
Designing Hybrids Targeting the Cholinergic System by Modulating the Muscarinic and Nicotinic Receptors: A Concept to Treat Alzheimer’s Disease


Abstract
1. Introduction
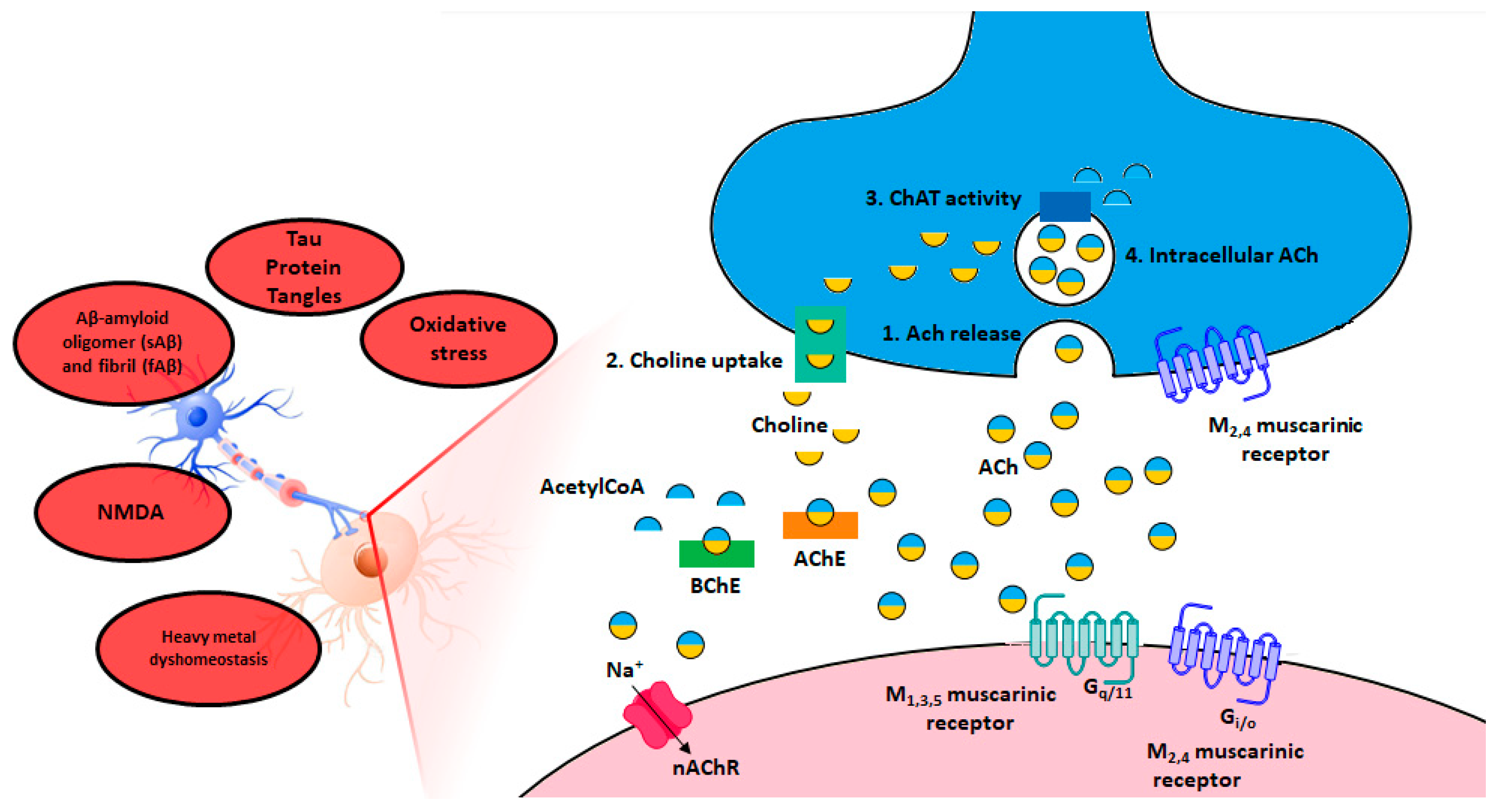
2. The Cholinergic Hypothesis of Memory Dysfunction
3. Hybrid Compounds as Drug Discovery Tools in AD
3.1. One Single Molecule—Different Targets
3.2. One Single Molecule—Different Binding Pockets of a Protein’s Surface
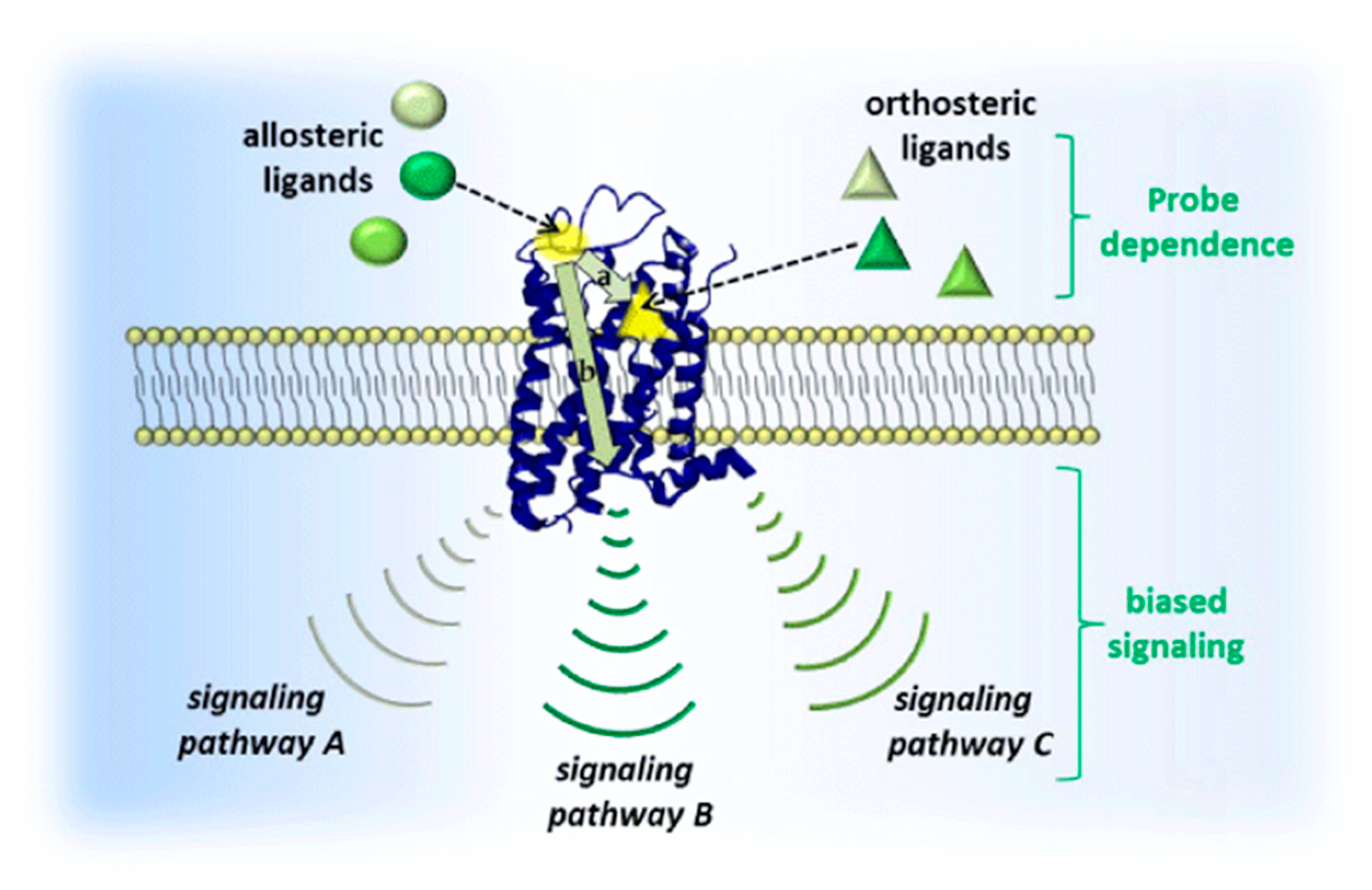
3.2.1. Allosteric Modulation
3.2.2. Bitopic Ligand Principle
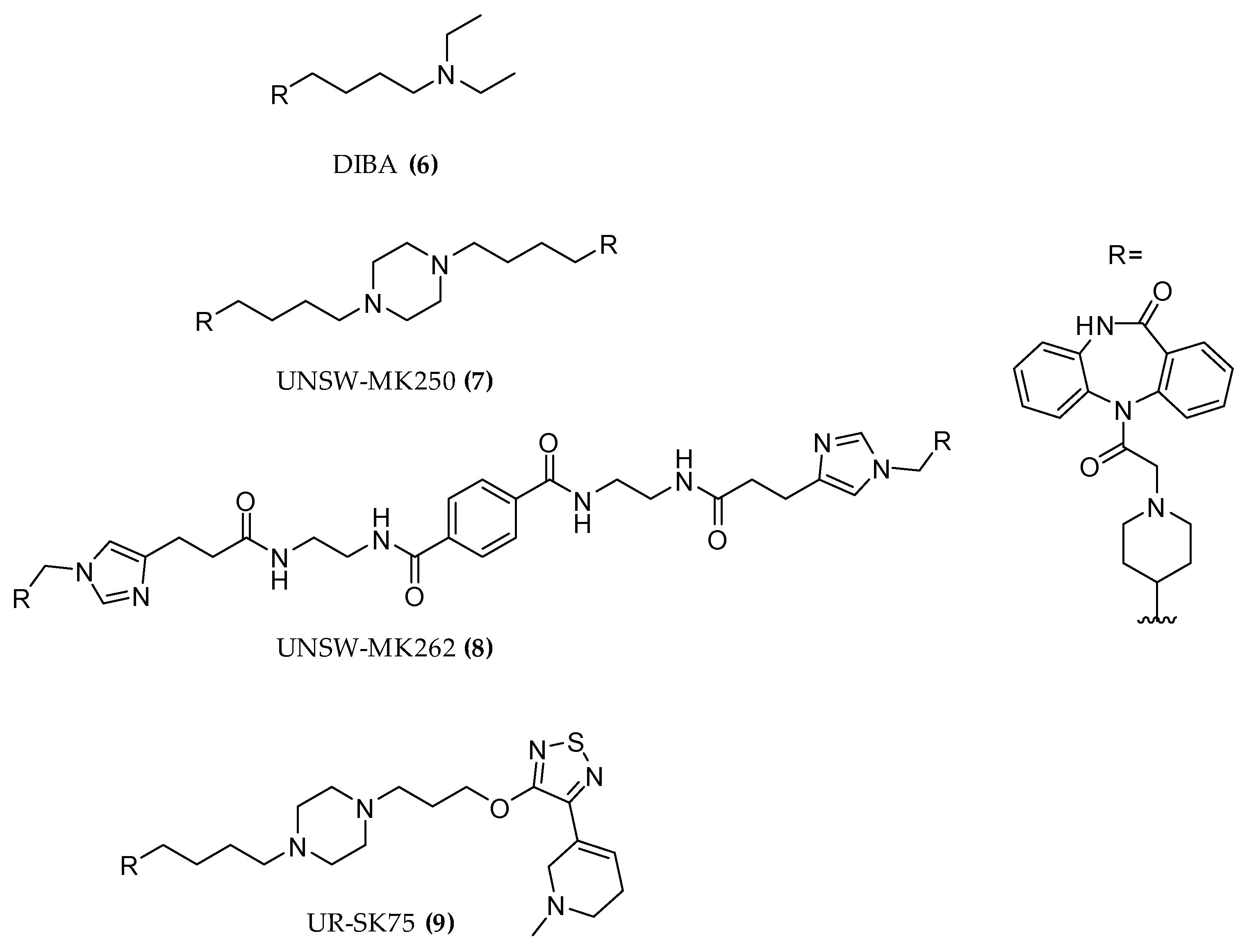
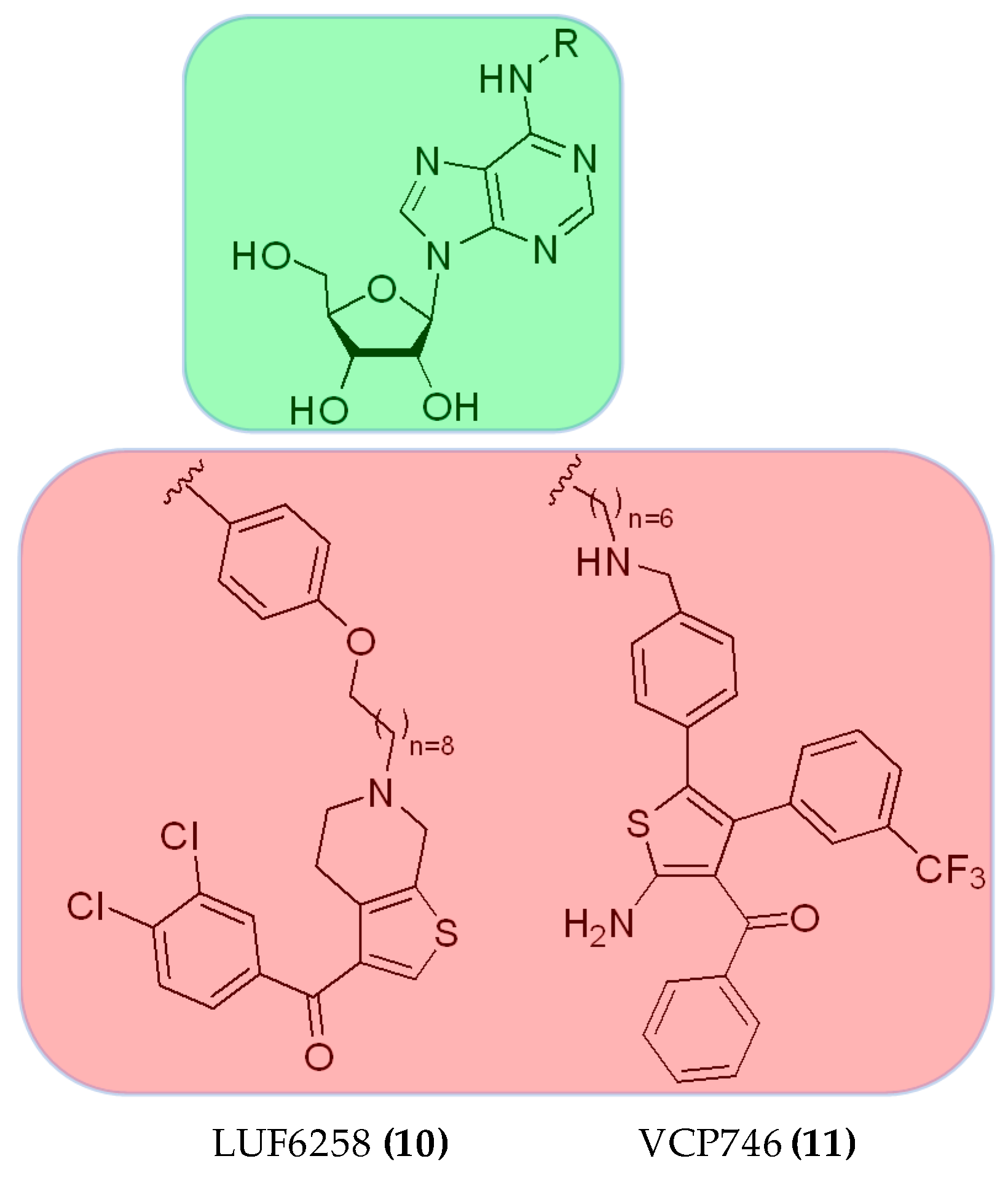
