New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology





Abstract
1. Introduction
2. Chemical Modifications of Synthetic Peptides
2.1. Peptide Cyclization and Insertion of Non-Natural Amino Acids
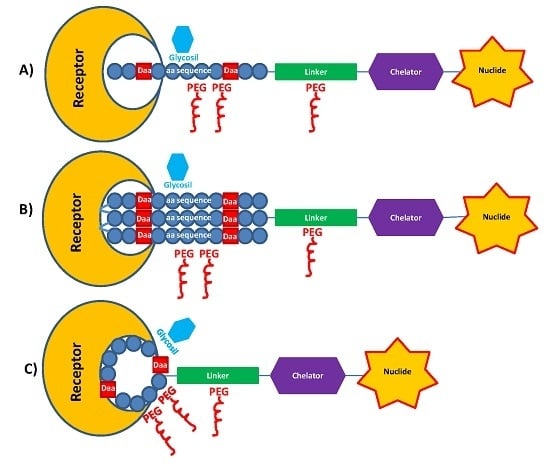
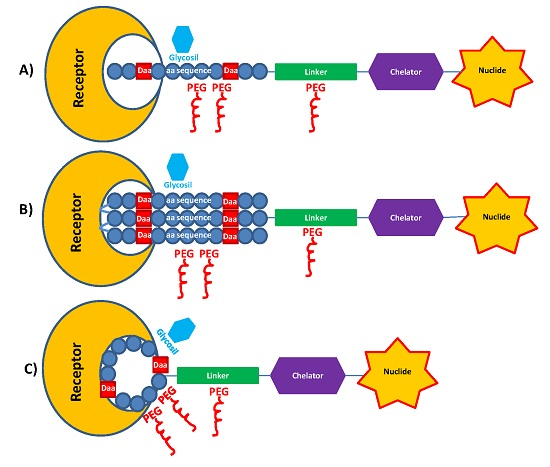
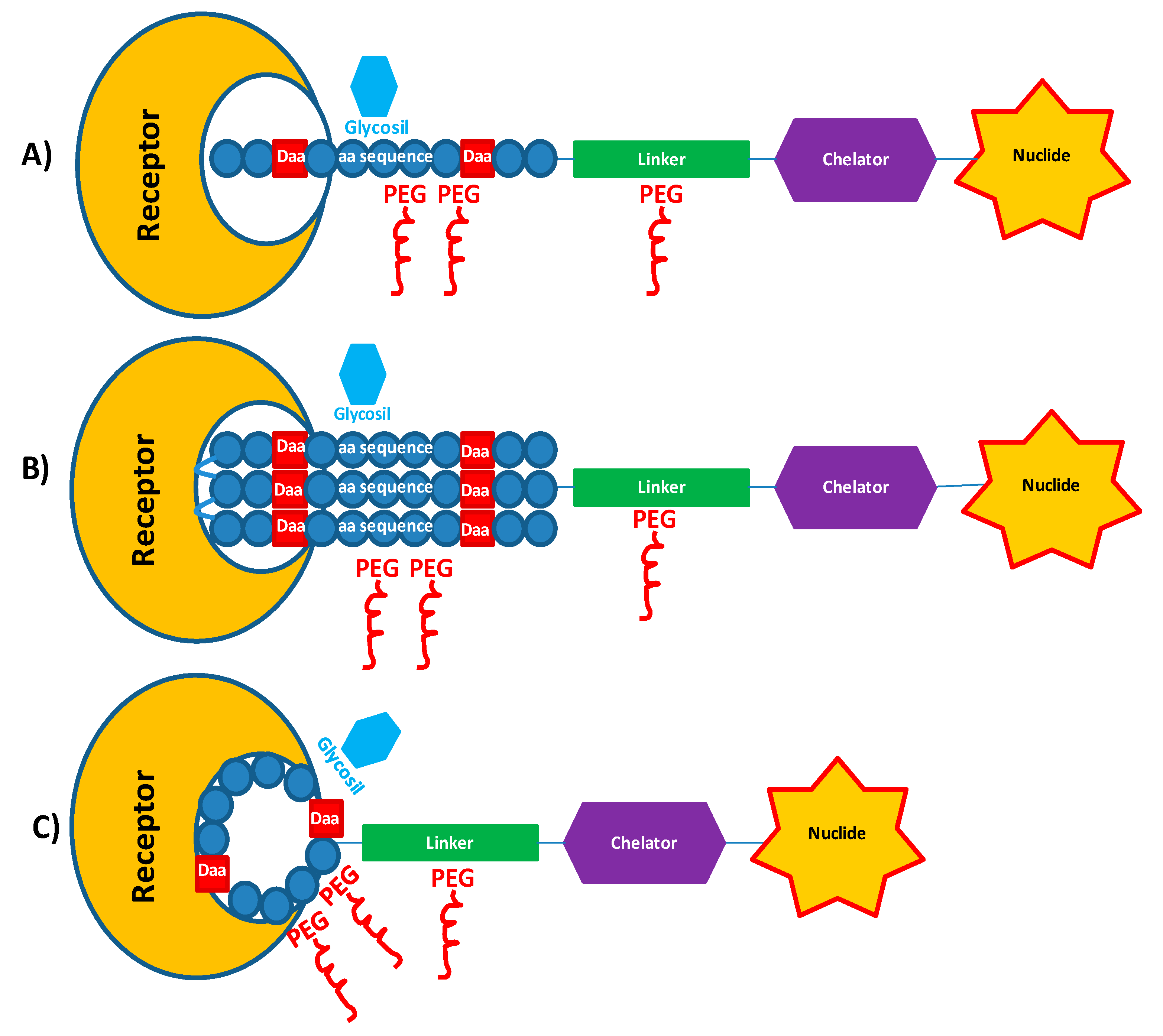
2.2. PEGylation and Glycosylation of Synthetic Peptides
3. Spacers, Chelators and Radionuclides
3.1. Spacers
3.2. Radionuclides
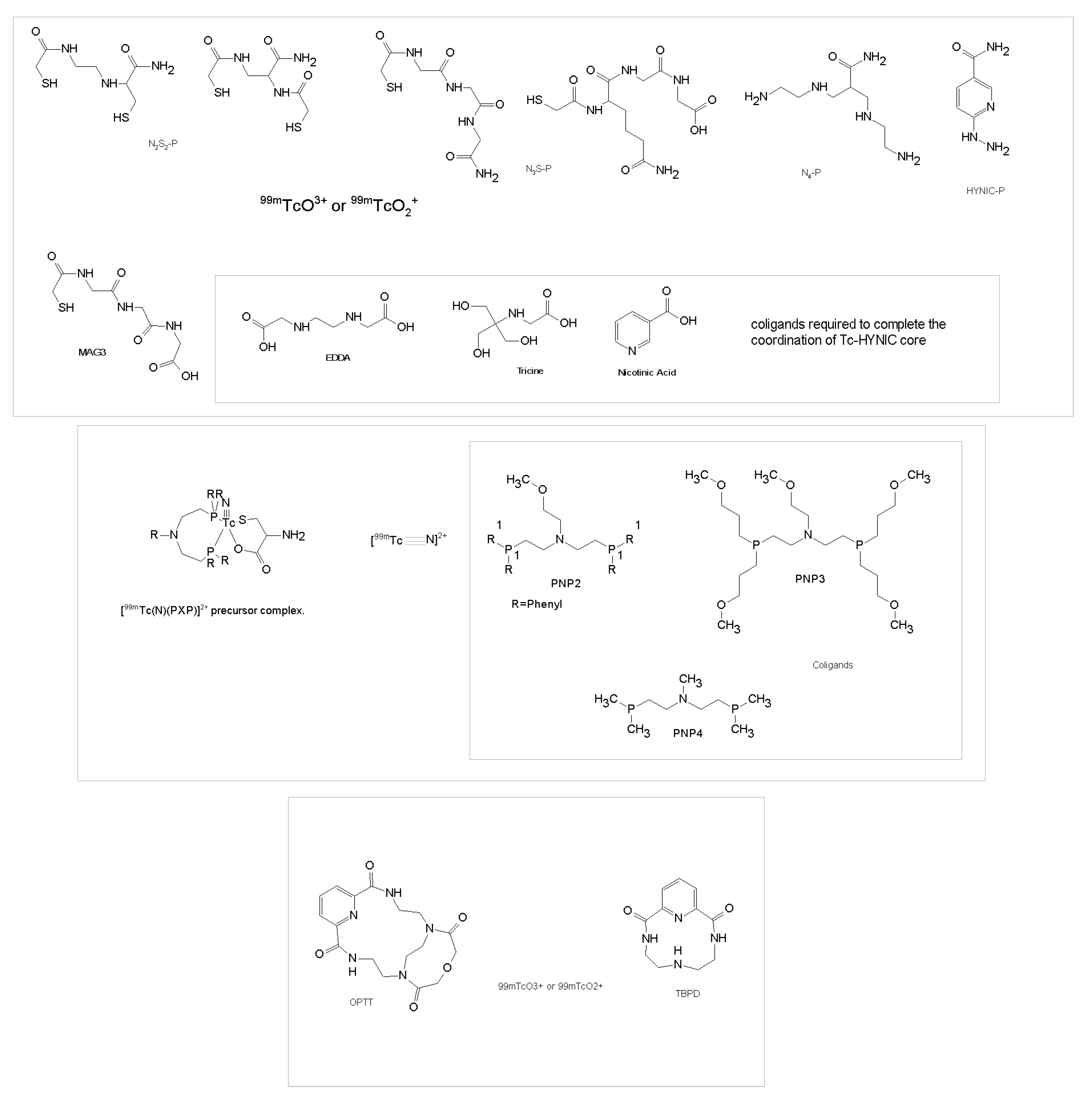
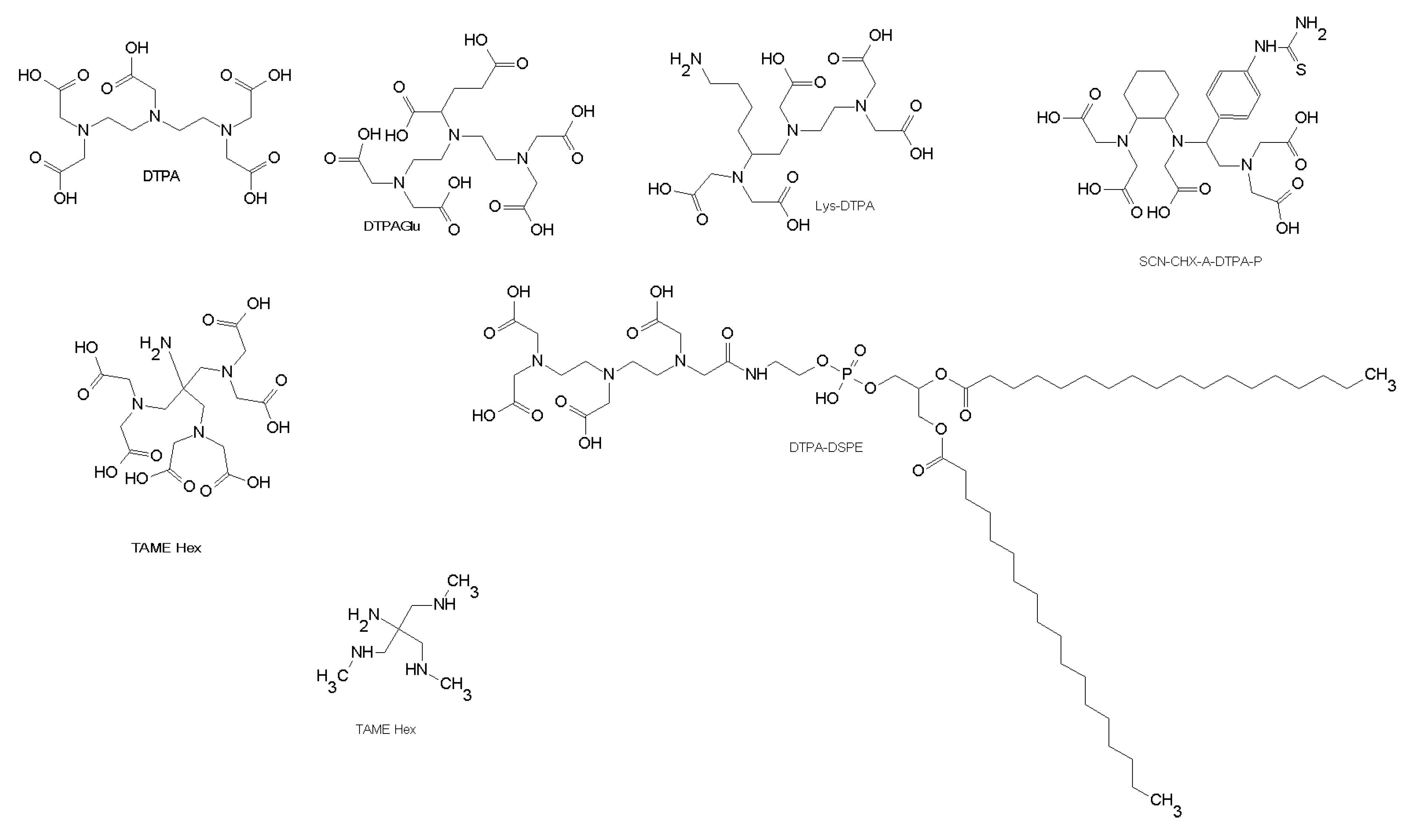
3.2.1. Technetium Chelators
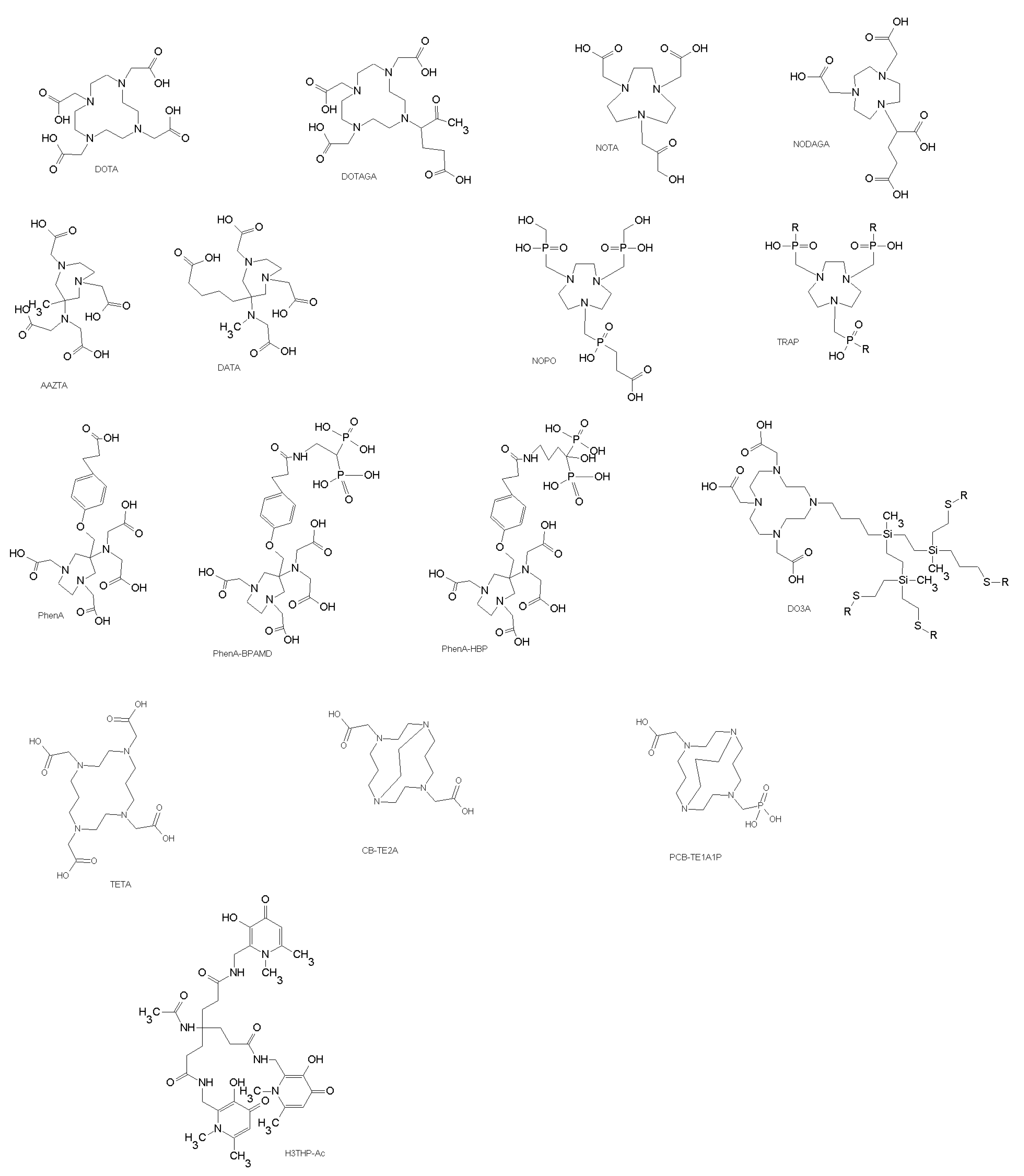
3.2.2. Yttrium, Indium, Gallium, Lutetium, and Copper Chelators
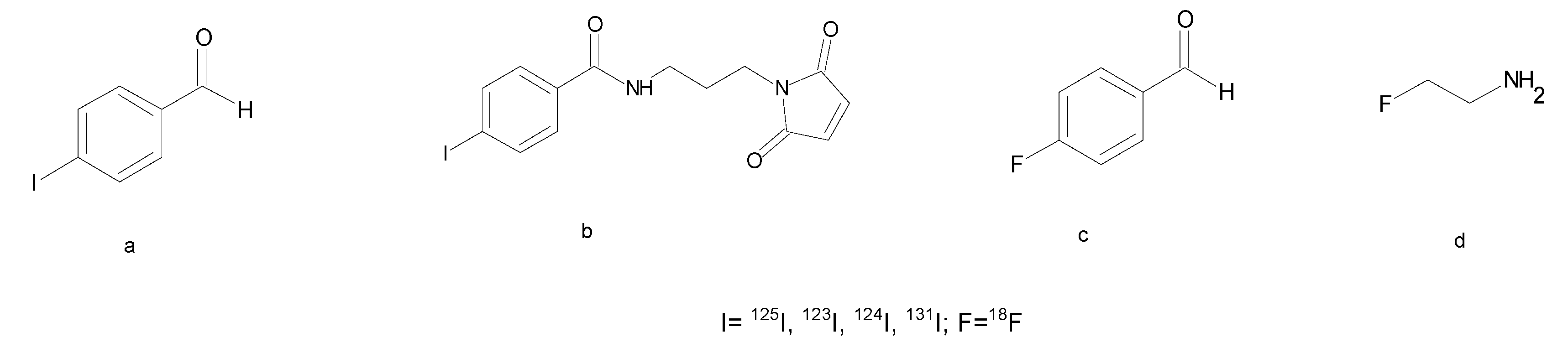
3.2.3. Fluorine and Iodine Prosthetic Groups
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. An overview of Bioactive Peptides for in vivo Imaging and Therapy in Human Diseases. Mini Rev. Med. Chem. 2017, 17, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Sultan, H.; Fesenkova, V.I.; Addis, D.; Fan, A.E.; Kumai, T.; Wu, J.; Salazar, A.M.; Celis, E. Designing therapeutic cancer vaccines by mimicking viral infections. Cancer Immunol. Immunother. 2017, 66, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Fischman, A.J.; Babich, J.W.; Strauss, H.W. A ticket to ride: Peptide radiopharmaceuticals. J. Nucl. Med. 1993, 34, 2253–2263. [Google Scholar] [PubMed]
- Lopez-Otin, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Maecke, H.R.; Okarvi, S.M. Radiolabeled peptides: Valuable tools for the detection and treatment of cancer. Theranostics 2012, 2, 481–501. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, A.L.; Sanseverino, M.; Buonaguro, F.M. Solid Phase Formylation of N-Terminus Peptides. Molecules 2016, 21, E736. [Google Scholar] [CrossRef] [PubMed]
- Sarko, D.; Eisenhut, M.; Haberkorn, U.; Mier, W. Bifunctional chelators in the design and application of radiopharmaceuticals for oncological diseases. Curr. Med. Chem. 2012, 19, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Hamman, J.H.; Enslin, G.M.; Kotze, A.F. Oral delivery of peptide drugs: Barriers and developments. BioDrugs 2005, 19, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Gentilucci, L.; De, M.R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef] [PubMed]
- Roxin, A.; Zheng, G. Flexible or fixed: A comparative review of linear and cyclic cancer-targeting peptides. Future Med. Chem. 2012, 4, 1601–1618. [Google Scholar] [CrossRef] [PubMed]
- Sabet, A.; Biersack, H.J.; Ezziddin, S. Advances in Peptide Receptor Radionuclide Therapy. Semin. Nucl. Med. 2016, 46, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.P.; Pictet, R.L.; Rutter, W.J. Human somatostatin I: Sequence of the cDNA. Proc. Natl. Acad. Sci. USA 1982, 79, 4575–4579. [Google Scholar] [CrossRef] [PubMed]
- de Herder, W.W.; van der Lely, A.J.; Lamberts, S.W. Somatostatin analogue treatment of neuroendocrine tumours. Postgrad. Med. J. 1996, 72, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Pless, J.; Bauer, W.; Briner, U.; Doepfner, W.; Marbach, P.; Maurer, R.; Petcher, T.J.; Reubi, J.C.; Vonderscher, J. Chemistry and pharmacology of SMS 201-995, a long-acting octapeptide analogue of somatostatin. Scand. J. Gastroenterol. Suppl. 1986, 119, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Virgolini, I.; Szilvasi, I.; Kurtaran, A.; Angelberger, P.; Raderer, M.; Havlik, E.; Vorbeck, F.; Bischof, C.; Leimer, M.; Dorner, G.; et al. Indium-111-DOTA-lanreotide: Biodistribution, safety and radiation absorbed dose in tumor patients. J. Nucl. Med. 1998, 39, 1928–1936. [Google Scholar] [PubMed]
