
Advances in the Synthesis of Lignan Natural Products


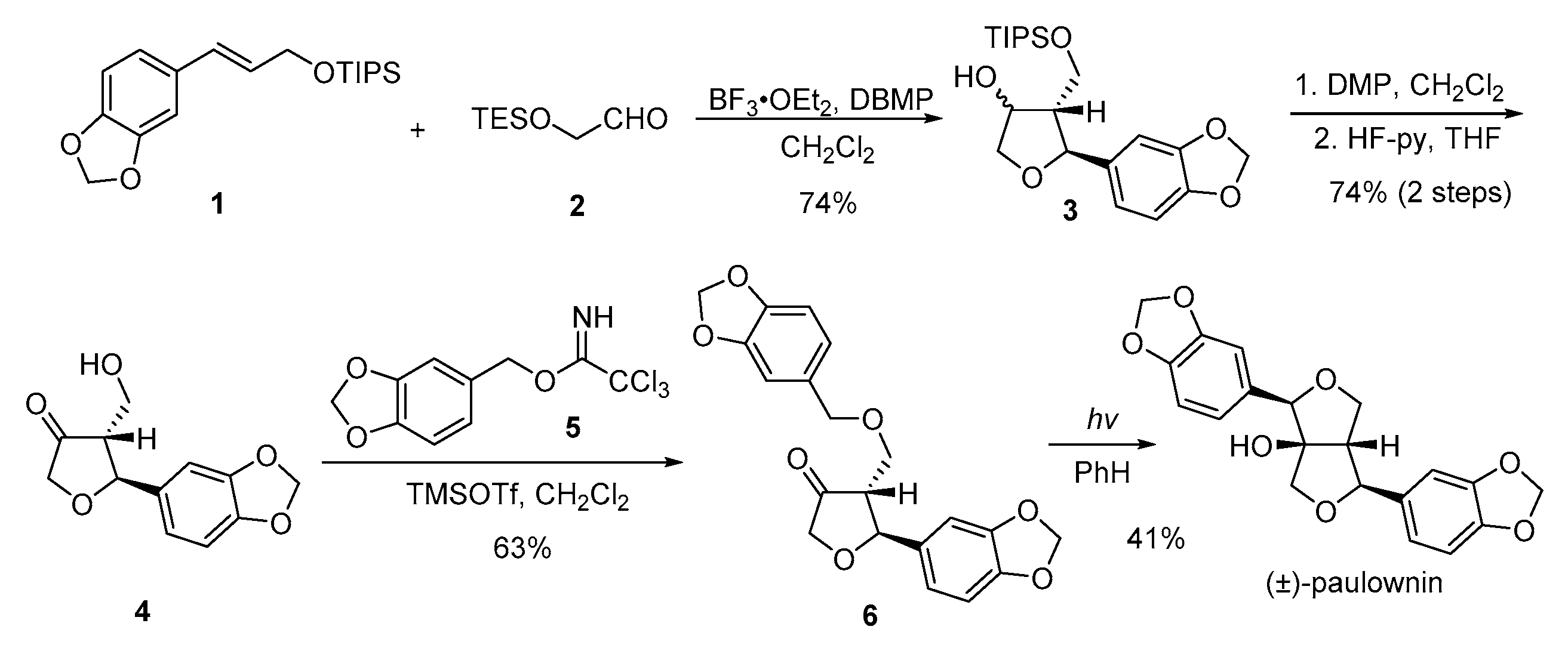{kind=link}
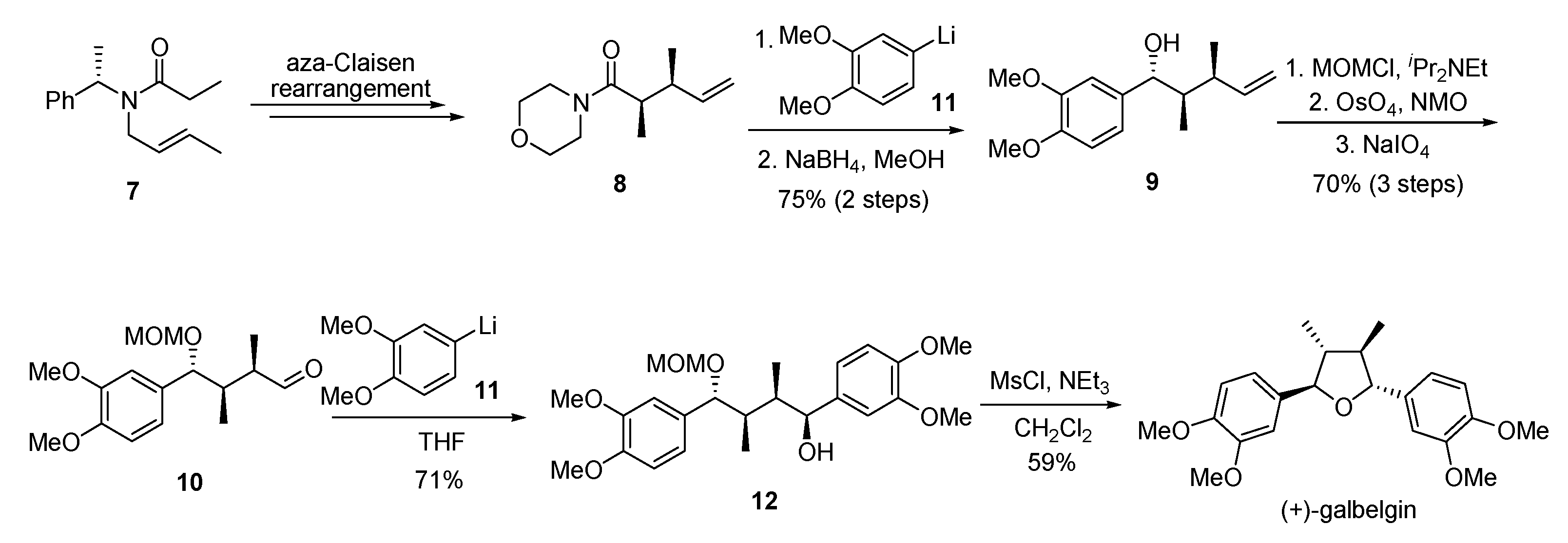{kind=link}
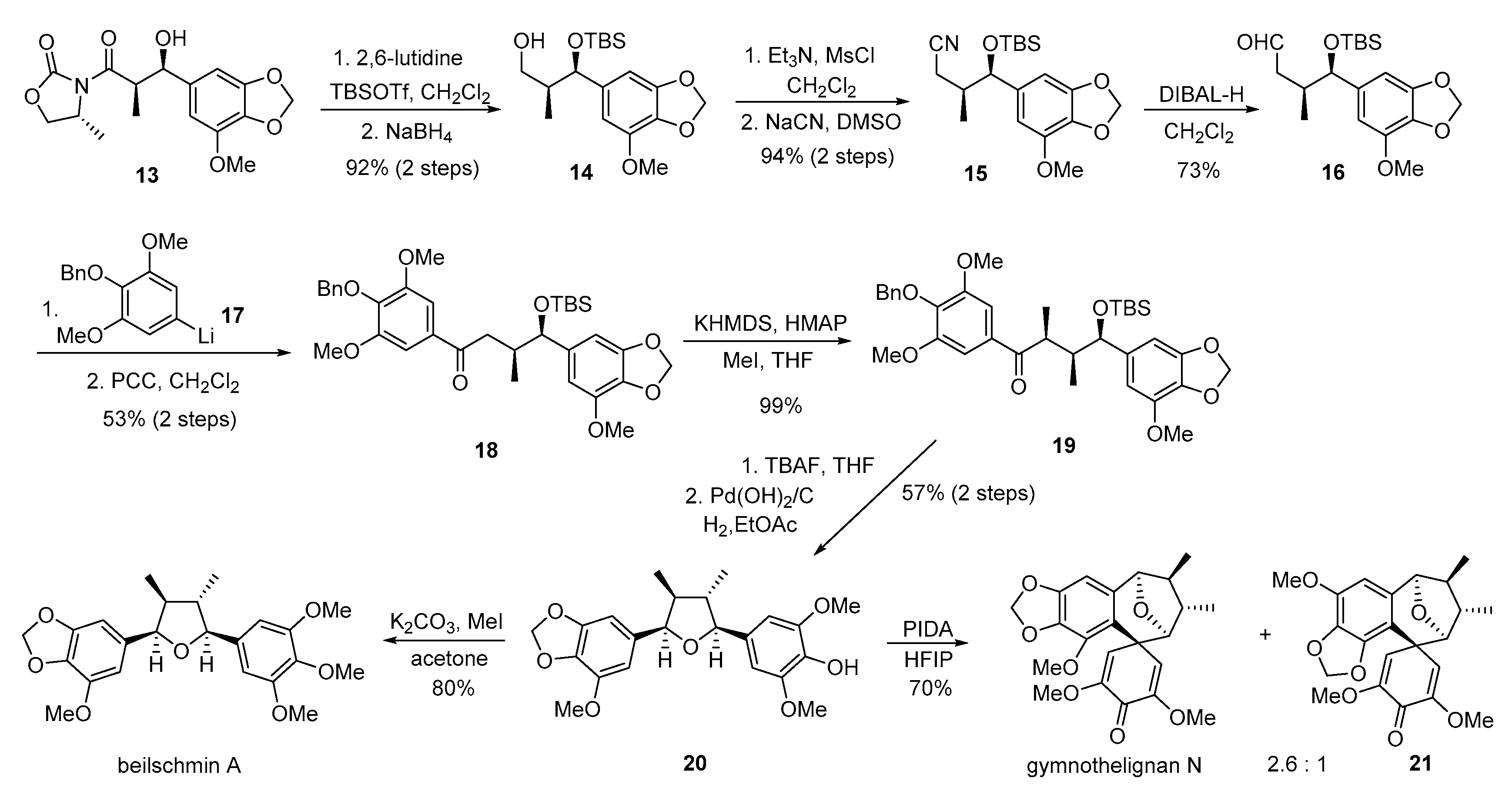{kind=link}
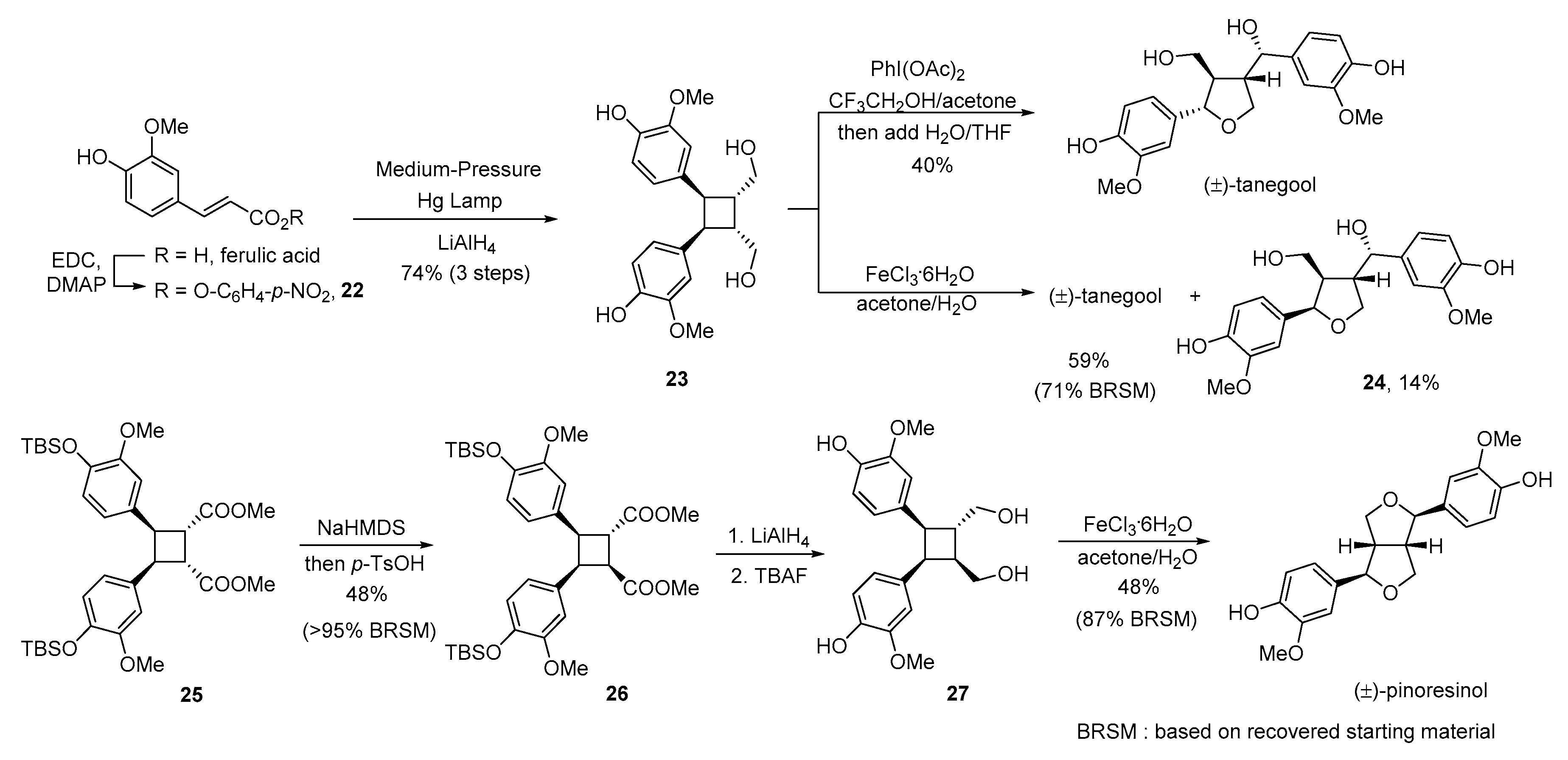{kind=link}
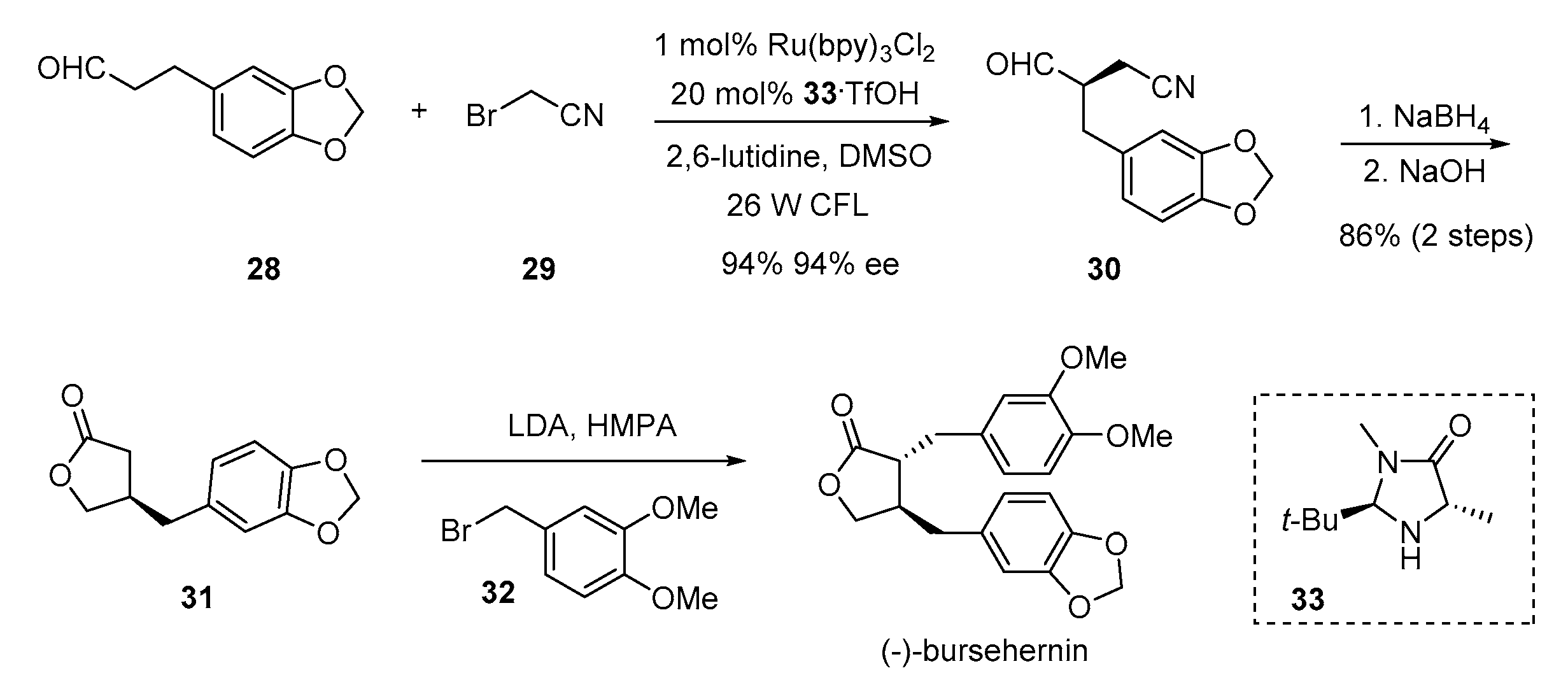{kind=link}
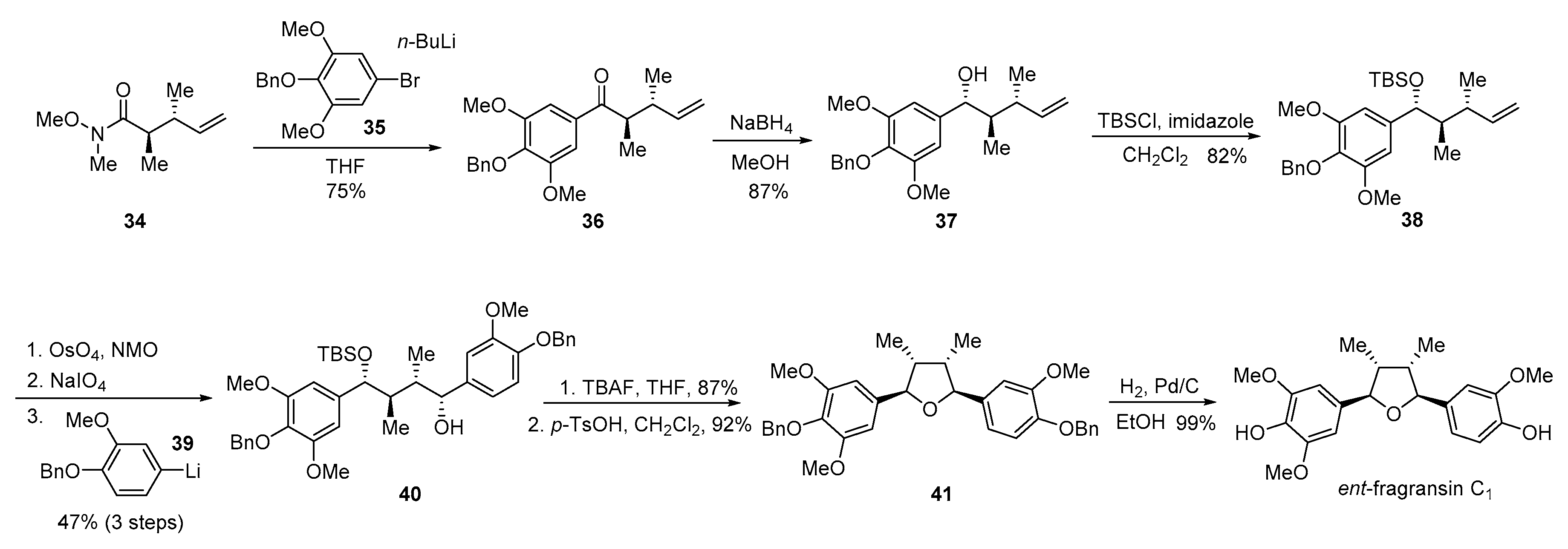{kind=link}
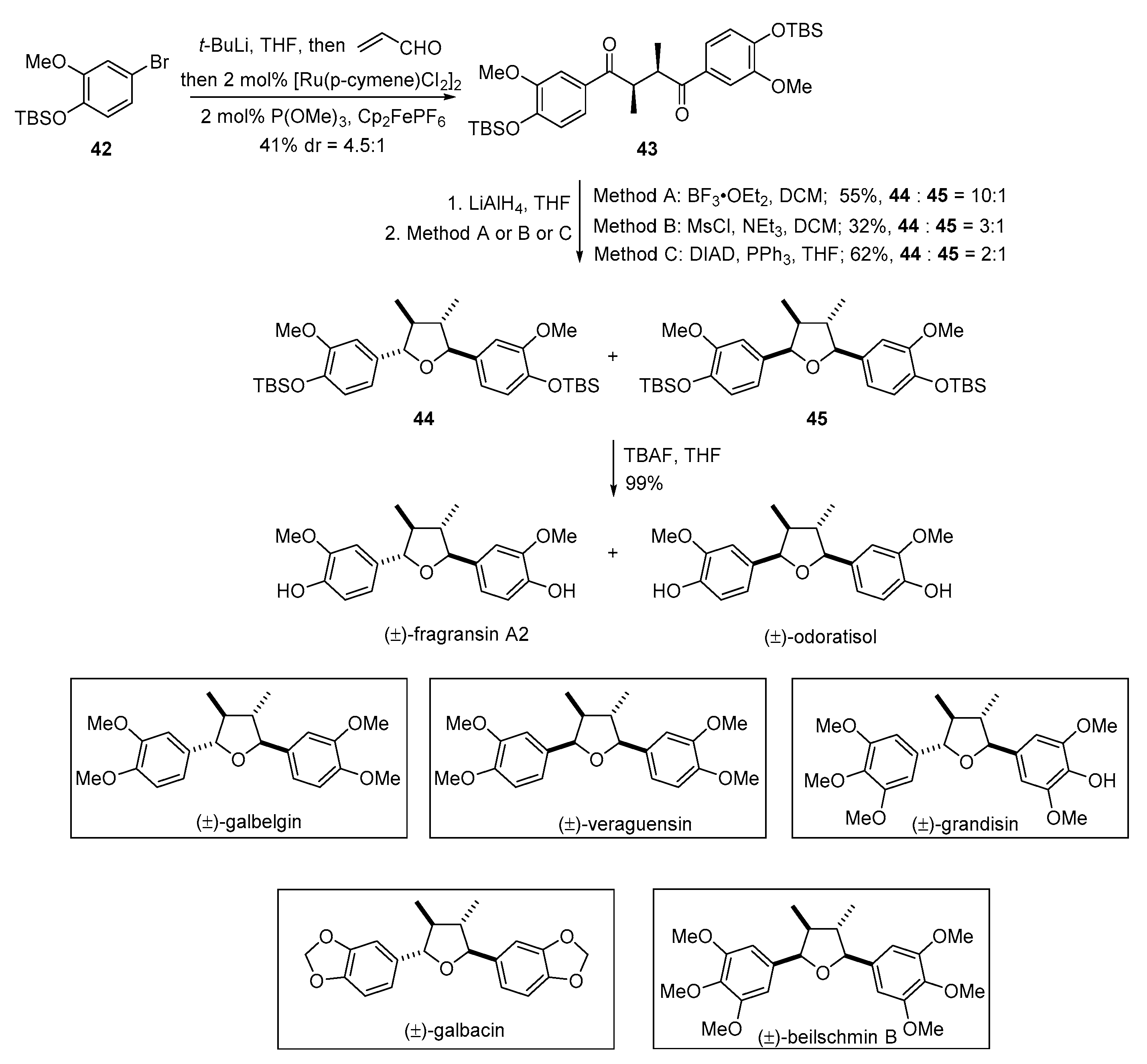{kind=link}
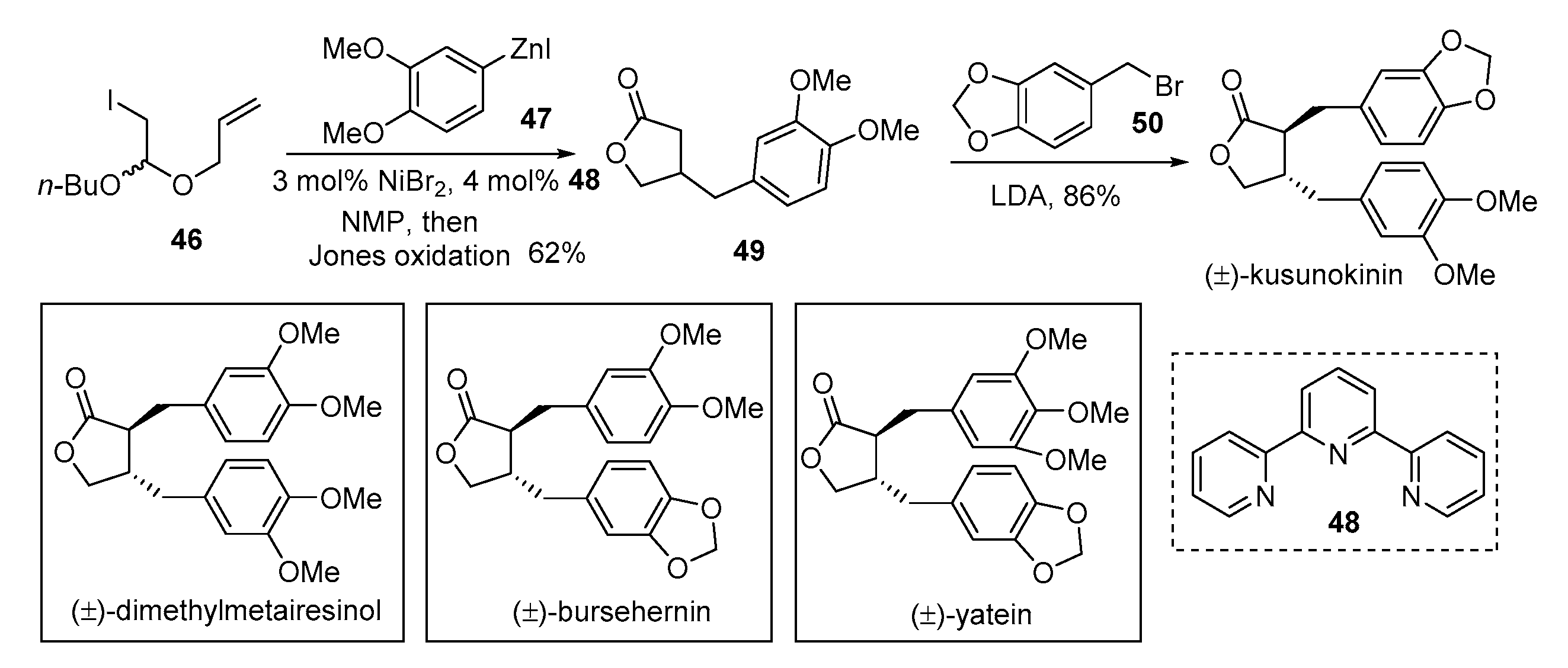{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Advances in the Synthesis of Acyclic Lignan Derivatives
3. Advances in Synthesis of Dibenzocyclooctadiene Serivatives
4. Advances in the Synthesis of Arylnaphthalene Derivatives
5. Conclusions
Funding
Conflicts of Interest
References
- Harworth, R.D. Chemistry of the lignan group of natural products. J. Chem. Soc. 1942, 448–456. [Google Scholar] [CrossRef]
- Lampe, J.W.; Martini, M.C.; Kurzer, M.S.; Adlercreutz, H.; Slavin, J.L. Urinary lignan and isoflavonoid excretion in premenopausal women consuming flaxseed powder. Am. J. Clin. Nutr. 1994, 60, 122–128. [Google Scholar] [CrossRef]
- Yamauchi, S.; Taniguchi, E. Synthesis and insecticidal activity of lignan analogs (1). Agric. Biol. Chem. 1991, 55, 3075–3084. [Google Scholar]
- Yamauchi, S.; Taniguchi, E. Synthesis and insecticidal activity of lignan analogs II. Biosci. Biotech. Biochem. 1992, 56, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, S.; Nagata, S.; Taniguchi, E. Synthesis and insecticidal activity of haedoxan analogs. (Part IV). Effect on insecticidal activity of substituents at the 1,4-benzodioxanyl moiety of haedoxan. Biosci. Biotech. Biochem. 1992, 56, 1193–1197. [Google Scholar] [CrossRef]
- Yamauchi, S.; Taniguchi, E. Synthesis and insecticidal activity of lignan analogs. Part 5. Influence on insecticidal activity of the 3-(3,4-methylenedioxyphenyl) group in the 1,4-benzodioxanyl moiety of haedoxan. Biosci. Biotech. Biochem. 1992, 56, 1744–1751. [Google Scholar] [CrossRef]
- Nitao, J.K.; Johnson, K.S.; Scriber, J.M.; Nair, M.G. Magnolia virginiana neolignan compounds as chemical barriers to swallowtail butterfly host use. J. Chem. Ecol. 1992, 18, 1661–1671. [Google Scholar] [CrossRef]
- Wang, C.T.; Makela, T.; Hase, H.; Adlercreutz, H.; Kurzer, M.S. Lignans and flavonoids inhibit aromatase enzyme in human preadipocytes. J. Steroid Biochem. Molec. Biol. 1994, 50, 205–212. [Google Scholar] [CrossRef]
- Kirkman, L.M.; Lampe, J.W.; Campbell, D.R.; Martini, M.C.; Slavin, J.L. Urinary lignan and isoflavonoid excretion in men and women consuming vegetable and soy diets. Nutr. Cancer 1995, 24, 1–12. [Google Scholar] [CrossRef]
- Makela, S.I.; Pylkkanen, L.H.; Santti, R.S.S.; Adlercreutz, H. Dietary soybean may be antiestrogenic in male mice. J. Nutr. 1995, 125, 437–445. [Google Scholar]
