
Preparation of 4-Flexible Amino-2-Arylethenyl-Quinoline Derivatives as Multi-Target Agents for the Treatment of Alzheimer’s Disease

Abstract
1. Introduction
2. Results and Discussion
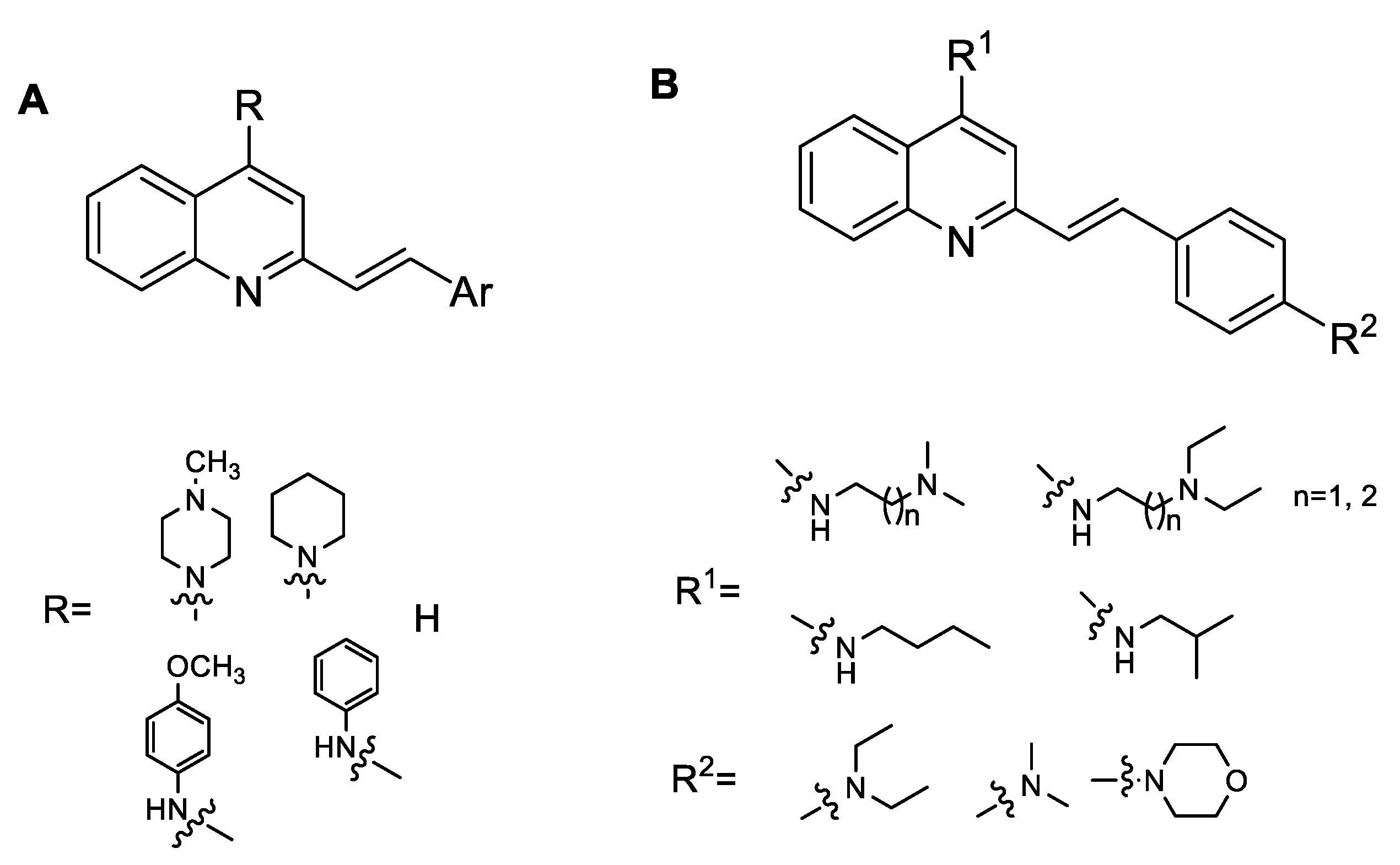
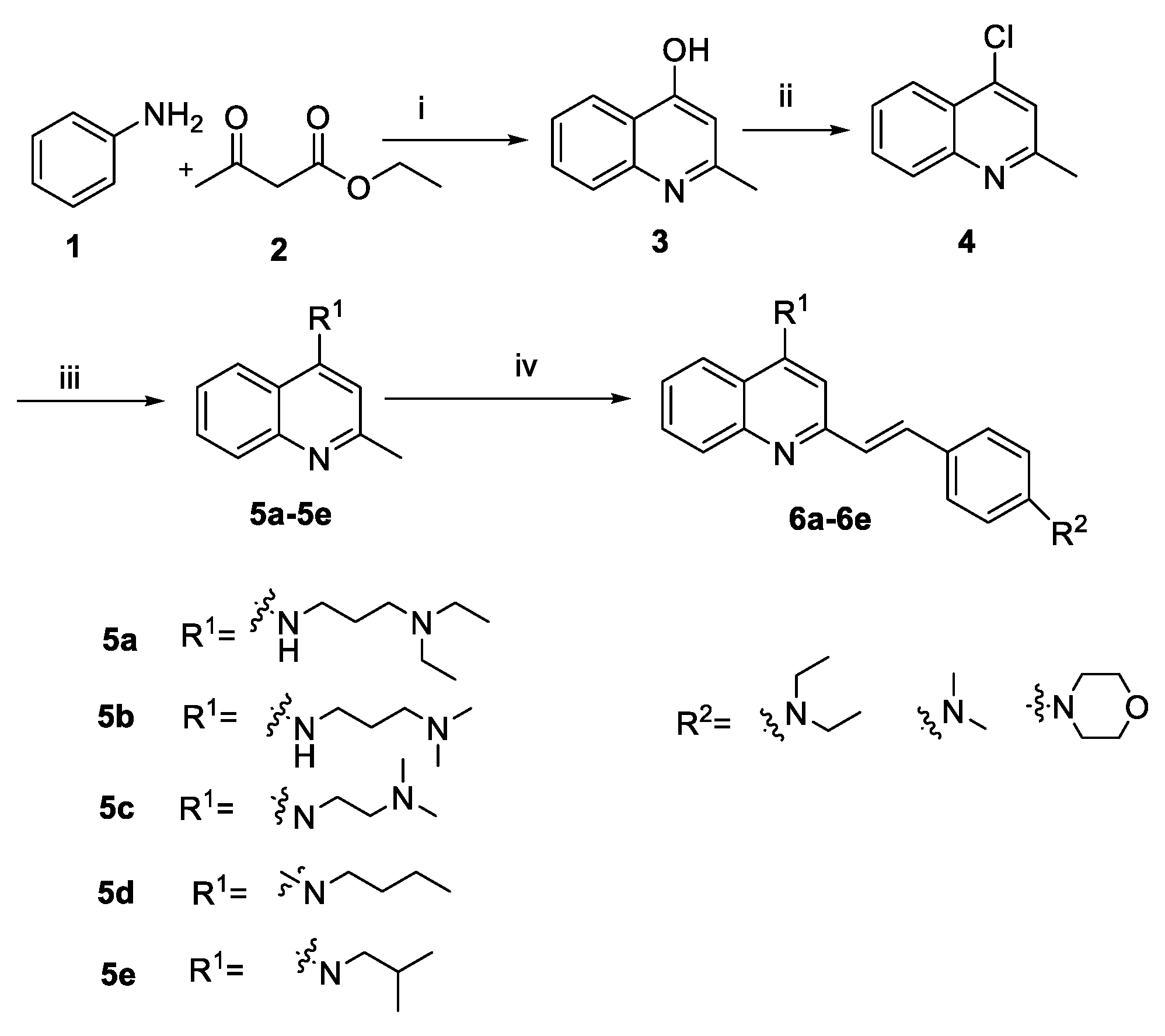
2.1. Chemistry
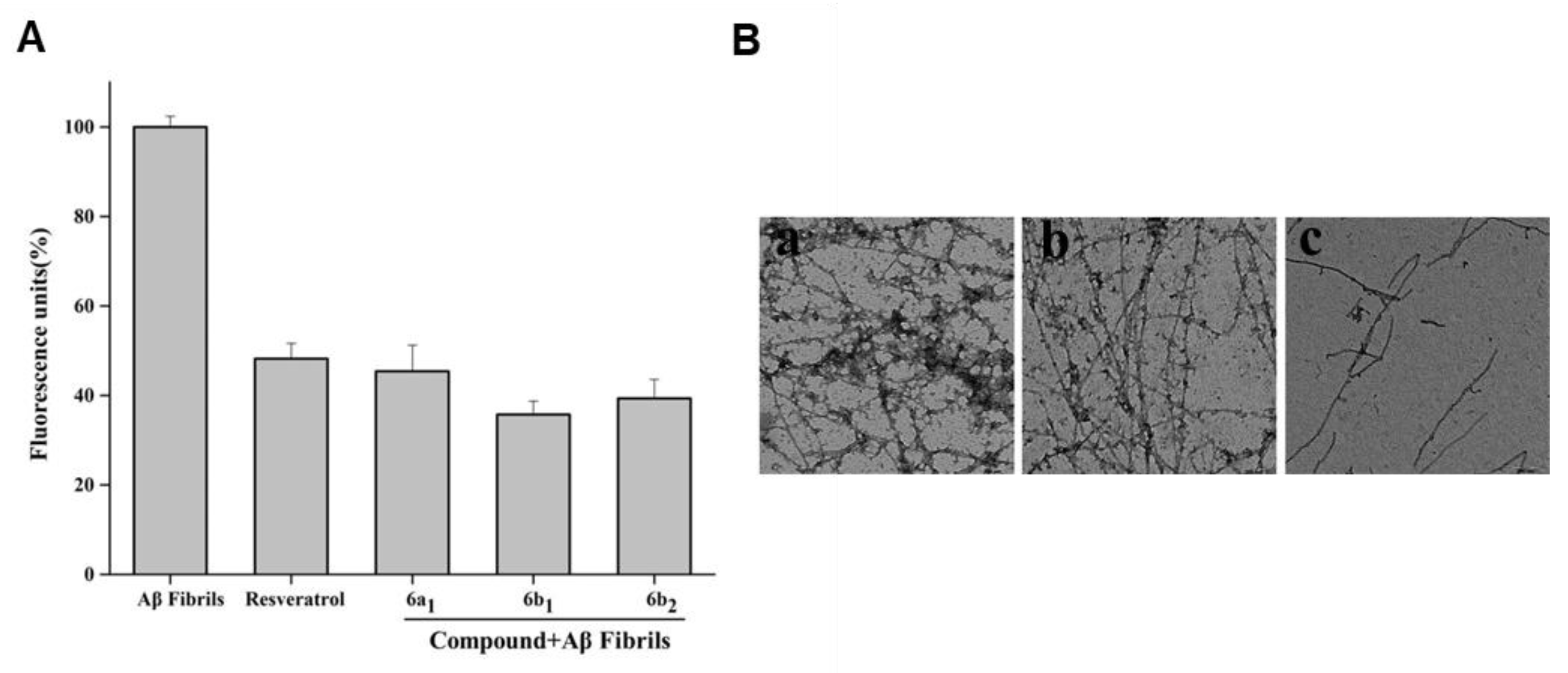
2.2. Inhibition of Aβ1–42 Self-Induced Aggregation
2.3. Antioxidant Activity In Vitro
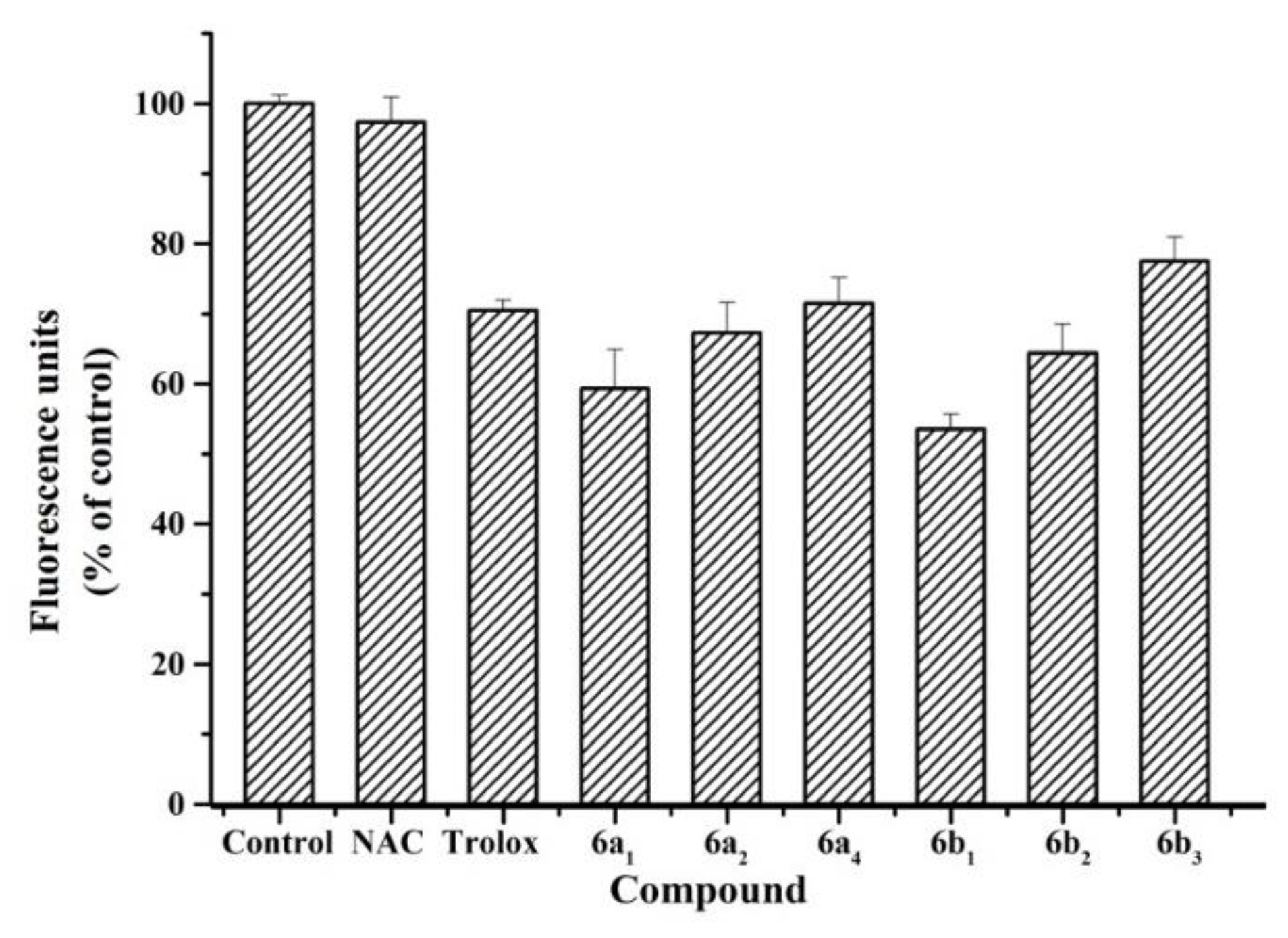
2.4. Antioxidant Activity in SH-SY5Y Cells
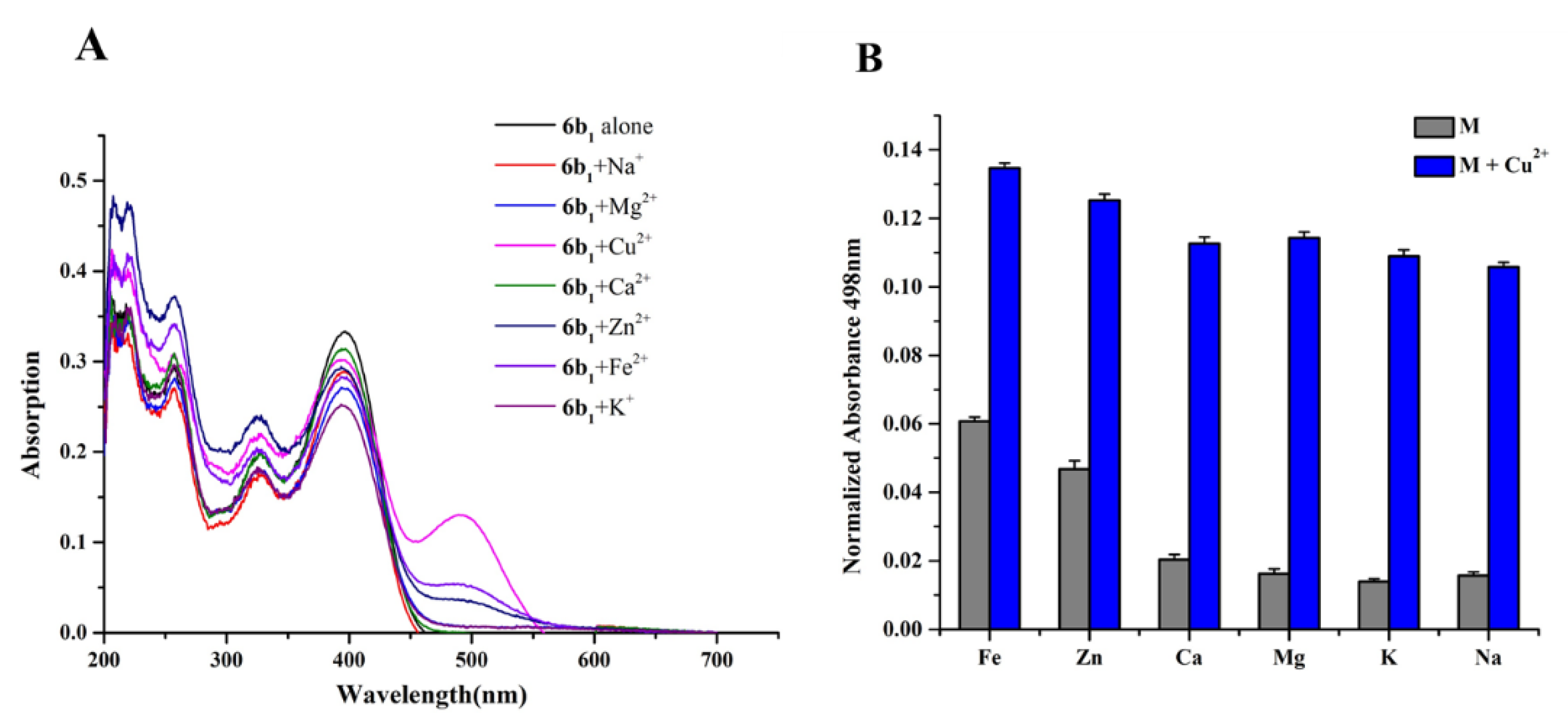
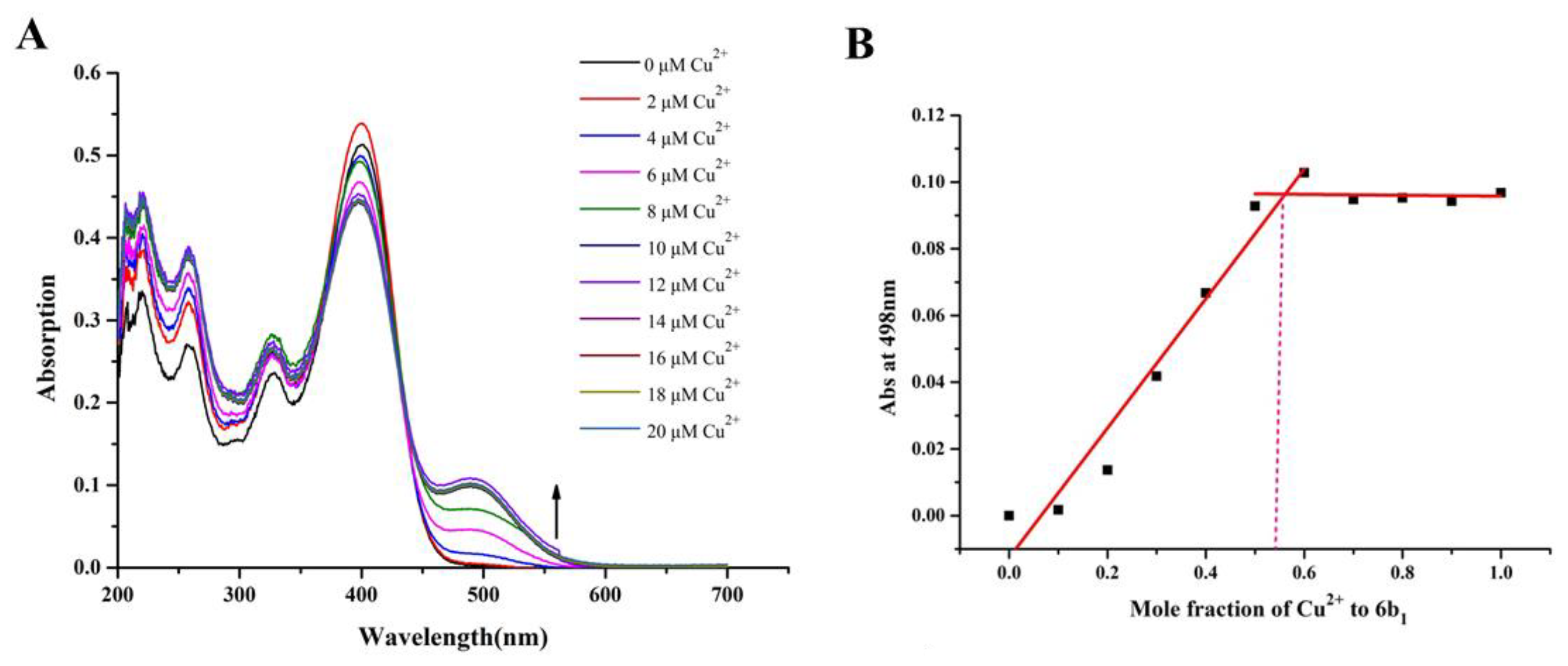
2.5. Metal Binding Properties of Compound 6b1
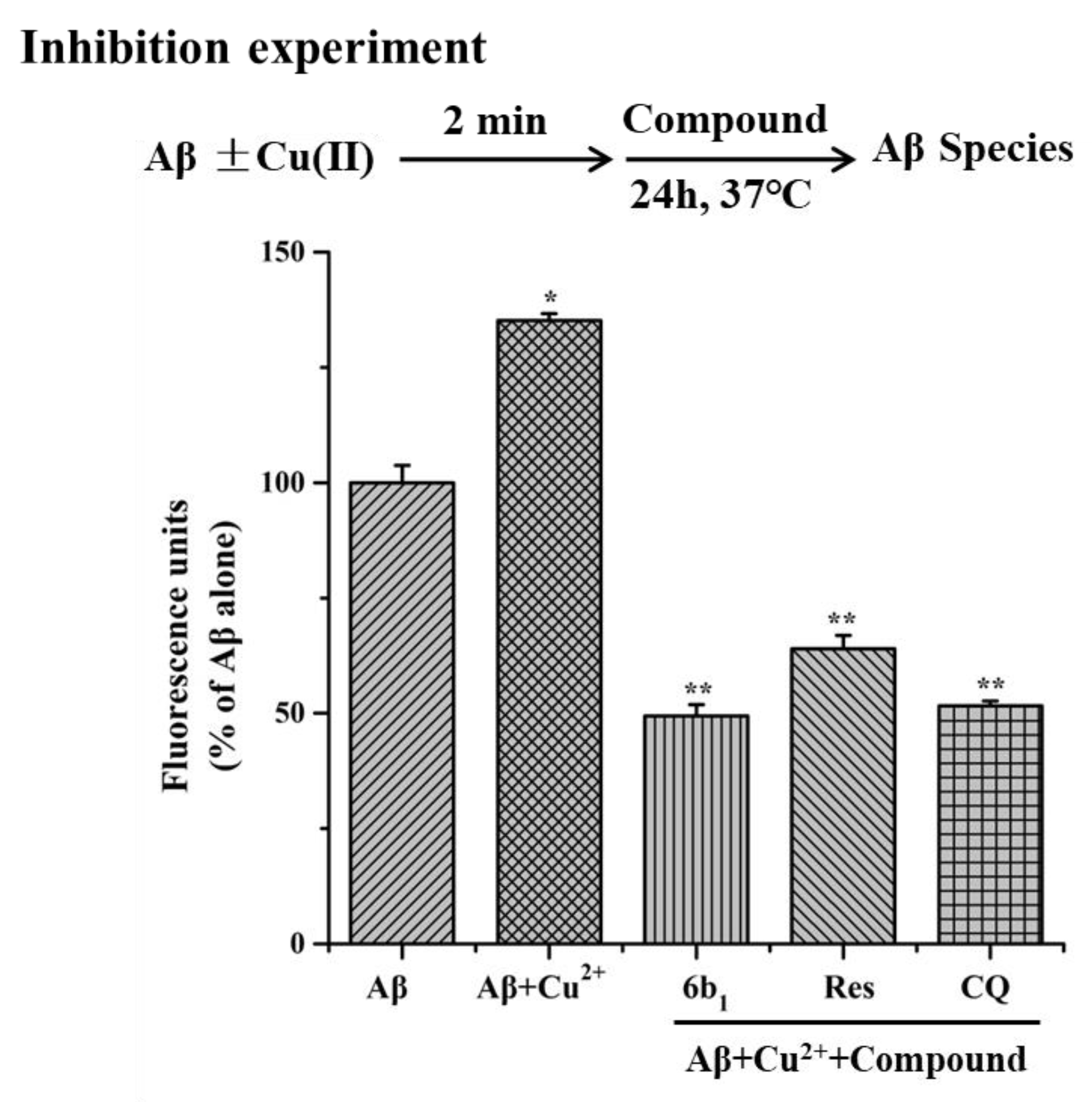
2.6. Inhibition of Cu2+-Induced Aβ1–42 Aggregation
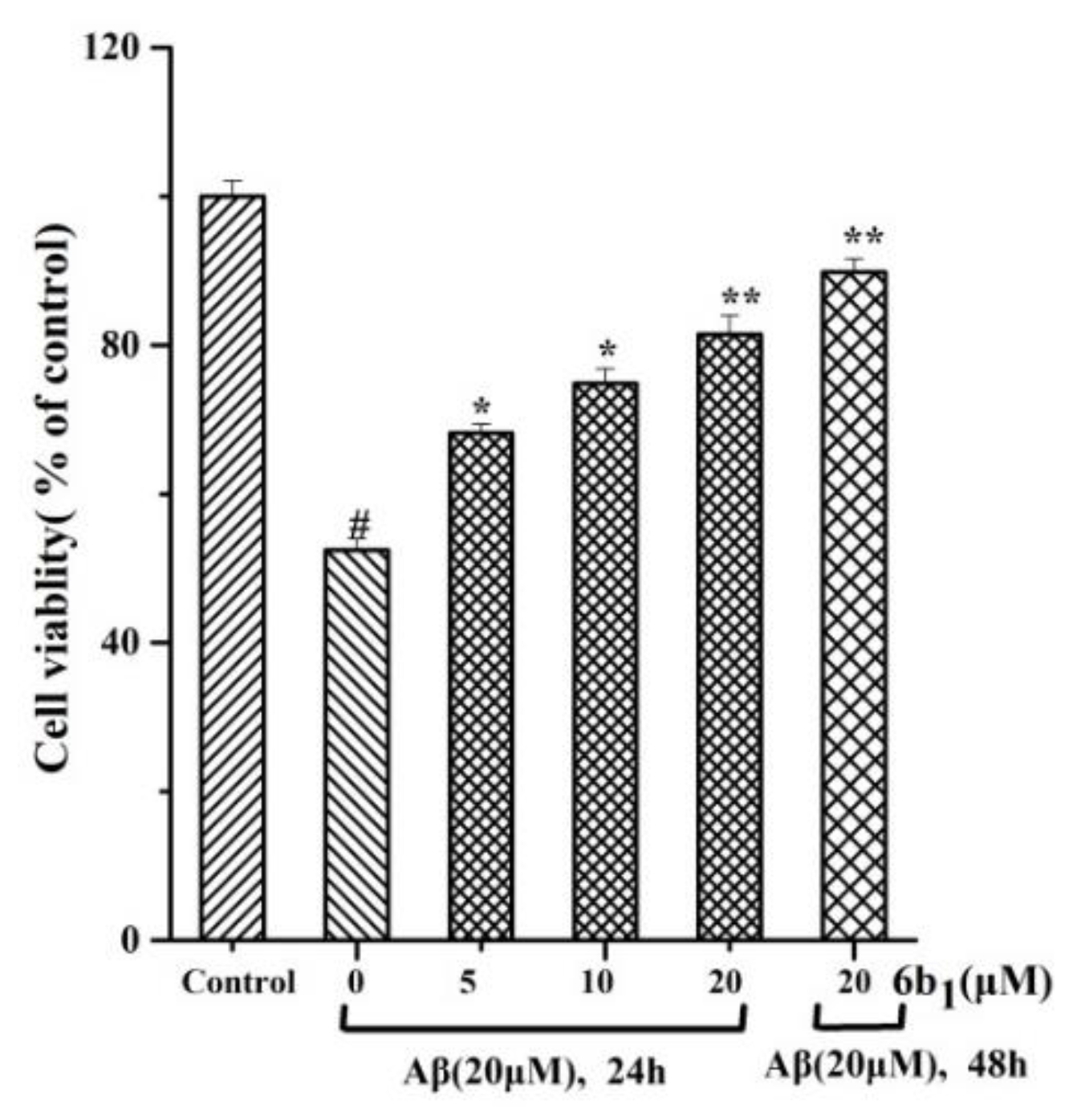
2.7. Cytotoxic Effect on SH-SY5Y
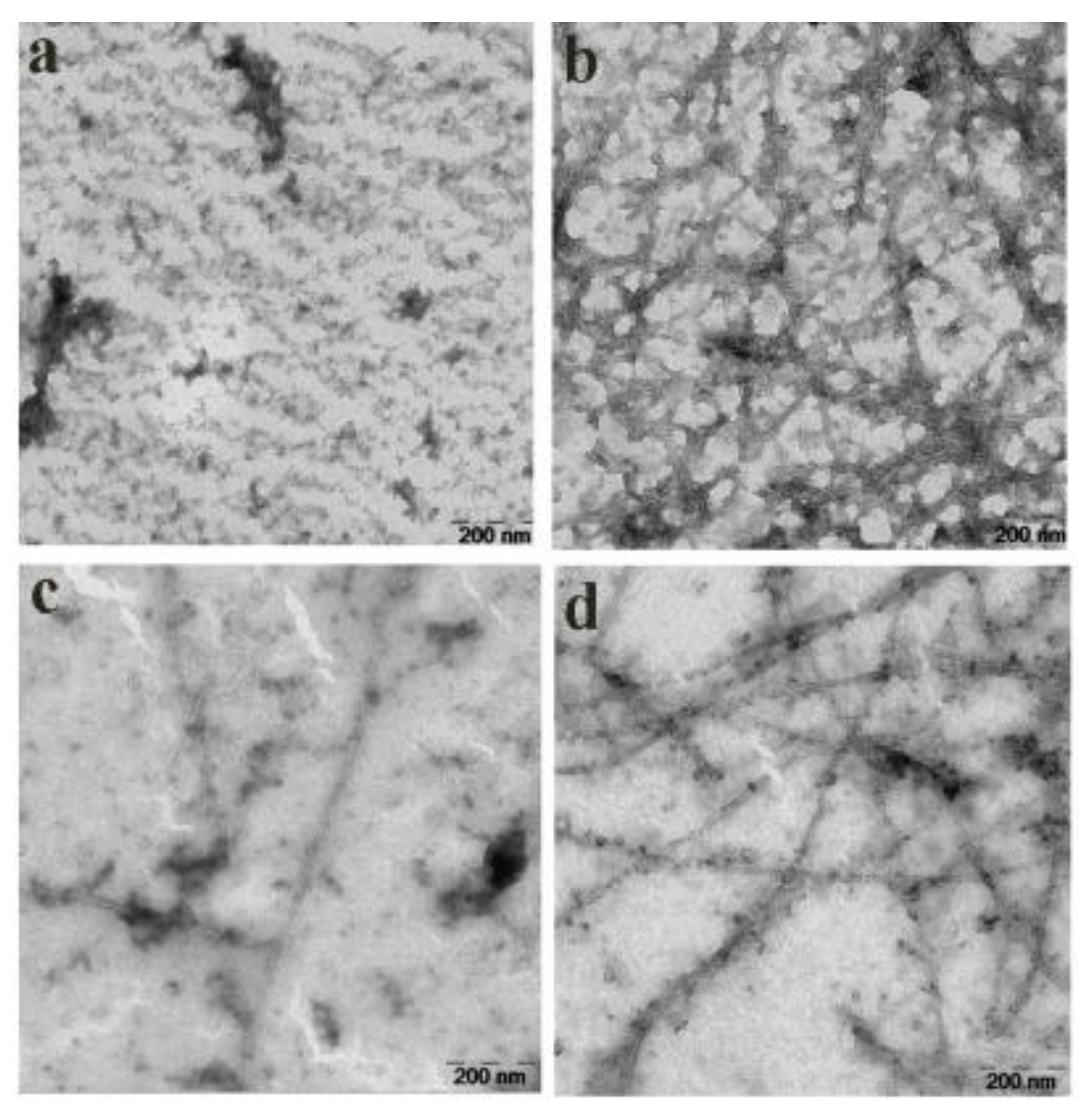
2.8. Effect of Compound 6b1 on Abundance of Aβ1–42 Fibrils
2.9. Disaggregation of Self-Induced Aβ1–42 Aggregation Fibrils by 6b1
2.10. In vitro Protective Effect of Compound 6b1 against Aβ1–42 Induced Toxicity in SH-SY5Y Human Neuroblastoma Cells
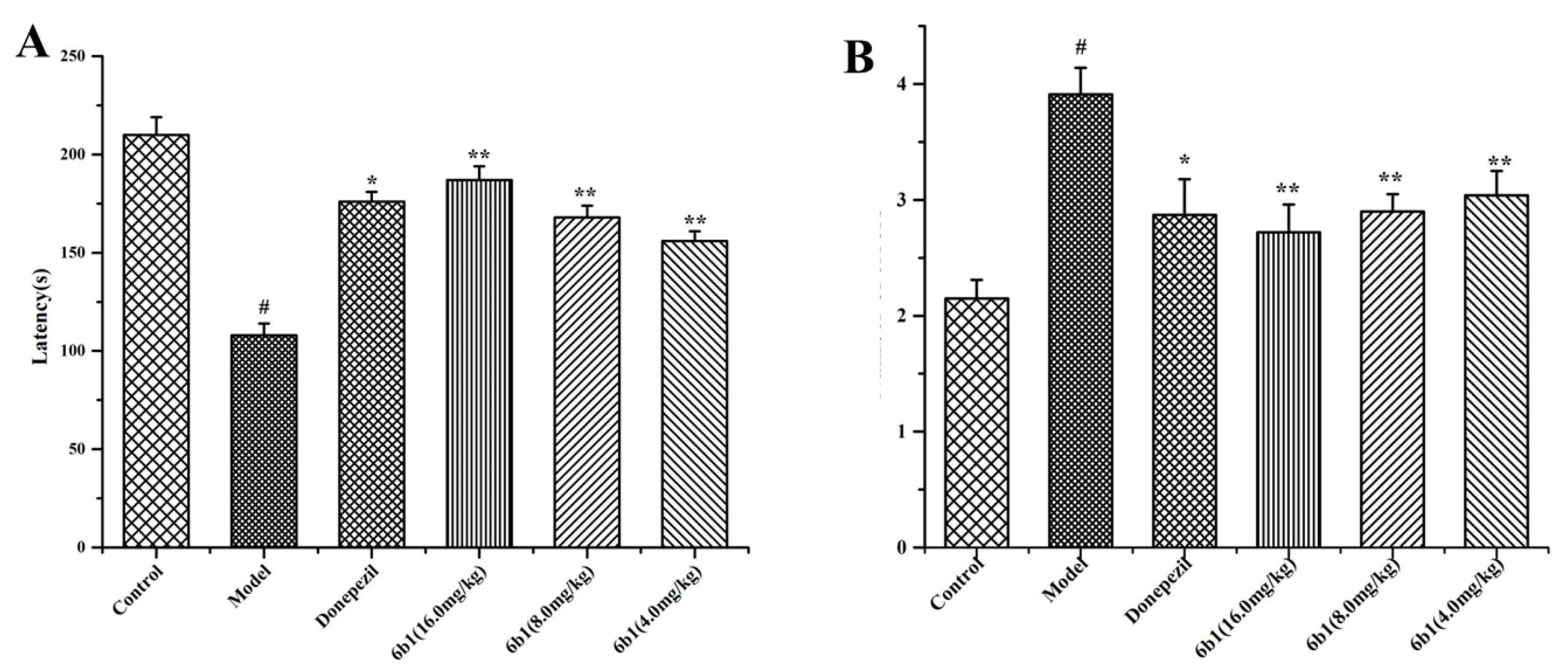
2.11. Step-Down Type Passive Avoidance Test
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. Synthesis of Intermediates
3.2.2. General Procedure A for the Synthesis of Compounds 6a–6e
3.3. Pharmacological Assay
3.3.1. ThT Assay
3.3.2. Antioxidant Activity Assay In Vitro
3.3.3. Antioxidant Activity in SH-SY5Y Cells
3.3.4. Metal Chelation
3.3.5. Inhibition of Cu2+-Induced Aβ1–42 Aggregation
3.3.6. Cytotoxicities of the Compounds on SH-SY5Y Cells
3.3.7. TEM Assay
3.3.8. Aβ1–42-Induced Neurotoxicity
3.3.9. Step-Down Type Passive Avoidance Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimer’s Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Wimo, A.; Guerchet, M.; Ail, G.C.; Wu, Y.T.; Prina, M. World Alzheimer Report 2015. The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; The Alzheimer’s Disease International: London, UK, 2015; pp. 1–88. [Google Scholar]
