Construction of Highly Stable Cytotoxic Nuclear-Directed Ribonucleases

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
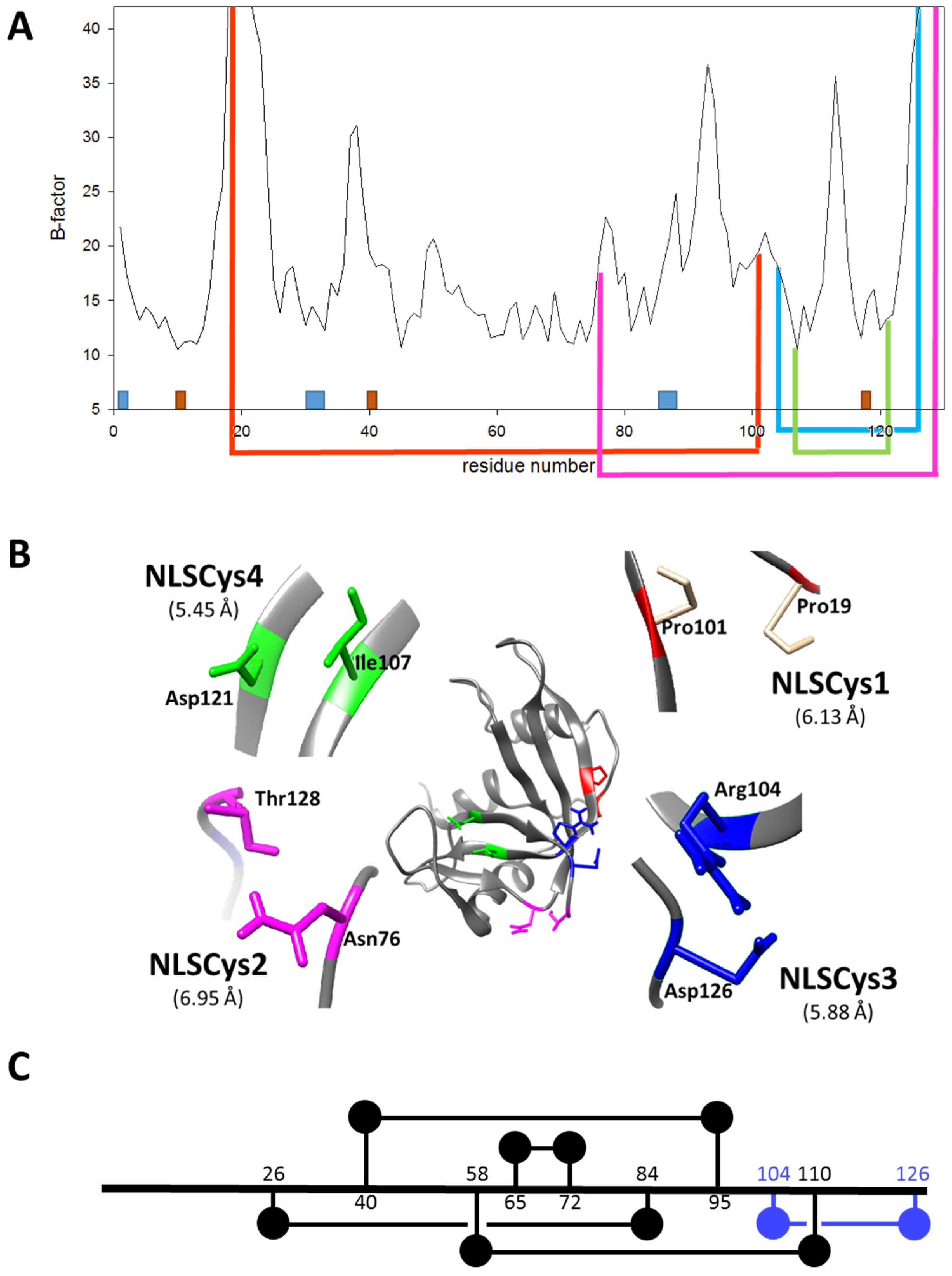
2.1. Design of ND-RNase Variants with Additional Disulfide Bonds
- (1)
- In order to be cytotoxic the ND-RNases must interact with α-importin [10], so we excluded those residues placed on the predicted zones of interaction of the ND-RNase with the importin; that is, around the N-terminus of α-helix 1 of the HP-RNase, the C-terminus of α-helix 2 and into the loop connecting β-strands 4 and 5. We also excluded those residues important for the catalytic activity of the RNase (see Figure 1A).