- Adlercreutz, H.; Goldin, B.R.; Gorbach, S.L.; Hockerstedt, K.A.V.; Watanabe, S.; Hamalainen, E.K.; Markkanen, M.H.; Makela, T.H.; Wahala, K.T.; Hase, T.A.; et al. Soybean phytoestrogen intake and cancer risk. J. Nutr. 1995, 125, 757–770. [Google Scholar]
- Xu, W.-H.; Zhao, P.; Wang, M.; Liang, Q. Naturally occurring furofuran lignans: Structural diversity and biological activities. Nat. Prod. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Teponno, R.B.; Kusari, S.; Spiteller, M. Recent advances in research on lignans and neolignans. Nat. Prod. Rep. 2016, 33, 1044–1092. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.-Y.; Chen, S.-L.; Yang, M.-H.; Wu, J.; Sinkkonen, J.; Zou, K. An update on lignans: Natural products and synthesis. Nat. Prod. Rep. 2009, 26, 1251–1292. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.S. Lignans, neolignans and related compounds. Nat. Prod. Rep. 1999, 16, 75–96. [Google Scholar] [CrossRef]
- Ward, R.S. Lignans, neolignans and related compounds. Nat. Prod. Rep. 1997, 14, 43–74. [Google Scholar] [CrossRef]
- Ward, R.S. Lignans, neolignans, and related compounds. Nat. Prod. Rep. 1995, 12, 183–205. [Google Scholar] [CrossRef]
- Ward, R.S. Lignans, neolignans, and related compounds. Nat. Prod. Rep. 1993, 10, 1–28. [Google Scholar] [CrossRef]
- Whiting, D.A. Lignans and neolignans. Nat. Prod. Rep. 1985, 2, 191–211. [Google Scholar] [CrossRef]
- Kawazoe, K.; Yutani, A.; Tamemoto, K.; Yuasa, S.; Shibata, H.; Higuti, T.; Takaishi, Y. Phenylnaphthalene compounds from the subterranean part of Vitex rotundifolia and their antibacterial activity against methicillin-resistant Staphylococcus aureus. J. Nat. Prod. 2001, 64, 588–591. [Google Scholar] [CrossRef]
- Sagar, K.S.; Chang, C.C.; Wang, W.K.; Lin, J.Y.; Lee, S.S. Preparation and anti-HIV activities of retrojusticidin B analogs and azalignans. Bioorg. Med. Chem. 2004, 12, 4045–4054. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.; Li, Y.; Fu, L.; Zhu, J.L.; Gullen, E.A.; Dutschman, G.E.; Lee, Y.; Chung, R.; Huang, E.S.; Austin, D.J.; et al. Synthesis and antiviral activity of helioxanthin analogues. J. Med. Chem. 2005, 48, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, L.; Yeo, H.; Zhu, J.L.; Chou, C.K.; Kou, Y.H.; Yeh, S.F.; Gullen, E.; Austin, D.; Cheng, Y.C. Inhibition of hepatitis B virus gene expression and replication by helioxanthin and its derivative. Antiviral Chem. Chemother. 2005, 16, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Janmanchi, D.; Tseng, Y.P.; Wang, K.C.; Huang, R.L.; Lin, C.H.; Yeh, S.F. Synthesis and the biological evaluation of arylnaphthalene lignans as anti-hepatitis B virus agents. Bioorg. Med. Chem. 2010, 18, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- McDoniel, P.B.; Cole, J.R. Antitumor activity of Bursera schlechtendalii (Burseraceae). Isolation and structure determination of two new lignans. J. Pharm. Sci. 1972, 61, 1992–1994. [Google Scholar] [CrossRef] [PubMed]