- Martin-Gago, P.; Rol, A.; Todorovski, T.; Aragon, E.; Martin-Malpartida, P.; Verdaguer, X.; Valles, M.M.; Fernandez-Carneado, J.; Ponsati, B.; Macias, M.J.; et al. Peptide aromatic interactions modulated by fluorinated residues: Synthesis, structure and biological activity of Somatostatin analogs containing 3-(3′,5′difluorophenyl)-alanine. Sci. Rep. 2016, 6, 27285. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczak, R.; Maecke, H.R. Radiopharmaceuticals for somatostatin receptor imaging. Nucl. Med. Rev. Cent. East Eur. 2016, 19, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Peitl, P.K.; Velikyan, I. Current status of Radiopharmaceuticals for the Theranostics of Neuroendocrine Neoplasms. Pharmaceuticals 2017, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Bass, R.T.; Buckwalter, B.L.; Patel, B.P.; Pausch, M.H.; Price, L.A.; Strnad, J.; Hadcock, J.R. Identification and characterization of novel somatostatin antagonists. Mol. Pharmacol. 1996, 50, 709–715. [Google Scholar] [PubMed]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Macke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Erchegyi, J.; Waser, B.; Piccand, V.; Maecke, H.R.; Rivier, J.E.; Reubi, J.C. Design and in vitro characterization of highly sst2-selective somatostatin antagonists suitable for radiotargeting. J. Med. Chem. 2008, 51, 4030–4037. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Braun, F.; Waser, B.; Beetschen, K.; Cescato, R.; Erchegyi, J.; Rivier, J.E.; Weber, W.A.; Maecke, H.R.; Reubi, J.C. Unexpected sensitivity of sst2 antagonists to N-terminal radiometal modifications. J. Nucl. Med. 2012, 53, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B.; Macke, H.; Rivier, J. Highly increased 125I-JR11 antagonist binding In Vitro reveals novel indications for sst2 targeting in human cancers. J. Nucl. Med. 2017, 58, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.P.; Mansi, R.; McDougall, L.; Kaufmann, J.; Bouterfa, H.; Wild, D.; Fani, M. Biodistribution, pharmacokinetics and dosimetry of 177Lu-, 90Y- and 111In-labeled somatostatin receptor antagonist OPS201 in comparison to the agonist 177Lu-DOTA-TATE: The mass effect. J. Nucl. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Chen, X.; Sievers, E.; Hou, Y.; Park, R.; Tohme, M.; Bart, R.; Bremner, R.; Bading, J.R.; Conti, P.S. Integrin alpha v beta 3-targeted imaging of lung cancer. Neoplasia 2005, 7, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Seftor, R.E.; Seftor, E.A.; Gehlsen, K.R.; Stetler-Stevenson, W.G.; Brown, P.D.; Ruoslahti, E.; Hendrix, M.J. Role of the alpha v beta 3 integrin in human melanoma cell invasion. Proc. Natl. Acad. Sci. USA 1992, 89, 1557–1561. [Google Scholar] [CrossRef] [PubMed]
- Gladson, C.L.; Cheresh, D.A. Glioblastoma expression of vitronectin and the alpha v beta 3 integrin. Adhesion mechanism for transformed glial cells. J. Clin. Investig. 1991, 88, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Carreiras, F.; Denoux, Y.; Staedel, C.; Lehmann, M.; Sichel, F.; Gauduchon, P. Expression and localization of alpha v integrins and their ligand vitronectin in normal ovarian epithelium and in ovarian carcinoma. Gynecol. Oncol. 1996, 62, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, F.; Liu, S. Radiolabeled cyclic RGD peptides as radiotracers for tumor imaging. Biophys. Rep. 2016, 2, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.O.; Otto, E.; Mas-Moruno, C.; Schiller, H.B.; Marinelli, L.; Cosconati, S.; Bochen, A.; Vossmeyer, D.; Zahn, G.; Stragies, R.; et al. Conformational control of integrin-subtype selectivity in isoDGR peptide motifs: A biological switch. Angew. Chem. Int. Ed. Engl. 2010, 49, 9278–9281. [Google Scholar] [CrossRef] [PubMed]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.J.; Mas-Moruno, C.; et al. A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, J.; Ovadia, O.; Zahn, G.; Marinelli, L.; Hoffman, A.; Gilon, C.; Kessler, H. Multiple N-methylation by a designed approach enhances receptor selectivity. J. Med. Chem. 2007, 50, 5878–5881. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, M.; Tangemann, K.; Muller, B.; Gurrath, M.; Muller, G.; Kessler, H.; Timpl, R.; Engel, J. Selective recognition of cyclic RGD peptides of NMR defined conformation by alpha IIb beta 3, alpha V beta 3, and alpha 5 beta 1 integrins. J. Biol. Chem. 1994, 269, 20233–20238. [Google Scholar] [PubMed]