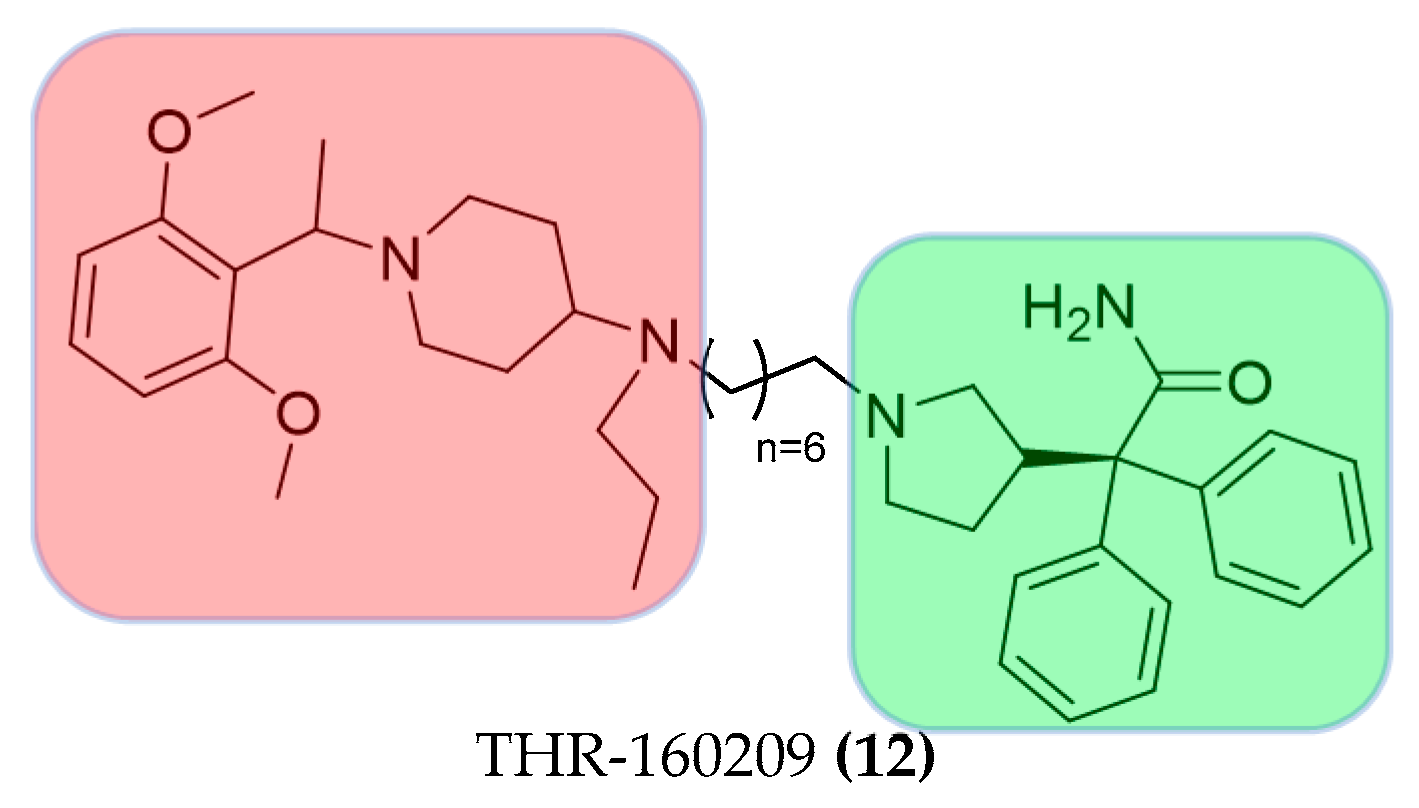
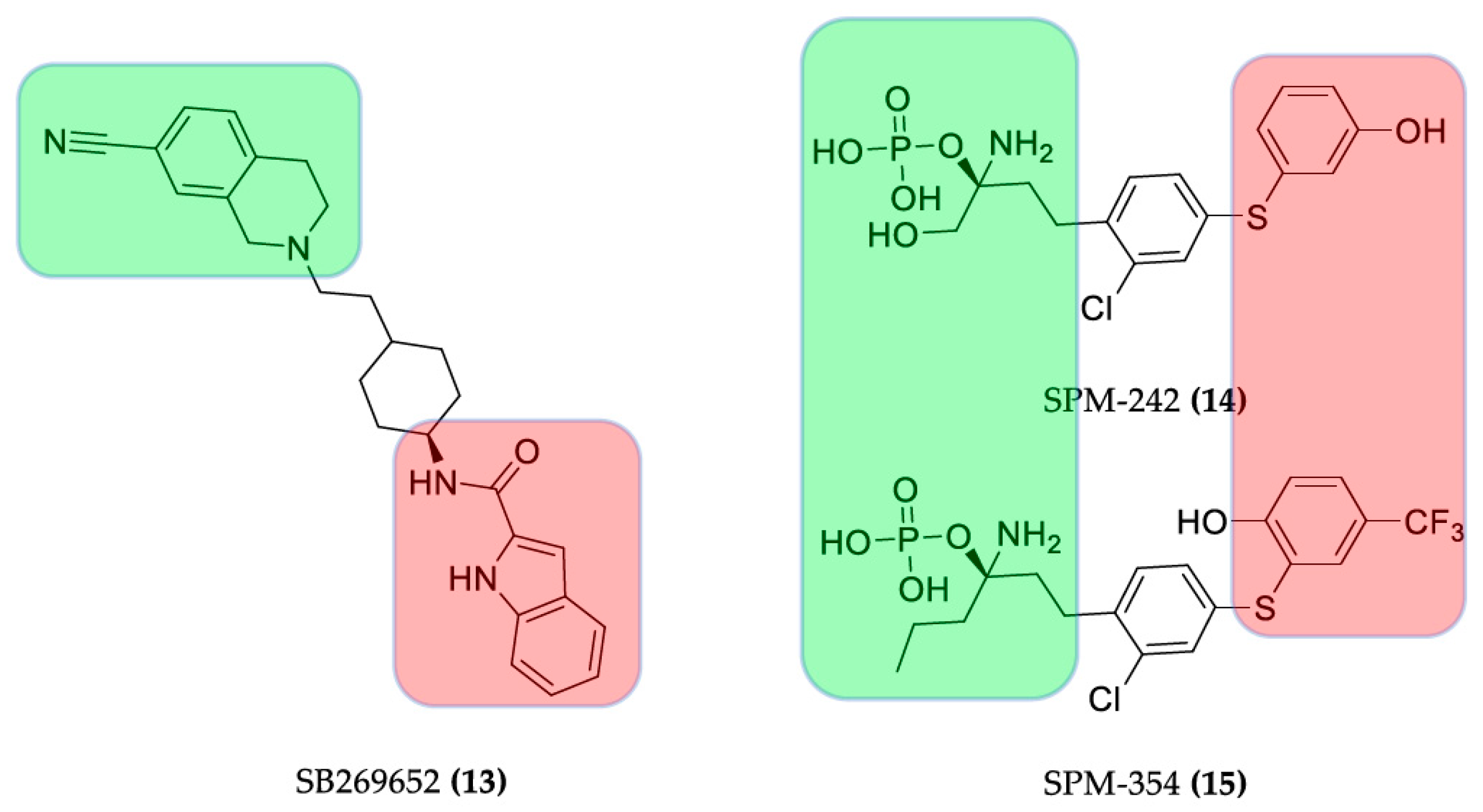
3.2.3. Hybrid Strategies Design
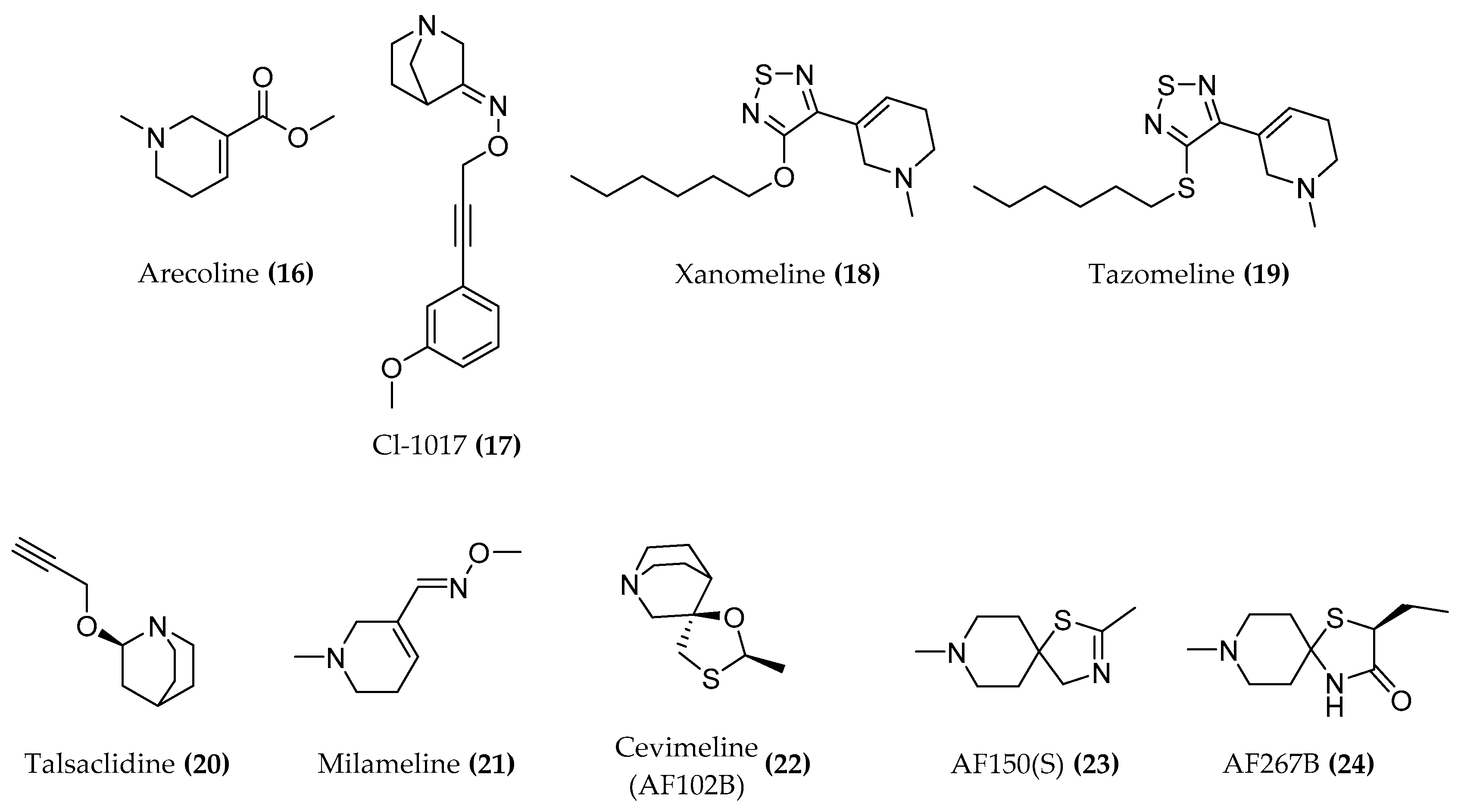
4. Muscarinic Receptor
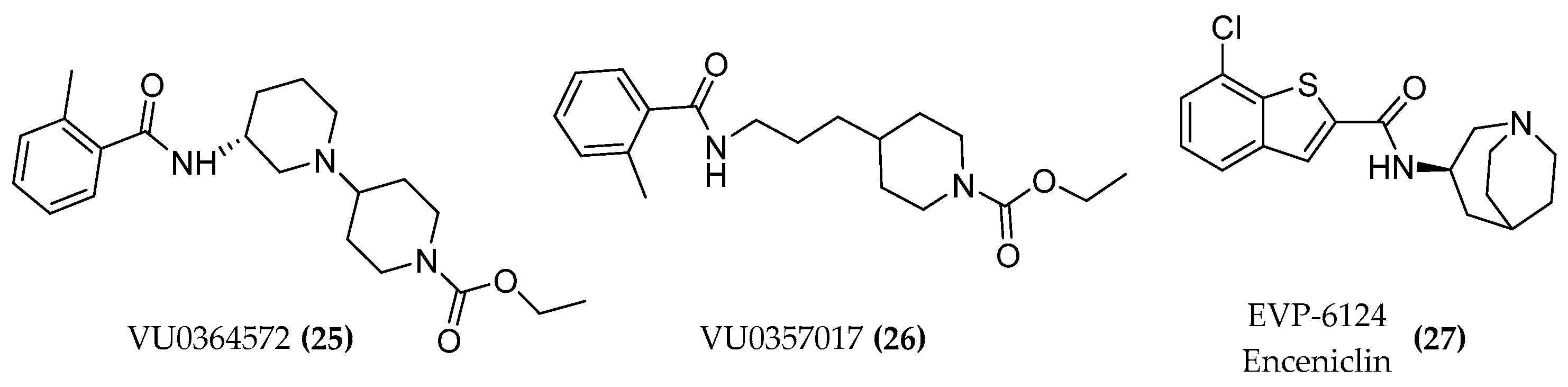
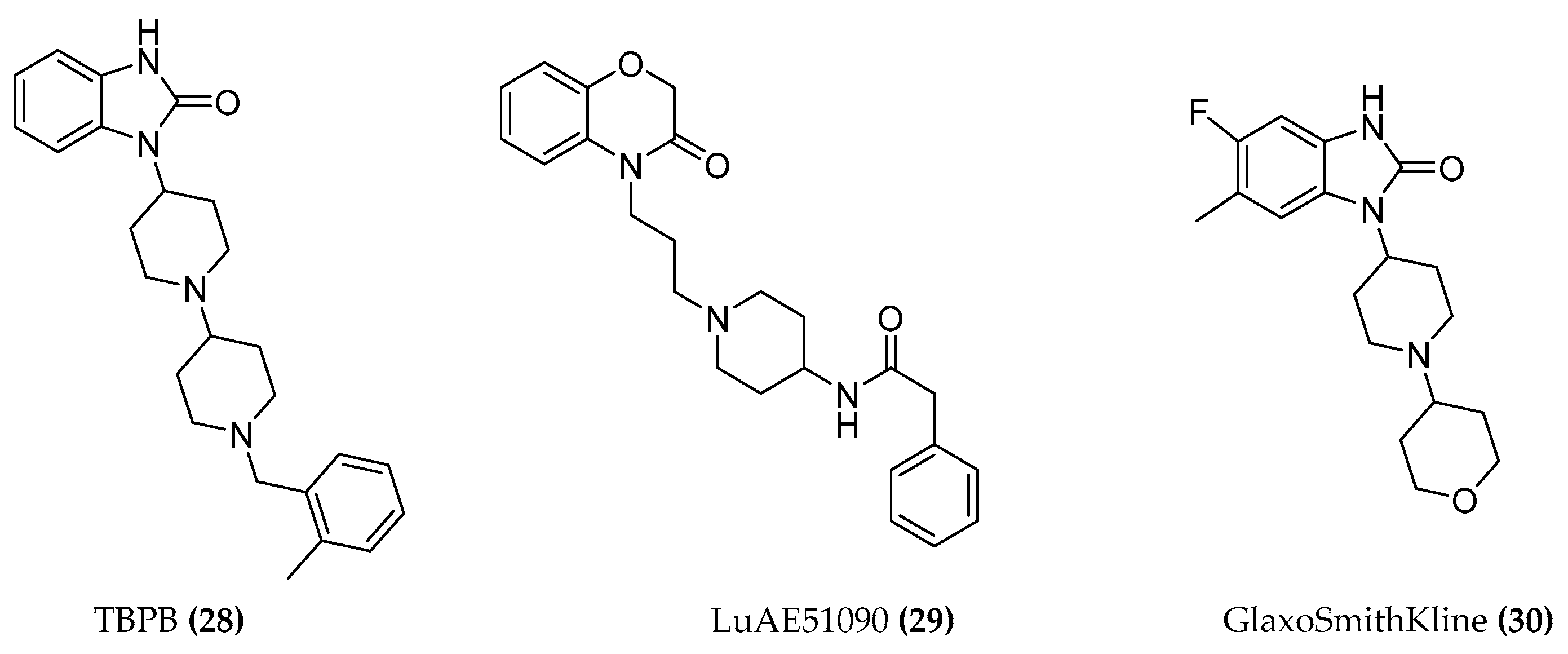
4.1. Design of Hybrid Compounds to Investigate the Muscarinic Receptor
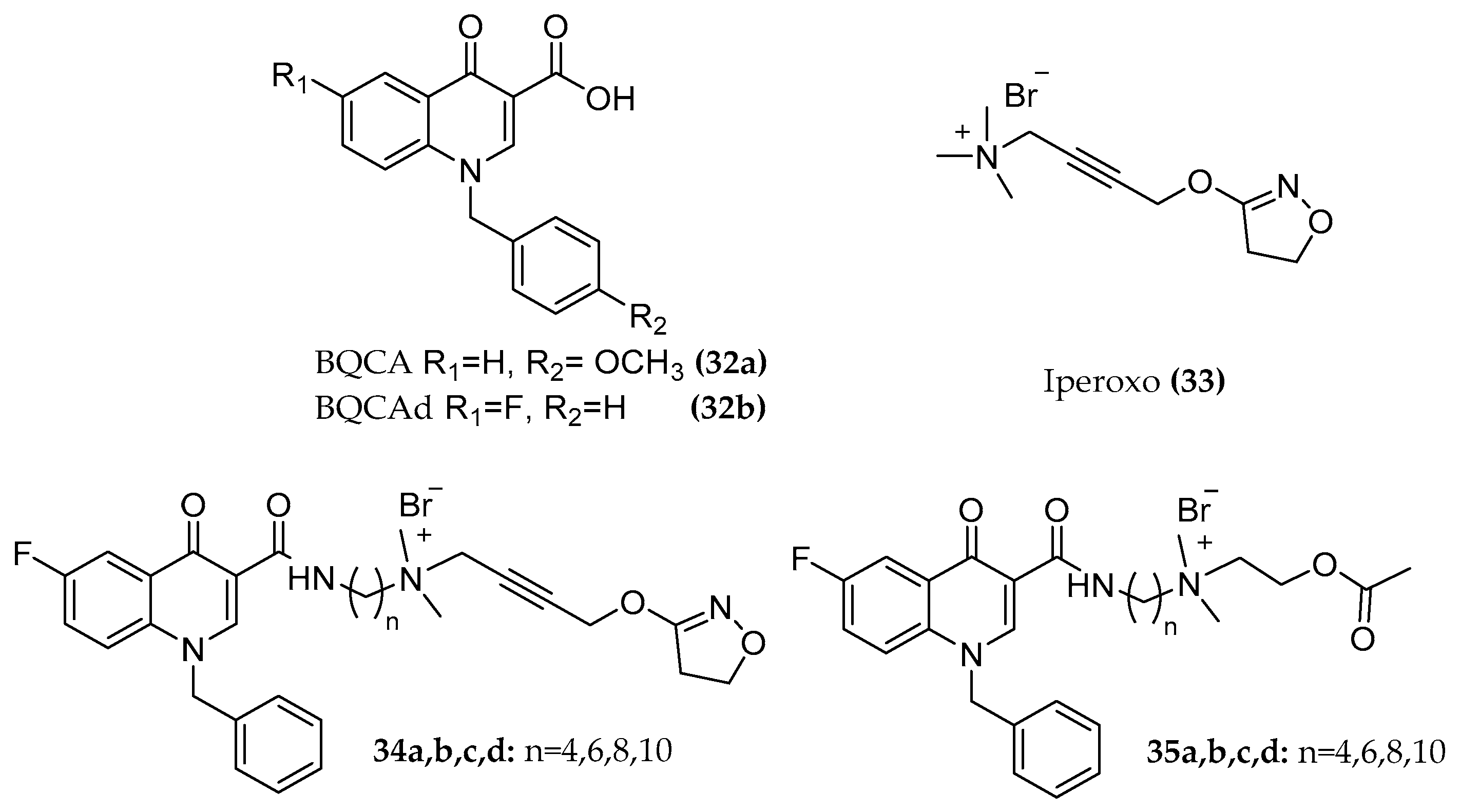
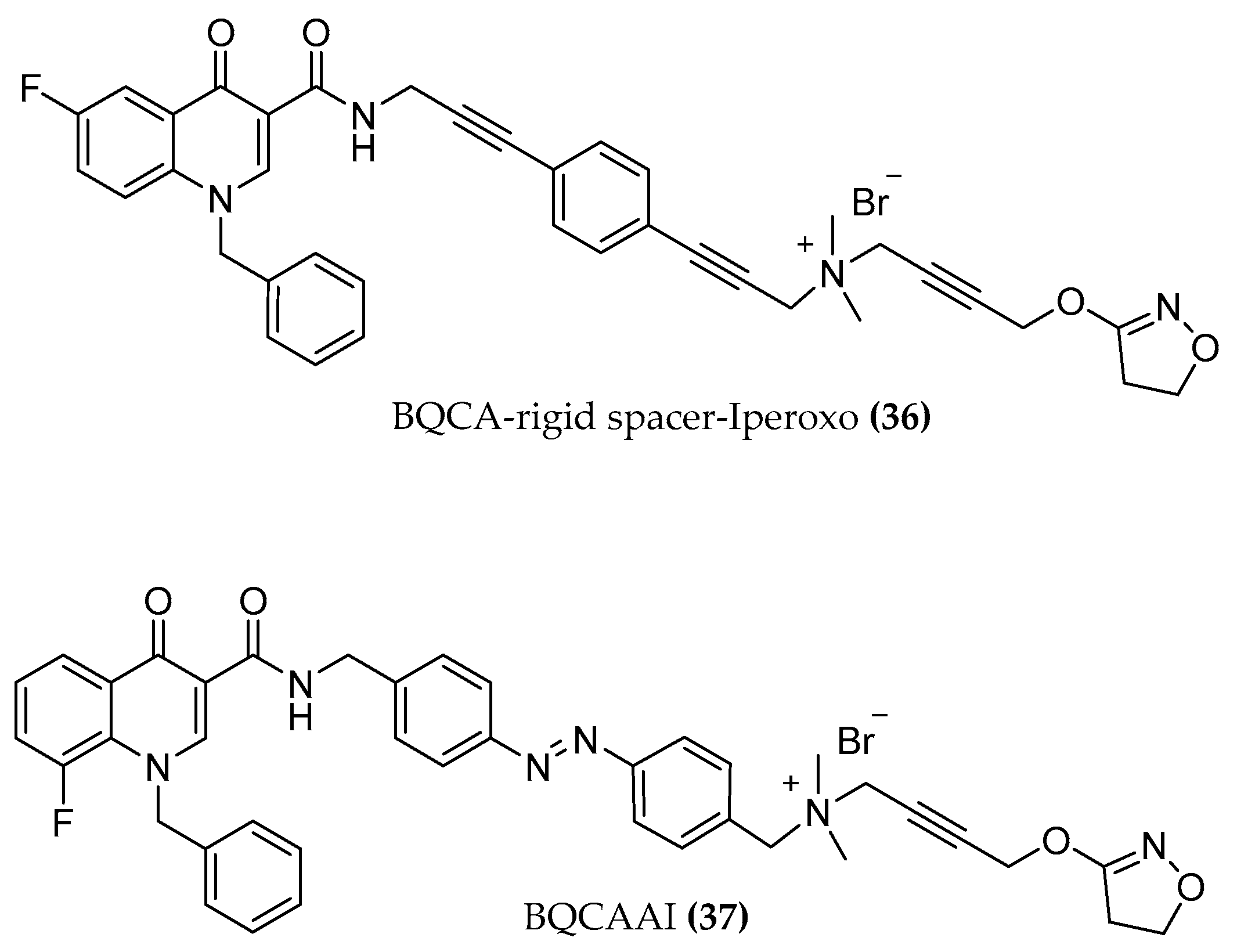
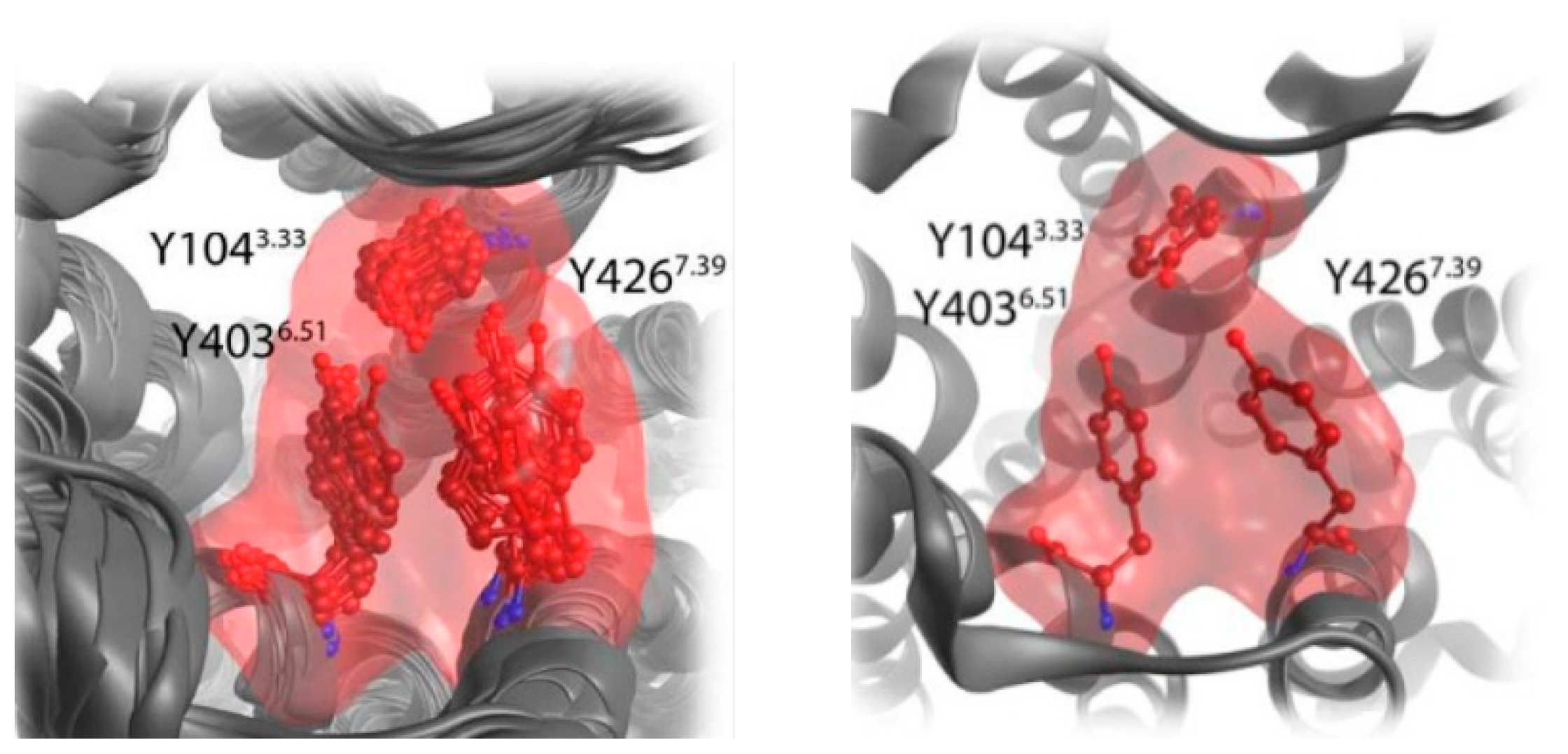
4.1.1. BQCAd Hybrid Derivatives
- subtype selectivity: receptor activation and signaling pathway studies performed with (dynamic mass redistribution) DMR in the presence and absence of specific Gi and Gq signaling blockers indicate a preferentially Gq mediated signal of the hybrids.
- partial agonism which may provide controlled activation without overdriving the M1 signaling and with possible improved side effect profile over the control of cognition [86].
- potency and efficacy dependency on the length of the chain and on the substitution pattern of the allosteric fragment. Elongating the chain from C4 to C6 switches the receptor fractional occupancy from the inactive, purely allosteric pose to the active binding pose which defines the efficacy of the dualsteric ligands [87].