- Pelter, A.; Ward, R.S.; Satyanarayana, P.; Collins, P. Synthesis of lignan lactones by conjugate addition of thioacetal carbanions to butanolide. J. Chem. Soc. Perkin Trans. 1983, 1983, 643–647. [Google Scholar] [CrossRef]
- Capilla, A.S.; Sánchez, I.; Caignard, D.H.; Renard, P.; Pujola, M.D. Antitumor agents. Synthesis and biological evaluation of new compounds related to podophyllotoxin, containing the 2,3-dihydro-1,4-benzodioxin system. Eur. J. Med. Chem. 2001, 36, 389–393. [Google Scholar] [CrossRef]
- Chen, C.C.; Hsin, W.C.; Ko, F.N.; Huang, Y.L.; Ou, J.C.; Teng, C.M. Antiplatelet arylnaphthalide lignans from Justicia procumbens. J. Nat. Prod. 1996, 59, 1149–1150. [Google Scholar] [CrossRef]
- Weng, J.R.; Ko, H.H.; Yeh, T.L.; Lin, H.C.; Lin, C.N. Two new arylnaphthalide lignans and antiplatelet constituents from Justicia procumbens. Arch. Pharm. 2004, 337, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Ukita, T.; Nakamura, Y.; Kubo, A.; Yamomoto, Y.; Takahashi, M.; Kotera, J.; Ikeo, T. Synthesis and in vitro pharmacology of a series of new chiral histamine H3-receptor ligands: 2-(R and S)-amino-3-(1H-imidazol-4(5)-yl)propyl ether derivatives. J. Med. Chem. 1999, 42, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Kondo, K.; Kuroda, T.; Moritani, Y.; Yamagata, S.; Sugiura, M.; Kikkawa, H.; Kaminuma, O.; Ikezawa, K. Novel selective PDE IV inhibitors as antiasthmatic agents. synthesis and biological activities of a series of 1-aryl-2,3-bis(hydroxymethyl)naphthalene lignans. J. Med. Chem. 1996, 39, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Thérien, M.; Fitzsimmons, B.J.; Scheigets, J.; Macdonald, D.; Choo, L.Y.; Guay, J.; Falgueyret, J.P.; Riendeau, D. Justicidin E: A new leukotriene biosynthesis inhibitor. Bioorg. Med. Chem. 1993, 3, 2063–2066. [Google Scholar] [CrossRef]
- Delorme, D.; Ducharme, Y.; Brideau, C.; Chan, C.C.; Chauret, N.; Desmarais, S.; Dubé, D.; Falgueyret, J.P.; Fortin, R.; Guay, J.; et al. Dioxabicyclooctanyl naphthalenenitriles as nonredox 5-lipoxygenase inhibitors: structure−activity relationship study directed toward the improvement of metabolic stability. J. Med. Chem. 1996, 39, 3951–3970. [Google Scholar] [CrossRef] [PubMed]
- Ducharme, Y.; Brideau, C.; Dubé, D.; Chan, C.C.; Falgueyret, J.P.; Gillard, J.W.; Guay, J.; Hutchinson, J.H.; McFarlane, C.S.; Riendeau, D.; et al. Naphthalenic lignan lactones as selective, nonredox 5-lipoxygenase inhibitors. Synthesis and biological activity of (methoxyalkyl)thiazole and methoxytetrahydropyran hybrids. J. Med. Chem. 1994, 37, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Lin, M.T.; Lee, S.-S.; Liu, K.C.S.C.; Hsu, F.-L.; Lin, J.-Y. Differential inhibition of reverse transcriptase and cellular DNA polymerase-α activities by lignans isolated from Chinese herbs, Phyllanthus myrtifolius Moon, and tannins from Lonicera japonica Thunb and Castanopsis hystrix. Antiviral Res. 1995, 27, 367–374. [Google Scholar] [CrossRef]