- Dementia Statistics, Alzheimer’s Disease International. Available online: http://www.alz.co.uk/research/statistics (accessed on 15 January 2018).
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Stoehr, J. Aβ propagation and strains: Implications for the phenotypic diversity in Alzheimer’s disease. Neurobiol. Dis. 2018, 109, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Scarpini, E.; Schelterns, P.; Feldman, H. Treatment of Alzheimer’s disease: Current status and new perspectives. Lancet Neurol. 2003, 2, 539–547. [Google Scholar] [CrossRef]
- Kepp, K.P. Bioinorganic chemistry of Alzheimer’s disease. Chem. Rev. 2012, 112, 5193–5239. [Google Scholar] [CrossRef] [PubMed]
- Trippier, P.C.; Jansen Labby, K.; Hawker, D.D.; Mataka, J.J.; Silverman, R.B. Target and mechanism-based therapeutics for neurodegenerative diseases: Strength in numbers. J. Med. Chem. 2013, 56, 3121–3147. [Google Scholar] [CrossRef]
- Savelieff, M.G.; DeToma, A.S.; Derrick, J.S.; Lim, M.H. The ongoing search for small molecules to study metal-associated amyloid-β species in Alzheimer’s disease. Acc. Chem. Res. 2014, 47, 2475–2482. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; EI Khoury, J.; Kummer, M.P.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Sun, X.; Chen, W.D.; Wang, Y.D. β-Amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Wogulis, M.; Wright, S.; Cunningham, D.; Chilcote, T.; Powell, K.; Rydel, R.E. Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J. Neurosci. 2005, 25, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiang, X.; Luo, L.; Yang, X.; Xiao, G.; Liu, Q.; Ai, J.; Tan, Z.; Deng, Y. Aurone Mannich base derivatives as promising multifunctional agents with acetylcholinesterase inhibition, anti-β-amyloid aggragation and neuroprotective properties for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 126, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Landreth, G.E. Microglial