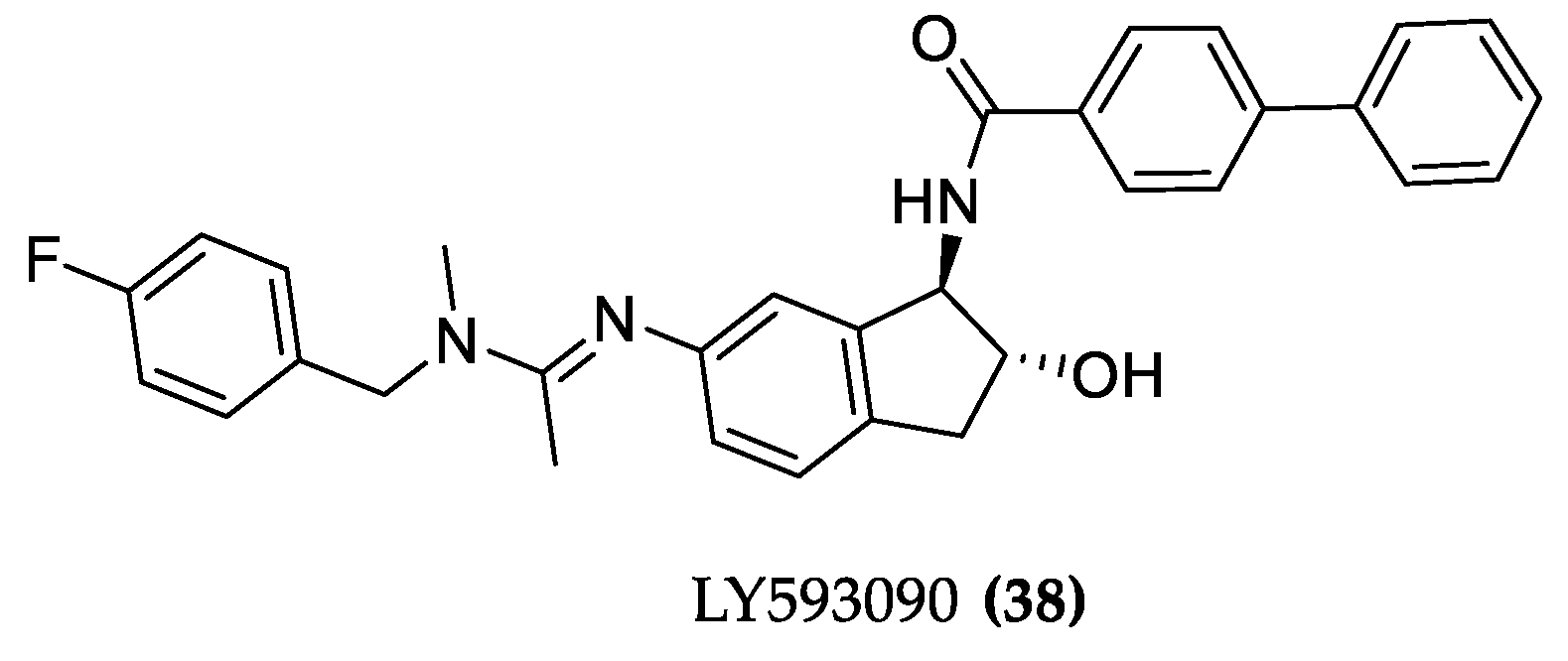
4.1.2. LY593093
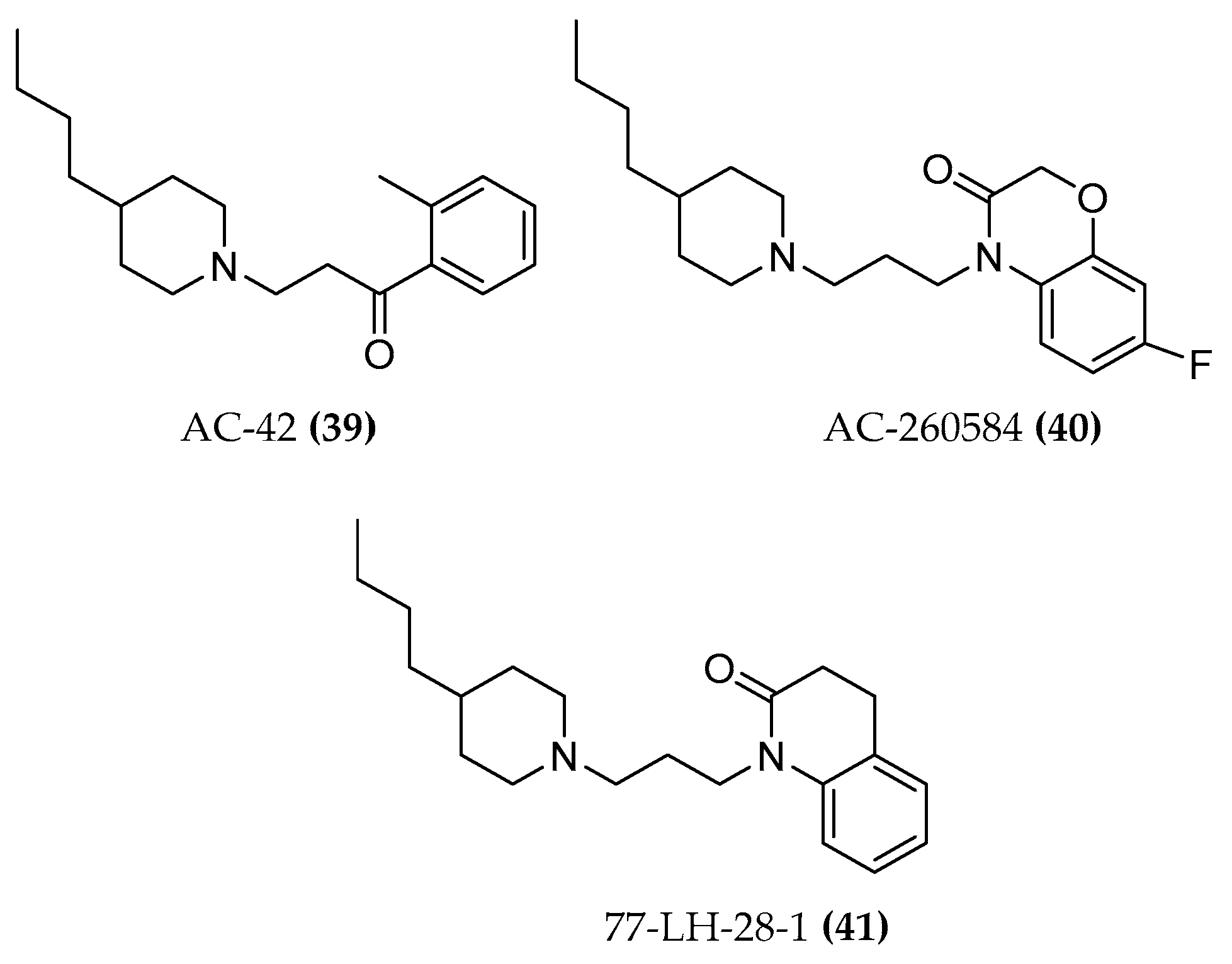
4.1.3. AC-42 and AC-260584
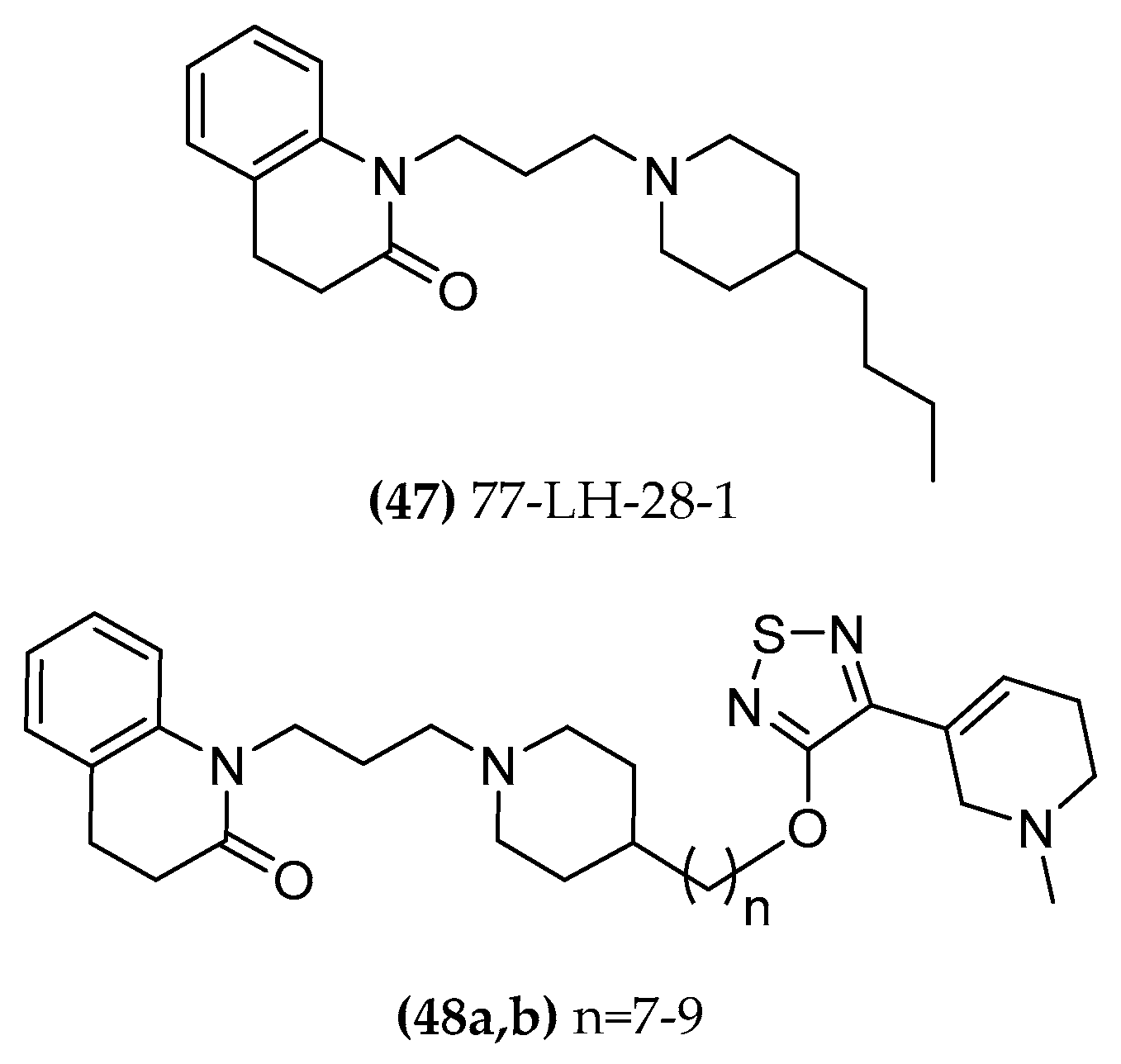
4.1.4. 77-LH-28-1
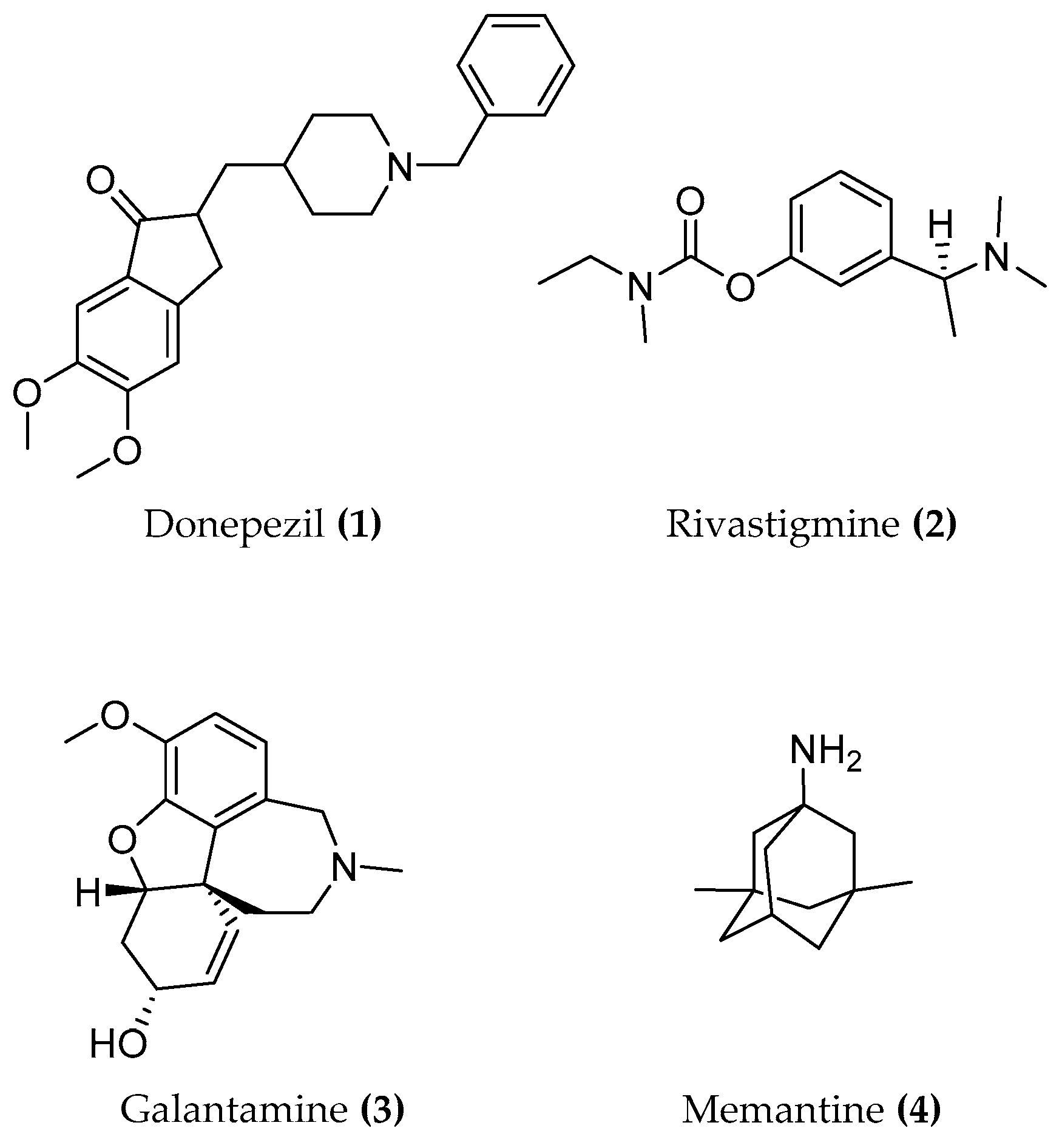
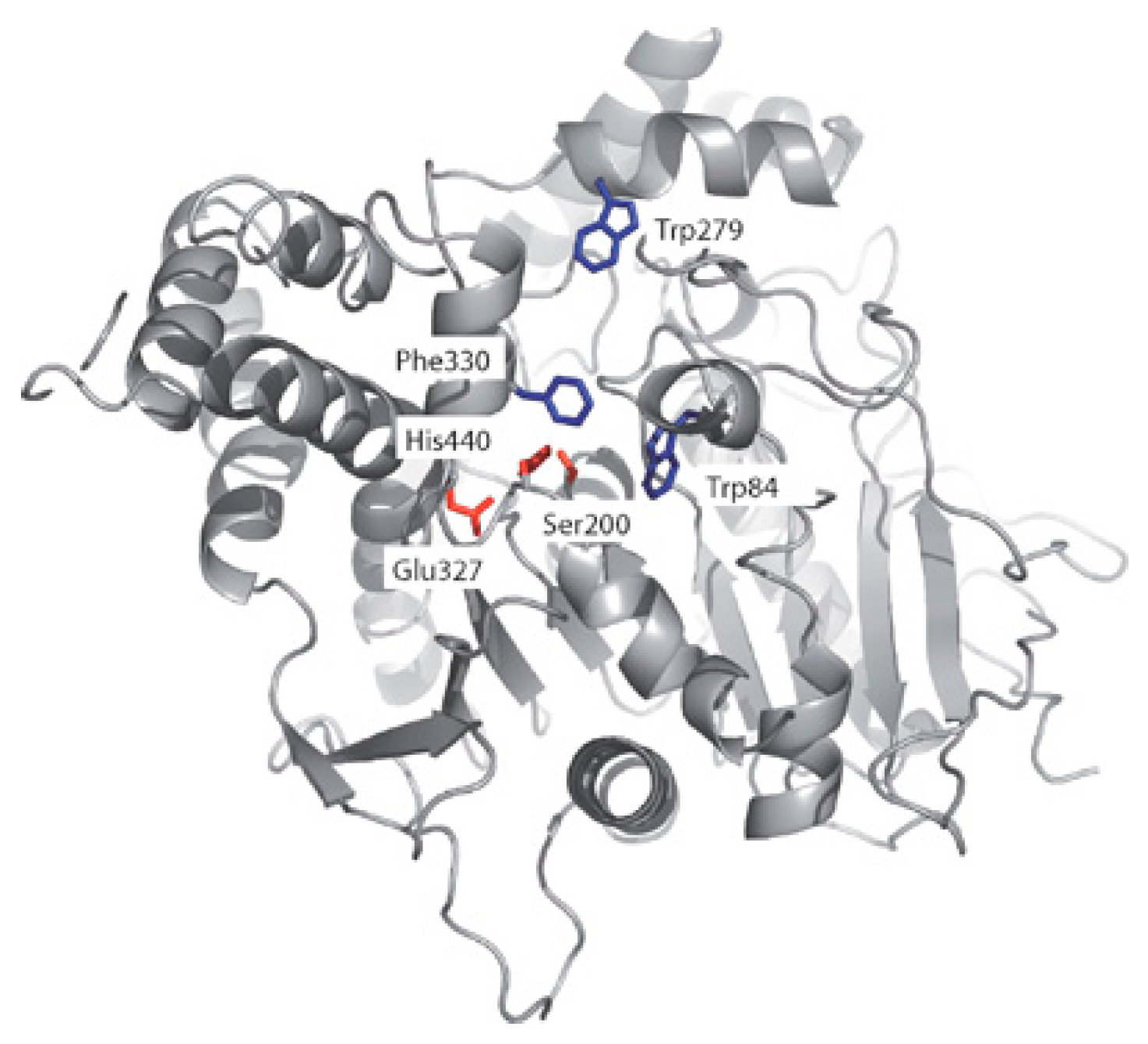
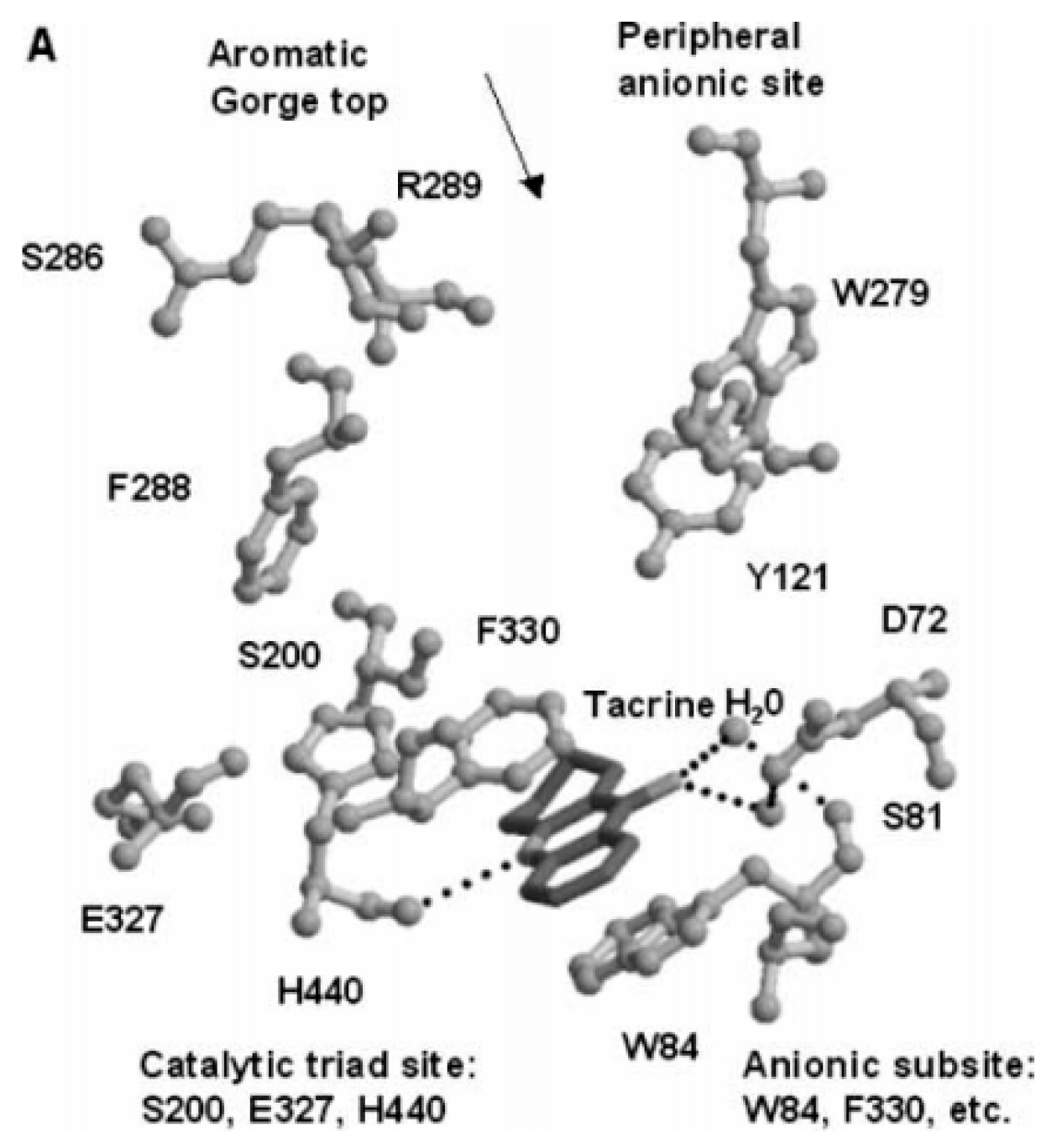
5. Acetylcholinesterase (AChE)
5.1. Design of Hybrid Compounds Inhibiting the Cholinesterase
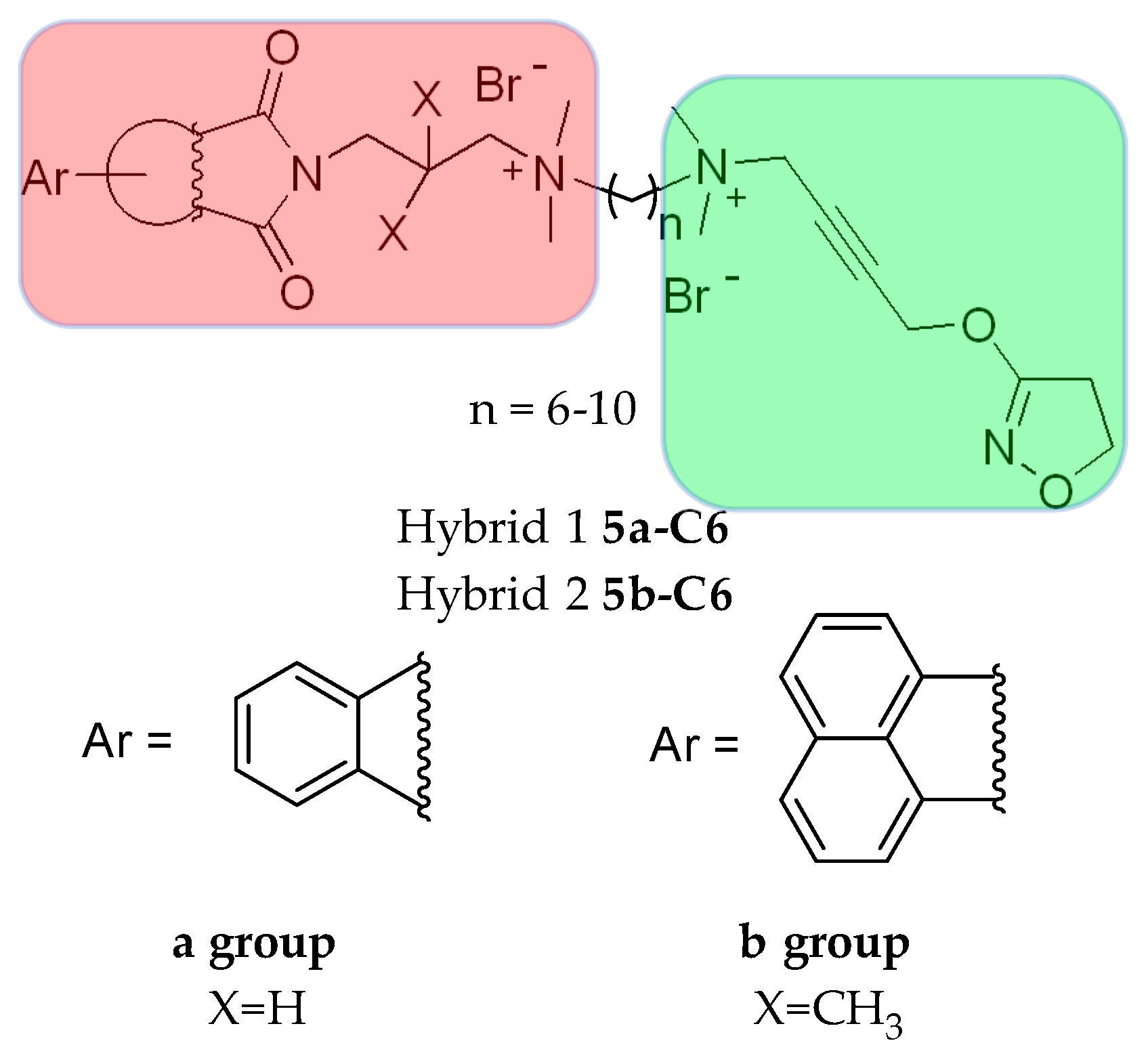
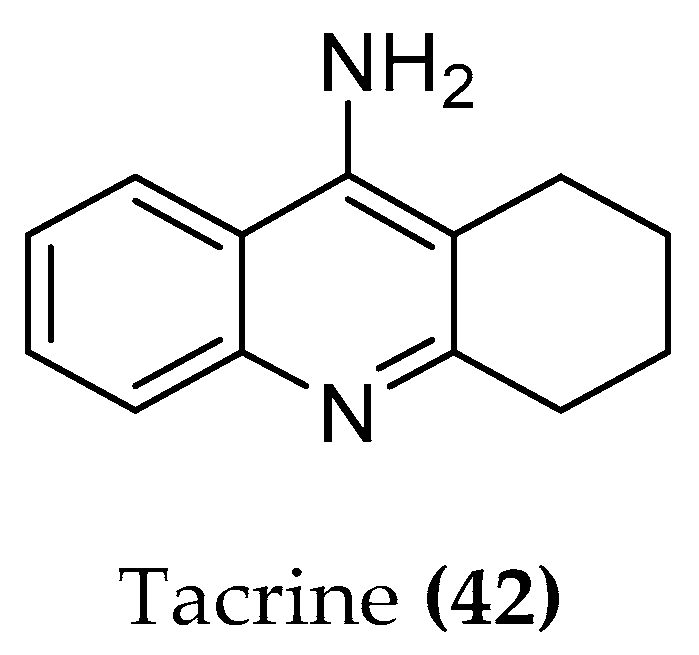
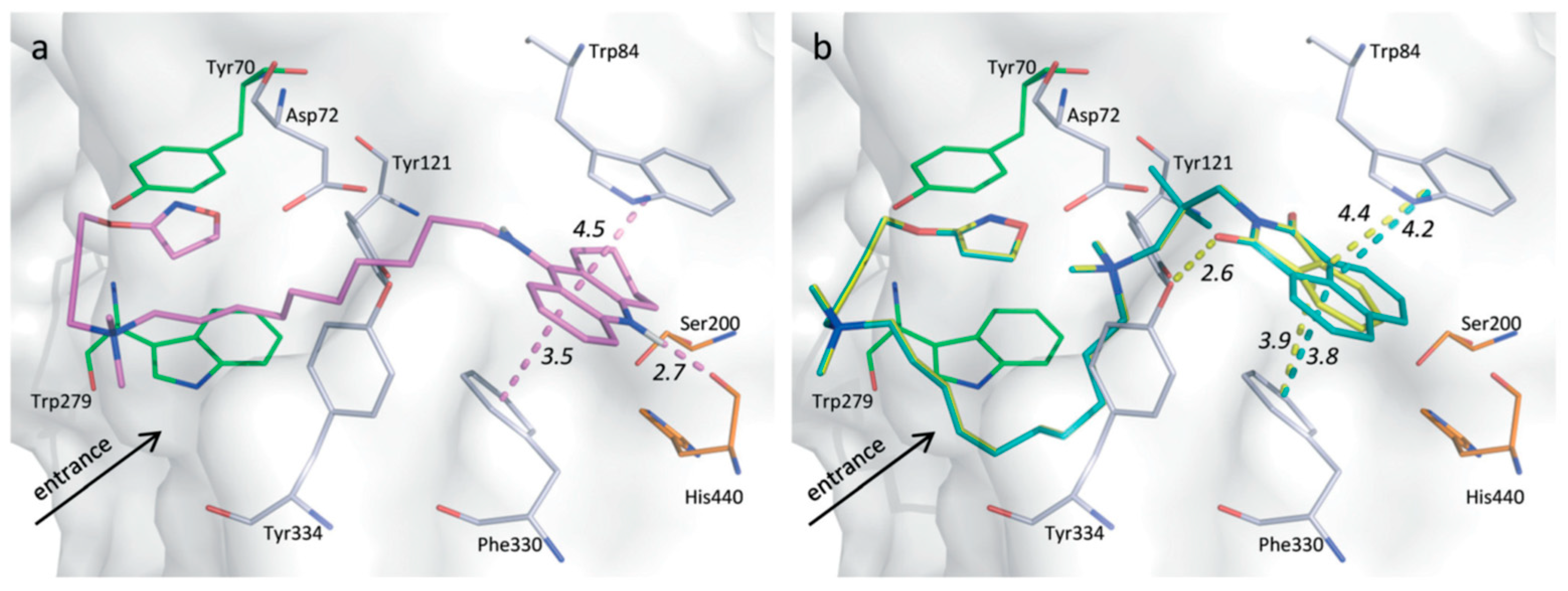
5.1.1. Tacrine Hybrids
5.1.2. Tacrine/Iperoxo hybrids
5.1.3. Tacrine/Xanomeline Hybrids
5.1.4. 77-LH-28-1/Xanomeline Hybrids
6. Nicotinic Receptor
6.1. Design of Hybrid Compounds to Investigate the Nicotinic Receptor
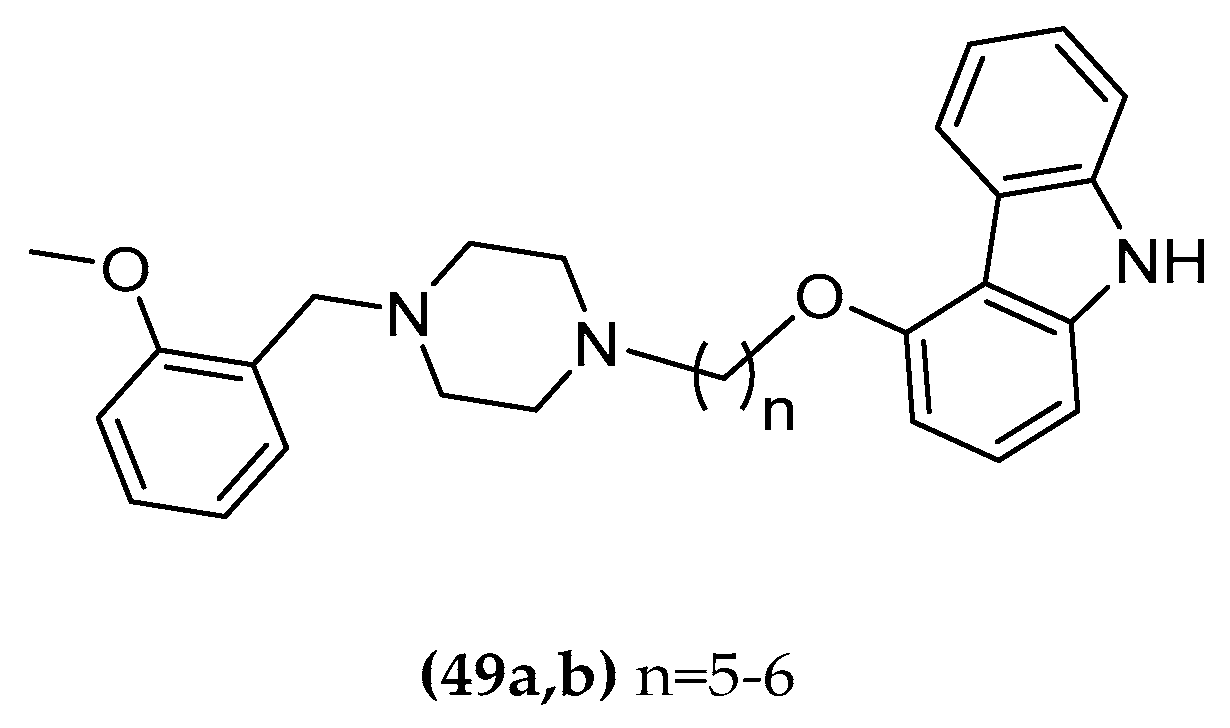
1-(2-Methoxybenzyl)-Piperazine-Carbazole Hybrids
7. Looking to the Future for AD Hybrids Tools
Funding
Conflicts of Interest
References
- Anand, A.; Patience, A.A.; Sharma, N.; Khurana, N. The present and future of pharmacotherapy of Alzheimer’s disease: A comprehensive review. Eur. J. Pharmacol. 2017, 815, 364–375. [Google Scholar] [CrossRef]
- Davie, B.J.; Christopoulos, A.; Scammells, P.J. Development of M1 mAChR allosteric and bitopic ligands: Prospective therapeutics for the treatment of cognitive deficits. ACS Chem. Neurosci. 2013, 4, 1026–1048. [Google Scholar] [CrossRef]
- Verma, S.; Kumar, A.; Tripathi, T.; Kumar, A. Muscarinic and nicotinic acetylcholine receptor agonists: Current scenario in Alzheimer’s disease therapy. J. Pharm. Pharmacol. 2018, 70, 985–993. [Google Scholar] [CrossRef]
- Li, J.J.; Dolios, G.; Wang, R.; Liao, F.F. Soluble beta-amyloid peptides, but not insoluble fibrils, have specific effect on neuronal microRNA expression. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Langmead, C.J.; Watson, J.; Reavill, C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 2008, 117, 232–243. [Google Scholar] [CrossRef]
- Mikiciuk-Olasik, E.; Szymański, P.; Zurek, E. Diagnostics and therapy of Alzheimer’s disease. Indian J. Exp. Biol. 2007, 45, 315–325. [Google Scholar]
- Hassan, M.; Abbas, Q.; Seo, S.Y.; Shahzadi, S.; Ashwal, H.; Zaki, N.; Iqbal, Z.; Moustafa, A.A. Computational modeling and biomarker studies of pharmacological treatment of Alzheimer’s disease (Review). Mol. Med. Rep. 2018, 18, 639–655. [Google Scholar] [CrossRef]
- Francis, P.T. The Interplay of Neurotransmitters in Alzheimer’s Disease. CNS Spectr. 2005, 10, 6–9. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinergic function and Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18. [Google Scholar] [CrossRef]
- Decker, M. Design of Hybrid Molecules for Drug Development; Elsevier: Cambridge, MA, USA, 2017. [Google Scholar]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef]
- Hamley, I.W. The amyloid beta peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- Wang, H.W.; Pasternak, J.F.; Kuo, H.; Ristic, H.; Lambert, M.P.; Chromy, B.; Viola, K.L.; Klein, W.L.; Stine, W.B.; Krafft, G.A.; et al. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002, 924, 133–140. [Google Scholar] [CrossRef]
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B.; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar] [CrossRef]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Rönicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 2012, 485, 651–655. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Esparza, T.J.; Wildburger, N.C.; Jiang, H.; Gangolli, M.; Cairns, N.J.; Bateman, R.J.; Brody, D.L. Soluble Amyloid-beta Aggregates from Human Alzheimer’s Disease Brains. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Tasker, A.; Perry, E.K.; Ballard, C.G. Butyrylcholinesterase: Impact on symptoms and progression of cognitive impairment. Expert Rev. Neurother. 2005, 5, 101–106. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol. Res. 2004, 50, 433–440. [Google Scholar] [CrossRef]
- Geula, C.; Darvesh, S. Butyrylcholinesterase, cholinergic neurotransmission and the pathology of Alzheimer’s disease. Drugs Today 2004, 40. [Google Scholar] [CrossRef]
- Auld, D.S.; Kar, S.; Quirion, R. β-Amyloid peptides as direct cholinergic neuromodulators: A missing link? Trends Neurosci. 1998, 21, 43–49. [Google Scholar] [CrossRef]
- Valant, C.; Robert Lane, J.; Sexton, P.M.; Christopoulos, A. The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 153–178. [Google Scholar] [CrossRef]
- Wermuth, C.G. Multitargeted drugs: The end of the ‘one-target-one-disease’ philosophy? Drug Discov. Today 2004, 9, 826–827. [Google Scholar] [CrossRef]
- Muñoz-Torrero, D. Multitarget Anti-Alzheimer Hybrid Compounds. Des. Hybrid Mol. Drug Dev. 2017, 167–192. [Google Scholar] [CrossRef]
- Gregory, K.J.; Sexton, P.M.; Christopoulos, A. Overview of receptor allosterism. Curr. Protoc. Pharmacol. 2010. [Google Scholar] [CrossRef]
- Burger, W.A.C.; Sexton, P.M.; Christopoulos, A.; Thal, D.M. Toward an understanding of the structural basis of allostery in muscarinic acetylcholine receptors. J. Gen. Physiol. 2018. [Google Scholar] [CrossRef]
- Bock, A.; Schrage, R.; Mohr, K. Allosteric modulators targeting CNS muscarinic receptors. Neuropharmacology 2017. [Google Scholar] [CrossRef]
- Mohr, K.; Schmitz, J.; Schrage, R.; Tränkle, C.; Holzgrabe, U. Molecular alliance-from orthosteric and allosteric ligands to dualsteric/bitopic agonists at G protein coupled receptors. Angew. Chem. 2013, 52, 508–516. [Google Scholar] [CrossRef]
- Conn, P.J.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar] [CrossRef]
- Kenakin, T.; Christopoulos, A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 2013, 12, 205–216. [Google Scholar] [CrossRef]
- Bock, A.; Kostenis, E.; Trankle, C.; Lohse, M.J.; Mohr, K. Pilot the pulse: Controlling the multiplicity of receptor dynamics. Trends Pharmacol. Sci. 2014, 35, 630–638. [Google Scholar] [CrossRef]
- Wild, C.; Cunningham, K.A.; Zhou, J. Allosteric Modulation of G Protein-Coupled Receptors: An Emerging Approach of Drug Discovery. Austin J. Pharmacol. Ther. 2014. [Google Scholar] [CrossRef]
- Ehlert, F.J. Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol. Pharmacol. 1988, 33, 187–194. [Google Scholar]