- Lee, S.S.; Lin, M.T.; Liu, C.L.; Lin, Y.Y.; Liu, K.C.S.C. Six lignans from Phyllanthus myrtifolius. J. Nat. Prod. 1996, 59, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Cow, C.; Leung, C.; Charlton, J.L. Antiviral activity of arylnaphthalene and aryldihydronaphthalene lignans. Can. J. Chem. 2000, 78, 553–561. [Google Scholar] [CrossRef]
- Wu, S.J.; Wu, T.S. Cytotoxic arylnaphthalene lignans from Phyllanthus oligospermus. Chem. Pharm. Bull. 2006, 54, 1223–1225. [Google Scholar] [CrossRef]
- Lu, H.; Liu, G.-T. Antioxidant activity of dibenzocyclooctene lignans isolated from Schisandraceae. Planta Med. 1992, 58, 311–313. [Google Scholar] [CrossRef]
- Hirano, T.; Wakasugi, A.; Oohara, M.; Oka, K.; Sashida, Y. Suppression of mitogen-induced proliferation of human peripheral blood lymphocytes by plant lignans. Planta Med. 1991, 57, 331–334. [Google Scholar] [CrossRef]
- Chang, J.; Reiner, J.; Xie, J. Progress on the chemistry of dibenzocyclooctadiene lignans. Chem. Rev. 2005, 105, 4581–4609. [Google Scholar] [CrossRef]
- Satake, H.; Koyama, T.; Bahabadi, S.E.; Matsumoto, E.; Ono, E.; Murata, J. Essences in metabolic engineering of lignan biosynthesis. Metabolites 2015, 5, 270–290. [Google Scholar] [CrossRef] [PubMed]
- Satake, H.; Ono, E.; Murata, J. Recent advances in the metabolic engineering of lignan biosynthesis pathways for the production of transgenic plant-based foods and supplements. J. Agric. Food Chem. 2013, 61, 11721–11729. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Umezawa, T. Biosynthesis of lignans and norlignans. J. Wood Sci. 2007, 53, 273–284. [Google Scholar] [CrossRef]
- Umezawa, T. Diversity in lignan biosynthesis. Phytochem. Rev. 2003, 2, 371–390. [Google Scholar] [CrossRef]
- Albertson, A.K.F.; Lumb, J.-P. A bio-inspired total synthesis of tetrahydrofuran lignans. Angew. Chem. Int. Ed. 2015, 54, 2204–2208. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Rakesh, K.P.; Mumtaz, S.; Moku, B.; Asiri, A.M.; Marwani, H.M.; Manukumar, H.M.; Qin, H.-L. Arylnaphthalene lactone analogues: Synthesis and development as excellent biological candidates for future drug discovery. RSC Adv. 2018, 8, 9487–9502. [Google Scholar] [CrossRef]
- Pohmakotr, M.; Kuhakarn, C.; Reutrakul, V.; Soorukram, D. Asymmetric synthesis of furofurans. Tetrahedron Lett. 2017, 58, 4740–4746. [Google Scholar] [CrossRef]
- Sun, J.-S.; Liu, H.; Guo, X.-H.; Liao, J.-X. The chemical synthesis of aryltetralin glycosides. Org. Biomol. Chem. 2016, 14, 1188–1200. [Google Scholar] [CrossRef]