interaction with β-amyloid: Implications for the pathogenesis of Alzheimer’s disease. Microsc Res. Tech. 2001, 54, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.H.; Savage, M.J.; Flood, D.G.; Thomas, J.M.; Levy, R.B.; Mahadomrongkul, V.; Shirao, T.; Aoki, C.; Huerta, P.T. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc. Natl. Acad. Sci. USA 2006, 10, 3410–3415. [Google Scholar] [CrossRef]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Finkelstein, D.; Hawco, N.J.; Rath, N.P.; Kim, J.; Mirica, L.M. Bifunctional Compounds for Controlling Metal-Mediated Aggregation of the Aβ42 Peptide. J. Am. Chem. Soc. 2012, 134, 6625–6636. [Google Scholar] [CrossRef]
- Viana da Silva, S.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Goncalves, N.; Gorlewicz, A.; Malezieux, M.; Goncalves, F.Q.; Grosjean, N.; et al. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 2016, 7, 11915. [Google Scholar] [CrossRef]
- Shiosaka, S.; Ishikawa, Y. Neuropsin—A possible modulator of synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.M.; Michikawa, M.; Kim, S.U.; Nagai, A. Lysophosphatidylcholine increases the neurotoxicity of Alzheimer’s amyloid β1–42 peptide: Role of oligomer formation. Neuroscience 2015, 292, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Mi, Z.; Ji, W.G.; Zhang, C.H.; Zhang, T.; Ren, S.C.; Zhu, Z.R. Rhynchophylline Protects Against the Amyloid β-Induced Increase of Spontaneous Discharges in the Hippocampal CA1 Region of Rats. Neurochem. Res. 2015, 40, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Munch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.; Du, X.G.; Geng, M.Y. Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease. Acta Pharmacol. Sin. 2011, 32, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Pepeu, G.; Giovannini, M.G. Cholinesterase inhibitors and beyond. Curr. Alzheimer Res. 2009, 6, 86–96. [Google Scholar] [CrossRef]