- Lazareno, S.; Dolezal, V.; Popham, A.; Birdsall, N.J.M. Thiochrome enhances acetylcholine affinity at muscarinic M4 receptors: Receptor subtype selectivity via cooperativity rather than affinity. Mol. Pharmacol. 2004, 65, 257–266. [Google Scholar] [CrossRef]
- Decker, M.; Holzgrabe, U. M1 muscarinic Acetylcholine receptor allosteric modulators as potential therapeutic opportunities for treating Alzheimer’s disease. Med. Chem. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Mohr, K.; Tränkle, C.; Kostenis, E.; Barocelli, E.; Amici, M.; Holzgrabe, U. Rational design of dualsteric GPCR ligands: Quests and promise. Br. J. Pharmacol. 2010, 159, 997–1008. [Google Scholar] [CrossRef]
- Valant, C.; Sexton, P.M.; Christopoulos, A. Orthosteric/allosteric bitopic ligands: Going hybrid at GPCRs. Mol. Interv. 2009, 9, 125–135. [Google Scholar] [CrossRef]
- Schwyzer, R. Acth: A short Introductory review. Ann. N. Y. Acad. Sci. 1977, 297, 3–26. [Google Scholar] [CrossRef]
- Portoghese, P.S. Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol. Sci. 1989, 10, 230–235. [Google Scholar] [CrossRef]
- Matera, C.; Tata, A. Pharmacological Approaches to Targeting Muscarinic Acetylcholine Receptors. RPCN 2014, 9, 85–100. [Google Scholar] [CrossRef]
- Sánchez-Fernández, G.; Cabezudo, S.; García-Hoz, C.; Tobin, A.B.; Mayor, F.; Ribas, C. ERK5 activation by Gq-coupled muscarinic receptors is independent of receptor internalization and β-arrestin recruitment. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Kenakin, T. Signaling bias in drug discovery. Expert Opin. Drug Discov. 2017, 12, 321–333. [Google Scholar] [CrossRef]
- Keller, M.; Tränkle, C.; She, X.; Pegoli, A.; Bernhardt, G.; Buschauer, A.; Read, R.W. M2 Subtype preferring dibenzodiazepinone-type muscarinic receptor ligands: Effect of chemical homo-dimerization on orthosteric (and allosteric?) binding. Bioorg. Med. Chem. 2015, 23, 3970–3990. [Google Scholar] [CrossRef]
- Pegoli, A.; She, X.; Wifling, D.; Hübner, H.; Bernhardt, G.; Gmeiner, P.; Keller, M. Radiolabeled Dibenzodiazepinone-Type Antagonists Give Evidence of Dualsteric Binding at the M2 Muscarinic Acetylcholine Receptor. J. Med. Chem. 2017, 60, 3314–3334. [Google Scholar] [CrossRef]
- She, X.; Pegoli, A.; Mayr, J.; Hübner, H.; Bernhardt, G.; Gmeiner, P.; Keller, M. Heterodimerization of Dibenzodiazepinone-Type Muscarinic Acetylcholine Receptor Ligands Leads to Increased M 2 R Affinity and Selectivity. ACS Omega 2017, 2, 6741–6754. [Google Scholar] [CrossRef]
- Narlawar, R.; Lane, J.R.; Doddareddy, M.; Lin, J.; Brussee, J.; Ijzerman, A.P. Hybrid ortho/allosteric ligands for the adenosine A (1) receptor. J. Med. Chem. 2010, 53, 3028–3037. [Google Scholar] [CrossRef]
- Aurelio, L.; Valant, C.; Flynn, B.L.; Sexton, P.M.; Christopoulos, A.; Scammells, P.J. Allosteric modulators of the adenosine A1 receptor: Synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J. Med. Chem. 2009, 52, 4543–4547. [Google Scholar] [CrossRef]
- Steinfeld, T.; Mammen, M.; Smith, J.A.M.; Wilson, R.D.; Jasper, J.R. A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol. Pharmacol. 2007, 72, 291–302. [Google Scholar] [CrossRef]
- Lane, J.R.; Donthamsetti, P.; Shonberg, J.; Draper-Joyce, C.J.; Dentry, S.; Michino, M.; Shi, L.; López, L.; Scammells, P.J.; Capuano, B.; et al. A new mechanism of allostery in a G protein-coupled receptor dimer. Nat. Chem. Biol. 2014, 10, 745–752. [Google Scholar] [CrossRef]
- Stemp, G.; Ashmeade, T.; Branch, C.L.; Hadley, M.S.; Hunter, A.J.; Johnson, C.N.; Nash, D.J.; Thewlis, K.M.; Vong, A.K.K.; Austin, N.E.; et al. Design and Synthesis of trans—N -[4-[2-(6-Cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide (SB-277011): A Potent and Selective Dopamine D 3 Receptor Antagonist with High Oral Bioavailability and CNS Penetration in the Rat. J. Med. Chem. 2000, 43, 1878–1885. [Google Scholar] [CrossRef]
- Rossi, M.; Fasciani, I.; Marampon, F.; Maggio, R.; Scarselli, M. The First Negative Allosteric Modulator for Dopamine D2 and D3 Receptors, SB269652 May Lead to a New Generation of Antipsychotic Drugs. Mol. Pharmacol. 2017, 91, 586–594. [Google Scholar] [CrossRef]
- Kopinathan, A.; Scammells, P.J.; Lane, J.R.; Capuano, B. Multivalent approaches and beyond: Novel tools for the investigation of dopamine D2 receptor pharmacology. Future Med. Chem. 2016, 8, 1349–1372. [Google Scholar] [CrossRef]
- Jo, E.; Bhhatarai, B.; Repetto, E.; Guerrero, M.; Riley, S.; Brown, S.J.; Kohno, Y.; Roberts, E.; Schürer, S.C.; Rosen, H. Novel selective allosteric and bitopic ligands for the S1P (3) receptor. ACS Chem. Biol. 2012, 7, 1975–1983. [Google Scholar] [CrossRef]
- Sanna, M.G.; Vincent, K.P.; Repetto, E.; Nguyen, N.; Brown, S.J.; Abgaryan, L.; Riley, S.W.; Leaf, N.B.; Cahalan, S.M.; Kiosses, W.B.; et al. Bitopic Sphingosine 1-Phosphate Receptor 3 (S1P3) Antagonist Rescue from Complete Heart Block: Pharmacological and Genetic Evidence for Direct S1P3 Regulation of Mouse Cardiac Conduction. Mol. Pharmacol. 2016, 89, 176–186. [Google Scholar] [CrossRef]
- Antony, J.; Kellershohn, K.; Mohr-Andra, M.; Kebig, A.; Prilla, S.; Muth, M.; Heller, E.; Disingrini, T.; Dallanoce, C.; Bertoni, S.; et al. Dualsteric GPCR targeting: A novel route to binding and signaling pathway selectivity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 442–450. [Google Scholar]
- Chen, X.; Klockner, J.; Holze, J.; Zimmermann, C.; Seemann, W.K.; Schrage, R.; Bock, A.; Mohr, K.; Tränkle, C.; Holzgrabe, U.; et al. Rational design of partial agonists for the muscarinic m1 acetylcholine receptor. J. Med. Chem. 2015, 58, 560–576. [Google Scholar] [CrossRef]
- Messerer, R.; Kauk, M.; Volpato, D.; Alonso Canizal, M.C.; Kloeckner, J.; Zabel, U.; Nuber, S.; Hoffmann, C.; Holzgrabe, U. FRET Studies of Quinolone-Based Bitopic Ligands and their Structural Analogues at the Muscarinic M1 Receptor. ACS Chem. Biol. 2017. [Google Scholar] [CrossRef]
- Schmitz, J.; van der Mey, D.; Bermudez, M.; Klockner, J.; Schrage, R.; Kostenis, E.; Tränkle, C.; Wolber, G.; Mohr, K.; Holzgrabe, U. Dualsteric muscarinic antagonists—Orthosteric binding pose controls allosteric subtype selectivity. J. Med. Chem. 2014, 57, 6739–6750. [Google Scholar] [CrossRef]