- Sellars, J.D.; Steel, P.G. Advances in the synthesis of aryltetralin lignan lactones. Eur. J. Org. Chem. 2007, 2007, 3815–3828. [Google Scholar] [CrossRef]
- Brown, R.C.D.; Swain, N.A. Synthesis of furofuran lignans. Synthesis 2004, 811–827. [Google Scholar] [CrossRef]
- Angle, S.R.; Choi, I.; Tham, F.S. Stereoselective synthesis of 3-alkyl-2-aryltetrahydrofuran-4-ols: Total synthesis of (±)-paulownin. J. Org. Chem. 2008, 73, 6268–6278. [Google Scholar] [CrossRef] [PubMed]
- Kraus, G.A.; Chen, L. A total synthesis of racemic paulownin using a type II photocyclization reaction. J. Am. Chem. Soc. 1990, 112, 3464–3466. [Google Scholar] [CrossRef]
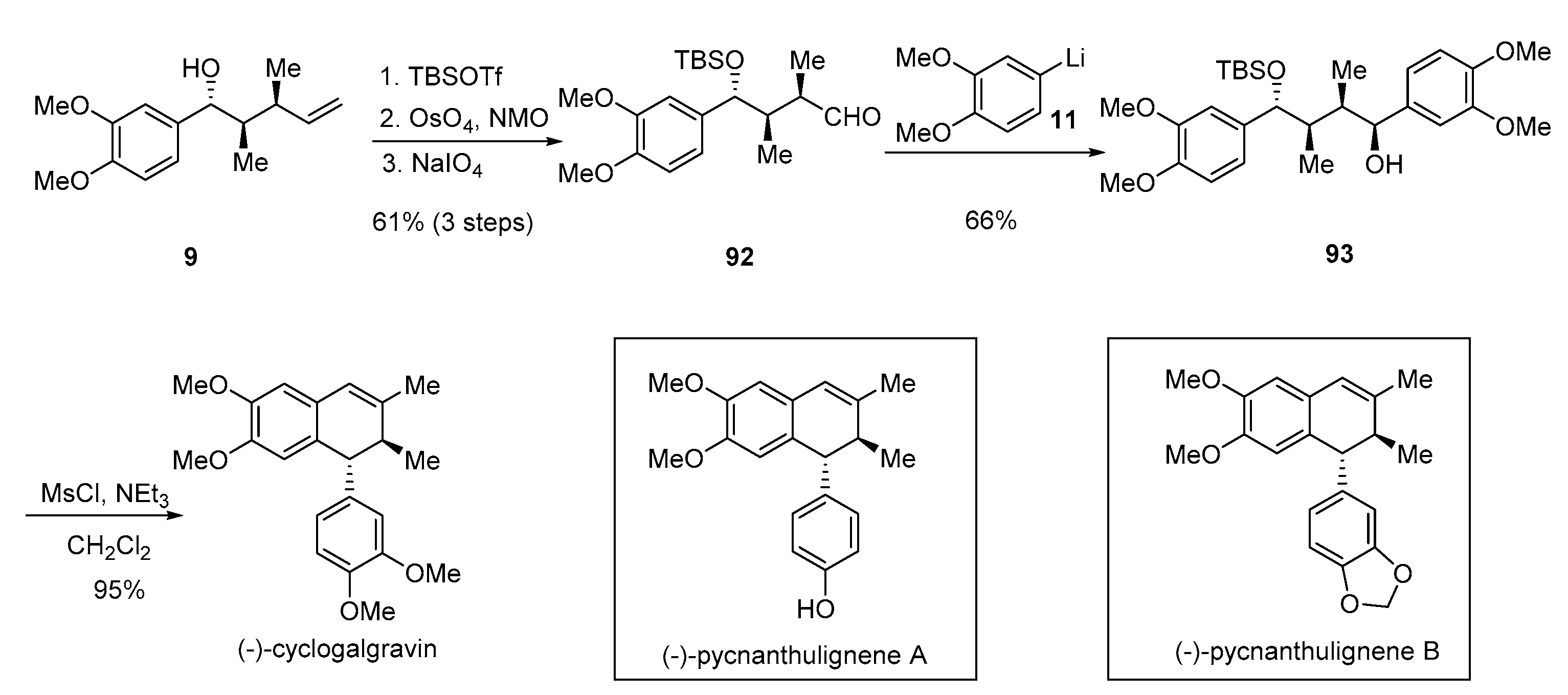
- Rye, C.E.; Barker, D. Asymmetric synthesis of (+)-galbelgin, (−)-kadangustin J, (−)-cyclogalgravin and (−)-pycnanthulignenes A and B, three structurally distinct lignan classes, using a common chiral precursor. J. Org. Chem. 2011, 76, 6636–6648. [Google Scholar] [CrossRef] [PubMed]
- Rye, C.E.; Barker, D. An acyl-Claisen approach to tetrasubstituted tetrahydrofuran lignans: Synthesis of fragransin A2, talaumidin, and lignan analogues. Synlett 2009, 2009, 3315–3319. [Google Scholar]
- Li, H.; Zhang, Y.; Xie, X.; Ma, H.; Zhao, C.; Zhao, G.; She, X. Bioinspired total synthesis of gymnothelignan N. Org. Lett. 2014, 16, 4440–4443. [Google Scholar] [CrossRef] [PubMed]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillian, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Tucher, J.W.; Stephenson, C.R.J. Shining light on photoredox catalysis: Theory and synthetic applications. J. Org. Chem. 2012, 77, 1617–1622. [Google Scholar] [CrossRef]
- Welin, E.R.; Warkentin, A.A.; Conrad, J.C.; MacMillian, D.W.C. Enantioselective α-alkylation of aldehydes by photoredox organocatalysis: Rapid access to pharmacophore fragments from β-cyanoaldehydes. Angew. Chem. Int. Ed. 2015, 54, 9668–9672. [Google Scholar] [CrossRef]
- Chaimanee, S.; Pohmakotr, M.; Kuhakarn, C.; Reutrakul, V.; Soorukram, D. Asymmetric synthesis of ent-fragransin C1. Org. Biomol. Chem. 2017, 15, 3985–3994. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.R.; Ford, L.; Deister, E.; Pohl, R.; Císařová, I.; Hodek, J.; Weber, J.; Mackman, R.; Bahador, G.; Jahn, U. Highly functionalized and potent antiviral cyclopentane derivatives formed by a tandem process consisting of organometallic, transition-metal-catalyzed, and radical reaction steps. Chem. Eur. J. 2014, 20, 10298–10304. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.R.; Císařová, I.; Jahn, U. Bioinspired total synthesis of tetrahydrofuran lignans by tandem nucleophilic addition/redox isomerization/oxidative coupling and cycloetherification reactions as key steps. Org. Biomol. Chem. 2018, 16, 750–755. [Google Scholar] [CrossRef]