- Rana, M.; Sharma, A.K. Cu and Zn interactions with Aβ peptides: Consequence of coordination on aggregation and formation of neurotoxic soluble Aβ oligomers. Metallomics 2018, in press. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-Peptide(1–42)-Induced Oxidative Stress in Alzheimer Disease: Importance in Disease Pathogenesis and Progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef]
- Peters, D.G.; Connor, J.R.; Meadowcroft, M.D. The relationship between iron dyshomeostasis and amyloidogenesis in Alzheimer’s disease: Two sides of the same coin. Neurobiol. Dis. 2015, 81, 49–65. [Google Scholar] [CrossRef]
- Bush, A.I. Drug development based on the metals hypothesis of Alzheimer’s Disease. J. Alzheimers. Dis. 2008, 15, 223–240. [Google Scholar] [CrossRef]
- Collin, F.; Cheignon, C.; Hureau, C. Oxidative stress as a biomarker for Alzheimer’s disease. Biomark. Med. 2018, 12, 201–203. [Google Scholar] [CrossRef]
- Perry, G.; Moreira, P.I.; Santos, M.S.; Oliveira, C.R.; Shenk, J.C.; Nunomura, A.; Smith, M.A.; Zhu, X. Alzheimer Disease and the Role of Free Radicals in the Pathogenesis of the Disease. CNS. Neurol. Disord. Drug Targets 2008, 7, 3–10. [Google Scholar] [CrossRef]
- Gura, T. Hope in Alzheimer’s fight emerges from unexpected places. Nat. Med. 2008, 14, 894. [Google Scholar] [CrossRef]
- Takeda, A.; Loveman, E.; Clegg, A.; Kirby, J.; Picot, J.; Payne, E.; Green, C. A systematic review of the clinical effectiveness of donepezil, rivastigmine and galantamine on cognition, quality of life and adverse events in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2006, 21, 17–28. [Google Scholar] [CrossRef]
- De Oliveira Pedrosa, M.; Duarte da Cruz, R.M.; de Oliveira Viana, J.; de Moura, R.O.; Ishiki, H.M.; Barbosa Filho, J.M.; Diniz, M.F.; Scotti, M.T.; Scotti, L.; Bezerra Mendonca, F.J. Hybrid Compounds as Direct Multitarget Ligands: A Review. Curr. Top. Med. Chem. 2017, 17, 1044–1079. [Google Scholar] [CrossRef]