- (2)
- We then inspected the modeled structure to find residue pairs with α-carbons placed at a distance between 5 and 7 Å. This range of distances is slightly higher than that found between the two α-carbon atoms of the disulfide bonds of the modeled structure (between 5.2 and 5.8 Å).
- (3)
- Among the residue pairs following the former criteria, we investigated the relative disposition of their side chains. We only took into account those residue pairs that had side chains facing each other or that were arranged in a parallel way, and we excluded those pairs that were facing away from each other on the structure.
- (4)
- We considered that it was possible the protein would have to rearrange locally when introducing a disulfide bond. Therefore, the introduction of the new disulfide bond between residues located at the most rigid regions of the protein was not a suitable strategy. This idea has been confirmed experimentally [21] and in silico [22]. Dani and colleagues [22] analyzed previously engineered disulfide bonds in a set of recombinant proteins where the effect on protein stability was known and the wild type crystal structure was available. They reported that stabilizing mutations were mostly found in regions of medium to high mobility and near the protein surface in loops larger than 25 residues. Here, we only considered those residue pairs in which at least one of them was placed in a highly mobile region. In the HP-RNase structure, the C-terminal region is highly mobile (Figure 1A). Likewise, the hinge loop connecting the first alpha helix with the rest of the protein (residues 16 to 24) also presents high mobility and indeed in some structures could not be solved (for a review, see [23]). Therefore, residues present in these regions were preferably considered.
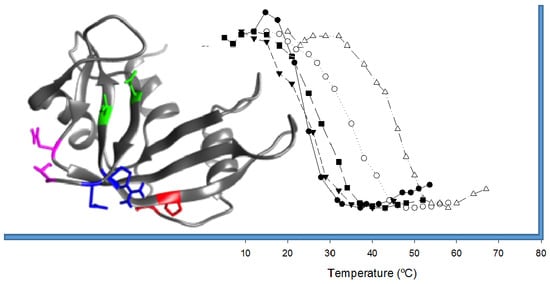
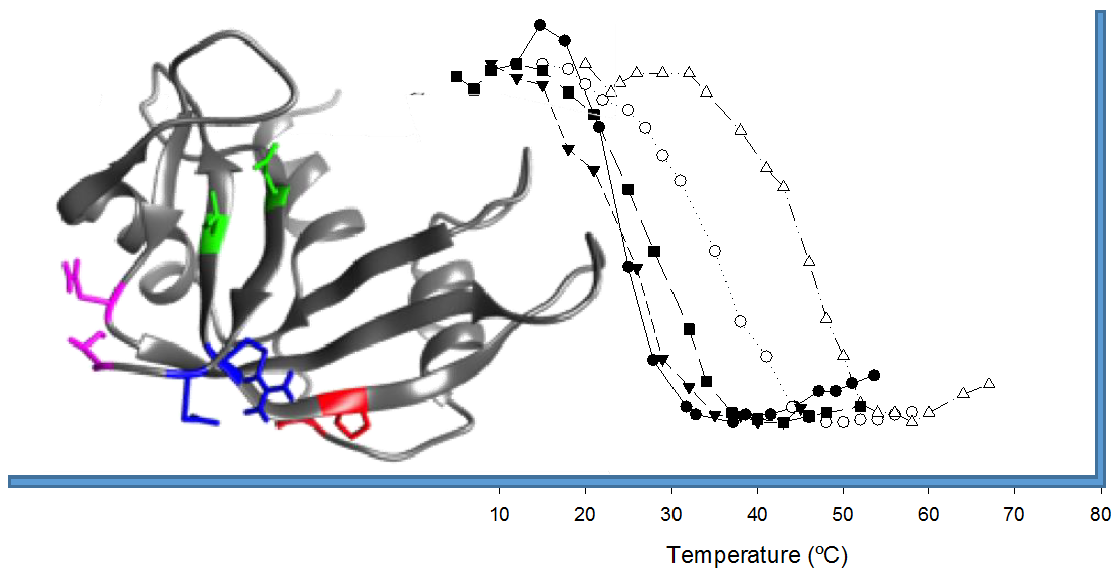
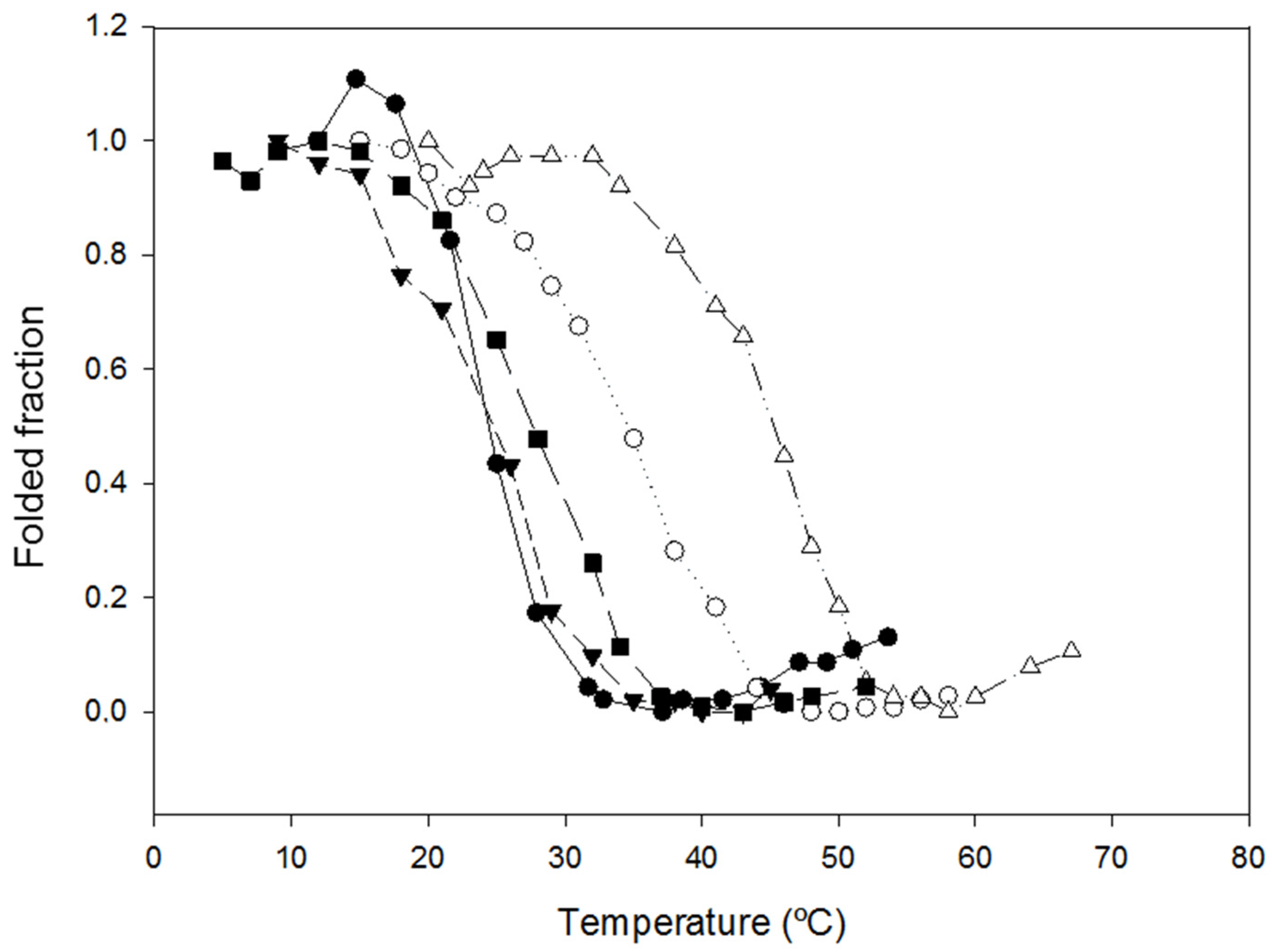