- Becker, A.; von, R.O.; Kovar, A.; Scheible, H.; van Lier, J.J.; Johne, A. Metabolism and disposition of the alphav-integrin ss3/ss5 receptor antagonist cilengitide, a cyclic polypeptide, in humans. J. Clin. Pharmacol. 2015, 55, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, I.; Kruijtzer, J.A.; Liu, S.; Soede, A.C.; Oyen, W.J.; Corstens, F.H.; Liskamp, R.M.; Boerman, O.C. Improved targeting of the alpha(v)beta (3) integrin by multimerisation of RGD peptides. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ruzza, P.; Marchiani, A.; Antolini, N.; Calderan, A. Peptide-receptor ligands and multivalent approach. Anticancer Agents Med. Chem. 2012, 12, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Vogetseder, A.; Thies, S.; Ingold, B.; Roth, P.; Weller, M.; Schraml, P.; Goodman, S.L.; Moch, H. Alphav-integrin isoform expression in primary human tumors and brain metastases. Int. J. Cancer 2013, 133, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Reich, D.; Maltsev, O.V.; Kapp, T.G.; Steiger, K.; Hoffmann, F.; Esposito, I.; Weichert, W.; Kessler, H.; Wester, H.J. In Vivo PET imaging of the cancer integrin alphavbeta6 using 68Ga-labeled cyclic RGD nonapeptides. J. Nucl. Med. 2017, 58, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Liao, Z.; Mitsios, J.V.; Wang, H.Y.; Deryugina, E.I.; Varner, J.A.; Quigley, J.P.; Shattil, S.J. The primacy of beta1 integrin activation in the metastatic cascade. PLoS ONE 2012, 7, e46576. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandria, C.; Pohle, K.; Rechenmacher, F.; Neubauer, S.; Notni, J.; Wester, H.J.; Schwaiger, M.; Kessler, H.; Beer, A.J. In vivo biokinetic and metabolic characterization of the 68Ga-labelled alpha5beta1-selective peptidomimetic FR366. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Rehfeld, J.F.; Friis-Hansen, L.; Goetze, J.P.; Hansen, T.V. The biology of cholecystokinin and gastrin peptides. Curr. Top. Med. Chem. 2007, 7, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Fonkoua, L.K.; Moody, T.W. The role of gastrin and CCK receptors in Pancreatic Cancer and other malignancies. Int. J. Biol. Sci. 2016, 12, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.H.; Lim, J.C.; Lee, S.Y.; Jung, S.H. An assessment tumor targeting ability of (177)Lu labeled cyclic CCK analogue peptide by binding with cholecystokinin receptor. J. Pharmacol. Sci. 2016, 131, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Tesauro, D.; Morelli, G.; Del, P.L.; Pedone, C.; Tornesello, A.L.; Benedetti, E. Supramolecular aggregates derivatized by CCK8 peptide as selective nanocarriers for drug delivery. Adv. Exp. Med. Biol. 2009, 611, 603–604. [Google Scholar] [PubMed]
- Feng, J.; Petersen, C.D.; Coy, D.H.; Jiang, J.K.; Thomas, C.J.; Pollak, M.R.; Wank, S.A. Calcium-sensing receptor is a physiologic multimodal chemosensor regulating gastric G-cell growth and gastrin secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 17791–17796. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, A.L.; Aurilio, M.; Accardo, A.; Tarallo, L.; Barbieri, A.; Arra, C.; Tesauro, D.; Morelli, G.; Aloj, L. Gastrin and cholecystokinin peptide-based radiopharmaceuticals: An in vivo and in vitro comparison. J. Pept. Sci. 2011, 17, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Kolenc, P.P.; Tamma, M.; Kroselj, M.; Braun, F.; Waser, B.; Reubi, J.C.; Sollner, D.M.; Maecke, H.R.; Mansi, R. Stereochemistry of amino acid spacers determines the pharmacokinetics of (111)In-DOTA-minigastrin analogues for targeting the CCK2/gastrin receptor. Bioconjug. Chem. 2015, 26, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Kaloudi, A.; Nock, B.A.; Lymperis, E.; Krenning, E.P.; de, J.M.; Maina, T. Improving the In Vivo profile of minigastrin radiotracers: A comparative study involving the neutral endopeptidase inhibitor phosphoramidon. Cancer Biother. Radiopharm. 2016, 31, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Alvarez, I.; Moreno, P.; Mantey, S.A.; Nakamura, T.; Nuche-Berenguer, B.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Insights into bombesin receptors and ligands: Highlighting recent advances. Peptides 2015, 72, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, N.; Czepielewski, R.S.; Bagatini, M.; Porto, B.N.; Bonorino, C. Neuropeptide gastrin-releasing peptide induces PI3K/reactive oxygen species-dependent migration in lung adenocarcinoma cells. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.C. Regulation and signaling of human bombesin receptors and their biological effects. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Brans, L.; Garcia-Garayoa, E.; Schweinsberg, C.; Maes, V.; Struthers, H.; Schibli, R.; Tourwe, D. Synthesis and evaluation of bombesin analogues conjugated to two different triazolyl-derived chelators for (99m)Tc labeling. ChemMedChem 2010, 5, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Galli, F.; Mansi, R.; Del, P.L.; Aurilio, M.; Morisco, A.; Ringhieri, P.; Signore, A.; Morelli, G.; Aloj, L. Pre-clinical evaluation of eight DOTA coupled gastrin-releasing peptide receptor (GRP-R) ligands for in vivo targeting of receptor-expressing tumors. EJNMMI Res. 2016, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Bryant, M.G. Vasoactive intestinal peptide (VIP). J. Clin. Pathol. Suppl. (Assoc. Clin. Pathol.) 1978, 8, 63–67. [Google Scholar] [CrossRef]
- Schwartz, C.J.; Kimberg, D.V.; Sheerin, H.E.; Field, M.; Said, S.I. Vasoactive intestinal peptide stimulation of adenylate cyclase and active electrolyte secretion in intestinal mucosa. J. Clin. Investig. 1974, 54, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Pallela, V.R.; Thakur, M.L.; Chakder, S.; Rattan, S. 99mTc-labeled vasoactive intestinal peptide receptor agonist: Functional studies. J. Nucl. Med. 1999, 40, 352–360. [Google Scholar] [PubMed]
- Li, J.; Wang, X.; Liu, H.; Li, H. In silico classification and prediction of VIP derivatives as VPAC1/ VPAC2 receptor agonists/antagonists. Comb. Chem. High Throughput Screen. 2015, 18, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Bodner, M.; Shani, Y.; Fridkin, M. Structure and expression of the vasoactive intestinal peptide (VIP) gene in a human tumor. Peptides 1986, 7, 1–6. [Google Scholar] [CrossRef]
- Cheng, D.; Liu, Y.; Shen, H.; Pang, L.; Yin, D.; Wang, Y.; Li, S.; Shi, H. F-18 labeled vasoactive intestinal peptide analogue in the PET imaging of colon carcinoma in nude mice. Biomed. Res. Int. 2013, 2013, 420480. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Yin, D.; Li, G.; Wang, M.; Li, S.; Zheng, M.; Cai, H.; Wang, Y. Radiolabeling and in vitro and in vivo characterization of [18F]FB-[R(8,15,21), L17]-VIP as a PET imaging agent for tumor overexpressed VIP receptors. Chem. Biol. Drug Des. 2006, 68, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Sihver, W.; Jurischka, C.; Bergmann, R.; Haase-Kohn, C.; Mosch, B.; Steinbach, J.; Carta, D.; Bolzati, C.; Calderan, A.; et al. Radiopharmacological characterization of (6)(4)Cu-labeled alpha-MSH analogs for potential use in imaging of malignant melanoma. Amino Acids 2016, 48, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, L.; Miao, Y. Effects of the Arg-Pro and Gly-Gly-Nle moieties on Melanocortin-1 receptor binding affinities of alpha-MSH Peptides. ACS Med. Chem. Lett. 2013, 4, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, R.; Besserer-Offroy, E.; Rene, A.; Cote, J.; Tetreault, P.; Collerette-Tremblay, J.; Longpre, J.M.; Leduc, R.; Martinez, J.; Sarret, P.; et al. Synthesis and characterization in Vitro and in Vivo of (l)-(Trimethylsilyl)alanine containing Neurotensin Analogues. J. Med. Chem. 2015, 58, 7785–7795. [Google Scholar] [CrossRef] [PubMed]
- Mascarin, A.; Valverde, I.E.; Mindt, T.L. Structure-activity relationship studies of Amino Acid Substitutions in Radiolabeled Neurotensin Conjugates. ChemMedChem 2016, 11, 102–107. [Google Scholar] [CrossRef] [PubMed]
- van Witteloostuijn, S.B.; Pedersen, S.L.; Jensen, K.J. Half-life extension of biopharmaceuticals using chemical methods: Alternatives to PEGylation. ChemMedChem 2016, 11, 2474–2495. [Google Scholar] [CrossRef] [PubMed]
