Exploring Protein Cavities through Rigidity Analysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Related Work
2. Results
2.1. Protein Selection
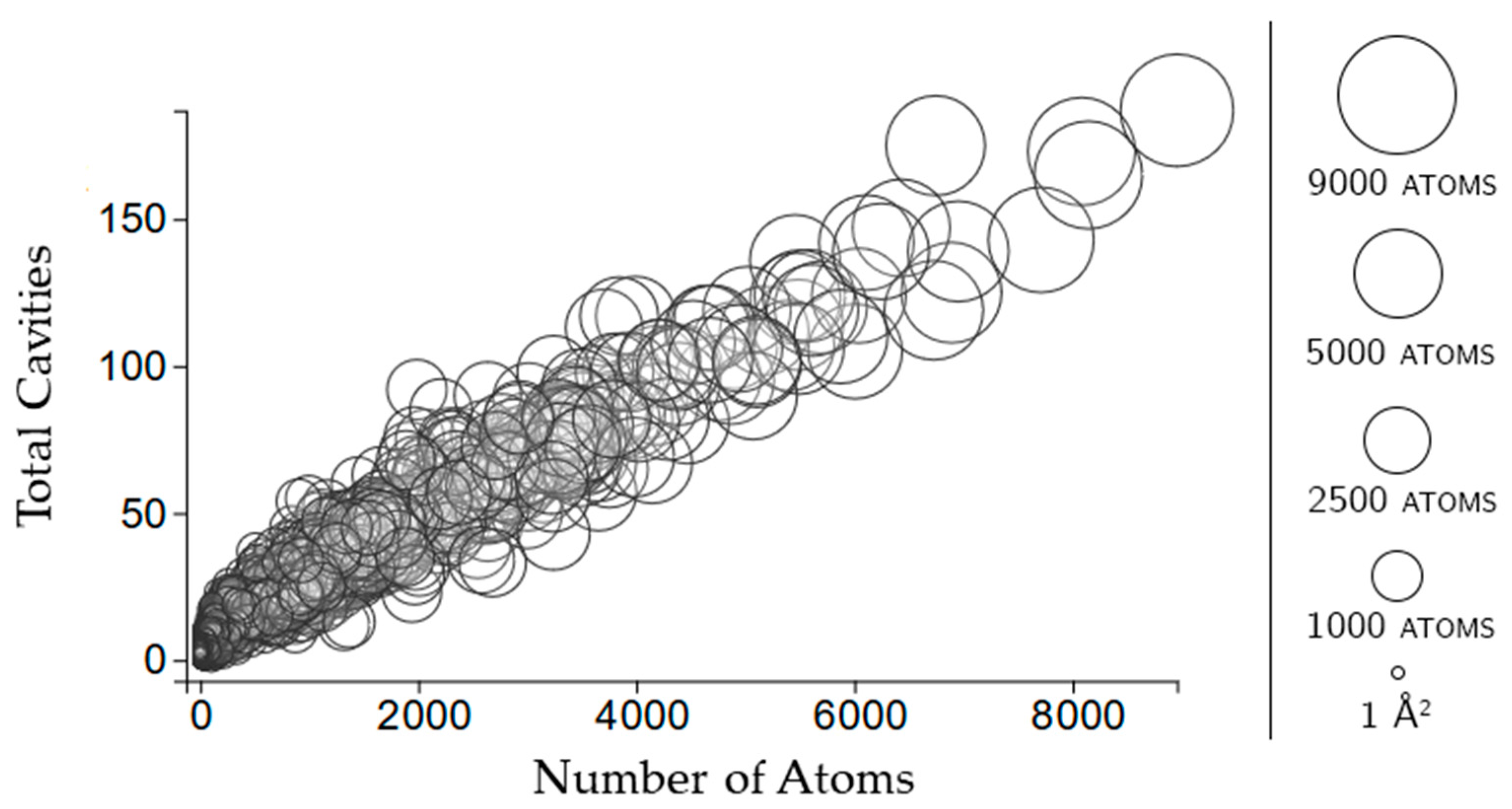
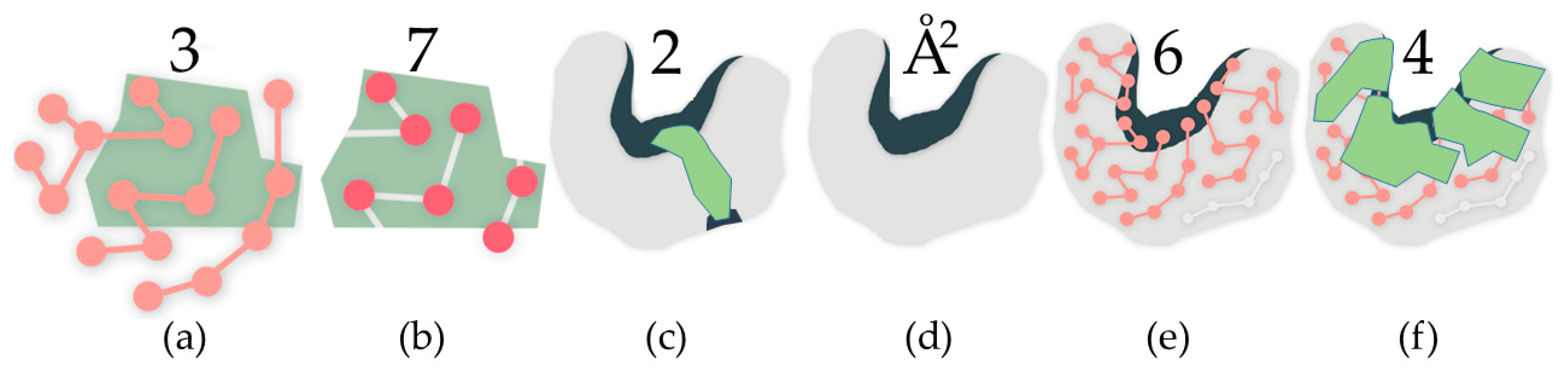
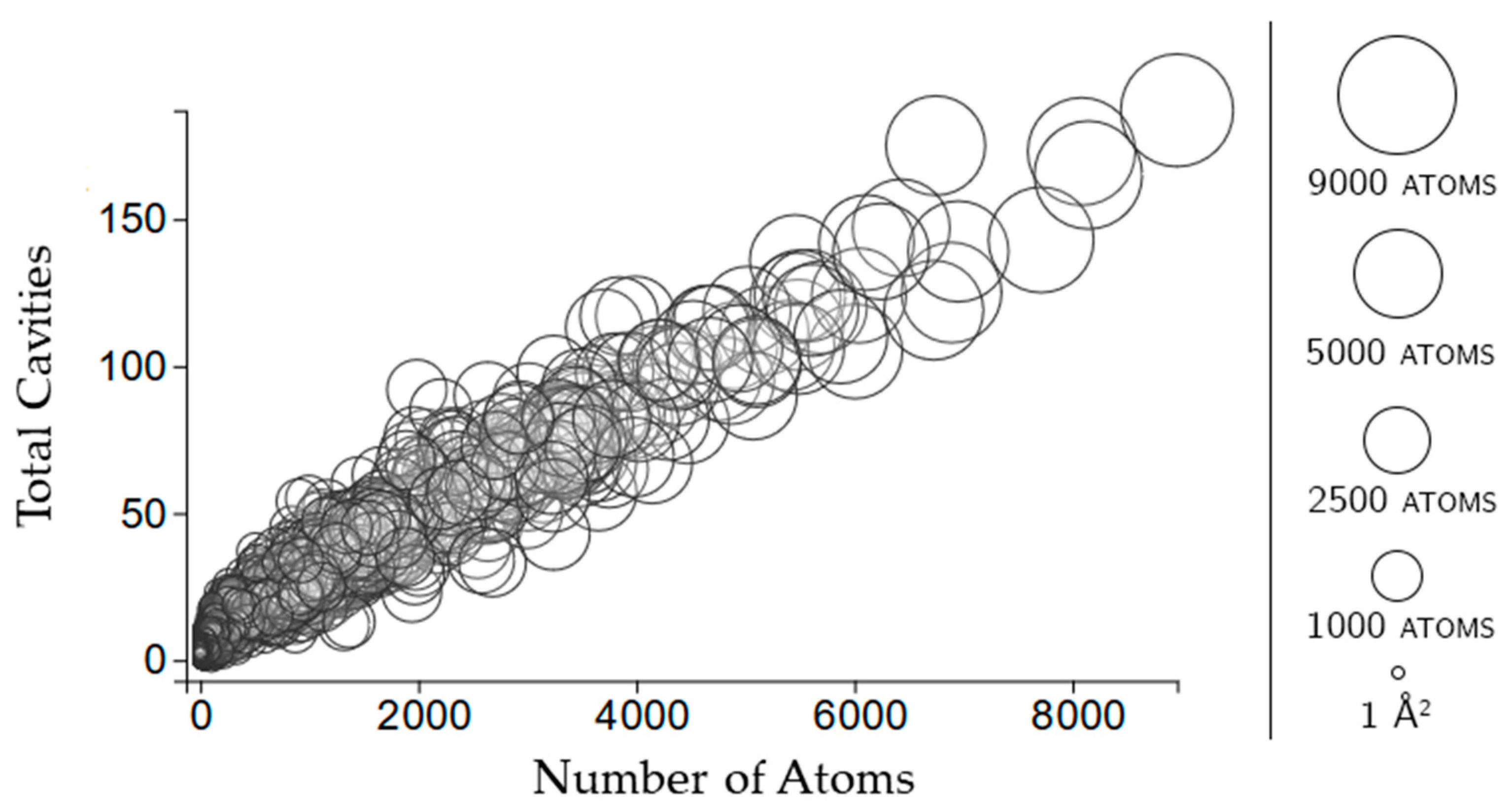
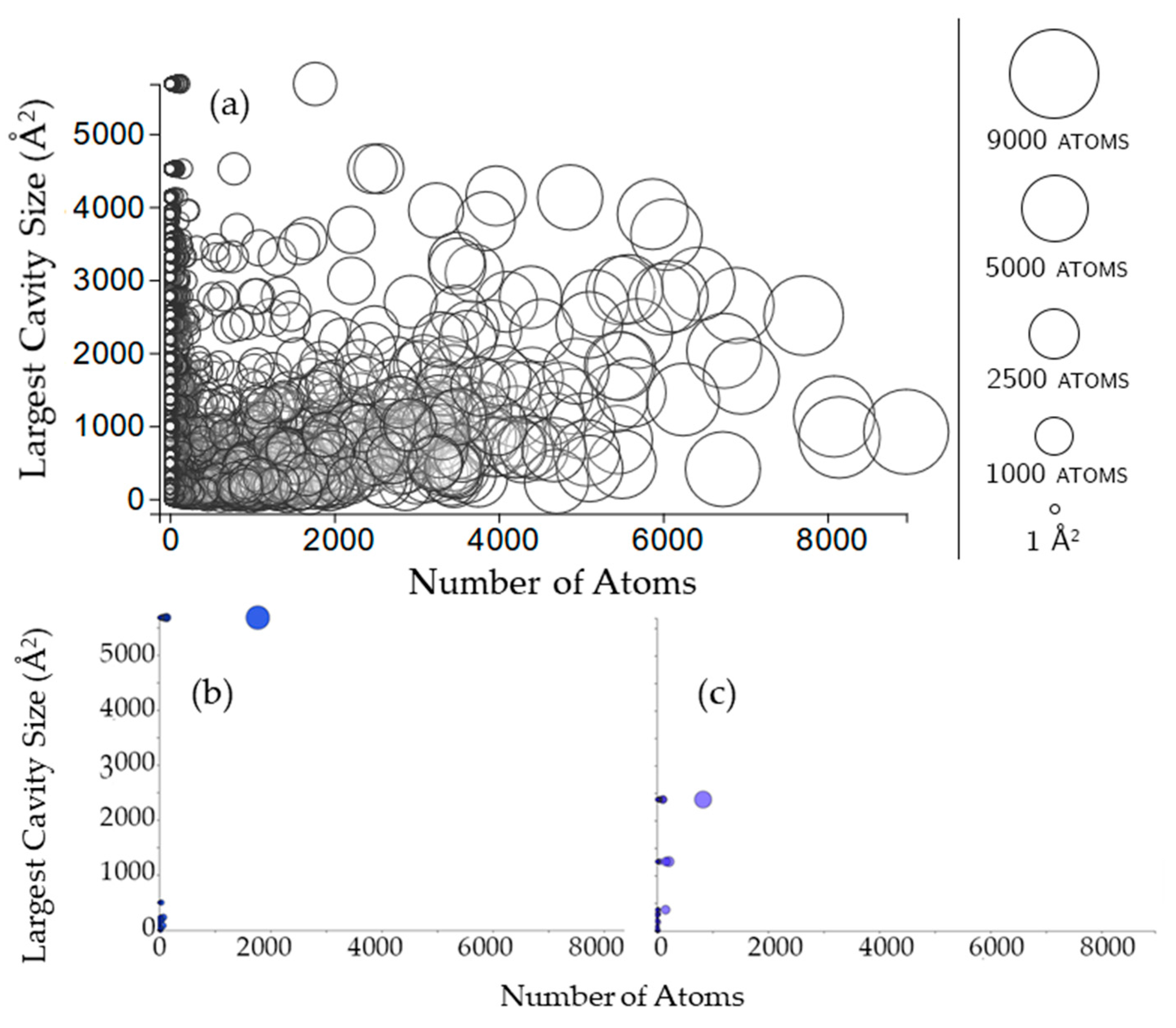
2.2. Properties of Cavities
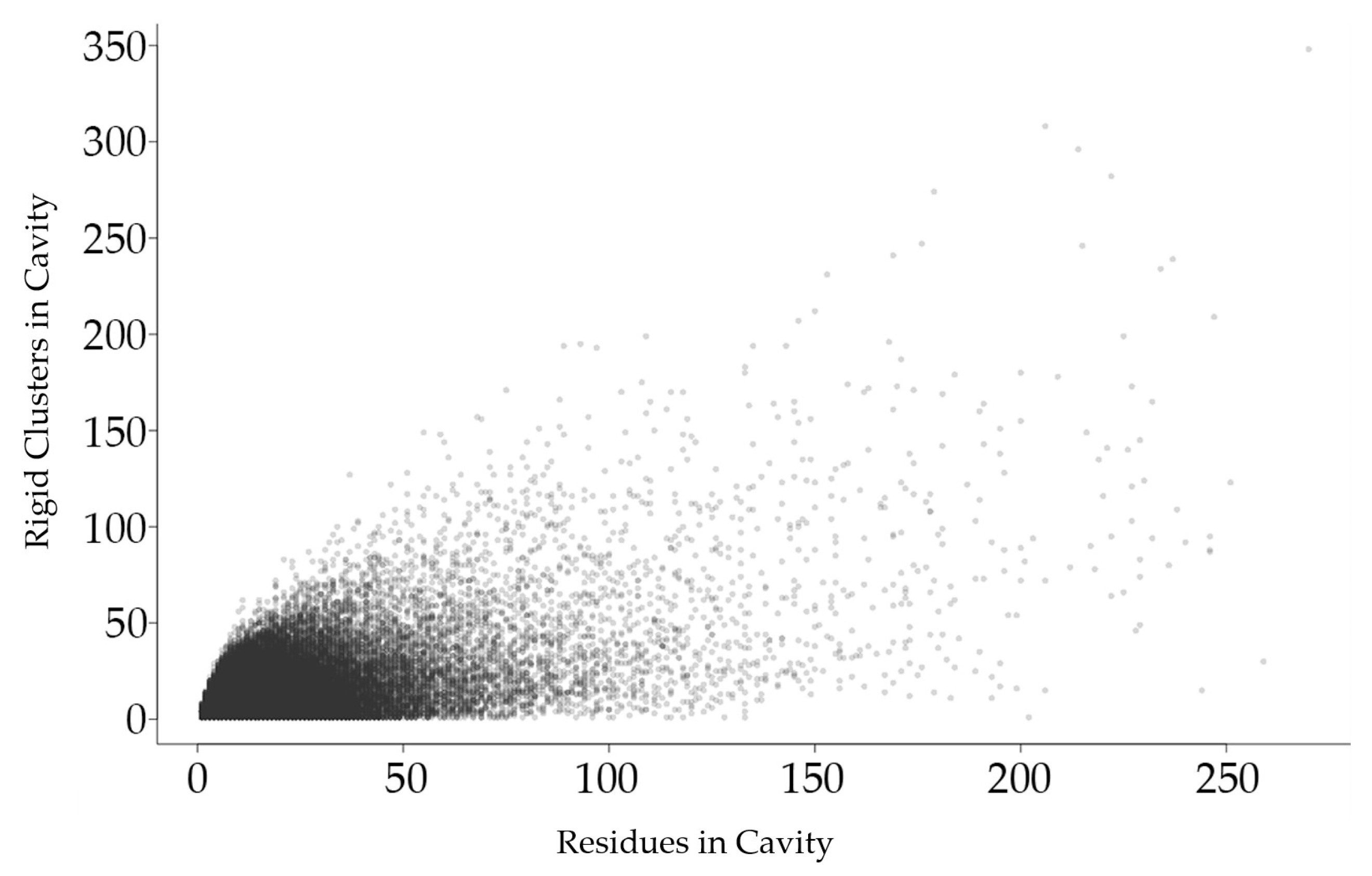
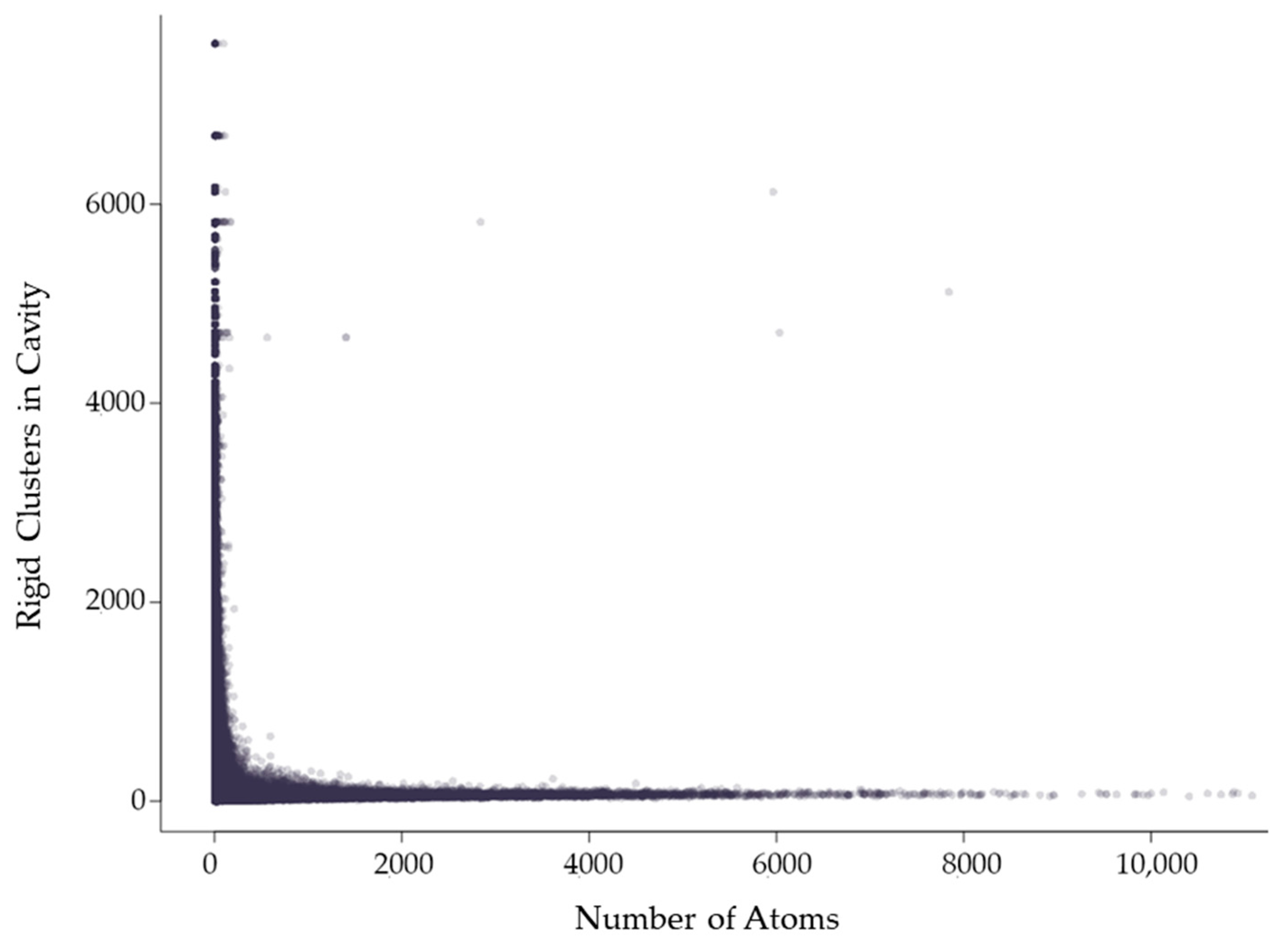
2.2.1. Relationship between Rigid Clusters and Residues
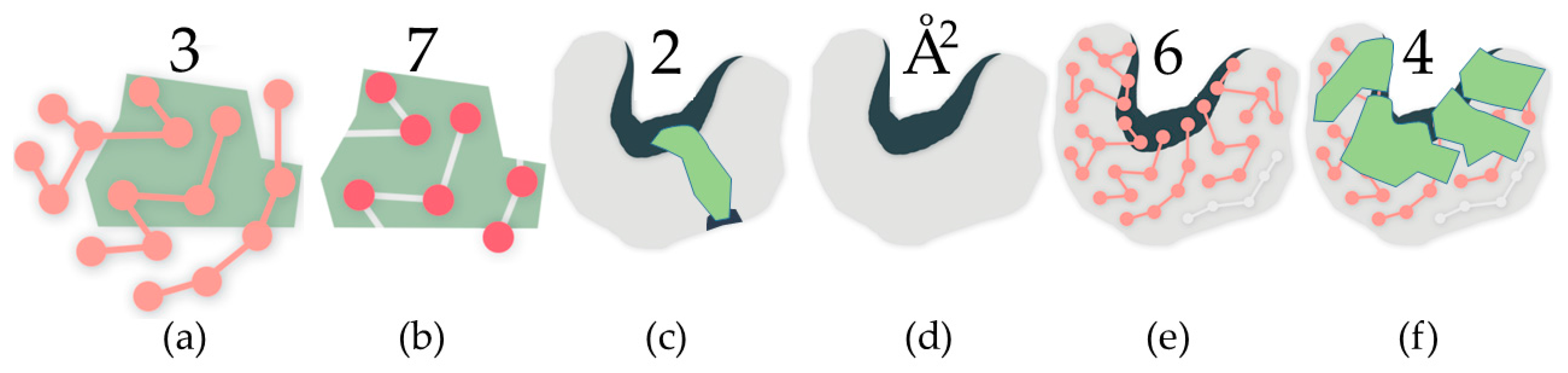
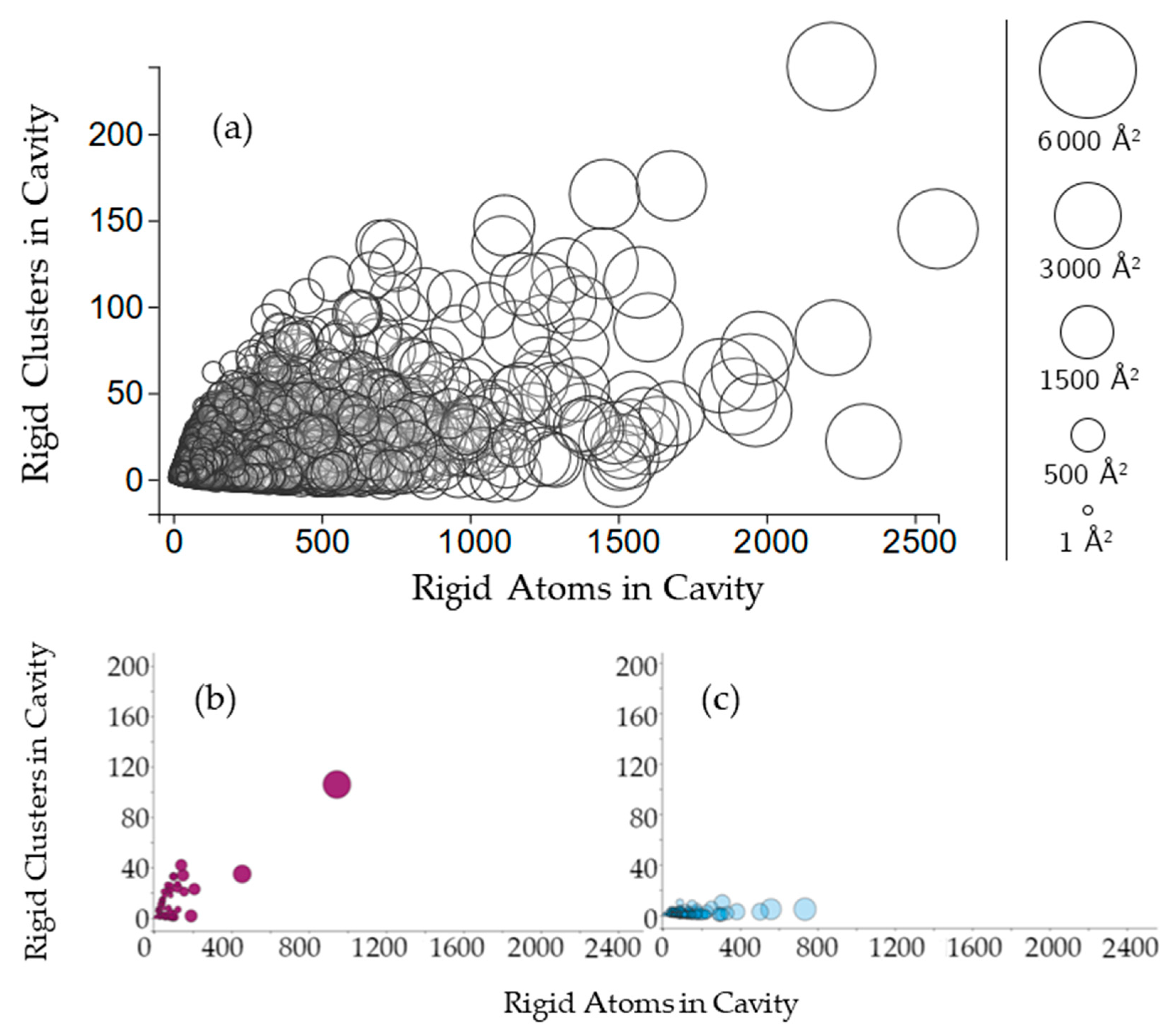
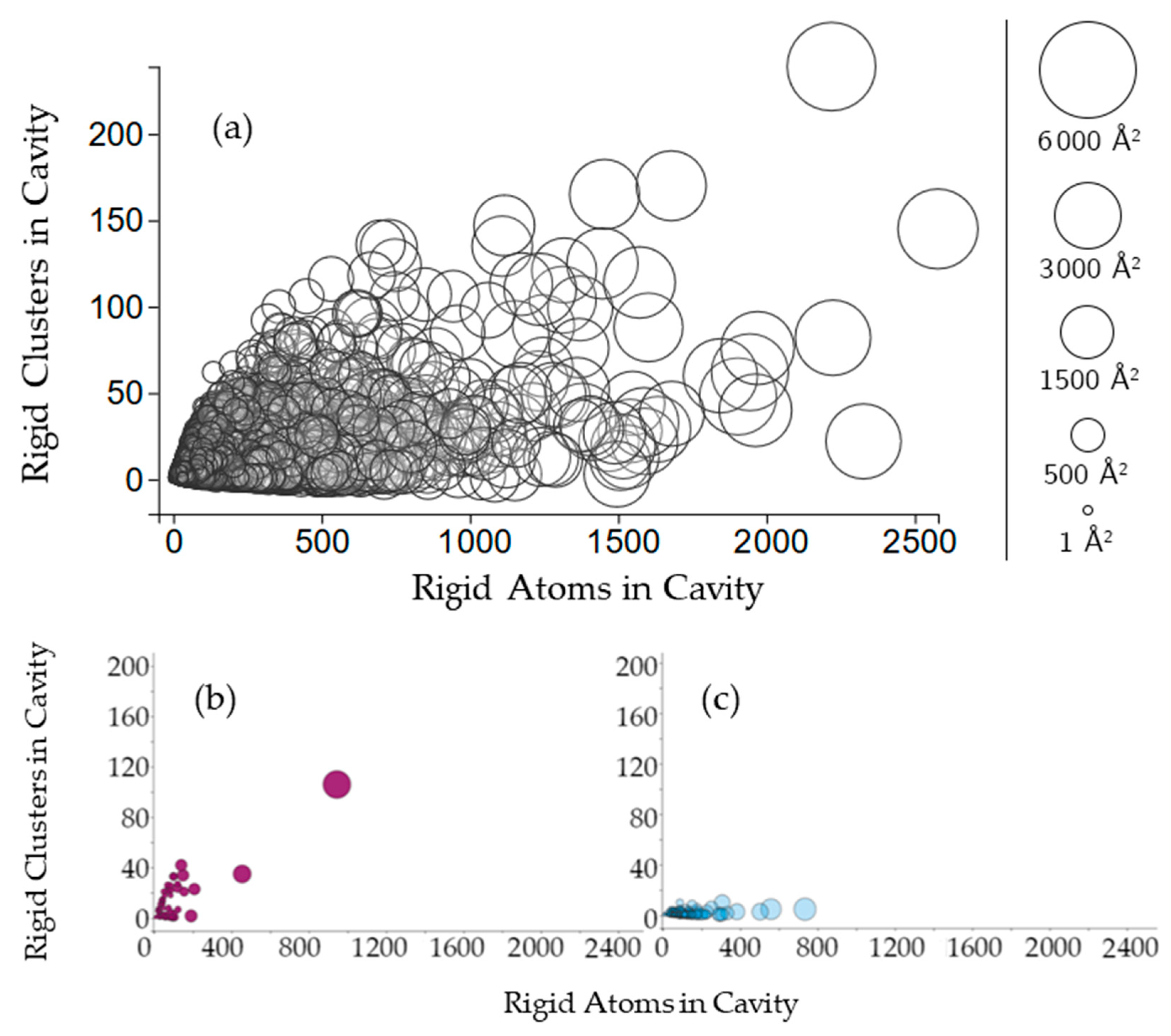
2.2.2. Protein Cavities: Rigid Clusters and Rigid Atoms
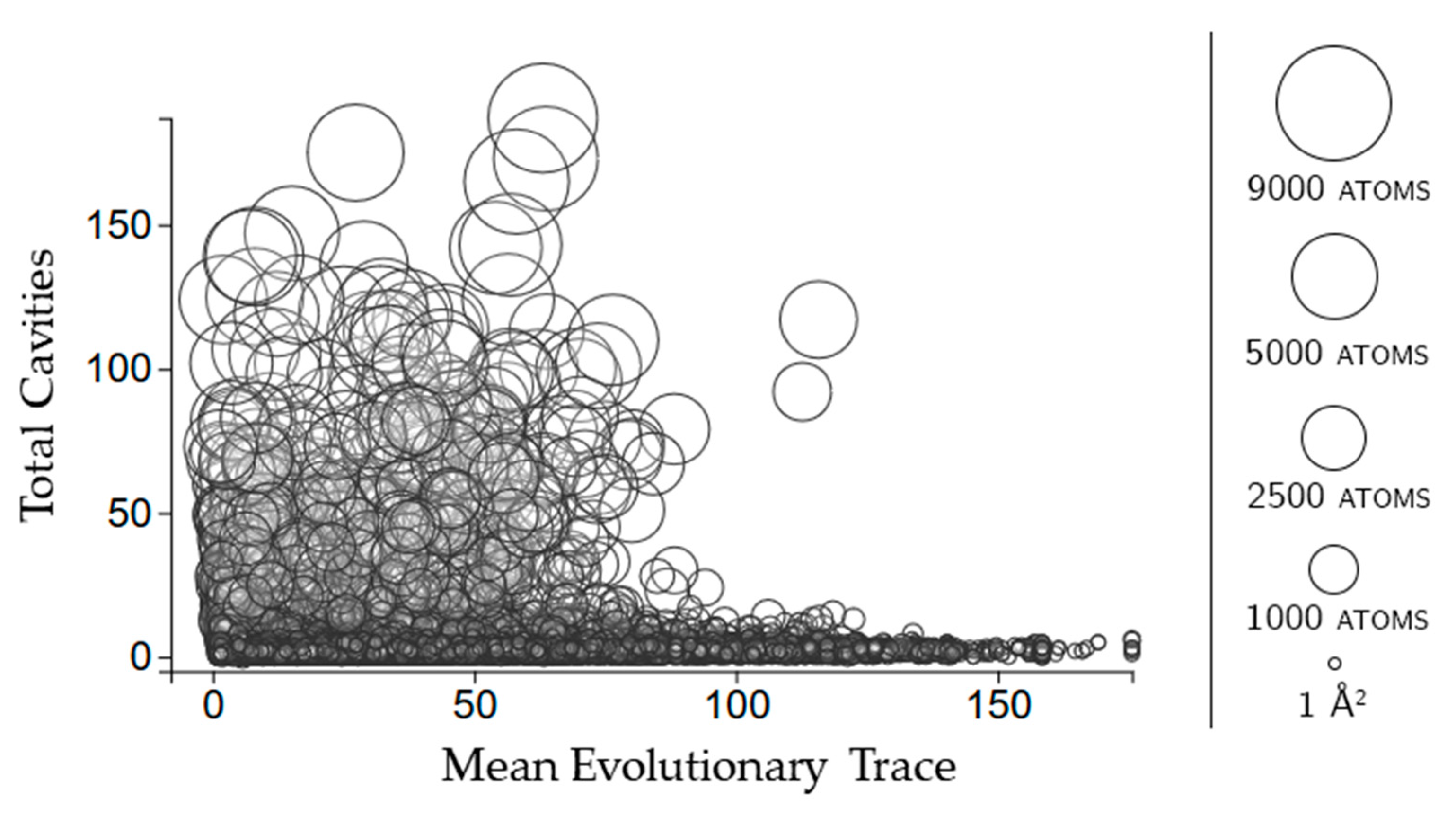
2.2.3. Protein Cavities: Evolutionary Significance
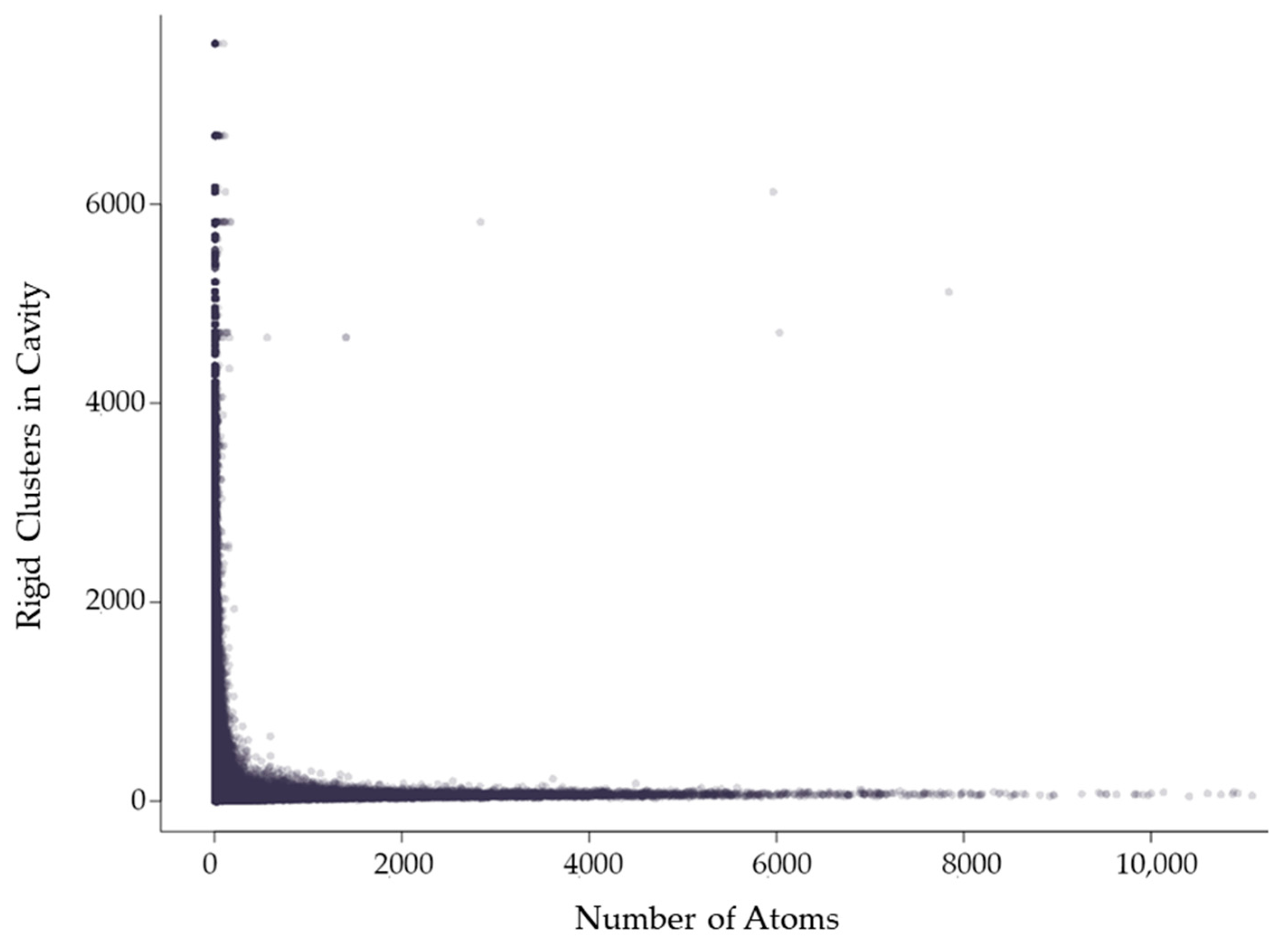
2.3. Properties of Rigid Clusters in Cavities
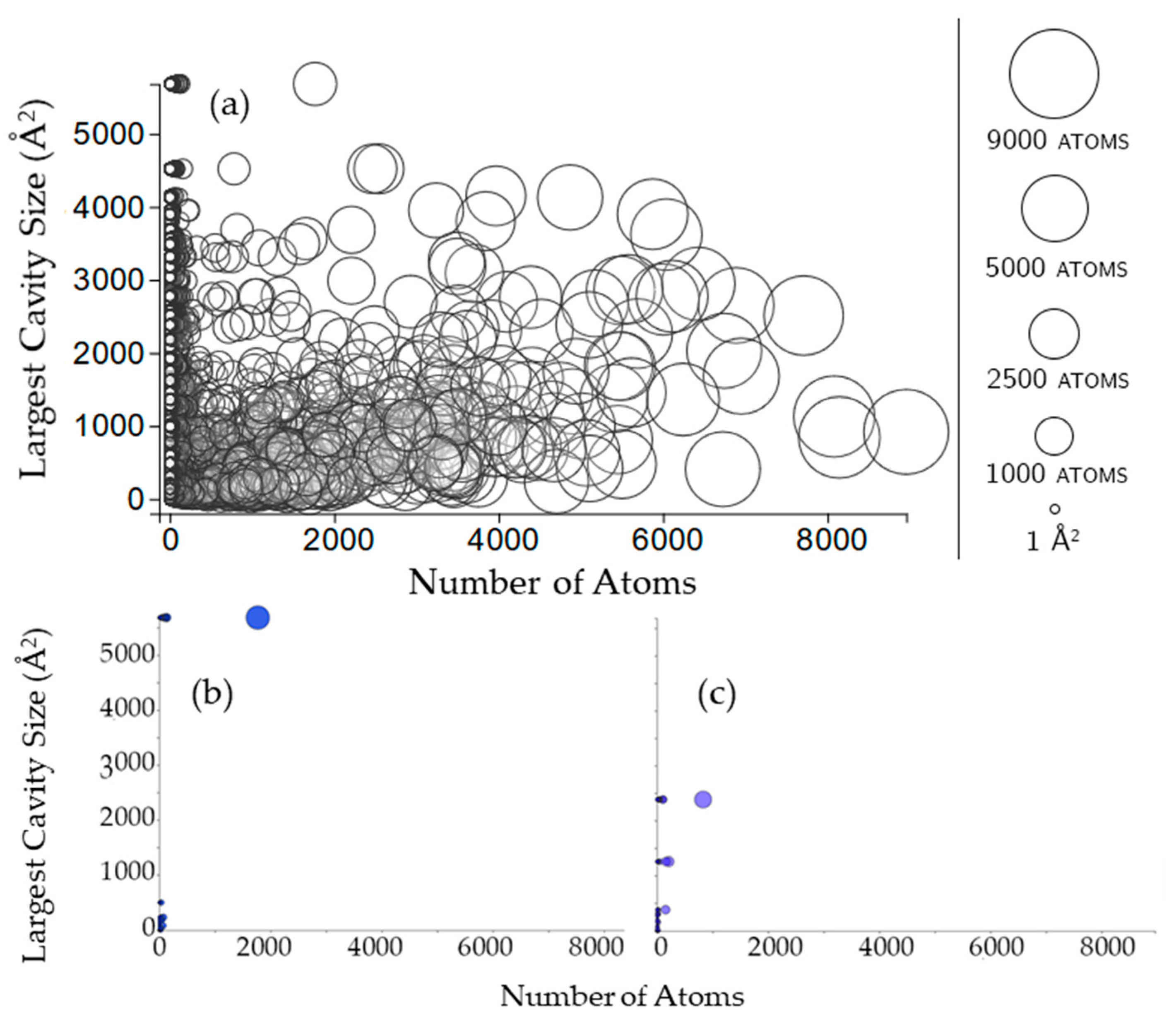
2.3.1. Rigidity Properties of Cavities: Cavity Size
