyl)-l-prolinamide | [31] |
| 2R2M | 2.10 | e2:N-[2-({[amino(imino)methyl]amino}oxy)ethyl]-2-{6-chloro-3-[(2,2-difluoro-2-phenylethyl)amino]-2-fluorophenyl}acetamide | [32] |
| 3LDX | 2.25 | f2: RWJ-671818 | |
| 3SV2 | 1.30 | g2: d-phenylalanyl-N-(pyridin-4-ylmethyl)-l-prolinamide | [33] |
| 1AHT | 1.60 | h2: (2S)-3-(4-carbamimidoylphenyl)-2-hydroxypropanoic acid | [34] |
| 4LXB | 1.61 | i2: 5-chloro-thiophene-2-carboxylic acid [(S)-2-[2-difluoromethoxy-3-(2-oxo-piperidin-1-yl)-benzenesulfonylamino]-3-((S)-3-dimethylamino-pyrrolidin-1-yl)-3-oxo-propyl]-amide | [35] |
| 3RM2 | 1.23 | j2:N-(benzylsulfonyl)-3-cyclohexyl-d-alanyl-N-(4-carbamimidoylbenzyl)-l-prolinamide | [36] |
| B-RAF protein kinase | |||
| PDB ID | Resolution (Å) | Ligand | Reference |
| 4XV9 | 2.00 | a3:N-{3-[(5-chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)carbonyl]-2,4-difluorophenyl}-4-(trifluoromethyl)benzenesulfonamide | [37] |
| 5CSX | 2.51 | b3: N-(3-{5-[(1-ethylpiperidin-4-yl)(methyl)amino]-3-(pyrimidin-5-yl)-1H-pyrrolo[3,2-b]pyridin-1-yl}-2,4-difluorophenyl)propane-1-sulfonamide | [38] |
| 4E26 | 2.55 | c3: 5-chloro-7-[(R)-furan-2-yl(pyridin-2-ylamino)methyl]quinolin-8-ol | [39] |
| 3C4C | 2.57 | d3:N-{3-[(5-chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)carbonyl]-2,4-difluorophenyl}propane-1-sulfonamide | [40] |
| 5CSW | 2.66 | e3: Dabrafenib | [38] |
| 4YHT | 3.05 | f3: 3-[(5-chloro-7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]-N-methyl-4-(morpholin-4-yl)benzenesulfonamide | [41] |
| 3D4Q | 2.80 | g3: (1E)-5-(1-piperidin-4-yl-3-pyridin-4-yl-1H-pyrazol-4-yl)-2,3-dihydro-1H-inden-1-one oxime | [42] |
| 2FB8 | 2.90 | h3: (1Z)-5-(2-{4-[2-(dimethylamino)ethoxy]phenyl}-5-pyridin-4-yl-1H-imidazol-4-yl)indan-1-one oxime | [43] |
| 3PRF | 2.90 | i3: 2-chloro-5-{[2-(pyrimidin-2-yl)furo[2,3-c]pyridin-3-yl]amino}phenol | [44] |
| 4KSP | 2.93 | j3:N-{7-cyano-6-[4-fluoro-3-({[3-(trifluoromethyl)phenyl]acetyl}amino)phenoxy]-1,3-benzothiazol-2-yl}cyclopropanecarboxamide | [45] |
| Protein Binding Site | Ligand | |||||
|---|---|---|---|---|---|---|
| PDB | Ligand | BSV (Å3) a | VD Value b | Averaged VD c | Number of Rotatable Angles | MW (g/mol) |
| MAO-B | ||||||
| 1S3E | a1 | 221 | 2485 | 11.2474 | 2 | 187.2 |
| 2V5Z | b1 | 237 | 3162 | 13.3418 | 6 | 300.3 |
| 1S3B | c1 | 110 | 1417 | 12.8848 | 2 | 201.2 |
| 1OJC | d1 | 270 | 2900 | 10.7420 | 3 | 199.6 |
| 2C65 | e1 | 276 | 2974 | 10.7754 | 5 | 287.3 |
| 2VZ2 | f1 | 116 | 1555 | 13.4080 | 4 | 177.2 |
| 4A79 | g1 | 97 | 1274 | 13.1409 | 7 | 356.4 |
| 1S2Y | h1 | 132 | 1677 | 12.7071 | 2 | 171.2 |
| 2BYB | i1 | 286 | 3097 | 10.8298 | 5 | 189.3 |
| 1OJ9 | j1 | 289 | 3285 | 11.3691 | 4 | 208.3 |
| Thrombin | ||||||
| 1T4U | a2 | 286 | 670 | 2.34499 | 10 | 483.5 |
| 1T4V | b2 | 388 | 1054 | 2.71649 | 10 | 394.9 |
| 3C27 | c2 | 366 | 937 | 2.56011 | 10 | 435.4 |
| 4UDW | d2 | 172 | 486 | 2.82946 | 6 | 420.3 |
| 2R2M | e2 | 333 | 796 | 2.39239 | 10 | 443.8 |
| 3LDX | f2 | 336 | 805 | 2.39583 | 10 | 423.4 |
| 3SV2 | g2 | 297 | 835 | 2.81145 | 6 | 352.4 |
| 1AHT | h2 | 321 | 852 | 2.65628 | 4 | 208.2 |
| 4LXB | i2 | 334 | 790 | 2.36727 | 11 | 648.1 |
| 3RM2 | j2 | 308 | 724 | 2.35281 | 11 | 553.7 |
| B-RAF | ||||||
| 4XV9 | a3 | 359 | 1432 | 3.98886 | 6 | 515.8 |
| 5CSX | b3 | 438 | 2834 | 6.47108 | 9 | 569.6 |
| 4E26 | c3 | 316 | 1030 | 3.26160 | 4 | 351.7 |
| 3C4C | d3 | 311 | 925 | 2.97428 | 6 | 413.8 |
| 5CSW | e3 | 180 | 909 | 5.05000 | 6 | 519.5 |
| 4YHT | f3 | 185 | 662 | 3.58198 | 5 | 422.9 |
| 3D4Q | g3 | 216 | 815 | 3.77623 | 3 | 377.4 |
| 2FB8 | h3 | 202 | 708 | 3.50495 | 8 | 453.5 |
| 3PRF | i3 | 211 | 687 | 3.25750 | 2 | 338.7 |
| 4KSP | j3 | 444 | 1415 | 3.18769 | 9 | 556.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. https://doi.org/10.3390/molecules23051038
Ramírez D, Caballero J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules. 2018; 23(5):1038. https://doi.org/10.3390/molecules23051038
Chicago/Turabian StyleRamírez, David, and Julio Caballero. 2018. "Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data?" Molecules 23, no. 5: 1038. https://doi.org/10.3390/molecules23051038
APA StyleRamírez, D., & Caballero, J. (2018). Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules, 23(5), 1038. https://doi.org/10.3390/molecules23051038