- Dapp, S.; Garcia, G.E.; Maes, V.; Brans, L.; Tourwe, D.A.; Muller, C.; Schibli, R. PEGylation of (99m)Tc-labeled bombesin analogues improves their pharmacokinetic properties. Nucl. Med. Biol. 2011, 38, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, V.; Dadey, D.Y.; Nguyen, K.; Wildman, S.A.; Hoye, K.; Khudanyan, A.; Bandara, N.; Rogers, B.E.; Thotala, D.; Hallahan, D.E. Tumor-specific binding of Radiolabeled PEGylated GIRLRG Peptide: A novel agent for targeting cancers. J. Nucl. Med. 2016, 57, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Hausner, S.H.; Bauer, N.; Hu, L.Y.; Knight, L.M.; Sutcliffe, J.L. The effect of Bi-Terminal PEGylation of an integrin alphavbeta(6)-targeted (1)(8)F Peptide on pharmacokinetics and tumor uptake. J. Nucl. Med. 2015, 56, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Simecek, J.; Notni, J.; Kapp, T.G.; Kessler, H.; Wester, H.J. Benefits of NOPO as chelator in gallium-68 peptides, exemplified by preclinical characterization of (68)Ga-NOPO-c(RGDfK). Mol. Pharm. 2014, 11, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Laverman, P.; Sosabowski, J.K.; Boerman, O.C.; Oyen, W.J. Radiolabelled peptides for oncological diagnosis. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, S78–S92. [Google Scholar] [CrossRef] [PubMed]
- Liese, S.; Netz, R.R. Influence of length and flexibility of spacers on the binding affinity of divalent ligands. Beilstein J. Org. Chem. 2015, 11, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Pallarola, D.; Bochen, A.; Boehm, H.; Rechenmacher, F.; Sobahi, T.R.; Spatz, J.P.; Kessler, H. Interface immobilization chemistry of cRGD-based peptides regulates integrin mediated cell adhesion. Adv. Funct. Mater. 2014, 24, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, T.J.; Gali, H.; Smith, C.J.; Sieckman, G.L.; Hayes, D.L.; Owen, N.K.; Volkert, W.A. Novel series of 111In-labeled bombesin analogs as potential radiopharmaceuticals for specific targeting of gastrin-releasing peptide receptors expressed on human prostate cancer cells. J. Nucl. Med. 2003, 44, 823–831. [Google Scholar] [PubMed]
- Garrison, J.C.; Rold, T.L.; Sieckman, G.L.; Naz, F.; Sublett, S.V.; Figueroa, S.D.; Volkert, W.A.; Hoffman, T.J. Evaluation of the pharmacokinetic effects of various linking group using the 111In-DOTA-X-BBN(7–14)NH2 structural paradigm in a prostate cancer model. Bioconjug. Chem. 2008, 19, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- Antunes, P.; Ginj, M.; Walter, M.A.; Chen, J.; Reubi, J.C.; Maecke, H.R. Influence of different spacers on the biological profile of a DOTA-somatostatin analogue. Bioconjug. Chem. 2007, 18, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Shi, W.; Zhou, Z.; Wagh, N.K.; Fan, W.; Brusnahan, S.K.; Garrison, J.C. Evaluation of DOTA-chelated neurotensin analogs with spacer-enhanced biological performance for neurotensin-receptor-1-positive tumor targeting. Nucl. Med. Biol. 2015, 42, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Altenbrunn, H.J. Radionuclide therapy of malignant tumors. Z. Gesamte Inn. Med. 1978, 33, 475–484. [Google Scholar] [PubMed]
- Abiraj, K.; Mansi, R.; Tamma, M.L.; Forrer, F.; Cescato, R.; Reubi, J.C.; Akyel, K.G.; Maecke, H.R. Tetraamine-derived bifunctional chelators for technetium-99m labelling: Synthesis, bioconjugation and evaluation as targeted SPECT imaging probes for GRP-receptor-positive tumours. Chemistry 2010, 16, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Nikolopoulou, A.; Reubi, J.C.; Maes, V.; Conrath, P.; Tourwe, D.; Maina, T. Toward stable N4-modified neurotensins for NTS1-receptor-targeted tumor imaging with 99mTc. J. Med. Chem. 2006, 49, 4767–4776. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.F.; Zhang, X.; Shah, M.; Newton-Northup, J.; Cabral, P.; Cerecetto, H.; Quinn, T. (99m)Tc-bioorthogonal click chemistry reagent for in vivo pretargeted imaging. Bioorgan. Med. Chem. 2016, 24, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Marostica, L.L.; de Barros, A.L.; Silva, J.O.; Lopes, S.C.; Salgado, B.S.; Chondrogiannis, S.; Rubello, D.; Cassali, G.D.; Schenkel, E.P.; Cardoso, V.N.; et al. Feasibility study with 99mTc-HYNIC-betaAla-Bombesin(7–14) as an agent to early visualization of lung tumour cells in nude mice. Nucl. Med. Commun. 2016, 37, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Maecke, H.R. Radiopharmaceutical development of radiolabelled peptides. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, S11–S30. [Google Scholar] [CrossRef] [PubMed]