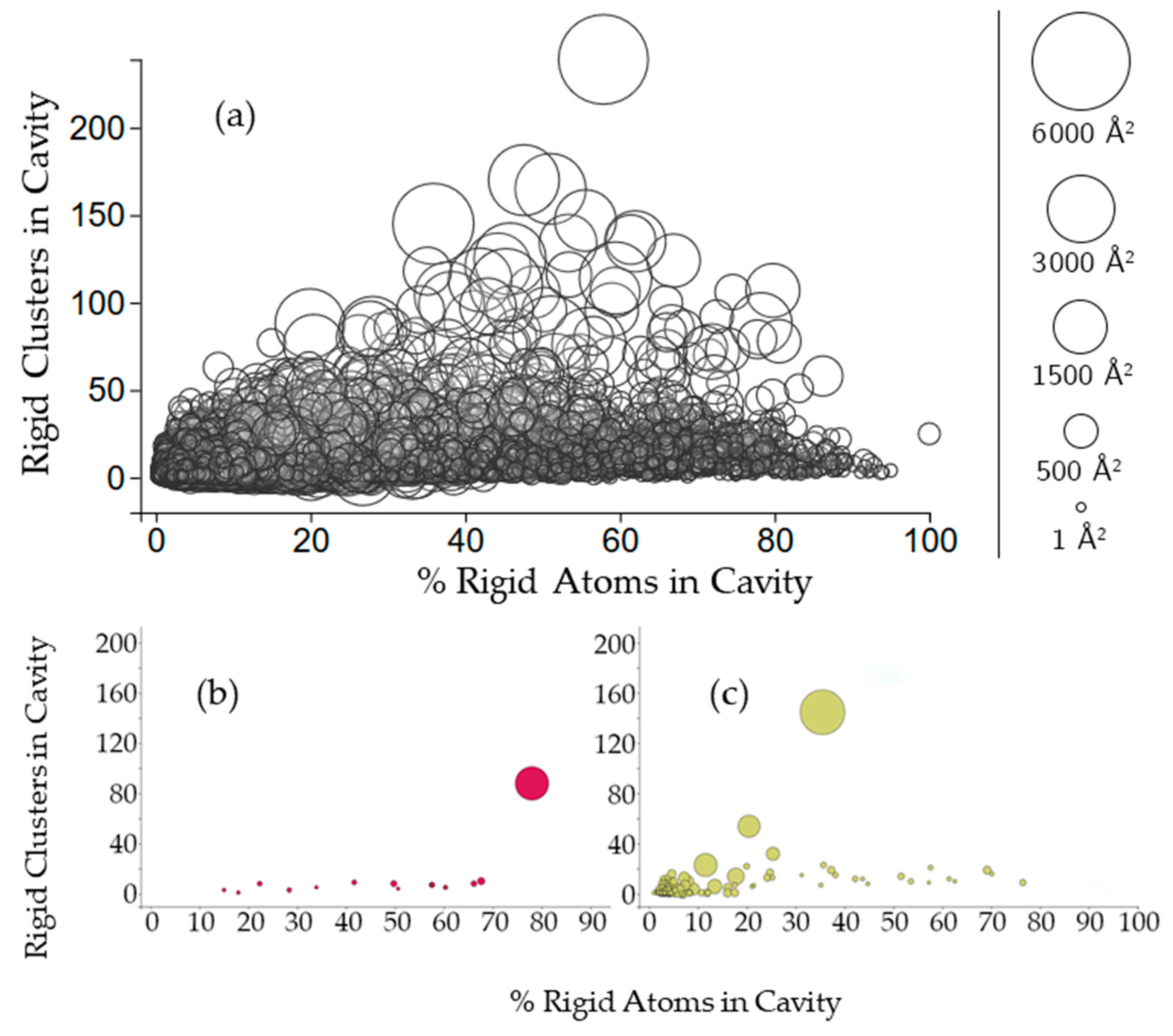
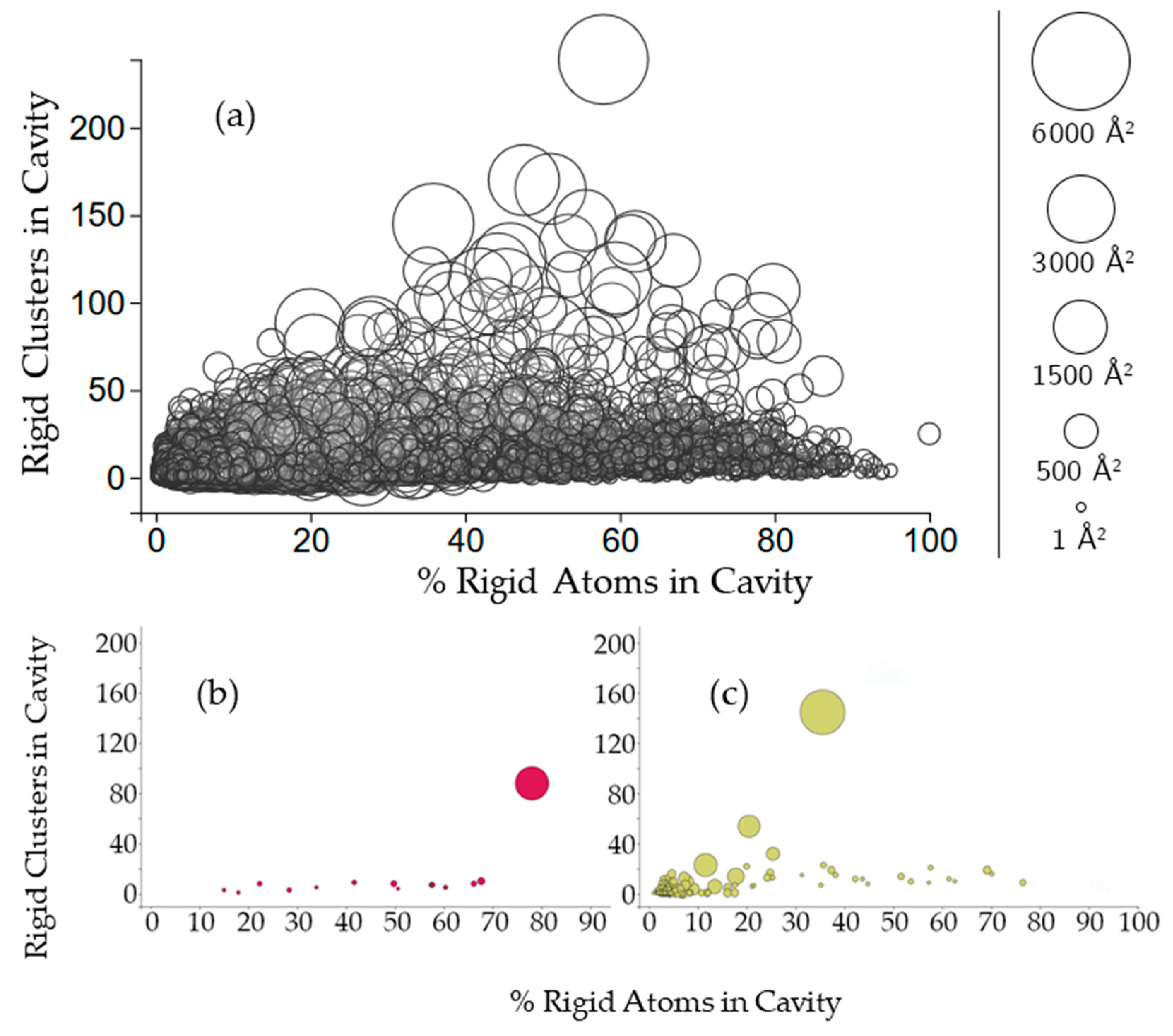
2.3.2. Rigidity Properties of Cavities: Cavity Participation
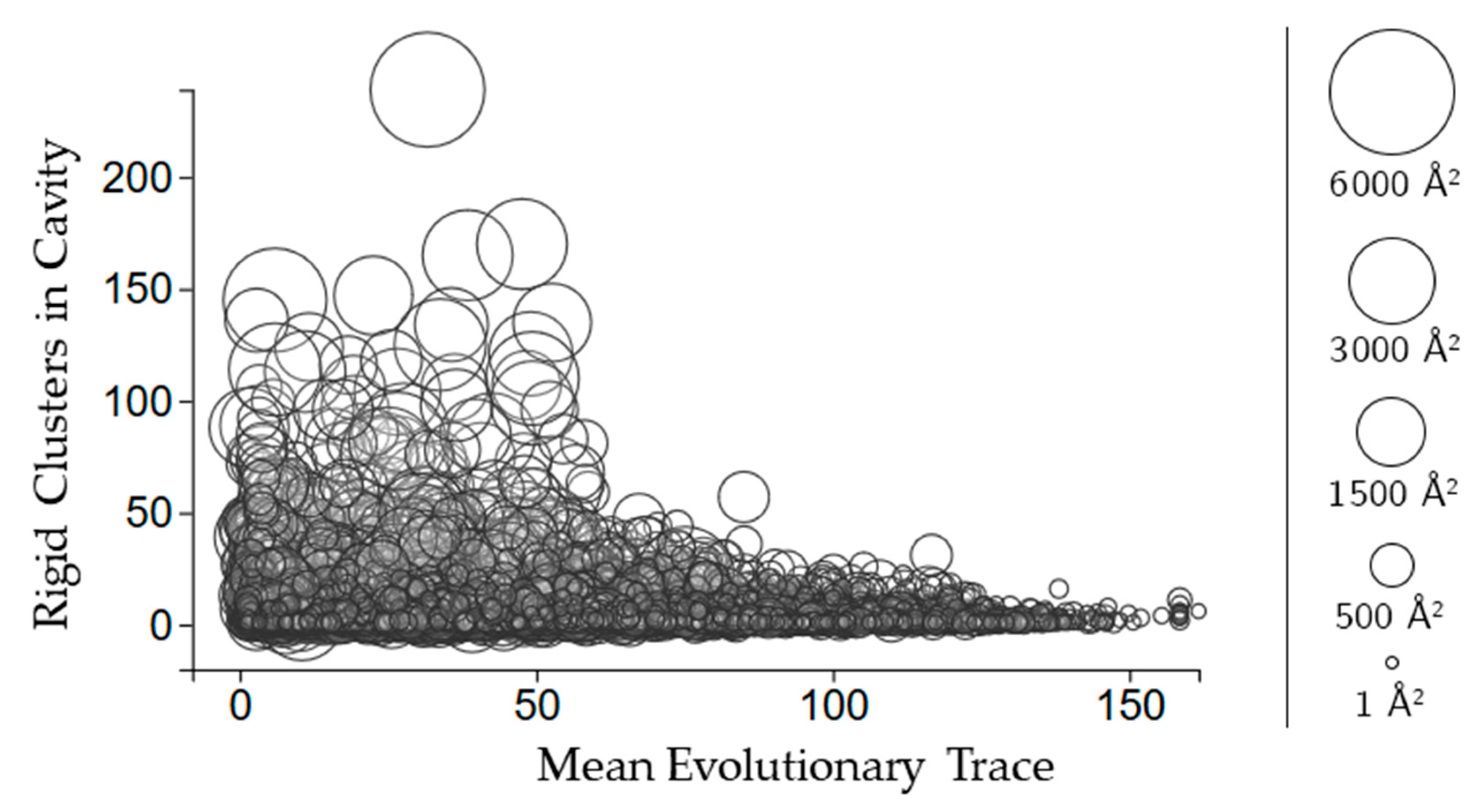
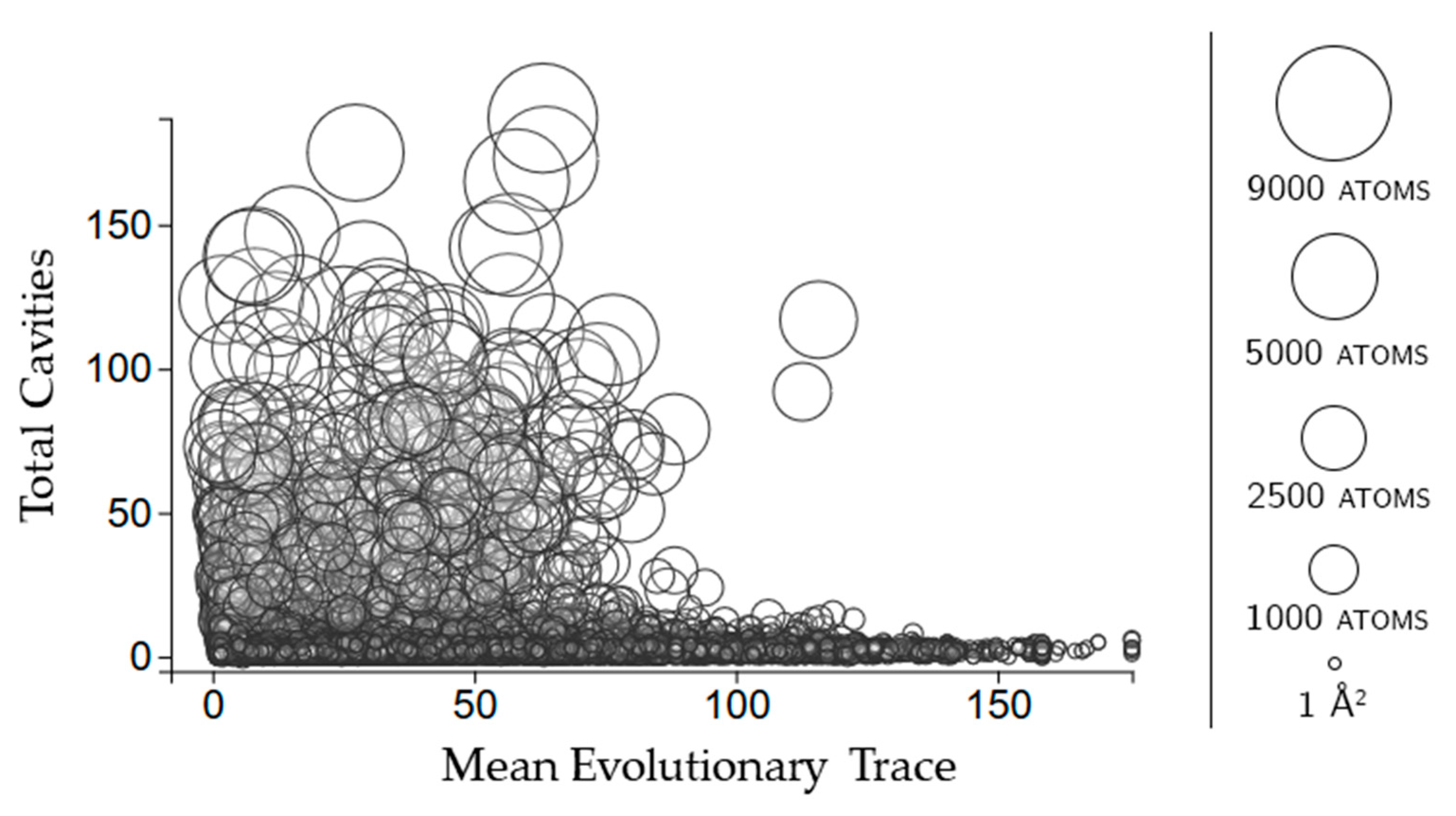
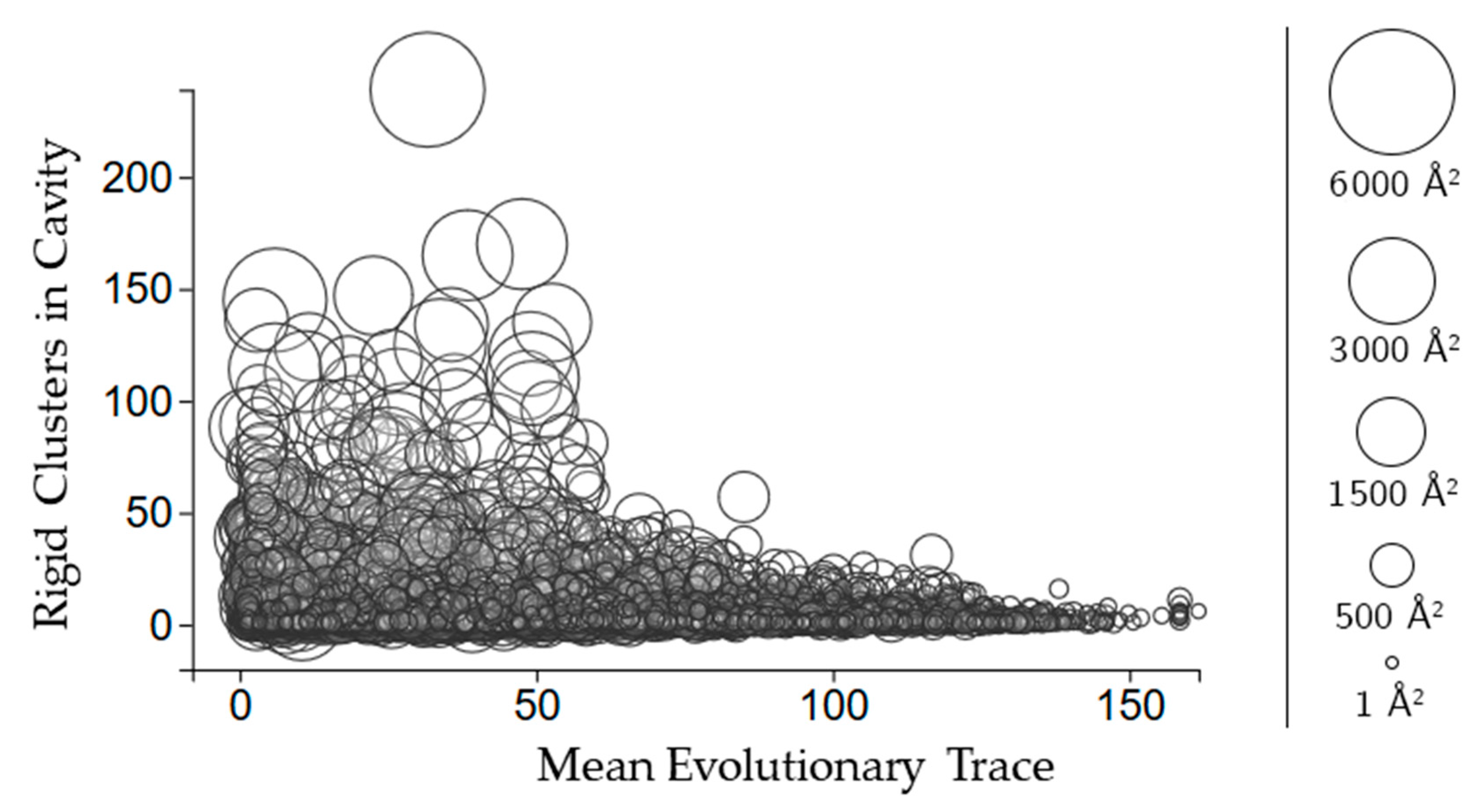
2.3.3. Rigidity Properties of Cavities: Evolutionary Trace Scores
3. Discussion and Conclusions
4. Materials and Methods
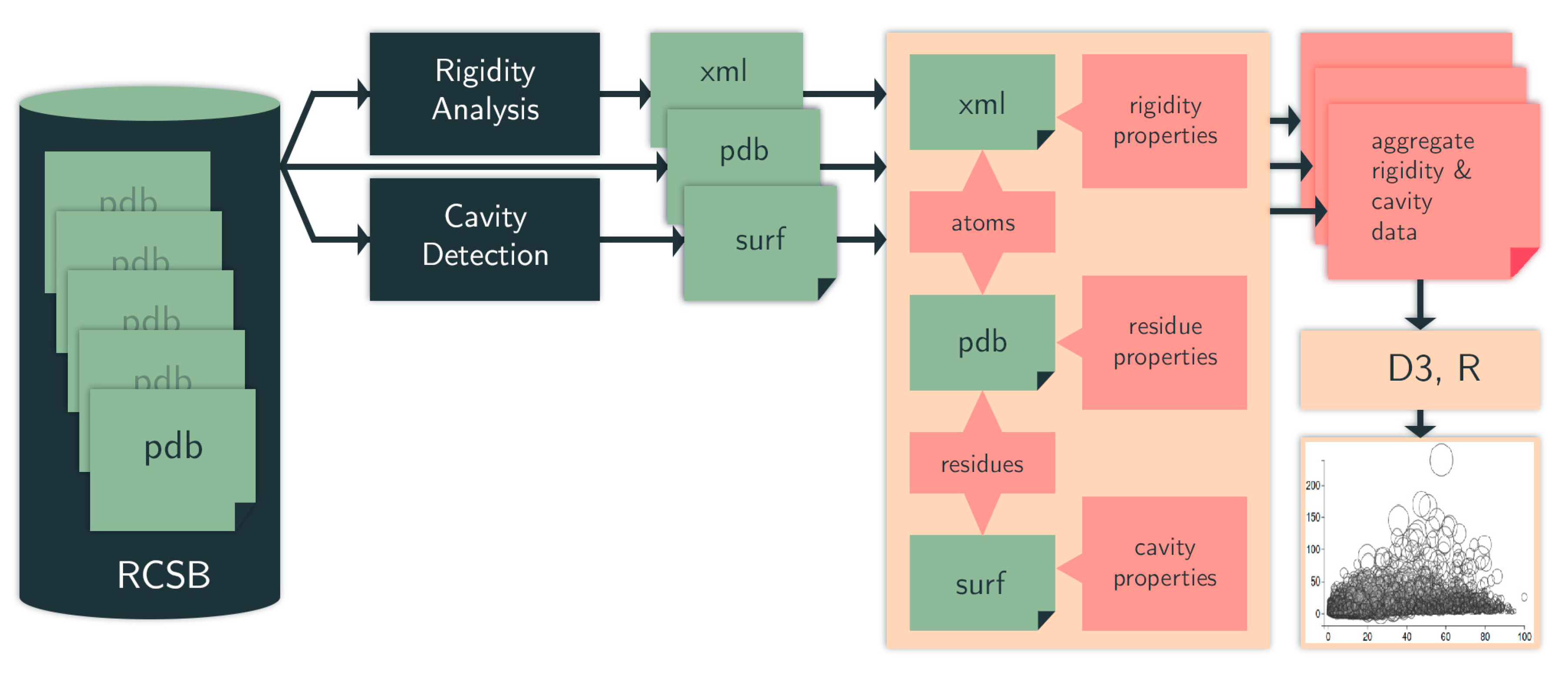
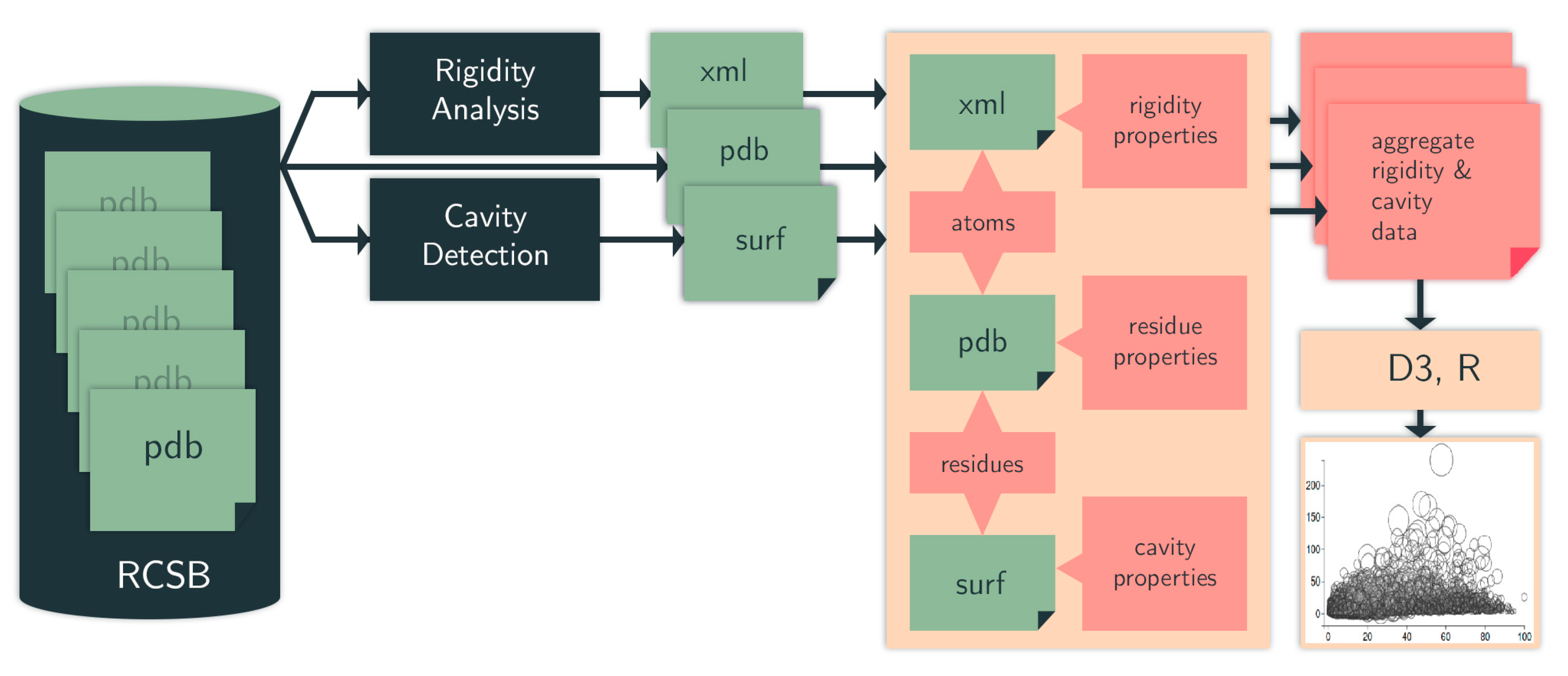
4.1. Computational Pipeline
4.1.1. Identifying Cavities
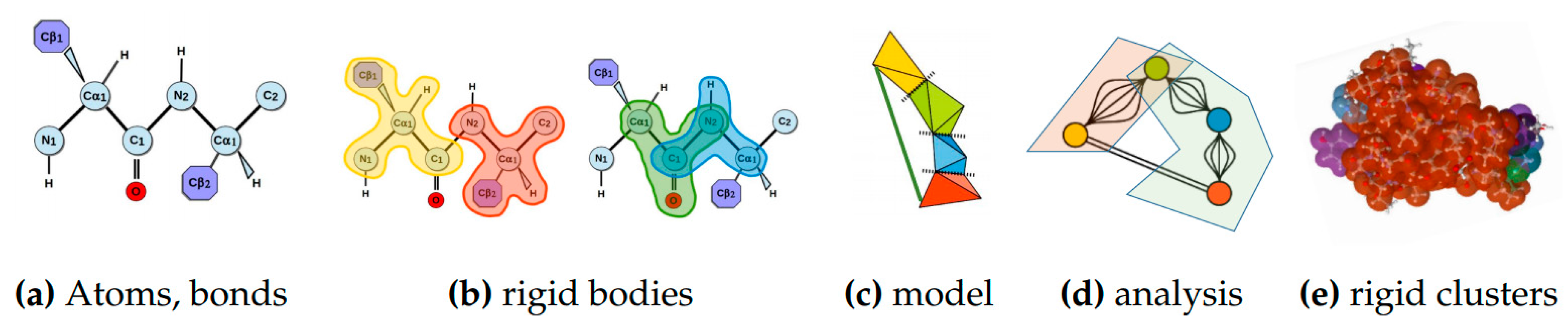
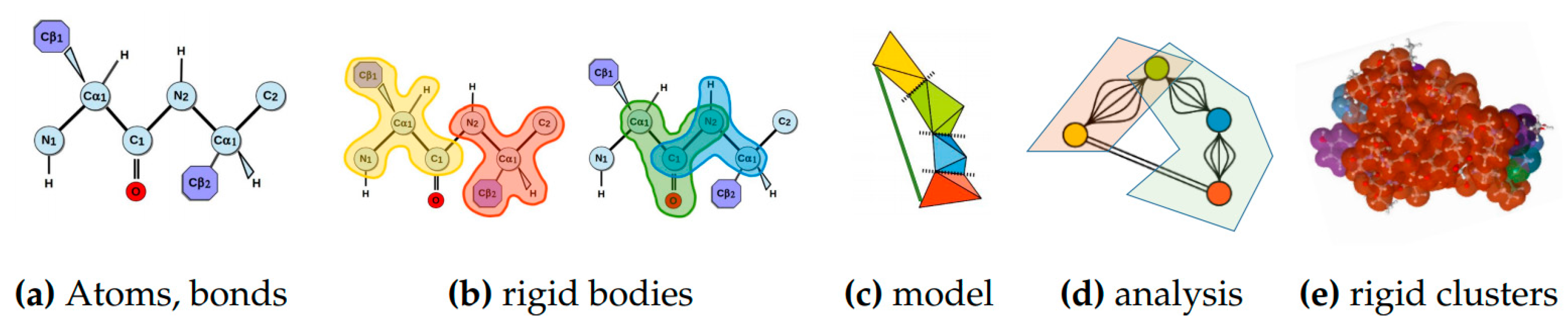
4.1.2. Rigidity Analysis
4.1.3. Evolutionary Trace
4.1.4. Data Selection, Aggregation, and Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liang, J.; Woodward, C.; Edelsbrunner, H. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. 1998, 7, 1884–1897. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Jacobson, M. Binding-site assessment by virtual fragment screening. PLoS ONE 2010, 5, e10109. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Luscombe, N.M.; Swindells, M.B.; Thornton, J.M. Protein clefts in molecular recognition and function. Protein Sci. A Publ. Protein Soc. 1996, 5, 2438. [Google Scholar]
- Chen, B.; Honig, B. VASP: A volumetric analysis of surface properties yields insights into protein-ligand binding specificity. PLoS Comput. Biol. 2010, 6, e1000881. [Google Scholar] [CrossRef] [PubMed]
- Glaser, F.; Morris, R.J.; Najmanovich, R.J.; Laskowski, R.A.; Thornton, J.M. A method for localizing ligand binding pockets in protein structures. Proteins Struct. Funct. Bioinform. 2006, 62, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.J.L. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007, 50, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Garzón, D.; Anselmi, C.; Bond, P.J.; Faraldo-Gómez, J.D. Dynamics of the Antigen-binding Grooves in CD1 Proteins: Reversible Hydrophobic Collapse in the Lipid-Free State. J. Biol. Chem. 2013, 288, 19528–19536. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.; Rader, A.; Thorpe, M.; Kuhn, L. Protein Flexibility Predictions Using Graph Theory. Proteins 2001, 44, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.; Hendrickson, B. An algorithm for two-dimensional rigidity percolation: The pebble game. J. Comput. Phys. 1997, 137, 346–365. [Google Scholar] [CrossRef]
- Fox, N.; Jagodzinski, F.; Streinu, I. KINARI-lib: A C++ library for pebble game rigidity analysis of mechanical models. In Proceedings of the Minisymposium on Publicly Available Geometric/Topological Software, Chapel Hill, NC, USA, 17–19 June 2012. [Google Scholar]