2.2. Characterization of the Effect of the Additional Disulfide Bond on the Thermal Stability of the Variants
2.3. Characterization of the Biological Activities of the Variants
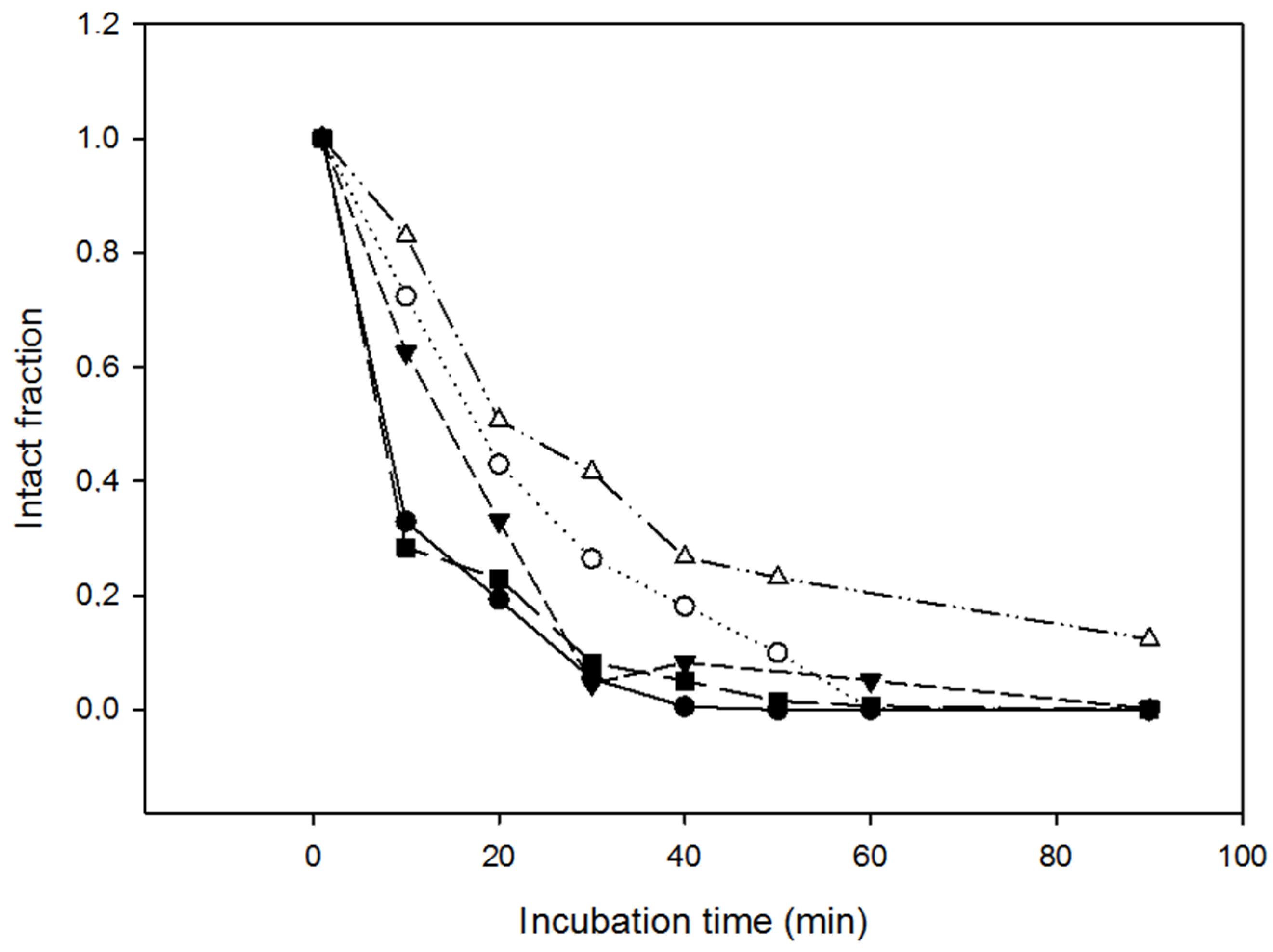
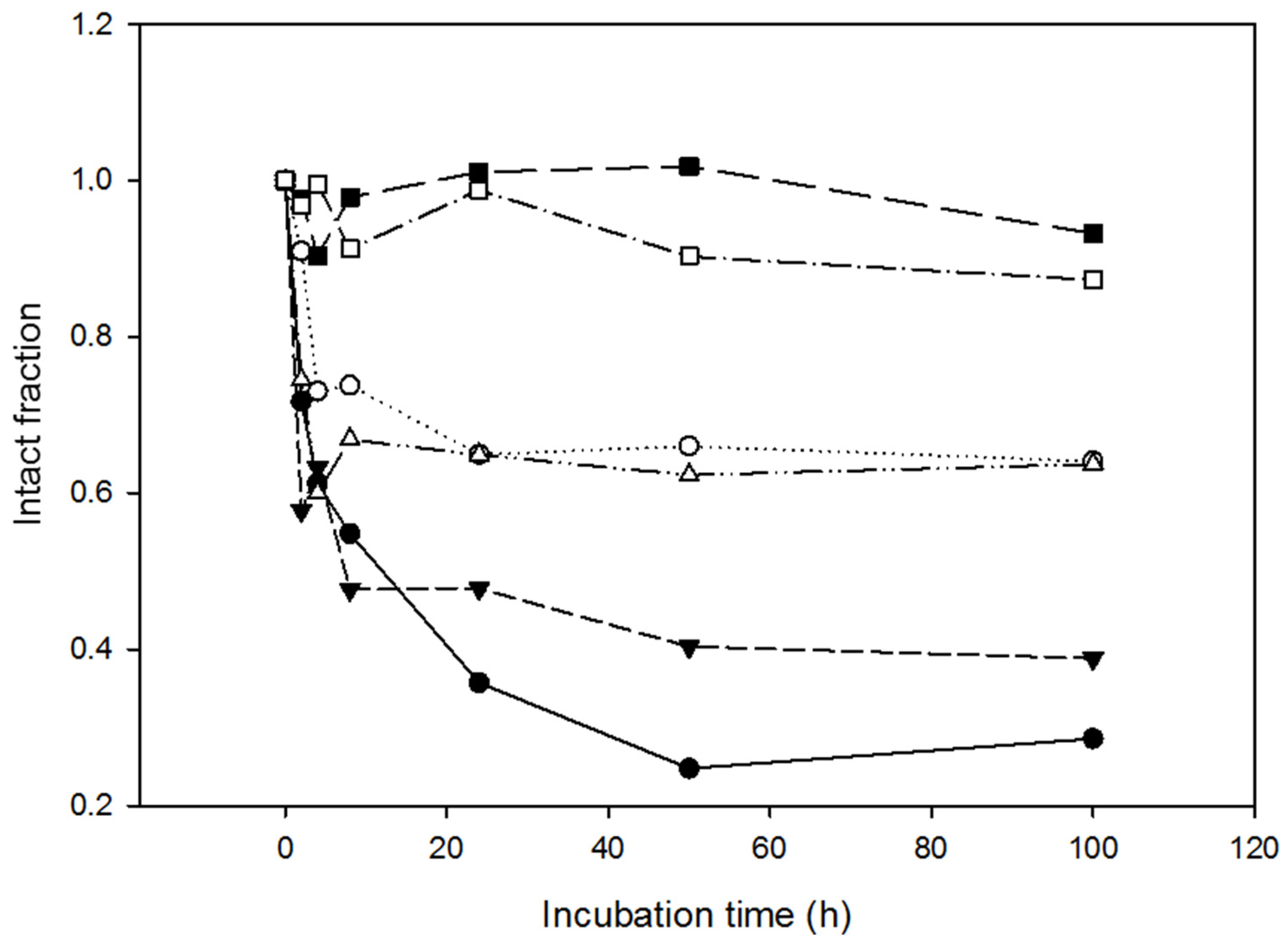
2.4. Resistance of the Variants to Proteolysis
3. Materials and Methods
3.1. Plasmid Construction
3.2. RNase Expression and Purification
3.3. Spectrophotometric Determination of Thermal Stability
3.4. Calorimetric Determination of Thermal Stability
3.5. Determination of Steady-State Kinetic Parameters
3.6. Cell Lines and Culture Conditions
3.7. Cell Proliferation Assays
3.8. Resistance to Proteinase K
3.9. Analysis of the Proteolysis Resistance in Serum
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Klink, T.A.; Raines, R.T. Conformational stability is a determinant of ribonuclease A cytotoxicity. J. Biol. Chem. 2000, 275, 17463–174637. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, P.; Gao, S.; Chu, X.; Wu, N.; Fan, Y. Enhanced thermostability of methyl parathion hydrolase from Ochrobactrum sp. M231 by rational engineering of a glycine to proline mutation. FEBS J. 2010, 277, 4901–4908. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, H.; Liu, C.; Liu, J.; Shen, Z. Improving stability of nitrile hydratase by bridging the salt-bridges in specific thermal-sensitive regions. J. Biotechnol. 2013, 164, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Benito, A.; Bosch, M.; Torrent, G.; Ribó, M.; Vilanova, M. Stabilization of human pancreatic ribonuclease through mutation at its N-terminal edge. Protein Eng. 2002, 15, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Benito, A.; Ribó, M.; Vilanova, M. On the track of antitumour ribonucleases. Mol. Biosyst. 2005, 1. [Google Scholar] [CrossRef]
- Benito, A.; Vilanova, M.; Ribó, M. Intracellular routing of cytotoxic pancreatic-type ribonucleases. Curr. Pharm. Biotech. 2008, 9, 169–179. [Google Scholar] [CrossRef]