- Agostini, S.; Bolzati, C.; Didone, E.; Cavazza-Ceccato, M.; Refosco, F.; Aloj, L.; Arra, C.; Aurilio, M.; Tornesello, A.L.; Tesauro, D.; et al. The [Tc(N)(PNP)]2+ metal fragment labeled cholecystokinin-8 (CCK8) peptide for CCK-2 receptors imaging: In vitro and in vivo studies. J. Pept. Sci. 2007, 13, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Salvarese, N.; Dolmella, A.; Refosco, F.; Bolzati, C. Reactivity of the [M(PS)22](+) building block (M = Re(III) and (99m)Tc(III); PS = phosphinothiolate) toward isopropylxanthate and pyridine-2-thiolate. Inorg. Chem. 2015, 54, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Salvarese, N.; Morellato, N.; Rosato, A.; Melendez-Alafort, L.; Refosco, F.; Bolzati, C. Novel [99mTcIII(PS)2(Ln)] mixed-ligand compounds (PS = phosphino-thiolate; L = dithiocarbamate) useful in design and development of TcIII-based agents: Synthesis, in vitro, and ex vivo biodistribution studies. J. Med. Chem. 2014, 57, 8960–8970. [Google Scholar] [CrossRef] [PubMed]
- Boschi, A.; Bolzati, C.; Benini, E.; Malago, E.; Uccelli, L.; Duatti, A.; Piffanelli, A.; Refosco, F.; Tisato, F. A novel approach to the high-specific-activity labeling of small peptides with the technetium-99m fragment [99mTc(N)(PXP)]2+ (PXP = diphosphine ligand). Bioconjug. Chem. 2001, 12, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Nick, H.; Acklin, P.; Lattmann, R.; Buehlmayer, P.; Hauffe, S.; Schupp, J.; Alberti, D. Development of tridentate iron chelators: From desferrithiocin to ICL670. Curr. Med. Chem. 2003, 10, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Boschi, A.; Cazzola, E.; Uccelli, L.; Pasquali, M.; Ferretti, V.; Bertolasi, V.; Duatti, A. Rhenium(V) and technetium(V) nitrido complexes with mixed tridentate pi-donor and monodentate pi-acceptor ligands. Inorg. Chem. 2012, 51, 3130–3137. [Google Scholar] [CrossRef] [PubMed]
- Boschi, A.; Uccelli, L.; Pasquali, M.; Pasqualini, R.; Guerrini, R.; Duatti, A. Mixed tridentate pi -donor and monodentate pi -acceptor ligands as chelating systems for rhenium-188 and technetium-99m nitrido radiopharmaceuticals. Curr. Radiopharm. 2013, 6, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, E.; Benini, E.; Pasquali, M.; Mirtschink, P.; Walther, M.; Pietzsch, H.J.; Uccelli, L.; Boschi, A.; Bolzati, C.; Duatti, A. Labeling of fatty acid ligands with the strong electrophilic metal fragment [99mTc(N)(PXP)]2+ (PNP = diphosphane ligand). Bioconjug. Chem. 2008, 19, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; Hnatowich, D.J. An improved synthesis of NHS-MAG3 for conjugation and radiolabeling of biomolecules with (99m)Tc at room temperature. Nat. Protoc. 2007, 2, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Chuttani, K.; Mishra, A.K.; Singh, B. Synthesis, characterization, and preclinical evaluation of (99m)Tc-Labeled macrobicyclic and tricyclic chelators as single photon emission computed tomography tracer. Chem. Biol. Drug Des. 2016, 87, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Gao, H.; Zhai, L.; Liu, X.; Jia, B.; Shi, J.; Wang, F. (99m)Tc-HisoDGR as a potential SPECT probe for orthotopic glioma detection via targeting of integrin alpha5beta1. Bioconjug. Chem. 2016, 27, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Milenic, D.E.; Garmestani, K.; Chappell, L.L.; Dadachova, E.; Yordanov, A.; Ma, D.; Schlom, J.; Brechbiel, M.W. In vivo comparison of macrocyclic and acyclic ligands for radiolabeling of monoclonal antibodies with 177Lu for radioimmunotherapeutic applications. Nucl. Med. Biol. 2002, 29, 431–442. [Google Scholar] [CrossRef]
- Arslantas, E.; Smith-Jones, P.M.; Ritter, G.; Schmidt, R.R. TAME-Hex A—A novel bifunctional chelating agent for radioimmunoimaging. Eur. J. Org. Chem. 2004, 19, 3979–3984. [Google Scholar] [CrossRef]
- Accardo, A.; Salsano, G.; Morisco, A.; Aurilio, M.; Parisi, A.; Maione, F.; Cicala, C.; Tesauro, D.; Aloj, L.; De, R.G.; et al. Peptide-modified liposomes for selective targeting of bombesin receptors overexpressed by cancer cells: A potential theranostic agent. Int. J. Nanomed. 2012, 7, 2007–2017. [Google Scholar]