- Lichtarge, O.; Bourne, H.R.; Cohen, F.E. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996, 257, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Mihalek, I.; Reš, I.; Lichtarge, O. A family of evolution–entropy hybrid methods for ranking protein residues by importance. J. Mol. Biol. 2004, 336, 1265–1282. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.H.; Kristensen, D.M.; Mittelman, D.; Lichtarge, O. ET viewer: An application for predicting and visualizing functional sites in protein structures. Bioinformatics 2006, 22, 2049–2050. [Google Scholar] [CrossRef] [PubMed]
- Musah, R.; Jensen, G.; Bunte, S.; Rosenfeld, R.; Goodin, D. Artificial protein cavities as specific ligand-binding templates: Characterization of an engineered heterocyclic cation-binding site that preserves the evolved specificity of the parent protein. J. Mol. Biol. 2002, 315, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Bade-Doeding, C.; Huyton, T.; Eiz-Vesper, B.; Blasczyk, R. The composition of the F pocket in HLA-A* 74 generates C-terminal promiscuity among its bound peptides. Tissue Antigens 2011, 78, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; Chutkowski, C.; Kim, P. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA 1998, 95, 15613–15617. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Newton-Vinson, P.; Hubálek, F.; Edmondson, D.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Mol. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Li, M.; Hubálek, F.; Restelli, N.; Edmondson, D.; Mattevi, A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.R.; Pei, X.; Dafforn, T.; Lomas, D. Topography of a 2.0 Å structure of α1-antitrypsin reveals targets for rational drug design to prevent conformational disease. Protein Sci. 2000, 9, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; DeBolt, S.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.A.