- Leon, R.; Garcia, A.G.; Marco-Contelles, J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. [Google Scholar] [CrossRef]
- Michalska, P.; Buendia, I.; Del Barrio, L.; Leon, R. Novel Multitarget Hybrid Compounds for the Treatment of Alzheimer’s Disease. Curr. Top. Med. Chem. 2017, 17, 1027–1043. [Google Scholar] [CrossRef]
- Wang, X.Q.; Xia, C.L.; Chen, S.B.; Tan, J.H.; Ou, T.M.; Huang, S.L.; Li, D.; Gu, L.Q.; Huang, Z.S. Design, synthesis, and biological evaluation of 2-arylethenylquinoline derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2015, 89, 349–361. [Google Scholar] [CrossRef]
- Wang, X.Q. Synthesis and Evaluation of 2-arylethenylquinoline Derivatives as Potential Agents for the Treatment of Alzheimer’s Disease. Ph.D. Thesis, Sun Yat-sen University, Guangzhou, China, 24 December 2014. (In Chinese). [Google Scholar]
- Wang, X.Q.; Cai, Y.H.; Xie, X.Y.; Huang, C.Y.; Li, J.Y.; Chen, W.N.; He, M.H.; Pan, W.J. Microwave-Assisted Efficient Synthesis of 4-Substituted Amino-2-methyl-quinolines Catalyzed by p-Toluenesulfonic Acid. Heterocycles 2016, 92, 1864–1874. [Google Scholar] [CrossRef]
- Panek, D.; Więckowska, A.; Pasieka, A.; Godyń, J.; Jończyk, J.; Bajda, M.; Knez, D.; Gobec, S.; Malawska, B. Design, Synthesis, and Biological Evaluation of 2-(Benzylamino-2-Hydroxyalkyl)Isoindoline-1,3-Diones Derivatives as Potential Disease-Modifying Multifunctional Anti-Alzheimer Agents. Molecules 2018, 23, 347. [Google Scholar] [CrossRef]
- Rodríguez-Franco, M.I.; Fernaandez-Bachiller, M.I.; Pearez, C.; Hernaandez-Ledesma, B.; Bartolomea, B. Novel tacrine-melatonin hybrids as dual-acting drugs for Alzheimer disease, with improved acetylcholinesterase inhibitory and antioxidant properties. J. Med. Chem. 2006, 49, 459–462. [Google Scholar] [CrossRef]
- Li, B.; Huang, A.L.; Zhang, Y.L.; Li, Z.; Ding, H.W.; Huang, C.; Meng, X.M.; Li, J. Design, Synthesis and Evaluation of Hesperetin Derivatives as Potential Multifunctional Anti-Alzheimer Agents. Molecules 2017, 22, 1067. [Google Scholar] [CrossRef]