- KC, S.; Basnet, P.; Thapa, S.; Shrestha, B.; Giri, R. Ni-catalyzed regioselective dicarbofunctionalization of unactivated olefins by tandem cyclization/cross-coupling and application to the concise synthesis of lignan natural products. J. Org. Chem. 2018, 83, 2920–2936. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-W.; Zhao, Q.; Xu, M.-H.; Lin, G.-Q. Nickel-catalyzed asymmetric Ullmann coupling for the synthesis of axially chiral tetra-ortho-substituted biaryl dials. Org. Lett. 2010, 12, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
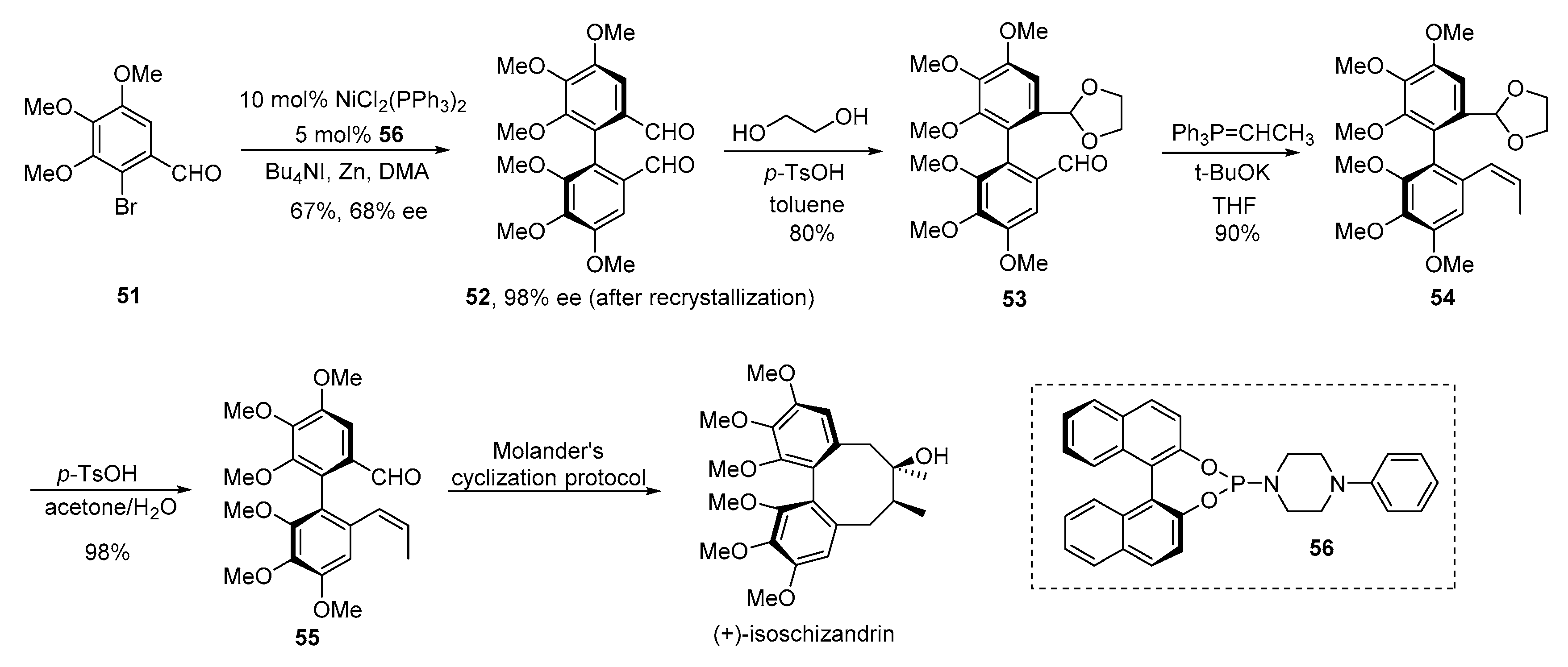
- Molander, G.A.; George, K.M.; Monovich, L.G. Total synthesis of (+)-isoschizandrin utilizing a samarium(II) iodide-promoted 8-endo ketyl-olefin cyclization. J. Org. Chem. 2003, 68, 9533–9540. [Google Scholar] [CrossRef] [PubMed]
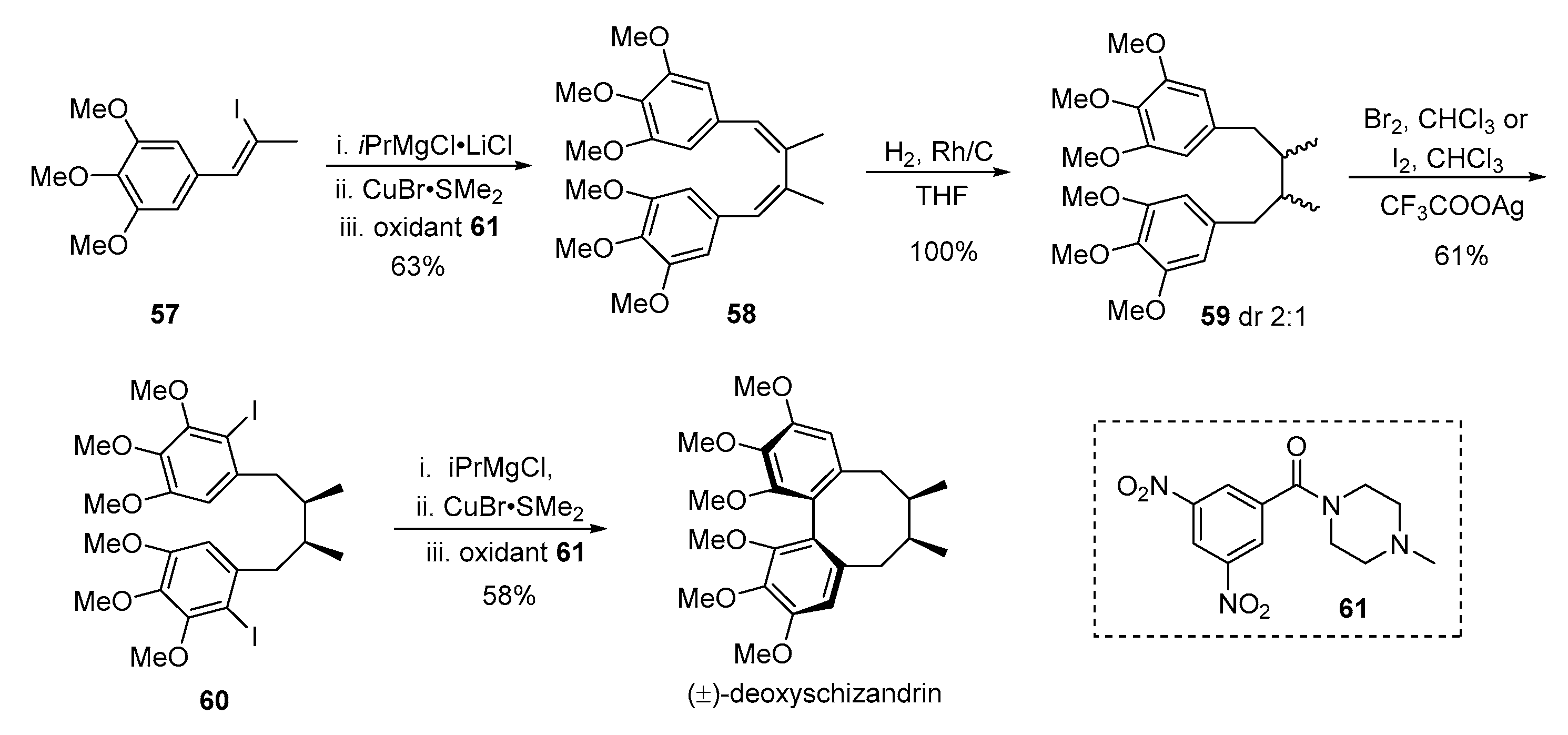
- Zheng, S.; Aves, S.J.; Laraia, L.; Galloway, W.R.J.D.; Pike, K.G.; Wu, W.; Spring, D.R. A concise total synthesis of deoxyschizandrin and exploration of its antiproliferative effects and those of structurally related derivatives. Chem. Eur. J. 2012, 18, 3193–3198. [Google Scholar] [CrossRef] [PubMed]
- Surry, D.S.; Su, X.B.; Fox, D.J.; Franckevicius, V.; Macdonald, S.J.F.; Spring, D.R. Synthesis of medium-ring and iodinated biaryl compounds by organocuprate oxidation. Angew. Chem. Int. Ed. 2005, 44, 1870–1873. [Google Scholar] [CrossRef]
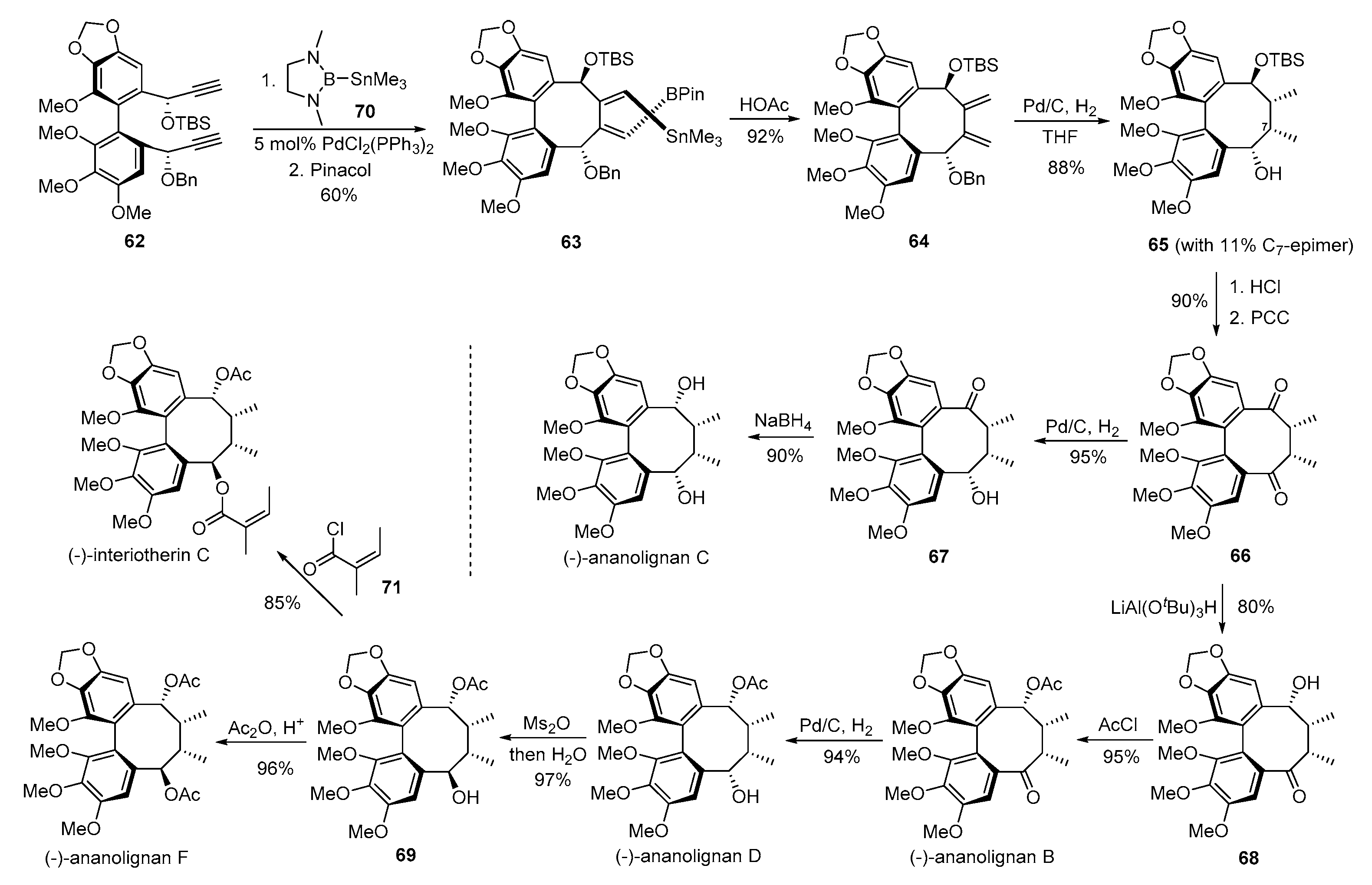
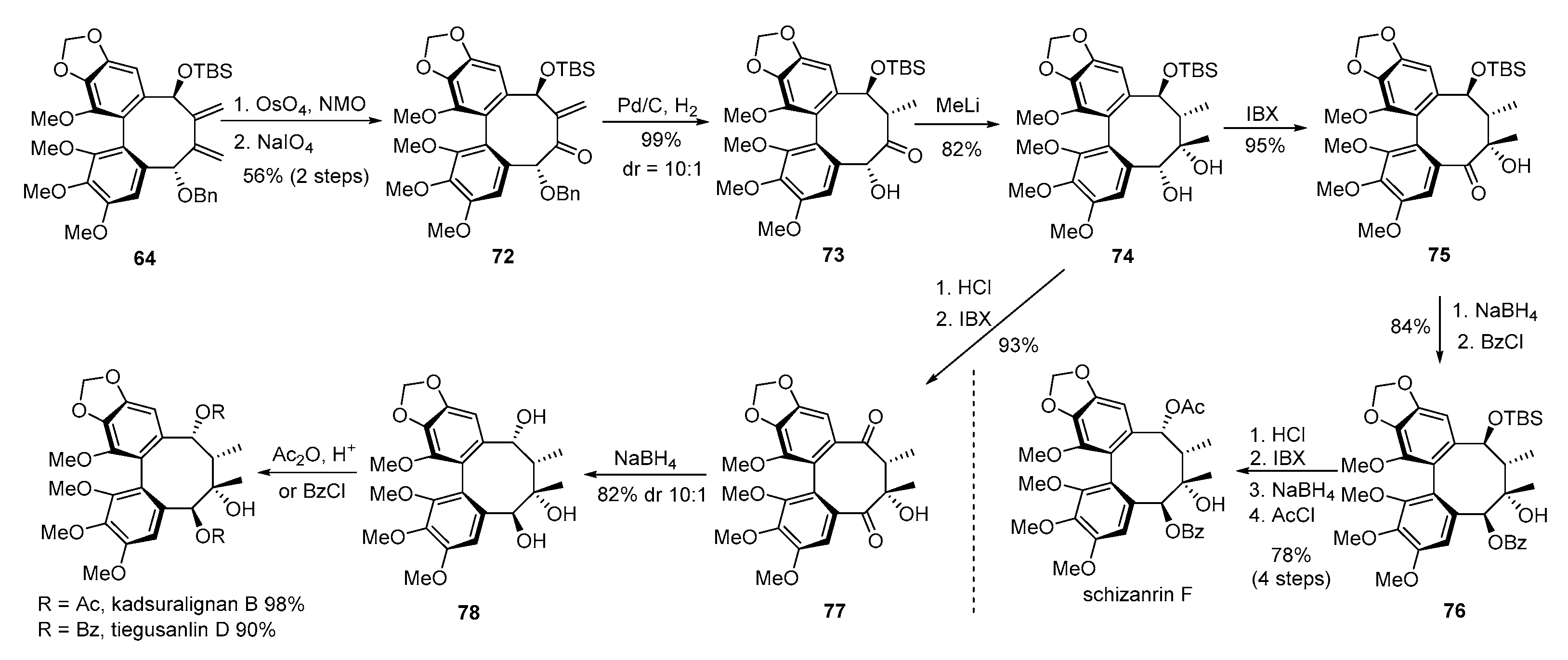
- Gong, W.; RajanBabu, T.V. Conformation and reactivity in dibenzocyclooctadienes (DBCOD). A general approach to the total synthesis of fully substituted DBCOD lignans via borostannylative cyclization of α,ω-diynes. Chem. Sci. 2013, 4, 3979–3985. [Google Scholar] [CrossRef]
- Gong, W.; Singidi, R.R.; Gallucci, J.C.; RajanBabu, T.V. On the stereochemistry of acetylide additions to highly functionalized biphenylcarbaldehydes and multi-component cyclization of 1,n-diynes. Syntheses of dibenzocyclooctadiene lignans. Chem. Sci. 2012, 3, 1221–1230. [Google Scholar] [CrossRef]
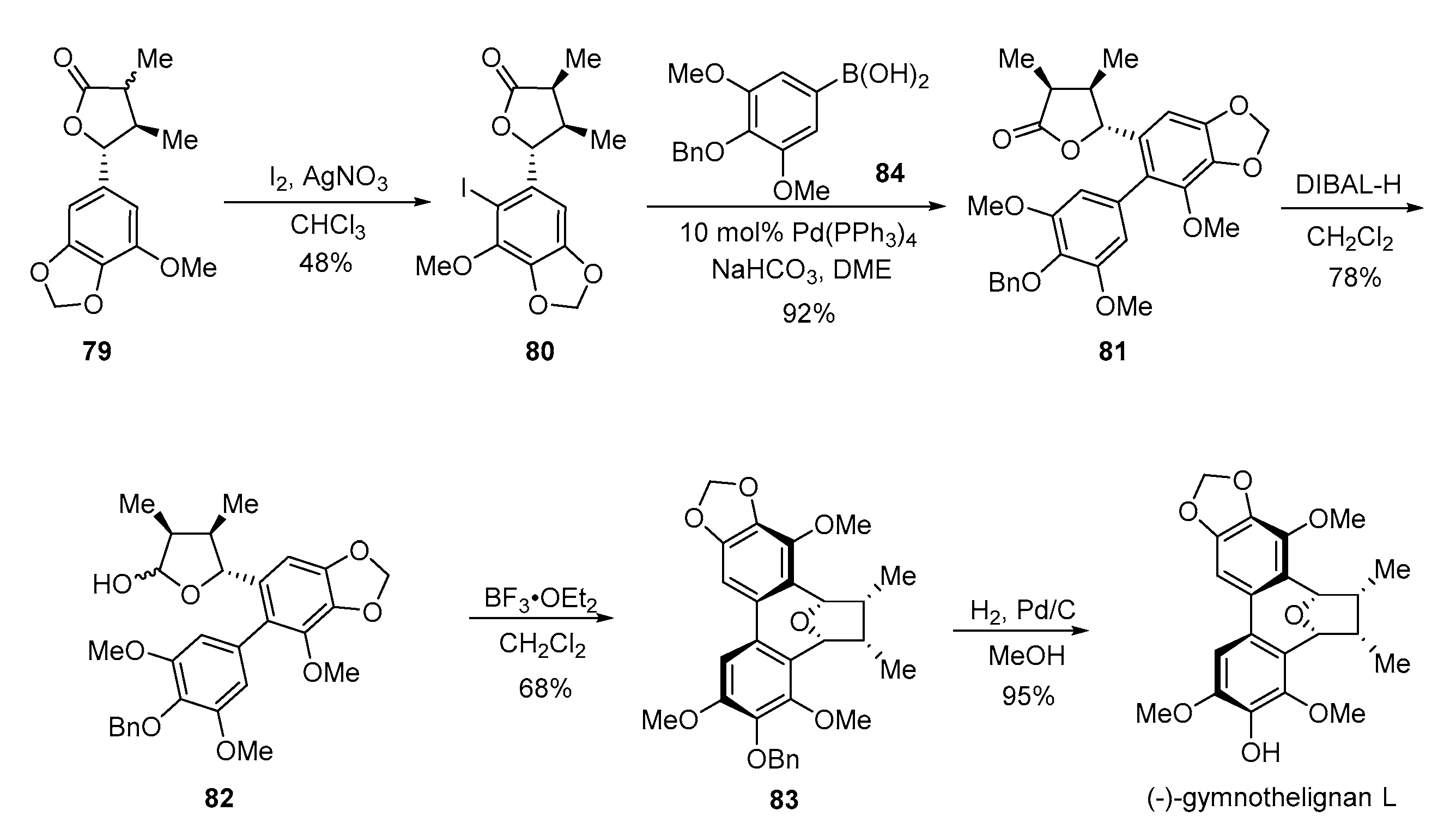
- Chen, P.; Huo, L.; Li, H.; Liu, L.; Yuan, Z.; Zhang, H.; Feng, S.; Xie, X.; Wang, X.; She, X. Bioinspired total synthesis of (−)-gymnothelignan L. Org. Chem. Front. 2018, 5, 1124–1128. [Google Scholar] [CrossRef]
- Soorukram, D.; Pohmakotr, M.; Kuhakarn, C.; Reutrakul, V. Bioinspired asymmetric synthesis of (−)-gymnothelignan V. J. Org. Chem. 2018, 83, 4173–4179. [Google Scholar] [CrossRef] [PubMed]
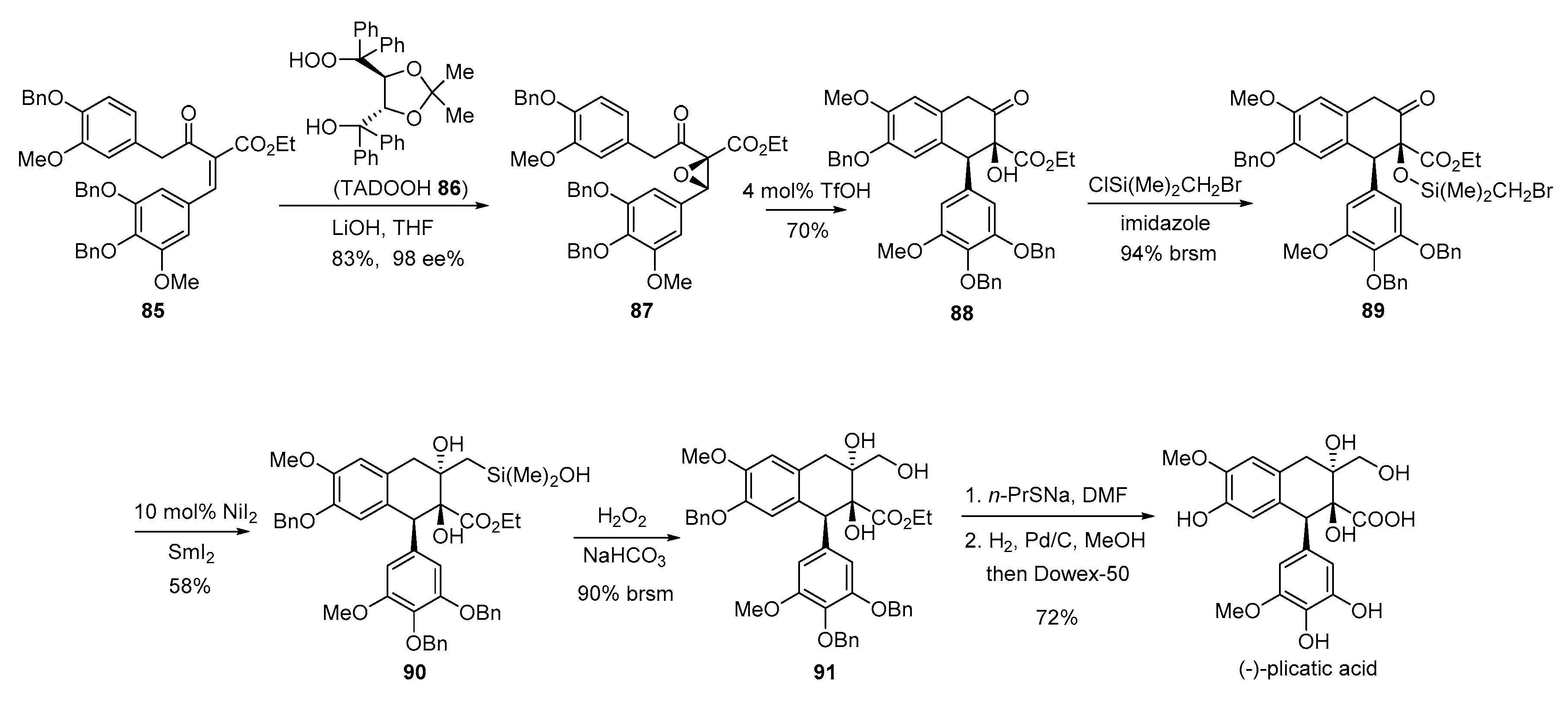
- Sun, B.-F.; Hong, R.; Kang, Y.-B.; Deng, L. Asymmetric total synthesis of (−)-plicatic acid via a highly enantioselective and diastereoselective nucleophilic epoxidation of acyclic trisubstitued olefins. J. Am. Chem. Soc. 2009, 131, 10384–10385. [Google Scholar] [CrossRef] [PubMed]