- Dickson, K.A.; Haigis, M.C.; Raines, R.T. Ribonuclease Inhibitor: Structure and Function. Prog. Nucleic Acid Res. Mol. Biol. 2005, 80, 349–374. [Google Scholar] [CrossRef]
- Furia, A.; Moscato, M.; Cali, G.; Pizzo, E.; Confalone, E.; Amoroso, M.R.; Esposito, F.; Nitsch, L.; D’Alessio, G. The ribonuclease/angiogenin inhibitor is also present in mitochondria and nuclei. FEBS Lett. 2011, 585, 613–617. [Google Scholar] [CrossRef]
- Bosch, M.; Benito, A.; Ribó, M.; Puig, T.; Beaumelle, B.; Vilanova, M. A Nuclear Localization Sequence Endows Human Pancreatic Ribonuclease with Cytotoxic Activity. Biochemistry 2004, 43, 2167–2177. [Google Scholar] [CrossRef]
- Rodríguez, M.; Benito, A.; Tubert, P.; Castro, J.; Ribó, M.; Beaumelle, B.; Vilanova, M. A Cytotoxic Ribonuclease Variant with a Discontinuous Nuclear Localization Signal Constituted by Basic Residues Scattered Over Three Areas of the Molecule. J. Mol. Biol. 2006, 360, 548–557. [Google Scholar] [CrossRef]
- Tubert, P.; Rodríguez, M.; Ribó, M.; Benito, A.; Vilanova, M. The nuclear transport capacity of a human-pancreatic ribonuclease variant is critical for its cytotoxicity. Investig. New Drugs 2011, 29, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Ribó, M.; Puig, T.; Colomer, R.; Vilanova, M.; Benito, A. A cytotoxic ribonuclease reduces the expression level of P-glycoprotein in multidrug-resistant cell lines. Investig. New Drugs 2012, 30. [Google Scholar] [CrossRef]
- Vert, A.; Castro, J.; Ribó, M.; Benito, A.; Vilanova, M. A nuclear-directed human pancreatic ribonuclease (PE5) targets the metabolic phenotype of cancer cells. Oncotarget 2016, 7, 18309–18324. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Ribó, M.; Benito, A.; Vilanova, M. Mini-Review: Nucleus-Targeted Ribonucleases As Antitumor Drugs. Curr. Med. Chem. 2013, 20, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Canals, A.; Ribó, M.; Benito, A.; Bosch, M.; Mombelli, E.; Vilanova, M. Production of engineered human pancreatic ribonucleases, solving expression and purification problems, and enhancing thermostability. Protein Expr. Purif. 