- Oyama, Y.; Hayashi, A.; Ueha, T.; Maekawa, K. Characterization of 2′,7′-dichloro-fluorescin fluorescence in dissociated mammalian brain neurons: Estimation on intra cellular content of hydrogen peroxide. Brain Res. 1994, 635, 113–117. [Google Scholar] [CrossRef]
- Huang, L.; Lu, C.; Sun, Y.; Mao, F.; Luo, Z.; Su, T.; Jiang, H.; Shan, W.; Li, X. Multitarget-directed benzylideneindanone derivatives: Anti-β-amyloid (Aβ) aggregation, antioxidant, metal chelation, and monoamine oxidase B (MAO-B) inhibition properties against Alzheimer’s disease. J. Med. Chem. 2012, 55, 8483–8492. [Google Scholar] [CrossRef]
- Martínez, A.; Zahran, M.; Gomez, M.; Cooper, C.; Guevara, J.; Ekengard, E.; Nordlander, E.; Alcendor, R.; Hambleton, S. Novel multi-target compounds in the quest for new chemotherapies against Alzheimer’s disease: An experimental and theoretical study. Bioorg. Med. Chem. 2018, 26, 4823–4840. [Google Scholar] [CrossRef]
- Li, S.Y.; Wang, X.B.; Kong, L.Y. Design, synthesis and biological evaluation of imine resveratrol derivatives as multi-targeted agents against Alzheimer’s disease. Eur. J. Med. Chem. 2014, 71, 36–45. [Google Scholar] [CrossRef]
- Hussein, W.; Sağlık, B.N.; Levent, S.; Korkut, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and Biological Evaluation of New Cholinesterase Inhibitors for Alzheimer’s Disease. Molecules 2018, 23, 2033. [Google Scholar] [CrossRef]
- Beck, M.W.; Derrick, J.S.; Suh, J.M.; Kim, M.; Korshavn, K.J.; Kerr, R.A.; Cho, W.J.; Larsen, S.D.; Ruotolo, B.T.; Ramamoorthy, A.; et al. Minor Structural Variations of Small Molecules Tune Regulatory Activities toward Pathological Factors in Alzheimer’s Disease. ChemMedChem. 2017, 12, 1828–1838. [Google Scholar] [CrossRef]
- Huang, L.; Miao, H.; Sun, Y.; Meng, F.; Li, X. Discovery of indanone derivatives as multi-target-directed ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2014, 87, 429–439. [Google Scholar] [CrossRef]