- Van der Geest, T.; Laverman, P.; Gerrits, D.; Franssen, G.M.; Metselaar, J.M.; Storm, G.; Boerman, O.C. Comparison of three remote radiolabelling methods for long-circulating liposomes. J. Controlled Release 2015, 220, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Kang, C.S.; Sin, I.; Ren, S.; Liu, D.; Ruthengael, V.C.; Lewis, M.R.; Chong, H.S. Promising bifunctional chelators for copper 64-PET imaging: Practical (64)Cu radiolabeling and high in vitro and in vivo complex stability. J. Biol. Inorg. Chem. 2016, 21, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Meckel, M.; Fellner, M.; Thieme, N.; Bergmann, R.; Kubicek, V.; Rosch, F. In vivo comparison of DOTA based 68Ga-labelled bisphosphonates for bone imaging in non-tumour models. Nucl. Med. Biol. 2013, 40, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Eisenwiener, K.P.; Prata, M.I.; Buschmann, I.; Zhang, H.W.; Santos, A.C.; Wenger, S.; Reubi, J.C.; Macke, H.R. NODAGATOC, a new chelator-coupled somatostatin analogue labeled with [67/68Ga] and [111In] for SPECT, PET, and targeted therapeutic applications of somatostatin receptor (hsst2) expressing tumors. Bioconjug. Chem. 2002, 13, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Pfister, J.; Summer, D.; Rangger, C.; Petrik, M.; von, G.E.; Minazzi, P.; Giovenzana, G.B.; Aloj, L.; Decristoforo, C. Influence of a novel, versatile bifunctional chelator on theranostic properties of a minigastrin analogue. EJNMMI Res. 2015, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zha, Z.; Choi, S.R.; Plossl, K.; Zhu, L.; Kung, H.F. New (68)Ga-PhenA bisphosphonates as potential bone imaging agents. Nucl. Med. Biol. 2016, 43, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Seemann, J.; Waldron, B.P.; Roesch, F.; Parker, D. Approaching ‘Kit-Type’ labelling with (68)Ga: The DATA chelators. ChemMedChem 2015, 10, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Seemann, J.; Waldron, B.; Parker, D.; Roesch, F. DATATOC: A novel conjugate for kit-type 68Ga labelling of TOC at ambient temperature. EJNMMI Radiopharm. Chem. 2016, 1, 1–12. [Google Scholar] [CrossRef]
- Parker, D.; Waldron, B.P. Conformational analysis and synthetic approaches to polydentate perhydro-diazepine ligands for the complexation of gallium(III). Org. Biomol. Chem. 2013, 11, 2827–2838. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Gallazzi, F.A.; Ferdani, R.; Anderson, C.J.; Quinn, T.P.; Deutscher, S.L. In vitro and in vivo evaluation of (6)(4)Cu-radiolabeled KCCYSL peptides for targeting epidermal growth factor receptor-2 in breast carcinomas. Cancer Biother. Radiopharm. 2010, 25, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Dale, A.V.; An, G.I.; Pandya, D.N.; Ha, Y.S.; Bhatt, N.; Soni, N.; Lee, H.; Ahn, H.; Sarkar, S.; Lee, W.; et al. Synthesis and evaluation of new generation cross-bridged bifunctional chelator for (64)Cu radiotracers. Inorg. Chem. 2015, 54, 8177–8186. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Ortega, P.; de la Mata, F.J.; Ottaviani, M.F.; Cangiotti, M.; Fattori, A.; Munoz-Fernandez, M.A.; Gomez, R. Bifunctional chelating agents based on ionic carbosilane dendrons with DO3A at the focal point and their complexation behavior with copper(II). Inorg. Chem. 2015, 54, 8943–8956. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Simecek, J.; Hermann, P.; Wester, H.J. TRAP, a powerful and versatile framework for gallium-68 radiopharmaceuticals. Chemistry 2011, 17, 14718–14722. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Pohle, K.; Wester, H.J. Be spoilt for choice with radiolabelled RGD peptides: Preclinical evaluation of (6)(8)Ga-TRAP(RGD)(3). Nucl. Med. Biol. 2013, 40, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.T.; Cullinane, C.; Imberti, C.; Baguna, T.J.; Terry, S.Y.; Roselt, P.; Hicks, R.J.; Blower, P.J. New Tris(hydroxypyridinone) bifunctional chelators containing isothiocyanate groups provide a versatile platform for rapid One-Step Labeling and PET Imaging with (68)Ga(3.). Bioconjug. Chem. 2016, 27, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Wuest, F. 18F-Labeled peptides: The future is bright. Molecules 2014, 19, 20536–20556. [Google Scholar] [CrossRef] [PubMed]