- Tamao, K.; Ishida, N.; Tanak, T.; Kunada, M. Silafunctional compounds in organic synthesis. Part 20. Hydrogen peroxide oxidation of the silicon-carbon bond in organoalkoxysilanes. Organometallics 1983, 2, 1694–1696. [Google Scholar] [CrossRef]
- Tamao, K.; Ishida, N.; Kunada, M. Chirality transfer in stereoselective synthesis. A highly stereoselective synthesis of optically active vitamin E side chains. J. Org. Chem. 1983, 48, 2122–2124. [Google Scholar]
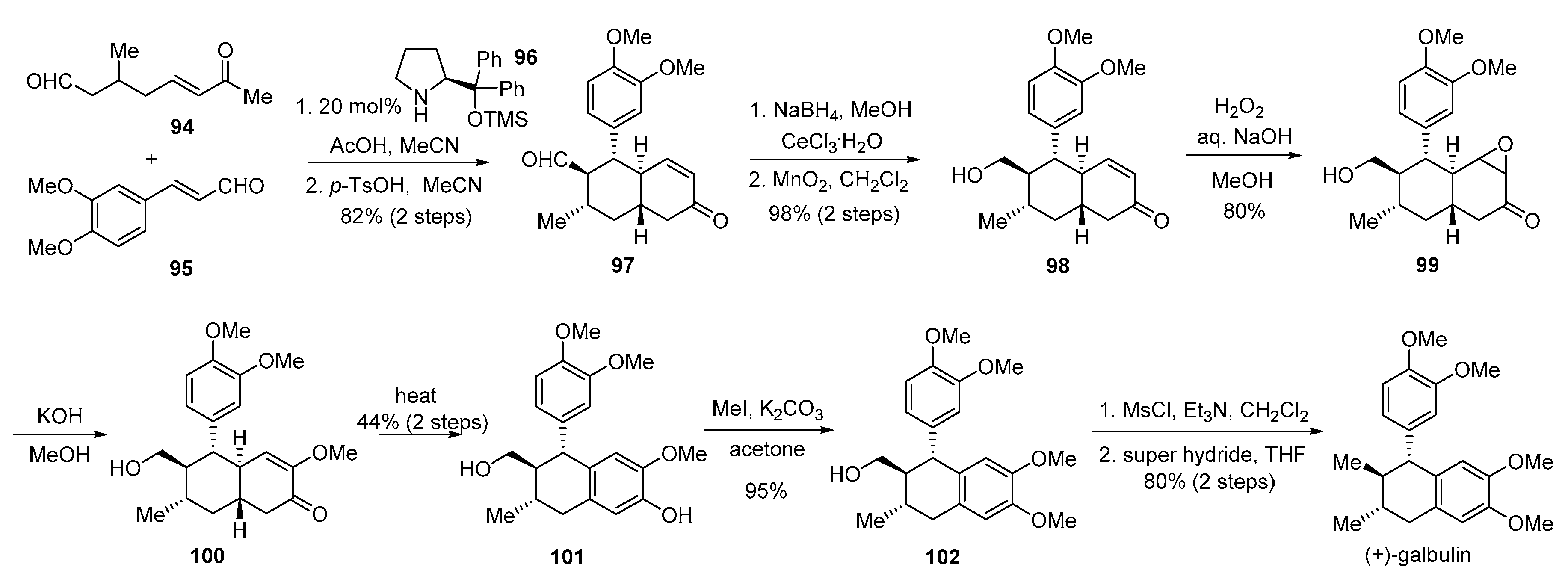
- Hong, B.-C.; Hsu, C.-S.; Lee, G.-H. Enantioselective total synthesis of (+)-galbulin via organocatalytic domino Michael–Michael–aldol condensation. Chem. Commun. 2012, 48, 2385–2387. [Google Scholar] [CrossRef]
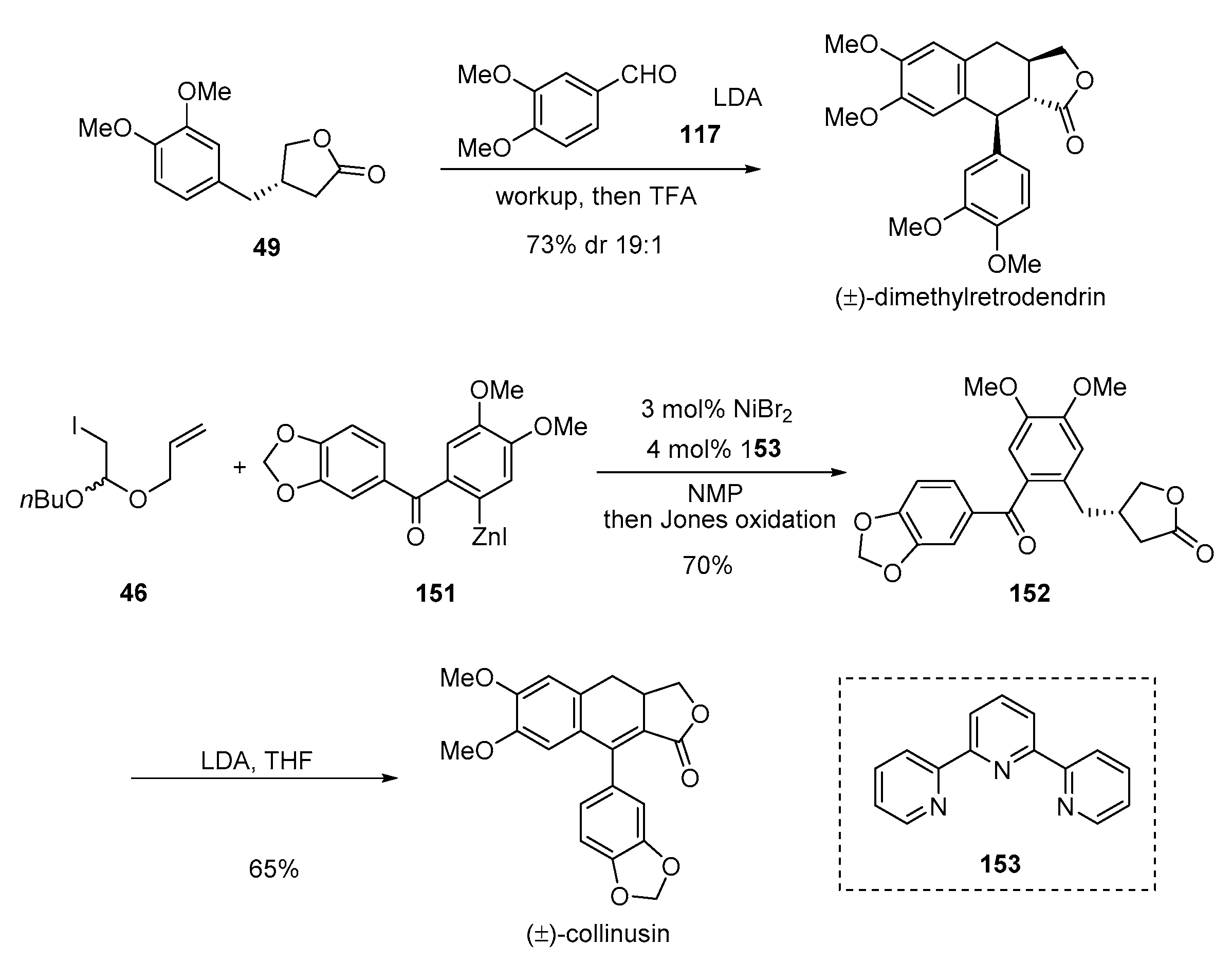
- Zhang, J.-J.; Yan, C.-S.; Peng, Y.; Luo, Z.-B.; Xu, X.-B.; Wang, Y.-W. Total synthesis of (±)-sacidumlignans D and A through Ueno–Stork radical cyclization reaction. Org. Biomol. Chem. 2013, 11, 2498–2513. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Luo, Z.-B.; Zhang, J.-J.; Luo, L.; Wang, Y.-W. Collective synthesis of several 2,7′-cyclolignans and their correlation by chemical transformations. Org. Biomol. Chem. 2013, 11, 7574–7586. [Google Scholar] [CrossRef]
- Patel, R.M.; Argade, N.P. Palladium-promoted [2 + 2 + 2] cocyclization of arynes and unsymmetrical conjugated dienes: Synthesis of justicidin B and retrojusticidin B. Org. Lett. 2013, 15, 14–17. [Google Scholar] [CrossRef]
- Kao, T.-T.; Lin, C.-C.; Shia, K.-S. The total synthesis of retrojusticidin B, justicidin E, and helioxanthin. J. Org. Chem. 2015, 80, 6708–6714. [Google Scholar] [CrossRef]
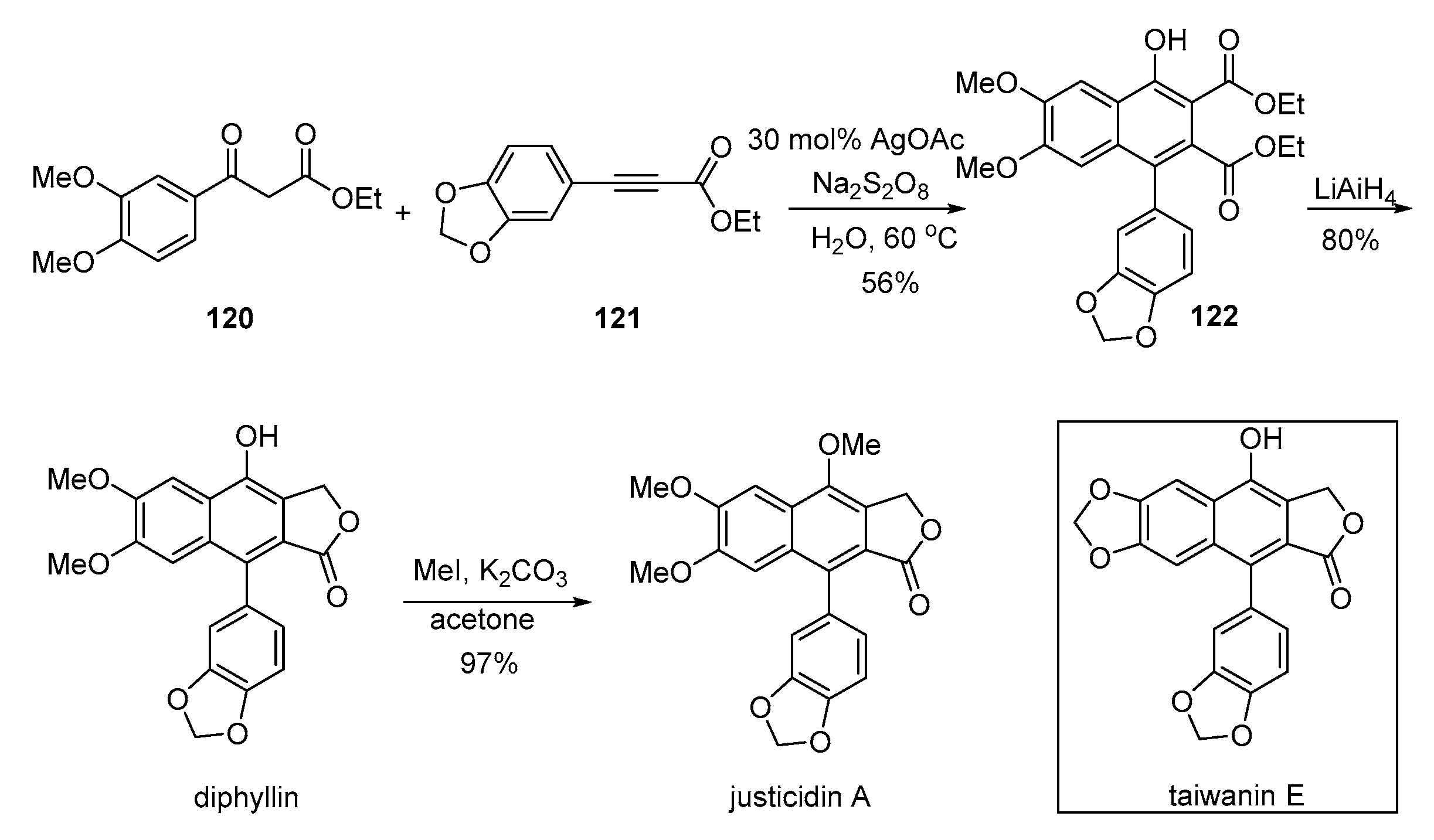
- Naresh, G.; Kant, R.; Narender, T. Silver(I)-Catalyzed regioselective construction of highly substituted α-naphthols and its application toward expeditious synthesis of lignan natural products. Org. Lett. 2015, 17, 3446–3449. [Google Scholar] [CrossRef] [PubMed]
- Singh, O.V.; Tapadiya, S.M.; Deshmukh, R.G. Process for the Synthesis of Cleistanthin A. WO 2010089778 A2, 12 August 2010. [Google Scholar]
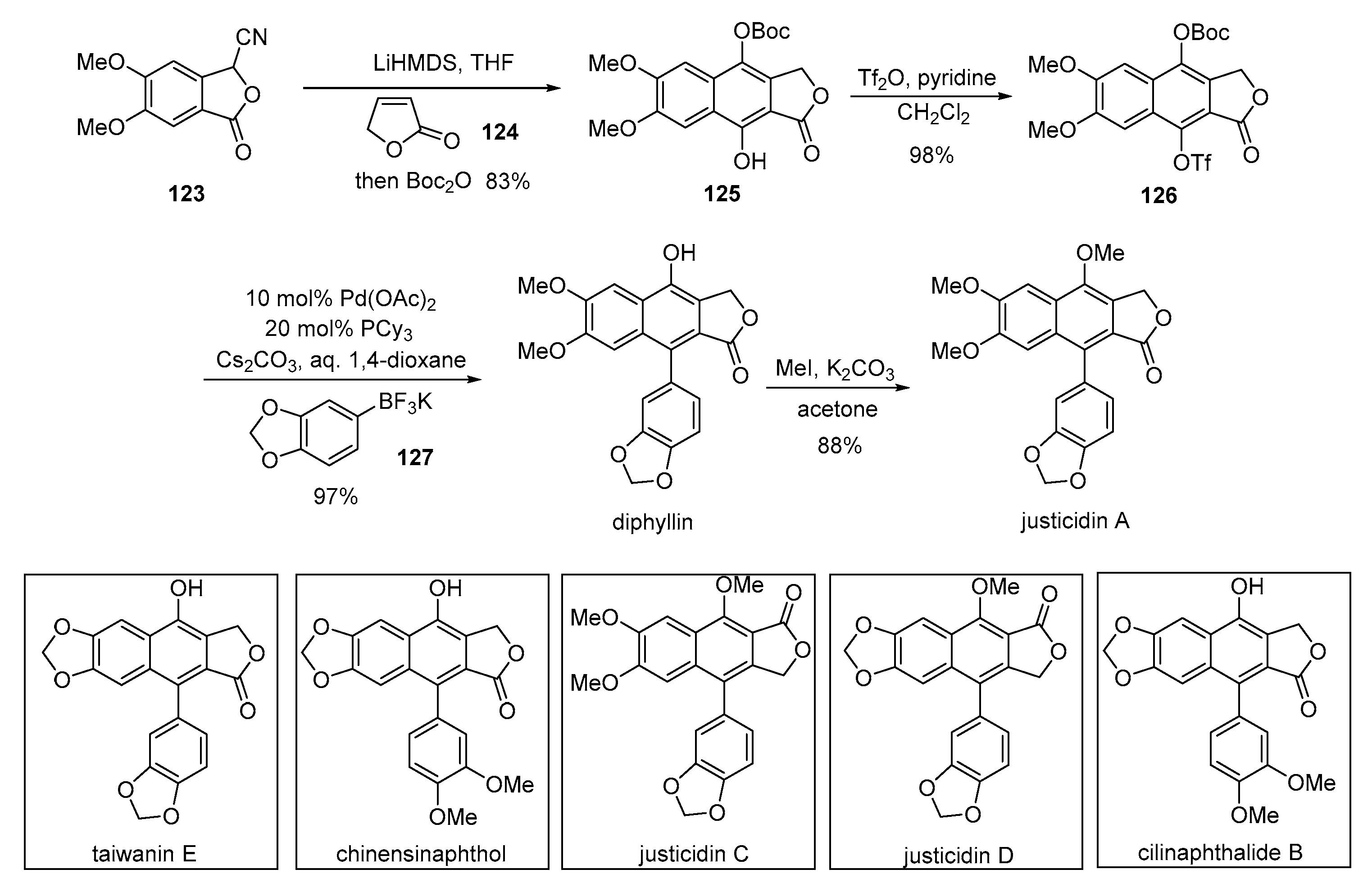
- Kim, T.; Jeong, K.H.; Kang, K.S.; Nakata, M.; Ham, J. An optimized and general synthetic strategy to prepare arylnaphthalene lactone natural products from cyanophthalides. Eur. J. Org. Chem. 2017, 2017, 1704–1712. [Google Scholar] [CrossRef]
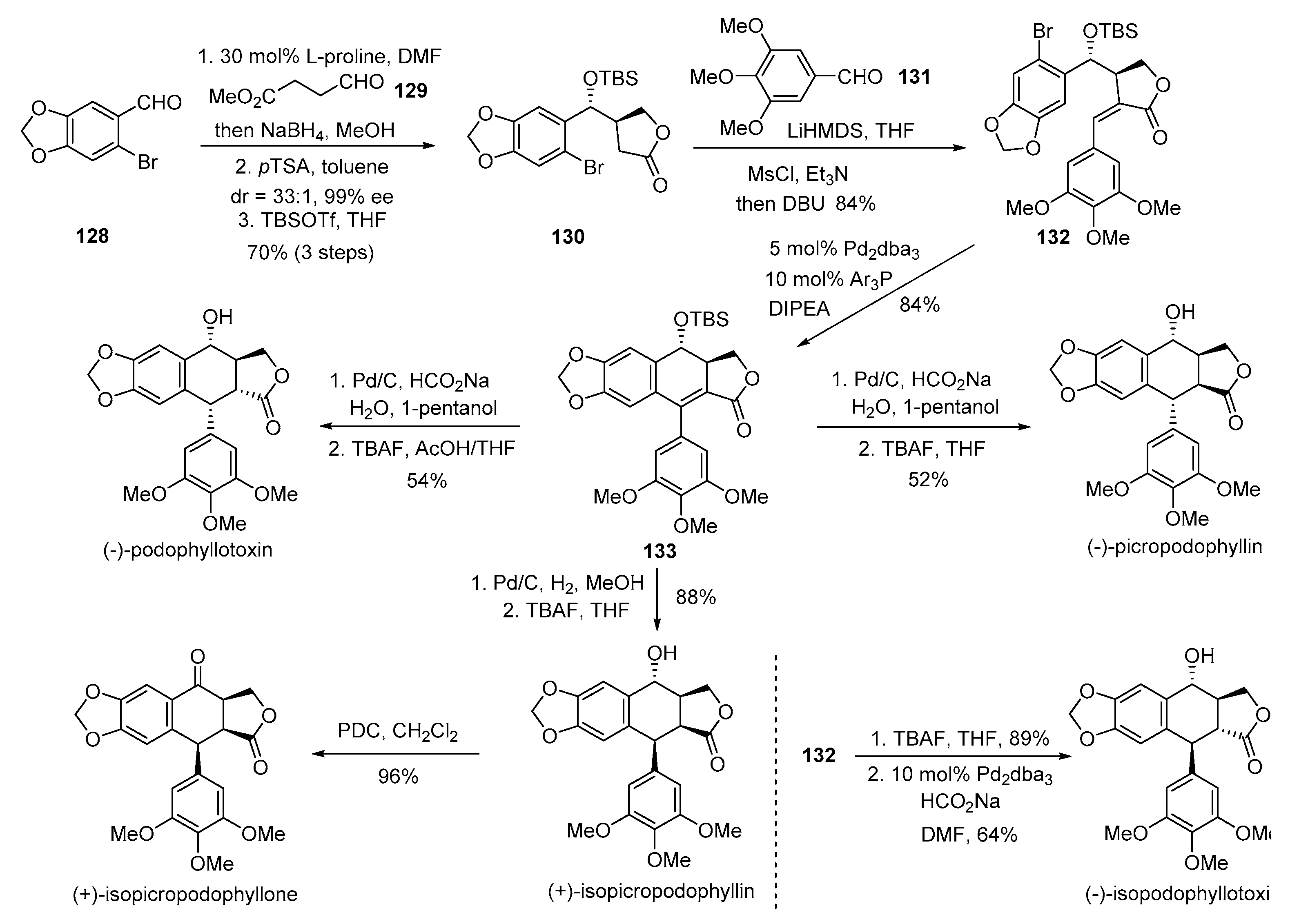
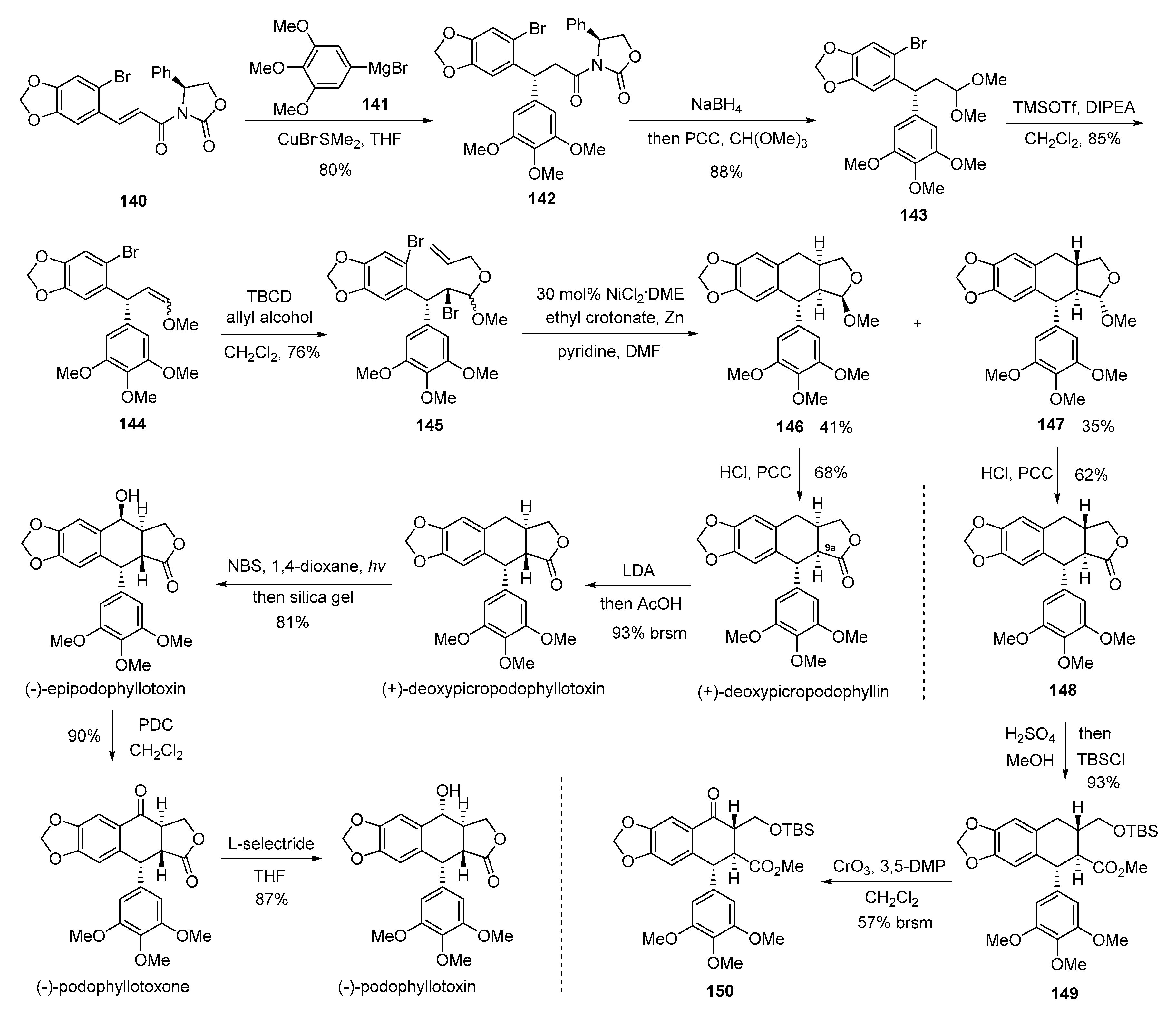
- Hjra, S.; Garai, S.; Hazra, S. Catalytic Enantioselective Synthesis of (−)-Podophyllotoxin. Org. Lett. 2017, 19, 6530–6533. [Google Scholar] [CrossRef] [PubMed]
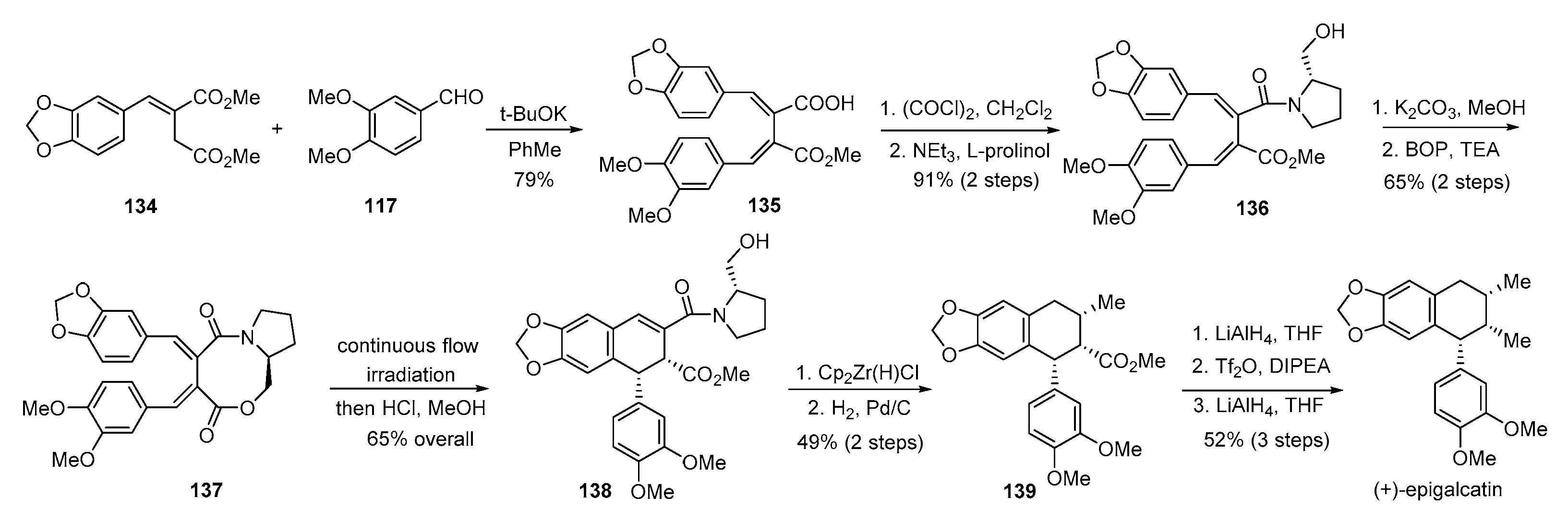