- Fronik, P.; Gaiser, B.I.; Sejer Pedersen, D. Bitopic Ligands and Metastable Binding Sites: Opportunities for G Protein-Coupled Receptor (GPCR) Medicinal Chemistry. J. Med. Chem. 2017, 60, 4126–4134. [Google Scholar] [CrossRef]
- Paoletta, S.; Sabbadin, D.; Kügelgen, I.; Hinz, S.; Katritch, V.; Hoffmann, K.; Abdelrahman, A.; Straburger, J.; Baqi, Y.; Zhao, Q.; et al. Modeling ligand recognition at the P2Y12 receptor in light of X-ray structural information. J. Comput. Aided Mol. Des. 2015, 29, 737–756. [Google Scholar] [CrossRef]
- Sabbadin, D.; Ciancetta, A.; Deganutti, G.; Cuzzolin, A.; Moro, S. Exploring the recognition pathway at the human A 2A adenosine receptor of the endogenous agonist adenosine using supervised molecular dynamics simulations. Med. Chem. Commun. 2015, 6, 1081–1085. [Google Scholar] [CrossRef]
- Sabbadin, D.; Ciancetta, A.; Moro, S. Bridging Molecular Docking to Membrane Molecular Dynamics to Investigate GPCR–Ligand Recognition: The Human A 2A Adenosine Receptor as a Key Study. J. Chem. Inf. Model. 2013, 54, 169–183. [Google Scholar] [CrossRef]
- Buch, I.; Giorgino, T.; Fabritiis, G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 10184–10189. [Google Scholar] [CrossRef]
- González, A.; Perez-Acle, T.; Pardo, L.; Deupi, X.; van Veen, H.W. Molecular Basis of Ligand Dissociation in β-Adrenergic Receptors. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef]
- Berque-Bestel, I.; Lezoualch, F.; Jockers, R. Bivalent Ligands as Specific Pharmacological Tools for G Protein-Coupled Receptor Dimers. CDDT 2008, 5, 312–318. [Google Scholar] [CrossRef]
- Disingrini, T.; Muth, M.; Dallanoce, C.; Barocelli, E.; Bertoni, S.; Kellershohn, K.; Mohr, K.; Amici, M.; Holzgrabe, U. Design, synthesis, and action of oxotremorine-related hybrid-type allosteric modulators of muscarinic acetylcholine receptors. J. Med. Chem. 2006, 49, 366–372. [Google Scholar] [CrossRef]
- Abrams, P.; Andersson, K.E.; Buccafusco, J.J.; Chapple, C.; Groat, W.C.; Fryer, A.D.; Kay, G.; Laties, A.; Nathanson, N.M.; Pasricha, P.J.; et al. Muscarinic receptors: Their distribution and function in body systems, and the implications for treating overactive bladder. Br. J. Pharmacol. 2006, 148, 565–578. [Google Scholar] [CrossRef]
- Bradley, S.J.; Bourgognon, J.M.; Sanger, H.E.; Verity, N.; Mogg, A.J.; White, D.J.; Butcher, A.J.; Moreno, J.A.; Molloy, C.; Macedo-Hatch, T.; et al. M1 muscarinic allosteric modulators slow prion neurodegeneration and restore memory loss. J. Clin. Investing. 2017, 127, 487–499. [Google Scholar] [CrossRef]
- Caccamo, A.; Oddo, S.; Billings, L.M.; Green, K.N.; Martinez-Coria, H.; Fisher, A.; LaFerla, F.M. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 2006, 49, 671–682. [Google Scholar] [CrossRef]
- Fisher, A. Cholinergic treatments with emphasis on m1 muscarinic agonists as potential disease-modifying agents for Alzheimer’s disease. Neurotherapeutics 2008, 5, 433–442. [Google Scholar] [CrossRef]
- Fisher, A.; Brandeis, R.; Bar-Ner, R.H.N.; Kliger-Spatz, M.; Natan, N.; Sonego, H.; Marcovitch, I.; Pittel, Z. AF150 (S) and AF267B: M1 muscarinic agonists as innovative therapies for Alzheimer’s disease. J. Mol. Neurosci. MN 2002, 19, 145–153. [Google Scholar] [CrossRef]
- Sams, A.G.; Hentzer, M.; Mikkelsen, G.K.; Larsen, K.; Bundgaard, C.; Plath, N.; Christoffersen, C.T.; Bang-Andersen, B. Discovery of N-{1-3-(3-oxo-2,3-dihydrobenzo1,4oxazin-4-yl)propylpiperidin-4-yl}-2-phenylacetamide (Lu AE51090): An allosteric muscarinic M1 receptor agonist with unprecedented selectivity and procognitive potential. J. Med. Chem. 2010, 53, 6386–6397. [Google Scholar] [CrossRef]
- Budzik, B.; Garzya, V.; Shi, D.; Foley, J.J.; Rivero, R.A.; Langmead, C.J.; Watson, J.; Wu, Z.; Forbes, I.T.; Jin, J. 2’biaryl amides as novel and subtype selective M1 agonists. Part I: Identification, synthesis, and initial SAR. Bioorg. Med. Chem. Lett. 2010, 20, 3540–3544. [Google Scholar] [CrossRef]
- Budzik, B.; Garzya, V.; Shi, D.; Walker, G.; Woolley-Roberts, M.; Pardoe, J.; Lucas, A.; Tehan, B.; Rivero, R.A.; Langmead, C.J.; et al. Novel N-Substituted Benzimidazolones as Potent, Selective, CNS-Penetrant, and Orally Active M1 mAChR Agonists. ACS Med. Chem. Lett. 2010, 1, 244–248. [Google Scholar] [CrossRef]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hubner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef]
- Matera, C.; Flammini, L.; Quadri, M.; Vivo, V.; Ballabeni, V.; Holzgrabe, U.; Mohr, K.; Amici, M.; Barocelli, E.; Bertoni, S.; et al. Bis(ammonio)alkane-type agonists of muscarinic acetylcholine receptors: Synthesis, in vitro functional characterization, and in vivo evaluation of their analgesic activity. Eur. J. Med. Chem. 2014, 75, 222–232. [Google Scholar] [CrossRef]
- Bock, A.; Merten, N.; Schrage, R.; Dallanoce, C.; Batz, J.; Klockner, J.; Schmitz, J.; Matera, C.; Simon, K.; Kebig, A.; et al. The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
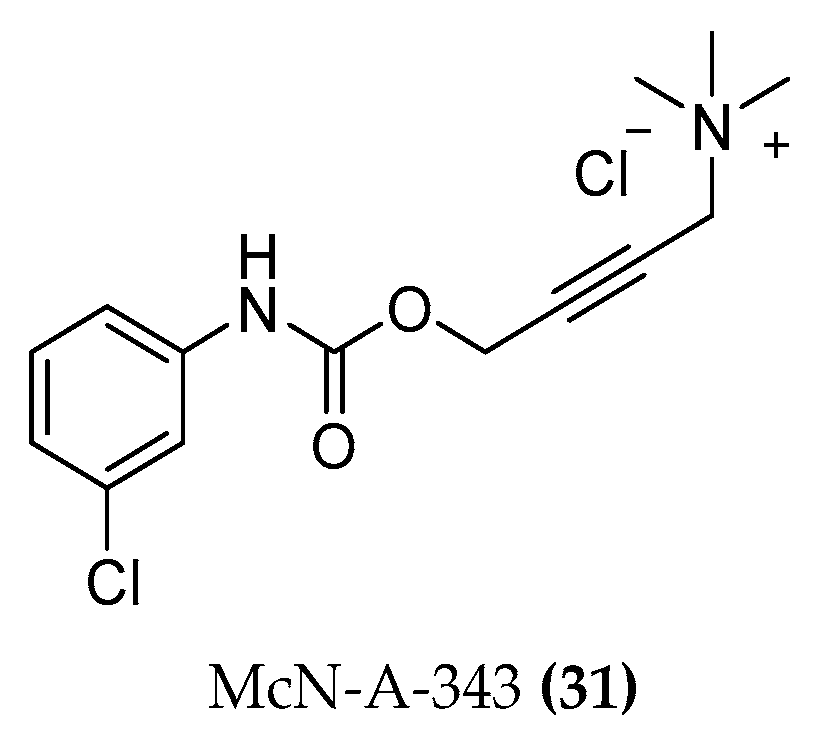
- Valant, C.; Gregory, K.J.; Hall, N.E.; Scammells, P.J.; Lew, M.J.; Sexton, P.M.; Christopoulos, A. A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J. Biol. Chem. 2008, 283, 29312–29321. [Google Scholar] [CrossRef]
- Bock, A.; Bermudez, M.; Krebs, F.; Matera, C.; Chirinda, B.; Sydow, D.; Dallanoce, C.; Holzgrabe, U.; Amici, M.; Lohse, M.J.; et al. Ligand Binding Ensembles Determine Graded Agonist Efficacies at a G Protein-coupled Receptor. J. Biol. Chem. 2016, 291, 16375–16389. [Google Scholar] [CrossRef]
- Ma, L.; Seager, M.A.; Seager, M.; Wittmann, M.; Jacobson, M.; Bickel, D.; Burno, M.; Jones, K.; Graufelds, V.K.; Xu, G.; et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 15950–15955. [Google Scholar] [CrossRef]
- Wess, J. Allosteric binding sites on muscarinic acetylcholine receptors. Mol. Pharmacol. 2005, 68, 1506–1509. [Google Scholar]
- Felder, C.C.; Goldsmith, P.J.; Jackson, K.; Sanger, H.E.; Evans, D.A.; Mogg, A.J.; Broad, L.M. Current status of muscarinic M1 and M4 receptors as drug targets for neurodegenerative diseases. Neuropharmacology 2018, 136, 449–458. [Google Scholar] [CrossRef]
- Bock, A.; Chirinda, B.; Krebs, F.; Messerer, R.; Batz, J.; Muth, M.; Dallanoce, C.; Klingenthal, D.; Tränkle, C.; Hoffmann, C.; et al. Dynamic ligand binding dictates partial agonism at a G protein-coupled receptor. Nat. Chem. Biol. 2014, 10, 18–20. [Google Scholar] [CrossRef]