- Purgatorio, R.; de Candia, M.; De Palma, A.; De Santis, F.; Pisani, L.; Campagna, F.; Cellamare, S.; Altomare, C.D.; Catto, M. Insights into Structure-Activity Relationships of 3-Arylhydrazonoindolin-2-One Derivatives for Their Multitarget Activity on β-Amyloid Aggregation and Neurotoxicity. Molecules 2018, 23, 1544. [Google Scholar] [CrossRef]
- Fu, Y.; Mu, Y.; Lei, H.; Wang, P.; Li, X.; Leng, Q.; Han, L.; Qu, X.; Wang, Z.; Huang, X. Design, Synthesis and Evaluation of Novel Tacrine-Ferulic Acid Hybrids as Multifunctional Drug Candidates against Alzheimer’s Disease. Molecules 2016, 21, 1338. [Google Scholar] [CrossRef]
- Sang, Z.; Pan, W.; Wang, K.; Ma, Q.; Yu, L.; Yang, Y.; Bai, P.; Leng, C.; Xu, Q.; Li, X.; et al. Design, synthesis and evaluation of novel ferulic acid-O-alkylamine derivativesas potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 130, 379–392. [Google Scholar] [CrossRef]
- Sang, Z.; Qiang, X.; Li, Y.; Xu, R.; Cao, Z.; Song, Q.; Wang, T.; Zhang, X.; Liu, H.; Tan, Z.; et al. Design, synthesis and evaluation of scutellarein-O-acetamidoalkyl-benzylamines as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 135, 307–323. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 6a–6e are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | General Structure | R1 | R2 | Aβ1–42 Aggregation Inhibition a (%) | ORAC b | |
|---|---|---|---|---|---|---|
| 5 μM | 1 μM | |||||
| 6a1 | A |  |  | 90.2 ± 1.3 | 6.85 ± 0.05 | 5.92 ± 0.03 |
| 6a2 | A | |  | 88.5 ± 1.5 | 6.28 ± 0.08 | 5.74 ± 0.09 |
| 6a3 | B | | _ | 84.3 ± 2.1 | 1.73 ±0.07 | 1.26 ± 0.06 |
| 6a4 | A | |  | 87.9 ± 1.1 | 4.52 ± 0.03 | 4.16 ± 0.04 |
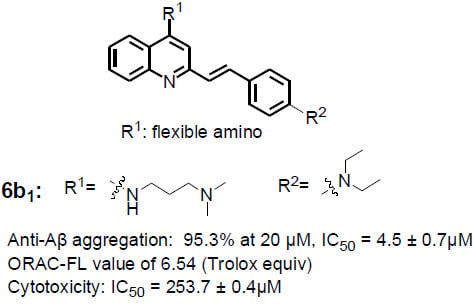
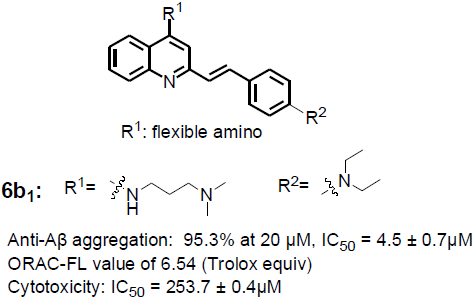
| 6b1 | A |  | | 95.3 ± 1.2 | 6.54 ± 0.07 | 6.23 ± 0.05 |
| 6b2 | A | | | 92.1 ± 1.3 | 5.88 ± 0.01 | 5.61 ± 0.09 |
| 6b3 | B | | _ | 87.2 ± 2.1 | 4.21 ± 0.08 | 3.96 ± 0.08 |