1999, 17, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Vert, A.; Castro, J.; Ruiz-Martínez, S.; Tubert, P.; Escribano, D.; Ribó, M.; Vilanova, M.; Benito, A. Generation of new cytotoxic human ribonuclease variants directed to the nucleus. Mol. Pharm. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.M.; Cowan, D.A.; Morgan, H.W.; Curran, M.P. A correlation between protein thermostability and resistance to proteolysis. Biochem. J. 1982, 207, 641–644. [Google Scholar] [CrossRef]
- Parsell, D.A.; Sauer, R.T. The Structural Stability of a Protein Is an Important Determinant of Its Proteolytic Susceptibility in Escherichia coli. J. Biol. Chem. 1989, 264, 7590–7595. [Google Scholar]
- Betz, S.F. Disulfide bonds and the stability of globular proteins. Protein Sci. 1993, 2, 1551–1558. [Google Scholar] [CrossRef]
- Yu, H.; Huang, H. Engineering proteins for thermostability through rigidifying flexible sites. Biotechnol. Adv. 2014, 32, 308–315. [Google Scholar] [CrossRef]
- Melnik, B.S.; Povarnitsyna, T.V.; Glukhov, A.S.; Melnik, T.N.; Uversky, V.N. Ss-stabilizing proteins rationally: Intrinsic disorder-based design of stabilizing disulphide bridges in gfp. J. Biomol. Struct. Dyn. 2012, 29, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Dani, V.S.; Ramakrishnan, C.; Varadarajan, R. MODIP revisited: Re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. Des. Sel. 2003, 16, 187–193. [Google Scholar] [CrossRef]
- Benito, A.; Laurents, D.V.; Ribó, M.; Vilanova, M. The structural determinants that lead to the formation of particular oligomeric structures in the pancreatic-type ribonuclease family. Curr. Protein Pept. Sci. 2008, 9, 370–393. [Google Scholar] [CrossRef] [PubMed]