- Choi, M.; Staus, D.P.; Wingler, L.M.; Ahn, S.; Pani, B.; Capel, W.D.; Lefkowitz, R.J. G protein-coupled receptor kinases (GRKs) orchestrate biased agonism at the β2-adrenergic receptor. Science Signal. 2018, 11. [Google Scholar] [CrossRef]
- Bonifazi, A.; Yano, H.; Del Bello, F.; Farande, A.; Quaglia, W.; Petrelli, R.; Matucci, R.; Nesi, M.; Vistoli, G.; Ferre, S.; et al. Synthesis and biological evaluation of a novel series of heterobivalent muscarinic ligands based on xanomeline and 1-3-(4-butylpiperidin-1-yl)propyl-1,2,3,4-tetrahydroquinolin-2-one (77-LH-28-1). J. Med. Chem. 2014, 57, 9065–9077. [Google Scholar] [CrossRef]
- Agnetta, L.; Kauk, M.; Canizal, M.C.A.; Messerer, R.; Holzgrabe, U.; Hoffmann, C.; Decker, M. A Photoswitchable Dualsteric Ligand Controlling Receptor Efficacy. Angew. Chem. 2017, 56, 7282–7287. [Google Scholar] [CrossRef]
- Watt, M.L.; Schober, D.A.; Hitchcock, S.; Liu, B.; Chesterfield, A.K.; McKinzie, D.; Felder, C.C. Pharmacological characterization of LY593093, an M1 muscarinic acetylcholine receptor-selective partial orthosteric agonist. J. Pharmacol. Exp. Ther. 2011, 338, 622–632. [Google Scholar] [CrossRef]
- Avlani, V.A.; Langmead, C.J.; Guida, E.; Wood, M.D.; Tehan, B.G.; Herdon, H.J.; Watson, J.M.; Sexton, P.M.; Christopoulos, A. Orthosteric and allosteric modes of interaction of novel selective agonists of the M1 muscarinic acetylcholine receptor. Mol. Pharmacol. 2010, 78, 94–104. [Google Scholar] [CrossRef]
- Langmead, C.J.; Fry, V.A.H.; Forbes, I.T.; Branch, C.L.; Christopoulos, A.; Wood, M.D.; Herdon, H.J. Probing the molecular mechanism of interaction between 4-n-butyl-1-4-(2-methylphenyl)-4-oxo-1-butyl-piperidine (AC-42) and the muscarinic M (1) receptor: Direct pharmacological evidence that AC-42 is an allosteric agonist. Mol. Pharmacol. 2006, 69, 236–246. [Google Scholar]
- Thomas, R.L.; Mistry, R.; Langmead, C.J.; Wood, M.D.; Challiss, R.A.J. G protein coupling and signaling pathway activation by m1 muscarinic acetylcholine receptor orthosteric and allosteric agonists. J. Pharmacol. Exp. Ther. 2008, 327, 365–374. [Google Scholar] [CrossRef]
- Spalding, T.A.; Ma, J.-N.; Ott, T.R.; Friberg, M.; Bajpai, A.; Bradley, S.R.; Davis, R.E.; Brann, M.R.; Burstein, E.S. Structural requirements of transmembrane domain 3 for activation by the M1 muscarinic receptor agonists AC-42, AC-260584, clozapine, and N-desmethylclozapine: Evidence for three distinct modes of receptor activation. Mol. Pharmacol. 2006, 70, 1974–1983. [Google Scholar] [CrossRef]
- Zhang, S.; Saathoff, J.M.; He, L. 8-Molecular Hybridization: An Emerging Tool for the Design of Novel Therapeutics for Alzheimer’s Disease. Des. Hybrid Mol. Drug Dev. 2017. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Inestrosa, N.C. Interactions of AChE with Aβ Aggregates in Alzheimer’s Brain: Therapeutic Relevance of IDN 5706. Front. Mol. Neurosci. 2011, 4. [Google Scholar] [CrossRef]
- Sussman, J.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Colletier, J.P.; Fournier, D.; Greenblatt, H.M.; Stojan, J.; Sussman, J.L.; Zaccai, G.; Silman, I.; Weik, M. Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J. 2006, 25, 2746–2756. [Google Scholar] [CrossRef]
- Zheng, Z.H.; Dong, Y.S.; Zhang, H.; Lu, X.H.; Ren, X.; Zhao, G.; He, J.G.; Si, S.Y. Isolation and characterization of N98-1272 A, B and C, selective acetylcholinesterase inhibitors from metabolites of an actinomycete strain. J. Enzyme Inhib. Med. Chem. 2007, 22, 43–49. [Google Scholar] [CrossRef]
- Bullock, R.; Bergman, H.; Touchon, J.; Gambina, G.; He, Y.; Nagel, J.; Lane, R. Effect of age on response to rivastigmine or donepezil in patients with Alzheimer’s disease. Curr. Med. Res. Opin. 2006, 22, 483–494. [Google Scholar] [CrossRef]
- Bullock, R.; Touchon, J.; Bergman, H.; Gambina, G.; He, Y.; Rapatz, G.; Nagel, J.; Lane, R. Rivastigmine and donepezil treatment in moderate to moderately-severe Alzheimer’s disease over a 2-year period. Curr. Med. Res. Opin. 2005, 21, 1317–1327. [Google Scholar] [CrossRef]
- Mueller, B.; Adler, G. Prevalence of Wild-Type Butyrylcholinesterase Genotype in Patients with Alzheimer’s Dementia. WJNS 2015, 05, 175–179. [Google Scholar] [CrossRef]
- Muñoz-Torrero, D. Acetylcholinesterase Inhibitors as Disease-Modifying Therapies for Alzheimers Disease. CMC 2008, 15, 2433–2455. [Google Scholar] [CrossRef]
- Mehta, M.; Adem, A.; Sabbagh, M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2012. [Google Scholar] [CrossRef]
- Stepankova, S.; Komers, K. Cholinesterases and Cholinesterase Inhibitors. CEI 2008, 4, 160–171. [Google Scholar] [CrossRef]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- McGleenon, B.M.; Dynan, P. Acetylcholinesterase inhibitors in Alzheimer’s disease. Br. J. Clin. Pharmacol. 1999, 48, 471–480. [Google Scholar] [CrossRef]
- Pang, Y.P.; Hong, F.; Quiram, P.; Jelacic, T.; Brimijoin, S. Synthesis of alkylene linked bis-THA and alkylene linked benzyl-THA as highly potent and selective inhibitors and molecular probes of acetylcholinesterase. J. Chem. Soc. Perkin Trans. 1997. [Google Scholar] [CrossRef]
- Messerer, R.; Dallanoce, C.; Matera, C.; Wehle, S.; Flammini, L.; Chirinda, B.; Bock, A.; Irmen, M.; Tränkle, C.; Barocelli, E.; et al. Novel bipharmacophoric inhibitors of the cholinesterases with affinity to the muscarinic receptors M 1 and M 2. Med. Chem. Commun. 2017, 8, 1346–1359. [Google Scholar] [CrossRef]
- Messerer, R. Synthesis of Dualsteric Ligands for Muscarinic Acetylcholine Receptors and Cholinesterase Inhibitors. Ph.D Thesis, Universität Würzburg, Würzburg, German, March 2017. [Google Scholar]
- Campiani, G.; Fattorusso, C.; Butini, S.; Gaeta, A.; Agnusdei, M.; Gemma, S.; Persico, M.; Catalanotti, B.; Savini, L.; Nacci, V.; et al. Development of molecular probes for the identification of extra interaction sites in the mid-gorge and peripheral sites of butyrylcholinesterase (BuChE). Rational design of novel, selective, and highly potent BuChE inhibitors. J. Med. Chem. 2005, 48, 1919–1929. [Google Scholar] [CrossRef]