| 6c1 | A |  | | 85.1 ± 1.4 | 3.24 ± 0.10 | 2.79 ± 0.04 |
| 6c2 | A | | | 83.9 ± 2.4 | 3.53 ± 0.07 | 3.28 ± 0.09 |
| 6d1 | A |  | | 80.5 ± 1.4 | 1.56 ± 0.05 | 1.38 ± 0.02 |
| 6d2 | A | | | 79.2 ± 2.3 | 0.95 ± 0.09 | 0.75 ± 0.06 |
| 6e1 | A |  | | 77.9 ± 1.3 | 1.92 ± 0.11 | 1.71 ± 0.05 |
| 6e2 | A | | | 77.5 ± 1.5 | 1.78 ± 0.05 | 1.68 ± 0.06 |
| 4b1 | A |  | | 83.5 ± 1.4 | 3.91 ± 0.11 | 3.70 ± 0.11 |
| 4b2 | A | | | 81.2 ± 0.8 | 3.12 ± 0.10 | 2.91 ± 0.11 |
| Res | _ | _ | _ | 77.2 ± 1.1 | 5.21 ± 0.50 | 5.10 ± 0.41 |
| Cur | _ | _ | _ | 49.3 ± 1.2 | 2.61 ± 0.12 | 2.11 ± 0.61 |
| Compound | IC50 (μM) a |
|---|---|
| 6a1 | 7.8 ± 0.4 |
| 6b1 | 4.5 ± 0.7 |
| 6b2 | 6.1 ± 0.8 |
| Res | 11.8 ± 0.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.-Q.; Zhao, C.-P.; Zhong, L.-C.; Zhu, D.-L.; Mai, D.-H.; Liang, M.-G.; He, M.-H. Preparation of 4-Flexible Amino-2-Arylethenyl-Quinoline Derivatives as Multi-Target Agents for the Treatment of Alzheimer’s Disease. Molecules 2018, 23, 3100. https://doi.org/10.3390/molecules23123100
Wang X-Q, Zhao C-P, Zhong L-C, Zhu D-L, Mai D-H, Liang M-G, He M-H. Preparation of 4-Flexible Amino-2-Arylethenyl-Quinoline Derivatives as Multi-Target Agents for the Treatment of Alzheimer’s Disease. Molecules. 2018; 23(12):3100. https://doi.org/10.3390/molecules23123100
Chicago/Turabian StyleWang, Xiao-Qin, Chu-Ping Zhao, Long-Cheng Zhong, De-Ling Zhu, De-Hao Mai, Mei-Gui Liang, and Ming-Hua He. 2018. "Preparation of 4-Flexible Amino-2-Arylethenyl-Quinoline Derivatives as Multi-Target Agents for the Treatment of Alzheimer’s Disease" Molecules 23, no. 12: 3100. https://doi.org/10.3390/molecules23123100
APA StyleWang, X.-Q., Zhao, C.-P., Zhong, L.-C., Zhu, D.-L., Mai, D.-H., Liang, M.-G., & He, M.-H. (2018). Preparation of 4-Flexible Amino-2-Arylethenyl-Quinoline Derivatives as Multi-Target Agents for the Treatment of Alzheimer’s Disease. Molecules, 23(12), 3100. https://doi.org/10.3390/molecules23123100