- Pous, J.; Canals, A.; Terzyan, S.S.; Guasch, A.; Benito, A.; Ribó, M.; Vilanova, M.; Coll, M. Three-dimensional structure of a human pancreatic ribonuclease variant, a step forward in the design of cytotoxic ribonucleases. J. Mol. Biol. 2000, 303, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Ribó, M.; Benito, A.; Canals, A.; Nogués, M.V.; Cuchillo, C.M.; Vilanova, M. Purification of engineered human pancreatic ribonuclease. Methods Enzymol. 2001, 341, 221–234. [Google Scholar] [CrossRef]
- Matthews, B.W.; Nicholson, H.; Becktel, W.J. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc. Natl. Acad. Sci. USA 1987, 84, 6663–6667. [Google Scholar] [CrossRef] [PubMed]
- Flory, P.J. Theory of Elastic Mechanisms in Fibrous Proteins. J. Am. Chem. Soc. 1956, 78, 5222–5235. [Google Scholar] [CrossRef]
- Lin, S.H.; Konishi, Y.; Denton, M.E.; Scheraga, H.A. Influence of an extrinsic cross-link on the folding pathway of ribonuclease A. Conformational and thermodynamic analysis of cross-linked (lysine7-lysine41)-ribonuclease A. Biochemistry 1984, 23, 5504–5512. [Google Scholar] [CrossRef]
- Doig, A.J.; Williams, D.H. Is the hydrophobic effect stabilizing or destabilizing in proteins? The contribution of disulphide bonds to protein stability. J. Mol. Biol. 1991, 217, 389–398. [Google Scholar] [CrossRef]
- Klink, T.A.; Woycechowsky, K.J.; Taylor, K.M.; Raines, R.T. Contribution of disulfide bonds to the conformational stability and catalytic activity of ribonuclease A. Eur. J. Biochem. 2000, 267, 566–572. [Google Scholar] [CrossRef]
- Pecher, P.; Arnold, U. The effect of additional disulfide bonds on the stability and folding of ribonuclease A. Biophys. Chem. 2009, 141, 21–28. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Baase, W.A.; Kuroki, R.; Matthews, B.W. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. USA 1995, 92, 452–456. [Google Scholar] [CrossRef]
- Futami, J.; Tada, H.; Seno, M.; Ishikami, S.; Enori Yamada, H. Stabilization of Human RNase 1 by Introduction of a Disulfide Bond between Residues 4 and 118. J. Biochem 2000, 128, 215–250. [Google Scholar] [CrossRef]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef]