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Nayal, M.; Honig, B. On the nature of cavities on protein surfaces: Application to the identification of drug-binding sites. Proteins Struct. Funct. Bioinform. 2006, 63, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Y.; Lin, B.; Schroeder, M.; Huang, B. Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics 2011, 27, 2083–2088. [Google Scholar] [CrossRef] [PubMed]
- Guilloux, V.L.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, B. VASP-E: Specificity annotation with a volumetric analysis of electrostatic isopotentials. PLoS Comput. Biol. 2014, 10, e1003792. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Richards, F. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55. [Google Scholar] [CrossRef]
- Tsang, I.; Tsang, I. Cluster size diversity, percolation, and complex systems. Phys. Rev. E 1999, 60. [Google Scholar] [CrossRef]
- Ladurner, A.; Fersht, A. Glutamine, alanine or glycine repeats inserted into the loop of a protein have 272 minimal effects on stability and folding rates. J. Mol. Biol. 1997, 273, 330–337. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mason, S.; Chen, B.Y.; Jagodzinski, F. Exploring Protein Cavities through Rigidity Analysis. Molecules 2018, 23, 351. https://doi.org/10.3390/molecules23020351
Mason S, Chen BY, Jagodzinski F. Exploring Protein Cavities through Rigidity Analysis. Molecules. 2018; 23(2):351. https://doi.org/10.3390/molecules23020351
Chicago/Turabian StyleMason, Stephanie, Brian Y. Chen, and Filip Jagodzinski. 2018. "Exploring Protein Cavities through Rigidity Analysis" Molecules 23, no. 2: 351. https://doi.org/10.3390/molecules23020351
APA StyleMason, S., Chen, B. Y., & Jagodzinski, F. (2018). Exploring Protein Cavities through Rigidity Analysis. Molecules, 23(2), 351. https://doi.org/10.3390/molecules23020351