- Lisiecki, K.; Czarnocki, Z. Flow photochemistry as a tool for the total synthesis of (+)-epigalcatin. Org. Lett. 2018, 20, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Lisiecki, K.; Krawczyk, K.K.; Roszkowski, P.; Maurin, J.K.; Czarnocki, Z. Formal synthesis of (−)-podophyllotoxin through the photocyclization of an axially chiral 3,4-bisbenzylidene succinate amide ester-a flow photochemistry approach. Org. Biomol. Chem. 2016, 14, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Cong, X.-W.; Yang, G.-Z.; Wang, Y.-W.; Peng, Y. Divergent asymmetric syntheses of podophyllotoxin and related family members via stereoselective reductive Ni-catalysis. Org. Lett. 2018, 20, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Cong, X.-W.; Yang, G.-Z.; Wang, Y.-W.; Peng, Y. Stereoselective synthesis of a Podophyllum lignan core by intramolecular reductive nickel-catalysis. Chem. Commun. 2018, 54, 2040–2043. [Google Scholar] [CrossRef] [PubMed]
- Long, R.; Huang, J.; Shao, W.; Liu, S.; Lan, Y.; Gong, J.; Yang, Z. Asymmetric total synthesis of (−)-lingzhiol via a Rh-catalysed [3 + 2] cycloaddition. Nat. Commun. 2014, 5, 5707. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.C.; Teague, S.J.; Meyers, A.I. Asymmetric total synthesis of (−)-podophyllotoxin. J. Am. Chem. Soc. 1988, 110, 7854–7858. [Google Scholar] [CrossRef]
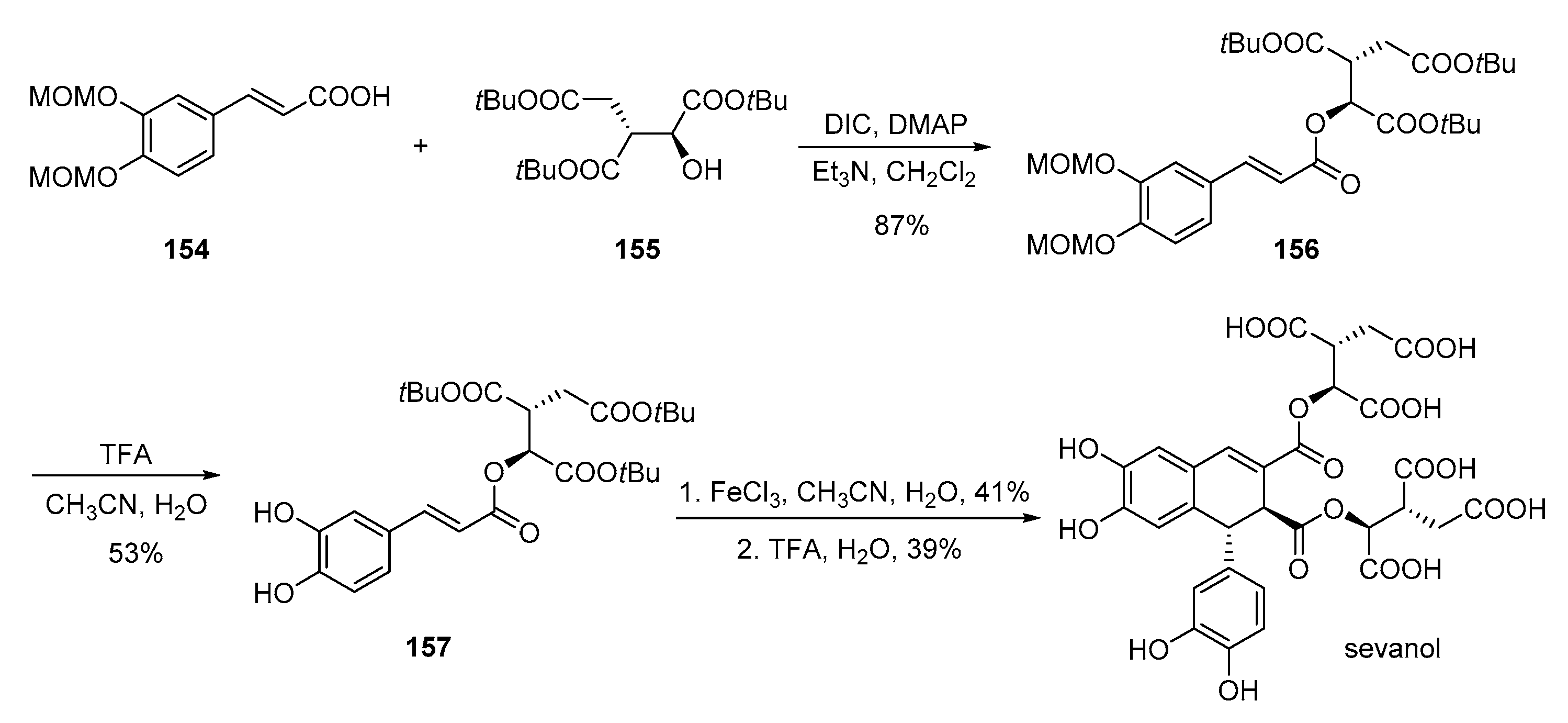
- Belozerova, O.A.; Deigin, V.I.; Khrushchev, A.Y.; Dubinnyi, M.A.; Kublitski, V.S. The total synthesis of sevanol, a novel lignan isolated from the thyme plant (Thymus armeniacus). Tetrahedron 2018, 74, 1449–1453. [Google Scholar] [CrossRef]
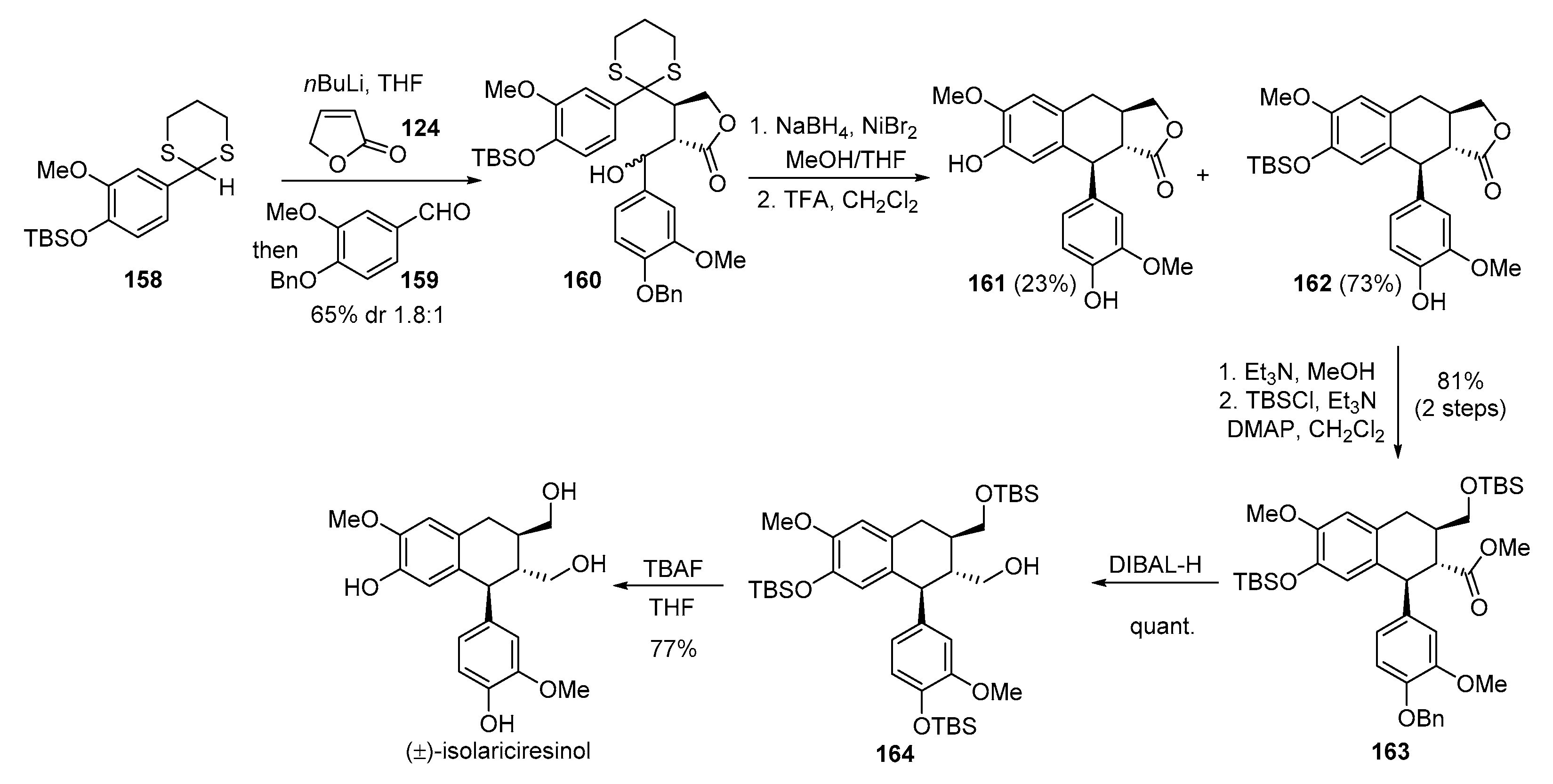
- Sampei, M.; Arai, M.A.; Ishibashi, M. Total syntheses of schizandriside, saracoside and (±)-isolariciresinol with antioxidant activities. J. Nat. Med. 2018, 72, 651–654. [Google Scholar] [CrossRef] [PubMed]
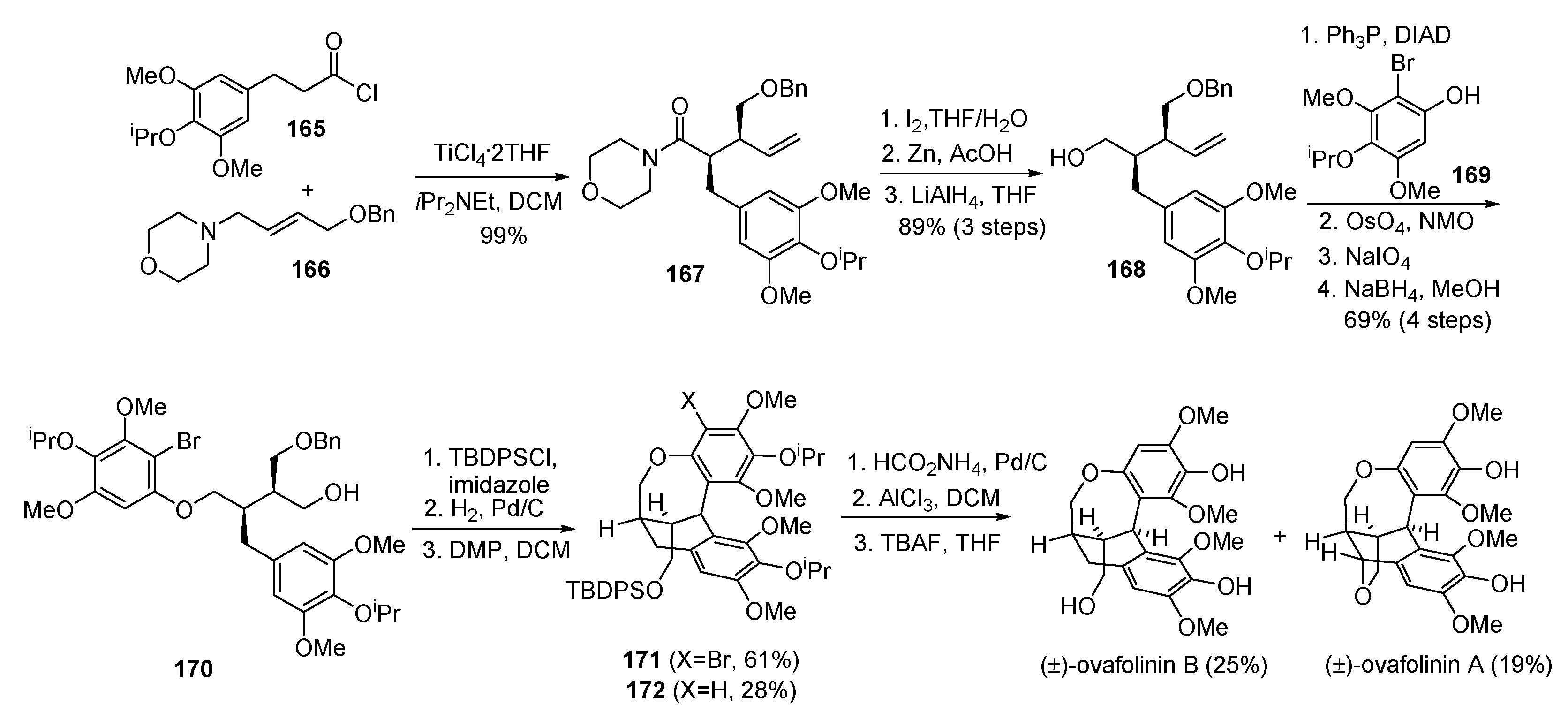
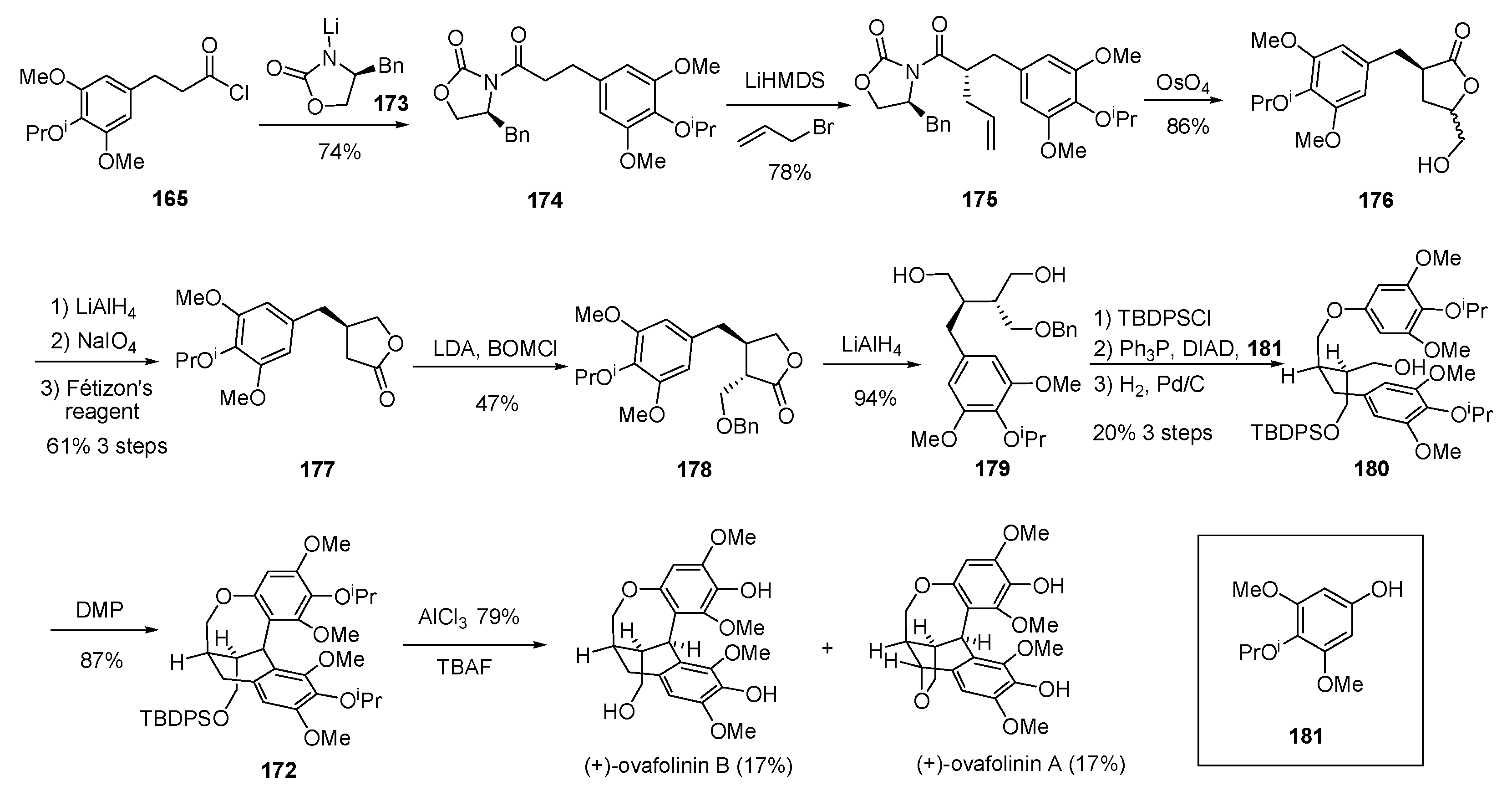
- Davidson, S.J.; Barker, D. Total synthesis of ovafolinins A and B: Unique poly-cyclic benzoxepin lignans through a cascade cyclization. Angew. Chem. Int. Ed. 2017, 56, 9483–9486. [Google Scholar] [CrossRef] [PubMed]
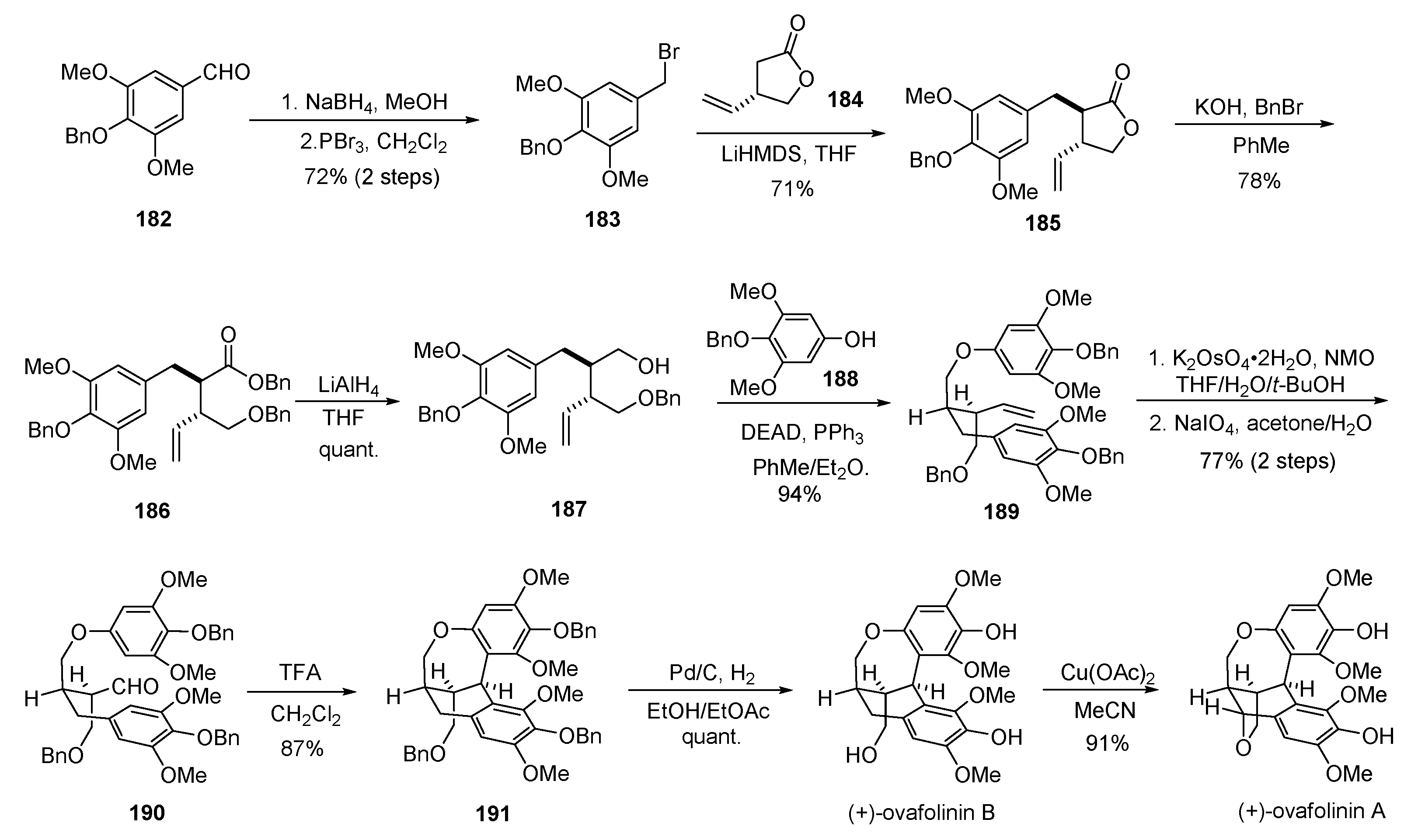
- Fang, X.; Shen, L.; Hu, X. Asymmetric total synthesis of (+)-ovafolinins A and B. Chem. Commun. 2018, 54, 7539–7541. [Google Scholar] [CrossRef] [PubMed]































© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, X.; Hu, X. Advances in the Synthesis of Lignan Natural Products. Molecules 2018, 23, 3385. https://doi.org/10.3390/molecules23123385
Fang X, Hu X. Advances in the Synthesis of Lignan Natural Products. Molecules. 2018; 23(12):3385. https://doi.org/10.3390/molecules23123385
Chicago/Turabian StyleFang, Xianhe, and Xiangdong Hu. 2018. "Advances in the Synthesis of Lignan Natural Products" Molecules 23, no. 12: 3385. https://doi.org/10.3390/molecules23123385
APA StyleFang, X., & Hu, X. (2018). Advances in the Synthesis of Lignan Natural Products. Molecules, 23(12), 3385. https://doi.org/10.3390/molecules23123385