- Font, J.; Benito, A.; Lange, R.; Ribó, M.; Vilanova, M. The contribution of the residues from the main hydrophobic core of ribonuclease A to its pressure-folding transition state. Protein Sci. 2006, 15, 1000–1009. [Google Scholar] [CrossRef]
- Torrent, J.; Connelly, J.P.; Coll, M.G.; Ribó, M.; Lange, R.; Vilanova, M. Pressure versus heat-induced unfolding of ribonuclease A: The case of hydrophobic interactions within a chain-folding initiation site. Biochemistry 1999, 38, 15952–15961. [Google Scholar] [CrossRef]
- Privalov, P.L.; Potekhin, S.A. Scanning microcalorimetry in studying temperature-induced changes in proteins. Methods Enzymol. 1986, 131, 4–51. [Google Scholar] [CrossRef]
- Boix, E.; Nogués, M.V.; Schein, C.H.; Benner, S.A.; Cuchillo, C.M. Reverse transphosphorylation by ribonuclease A needs an intact p2-binding site. Point mutations at Lys-7 and Arg-10 alter the catalytic properties of the enzyme. J. Biol. Chem. 1994, 269, 2529–2534. [Google Scholar]
- Castro, J.; Ribó, M.; Navarro, S.; Nogués, M.V.; Vilanova, M.; Benito, A. A human ribonuclease induces apoptosis associated with p21WAF1/CIP1 induction and JNK inactivation. BMC Cancer 2011, 11, 9. [Google Scholar] [CrossRef]
- Ruiz-Martínez, S.; Pantoja-Uceda, D.; Castro, J.; Vilanova, M.; Ribó, M.; Bruix, M.; Benito, A.; Laurents, D.V. Insights into the mechanism of Apoptin’s exquisitely selective anti-tumor action from atomic level characterization of its conformation and dynamics. Arch. Biochem. Biophys. 2017, 614, 53–64. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
Sample Availability: Samples of the ND-RNases are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Replacements | Yield 1 | T1/2 2 |
|---|---|---|---|
| NLSPE5 | - | 15 mg | 23.6 °C |
| NLSPE5Cys1 | Pro19–Pro101 | 15 mg | 38.7 °C |
| NLSPE5Cys2 | Asn76–Thr128 | 15 mg | 26.1 °C |
| NLSPE5Cys3 | Arg104–Asp126 | 30 mg | 49.9 °C |