- Thumshirn, G.; Hersel, U.; Goodman, S.L.; Kessler, H. Multimeric cyclic RGD peptides as potential tools for tumor targeting: Solid-phase peptide synthesis and chemoselective oxime ligation. Chemistry 2003, 9, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Bhojani, M.S.; Ranga, R.; Luker, G.D.; Rehemtulla, A.; Ross, B.D.; Van Dort, M.E. Synthesis and investigation of a radioiodinated F3 peptide analog as a SPECT tumor imaging radioligand. PLoS ONE 2011, 6, e22418. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Cancer | Peptide | |
|---|---|---|---|
| Somatostatin (sst1/sst2/sst3/sst4/sst5) | Neuroendocrine tumors | Agonists | |
| Somatostatin | AGCKNFFWKTFTSC (Cys-Cys cyclization) | ||
| Octreotide | (d-F)CF(d-W)KTCT-ol (Cys-Cys cyclization) | ||
| Y3-OC | (d-F)CY(d-W)KTCT-ol (Cys-Cys cyclization) | ||
| TATE | (d-F)CF(d-W)KTCT-OH (Cys-Cys cyclization) | ||
| Y3-TATE | (d-F)CY(d-W)KTCT-OH (Cys-Cys cyclization) | ||
| Lanreotide | (d-2-Nal)CY(d-W)KVCT-NH2 (Cys-Cys cyclization) | ||
| Depreotide | cyclo-[(N-Me)Phe-Y-d-Trp-KV-Hcy]CH2-CO.β-Dap-KCK-NH2 | ||
| Pep2 | AGCKNF(l-Dfp)(d-W)KTFTSC [l-Dfp7, d-Trp8,]-SRIF | ||
| Pep3 | AGCKNFF(d-Trp)KT(l-Dfp)TSC [d-Trp8, l-Dfp11]-SRIF | ||
| Antagonists | |||
| BASS | p-NO2-F((d-C)Y(d-W)KTC)(d-Y)-NH2 (Cys-Cys Cyclization) | ||
| LM3 | p-Cl-F((d-C)Y(d-Aph(Cbm)KTC)(d-Y)-NH2 (Cys-Cys Cyclization) | ||
| JR10 | p-NO2-F(d-C)Y(d-Aph(Cbm))KTC)(d-Y)-NH2 (Cys-Cys Cyclization) | ||
| JR11 | p-Cl-F((d-C)-Aph(Hor)-(d-Aph(Cbm))KTC)(d-Y)-NH2 (Cys-Cys Cyclization) | ||
| VPAC1/VPAC2 | Primary and metastatic breast, ovary, prostate, colon, bladder carcinomas, meningiomas | VIP | HSDAVFTDNYTRLRKQMAVKKYLNSILN |
| HSDAVFTRNYTRLRRQLAVKRYLNSILN-NH2 | |||
| VIP, [R8,15,21, L17]-VIP | |||
| CCK1/CCK2 | Gastrointestinal stromal tumor, stromal ovarian cancer, astrocytomas, medullary thyroid carcinomas | CCK analogs | DYMGWMDF-NH2 |
| DOTA-K(glucose)-GW-Nle-DF | |||
| DOTA-Nle-cyclo(EW-Nle-DFK)-NH2 | |||
| Minigastrin | (d-E)AYGWMDF-NH2 | ||
| l-(E)5-AYGWMDF-NH2 | |||
| (d-E)E(5)AYGWMDF-NH2 | |||
| BB1, BB2, BB3, BB4 | Prostate and breast cancer, glioma | Bombesin | pGlu-QRLNQWAVGHLM-NH2 |
| pGlu-QRLNQWAVGH-Cha-NLeu-NH2 | |||
| pGlu-QRLNQWAVG-Cha-Sta-Nleu-NH2 | |||
| pGlu-QRLNQWAV-Sta-NMeGly-Nleu-NH2 | |||
| pGlu-QRLNQWAVGH(d-Phe)M-NH2 | |||
| hMC-1R, hMC-3R, hMC-5R | Melanogenesis | α-MSH | Ac-SYSMEHFRWGKPV |
| Ac-GGNle-CCEH(d-F)RWC-NH2 | |||
| Ac-GGNle-CCEH(d-F)RWCRP-NH2 | |||
| Ac-CCEH(d-F)RWC-NleGG-NH2 | |||
| Ac-CCEH(d-F)RWCRP-NleGG-NH2 | |||
| NTR1, NTR2, NTR3 | Tumor progression (lung cancer, breast cancer, prostate cancer) | Neurotensin | ELYENKPRRPYIL |
| H-KKPYI-TMS-A-OH | |||
| RRPYIL | |||
| PEG4-RRPYIL | |||
| PEG4-RRPYIL | |||
| PEG4-RKPY-Tle-L | |||
| PEG4-KRPY-Tle-L | |||
| PEG4-KKPY-Tle-L | |||
| Integrins | angiogenesis | RGD analogs | RGD |
| RGDS | |||
| GRGDS | |||
| GRGDPS | |||
| GRGDSPK | |||
| c(RGDxX) x = d-Phe, d-Tyr, d-Trp; X = K, C, V | |||
| c(FRGDLAFp(NMe)K) | |||
| FR366 * | |||
| c(RGDfK) trimer | |||
| GRP78 | Cervix, esophagous, pancreas, lung and glioma tumors | GRP78 | GIRLRG |
| PEG-GIRLRG | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tornesello, A.L.; Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules 2017, 22, 1282. https://doi.org/10.3390/molecules22081282
Tornesello AL, Buonaguro L, Tornesello ML, Buonaguro FM. New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules. 2017; 22(8):1282. https://doi.org/10.3390/molecules22081282
Chicago/Turabian StyleTornesello, Anna Lucia, Luigi Buonaguro, Maria Lina Tornesello, and Franco Maria Buonaguro. 2017. "New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology" Molecules 22, no. 8: 1282. https://doi.org/10.3390/molecules22081282
APA StyleTornesello, A. L., Buonaguro, L., Tornesello, M. L., & Buonaguro, F. M. (2017). New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules, 22(8), 1282. https://doi.org/10.3390/molecules22081282