- Carlier, P.R.; Han, Y.F.; Chow, E.S.H.; Li, C.P.L.; Wang, H.; Lieu, T.X.; Wong, H.S.; Pang, Y.P. Evaluation of short-tether Bis-THA AChE inhibitors. A further test of the dual binding site hypothesis. Bioorg. Med. Chem. 1999, 7, 351–357. [Google Scholar] [CrossRef]
- Fang, L.; Jumpertz, S.; Zhang, Y.; Appenroth, D.; Fleck, C.; Mohr, K.; Tränkle, C.; Decker, M. Hybrid molecules from xanomeline and tacrine: Enhanced tacrine actions on cholinesterases and muscarinic M1 receptors. J. Med. Chem. 2010, 53, 2094–2103. [Google Scholar] [CrossRef]
- Bymaster, F.P.; Whitesitt, C.A.; Shannon, H.E.; DeLapp, N.; Ward, J.S.; Calligaro, D.O.; Shipley, L.A.; Buelke-Sam, J.L.; Bodick, N.C.; Farde, L.; et al. Xanomeline: A selective muscarinic agonist for the treatment of Alzheimer’s disease. Drug Dev. Res. 1997, 40, 158–170. [Google Scholar] [CrossRef]
- Farrell, M.; Roth, B.L. Allosteric antipsychotics: M4 muscarinic potentiators as novel treatments for schizophrenia. Neuropsychopharmacology 2010, 35, 851–852. [Google Scholar] [CrossRef]
- Mirza, N.R.; Peters, D.; Sparks, R.G. Xanomeline and the Antipsychotic Potential of Muscarinic Receptor Subtype Selective Agonists. CNS Drug Rev. 2003, 9, 159–186. [Google Scholar] [CrossRef]
- Jakubík, J.; El-Fakahany, E.E.; Dolezal, V. Differences in kinetics of xanomeline binding and selectivity of activation of G proteins at M (1) and M (2) muscarinic acetylcholine receptors. Mol. Pharmacol. 2006, 70, 656–666. [Google Scholar] [CrossRef]
- Jakubík, J.; Tucek, S.; El-Fakahany, E.E. Role of receptor protein and membrane lipids in xanomeline wash-resistant binding to muscarinic M1 receptors. J. Pharmacol. Exp. Ther. 2004, 308, 105–110. [Google Scholar] [CrossRef]
- Kane, B.E.; Grant, M.K.O.; El-Fakahany, E.E.; Ferguson, D.M. Synthesis and evaluation of xanomeline analogs—Probing the wash-resistant phenomenon at the M1 muscarinic acetylcholine receptor. Bioorg. Med. Chem. 2008, 16, 1376–1392. [Google Scholar] [CrossRef]
- Randáková, A.; Dolejší, E.; Rudajev, V.; Zimčík, P.; Doležal, V.; El-Fakahany, E.E.; Jakubík, J. Classical and atypical agonists activate M1 muscarinic acetylcholine receptors through common mechanisms. Pharmacol. Res. 2015, 97, 27–39. [Google Scholar] [CrossRef]
- Kruse, A.C.; Kobilka, B.K.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic acetylcholine receptors: Novel opportunities for drug development. Nat. Rev. Drug Discov. 2014, 13, 549–560. [Google Scholar] [CrossRef]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef]
- Miwa, J.M.; Freedman, R.; Lester, H.A. Neural systems governed by nicotinic acetylcholine receptors: Emerging hypotheses. Neuron 2011, 70, 20–33. [Google Scholar] [CrossRef]
- Simoni, E.; Bartolini, M.; Abu, I.F.; Blockley, A.; Gotti, C.; Bottegoni, G.; Caporaso, R.; Bergamini, C.; Andrisano, V.; Cavalli, A.; et al. Multitarget drug design strategy in Alzheimer’s disease: Focus on cholinergic transmission and amyloid-β aggregation. Future Med. Chem. 2017, 9, 953–963. [Google Scholar] [CrossRef]
- Haydar, S.N.; Ghiron, C.; Bettinetti, L.; Bothmann, H.; Comery, T.A.; Dunlop, J.; La Rosa, S.; Micco, I.; Pollastrini, M.; Quinn, J.; et al. SAR and biological evaluation of SEN12333/WAY-317538: Novel alpha 7 nicotinic acetylcholine receptor agonist. Bioorg. Med. Chem. 2009, 17, 5247–5258. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Capsoni, S.; Andrisano, V.; Bartolini, M.; Margotti, E.; Cattaneo, A.; Recanatini, M.; Melchiorre, C. A small molecule targeting the multifactorial nature of Alzheimer’s disease. Angew. Chem. 2007, 46, 3689–3692. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Bartolini, M.; Tarozzi, A.; Matera, R.; Milelli, A.; Hrelia, P.; Andrisano, V.; Bolognesi, M.L.; Melchiorre, C. Exploiting the lipoic acid structure in the search for novel multitarget ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 5435–5442. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Bartolini, M.; Tarozzi, A.; Morroni, F.; Lizzi, F.; Milelli, A.; Minarini, A.; Rosini, M.; Hrelia, P.; Andrisano, V.; et al. Multitargeted drugs discovery: Balancing anti-amyloid and anticholinesterase capacity in a single chemical entity. Bioorg. Med. Chem. Lett. 2011, 21, 2655–2658. [Google Scholar] [CrossRef]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | BChE pIC50 [M] | AChE pIC50 [M] | Cytotoxicity HEP G2 IC50 [μM] |
|---|---|---|---|
| Tacrine (42) | 8.57 | 7.60 | 111 (±2.28) |
| Phth-7-iper (5a-C7) | 5.19 | 4.83 | n.d. 1 |
| Phth-8-iper (5a-C8) | 5.13 | 4.89 | n.d. 1 |
| Phth-9-iper (5a-C9) | 4.88 | 4.80 | n.d. 1 |
| Phth-10-iper (5a-C10) | 5.37 | 5.26 | n.d. 1 |
| Naph-7-iper (5b-C7) | 6.69 | 6.04 | n.d. 1 |
| Naph-8-iper (5b-C8) | 6.46 | 6.10 | n.d. 1 |
| Naph-9-iper (5b-C9) | 6.49 | 6.44 | n.d. 1 |
| Naph-10-iper (5b-C10) | 6.99 | 6.50 | n.d. 1 |
| Tac-7-Tac (43-C7) | 9.14 | 10.48 | <1.38 (±0.08) |
| Tac-10-Tac (43-C10) | 9.34 | 9.00 | <1.25 (±0.00) |
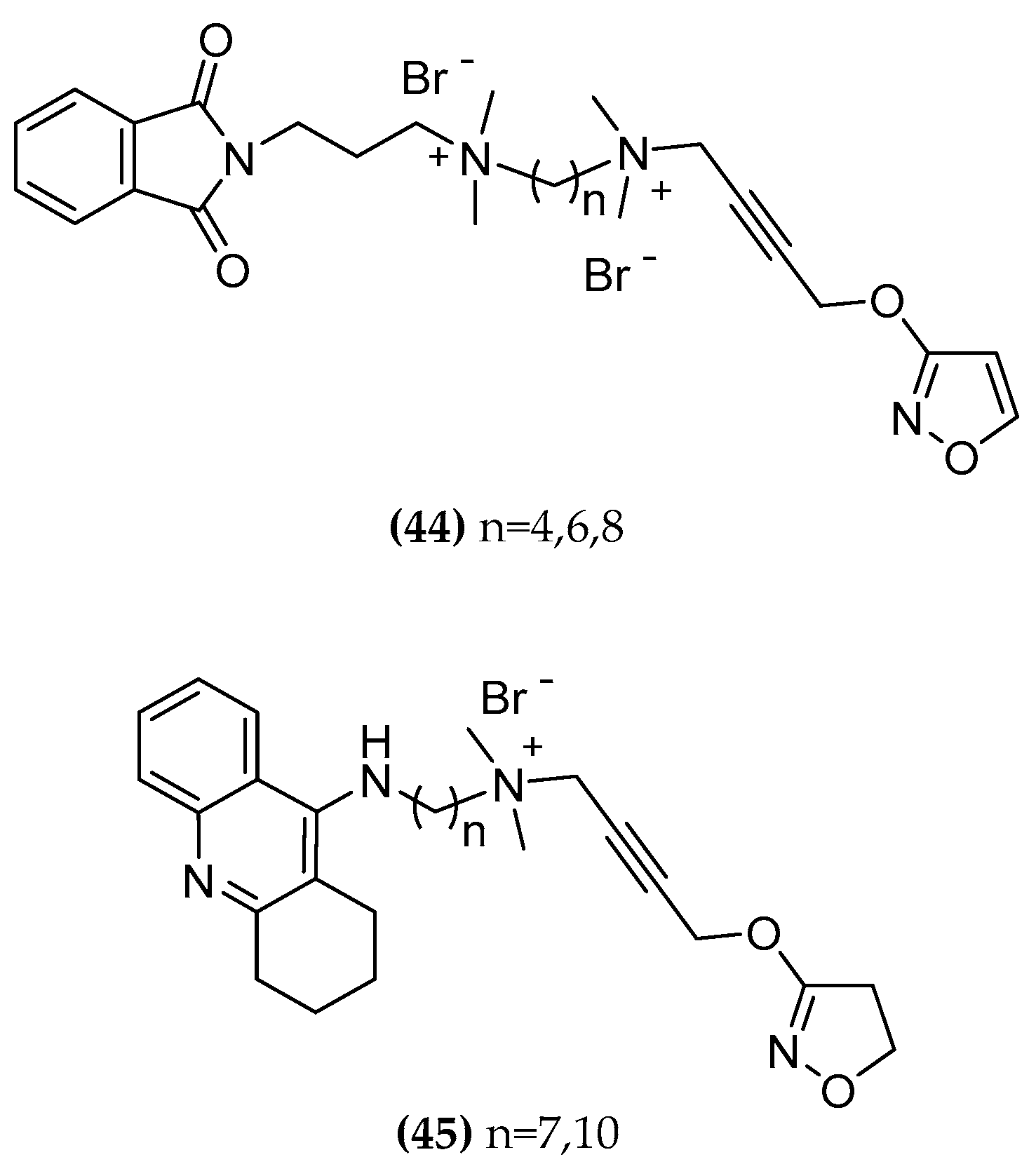
| Phth-4-isoxo (44-C4) | 4.07 | 19.12% inhib. at 100 μM | n.d. 1 |
| Phth-6-isoxo (44-C6) | 4.41 | 32.91% inhib. at 100 μM | n.d. 1 |
| Phtal-8-isoxo (44-C8) | 5.10 | 4.17 | n.d. 1 |
| Tac-7-iper (45-C7) | 8.29 | 8.76 | >160 (±0.00) |
| Tac-10-iper (45-C10) | 8.75 | 9.81 | 32.2 (±0.41) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volpato, D.; Holzgrabe, U. Designing Hybrids Targeting the Cholinergic System by Modulating the Muscarinic and Nicotinic Receptors: A Concept to Treat Alzheimer’s Disease. Molecules 2018, 23, 3230. https://doi.org/10.3390/molecules23123230
Volpato D, Holzgrabe U. Designing Hybrids Targeting the Cholinergic System by Modulating the Muscarinic and Nicotinic Receptors: A Concept to Treat Alzheimer’s Disease. Molecules. 2018; 23(12):3230. https://doi.org/10.3390/molecules23123230
Chicago/Turabian StyleVolpato, Daniela, and Ulrike Holzgrabe. 2018. "Designing Hybrids Targeting the Cholinergic System by Modulating the Muscarinic and Nicotinic Receptors: A Concept to Treat Alzheimer’s Disease" Molecules 23, no. 12: 3230. https://doi.org/10.3390/molecules23123230
APA StyleVolpato, D., & Holzgrabe, U. (2018). Designing Hybrids Targeting the Cholinergic System by Modulating the Muscarinic and Nicotinic Receptors: A Concept to Treat Alzheimer’s Disease. Molecules, 23(12), 3230. https://doi.org/10.3390/molecules23123230