| NLSPE5Cys4 | Ile107–Asp121 | 3 mg | 29.3 °C |
| NLSPE5 | NLSPE5Cys1 | NLSPE5Cys3 | |
|---|---|---|---|
| T1/2 (°C) | 48.3 ± 0.4 | 59.4 ± 0.2 | 64.9 ± 1.0 |
| ΔHcal (kJ/mol) | 241.1 ± 1.6 | 273.2 ± 0.1 | 308.3 ± 8.4 |
| ΔΔG (kJ/mol) | - | −8.35 ± 0.2 | −12.47 ± 0.35 |
| Parameter | ND-RNase Variant | ||||
|---|---|---|---|---|---|
| NLSPE5 | NLSPE5Cys1 | NLSPE5Cys2 | NLSPE5Cys3 | NLSPE5Cys4 | |
| Kcat (min−1) | 413 ± 191 | 695 ± 131 | 480 ± 48 | 573 ± 7 | 139 ± 41 |
| KM (mM) | 0.88 ± 0.53 | 1.34 ± 0.11 | 1.44 ± 0.27 | 0.65 ± 0.15 | 4.08 ± 0.51 |
| Relative Kcat/KM (%) | 100 ± 7.7 | 134.8 ± 14.6 | 88.9 ± 25.4 | 233.1 ± 49.3 | 8.8 ± 1.5 |
| Cell Line | NLSPE5 | NLSPE5Cys1 | NLSPE5Cys2 | NLSPE5Cys3 | NLSPE5Cys4 |
|---|---|---|---|---|---|
| OVCAR-8 | 0.2 ± 0.1 | 0.3 ± 0.2 | 0.4 ± 0.2 | 0.2 ± 0.1 | 6.9 ± 3.8 |
| NCI-H460 | 0.2 ± 0.1 | 0.3 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.1 | 9.2 ± 4.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roura Padrosa, D.; Castro, J.; Romero-Casañas, A.; Ribó, M.; Vilanova, M.; Benito, A. Construction of Highly Stable Cytotoxic Nuclear-Directed Ribonucleases. Molecules 2018, 23, 3273. https://doi.org/10.3390/molecules23123273
Roura Padrosa D, Castro J, Romero-Casañas A, Ribó M, Vilanova M, Benito A. Construction of Highly Stable Cytotoxic Nuclear-Directed Ribonucleases. Molecules. 2018; 23(12):3273. https://doi.org/10.3390/molecules23123273
Chicago/Turabian StyleRoura Padrosa, David, Jessica Castro, Alejandro Romero-Casañas, Marc Ribó, Maria Vilanova, and Antoni Benito. 2018. "Construction of Highly Stable Cytotoxic Nuclear-Directed Ribonucleases" Molecules 23, no. 12: 3273. https://doi.org/10.3390/molecules23123273
APA StyleRoura Padrosa, D., Castro, J., Romero-Casañas, A., Ribó, M., Vilanova, M., & Benito, A. (2018). Construction of Highly Stable Cytotoxic Nuclear-Directed Ribonucleases. Molecules, 23(12), 3273. https://doi.org/10.3390/molecules23123273