Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of Human Defensin Mimetics

 ,
,  ,
,  ,
, 
 and
and 
Abstract
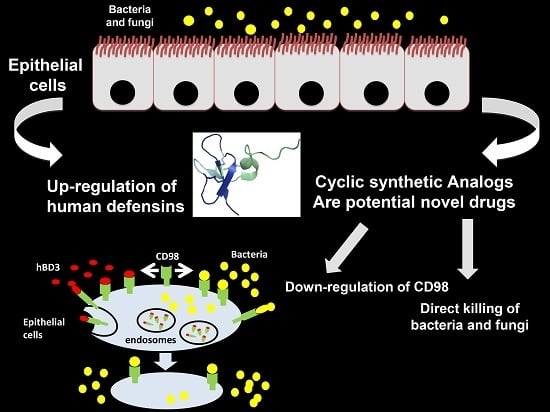
1. Introduction
2. Structure and Function Relationship: A Tool to Develop Antimicrobial Drugs
3. Design and Structure of Cyclic Analogs
4. Defensins
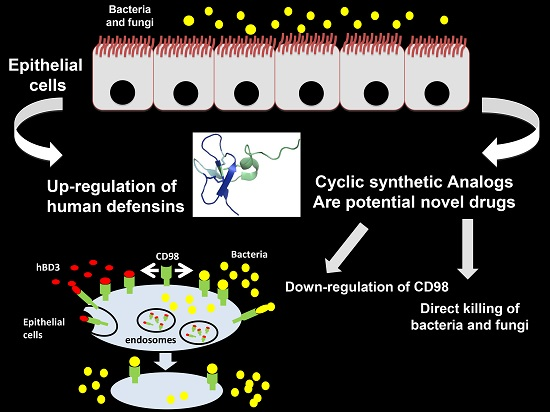
4.1. Human α-Defensins
- The antimicrobial mechanism of action is almost common between α- and β-defensins; several modes of action have been speculated, but it seems ascertained that the initial phase consists of the interactions between the positive charges of peptides and negative membranes of bacteria and viruses. The feature of human cell membranes being neutral guarantees the selective contact of defensins with host pathogens. Once interacted with membranes, defensins amass into the membrane of microbes and cause depolarization, which would finally induce death. The capacity to assemble in dimers, and maybe in oligomers, assists this process in determining the pores in the membranes. In addition to this well-described mechanism, both α- and β-defensins can also avoid pathogen cellular internalization by interacting with membrane/envelope glycoproteins. Specifically for α-defensins, it has been anticipated that a further antiviral mechanism exists; accordingly, α-defensins, by interacting with human cells, can shrink virus replication and transcription.
- Thanks to their chemotactic activity, α-defensins are also considered molecules able to activate the immune system. Indeed, in vitro studies demonstrated that HNP1, HNP3, and HD-5 increase the migration of macrophages, T cells, and immature mast cells.
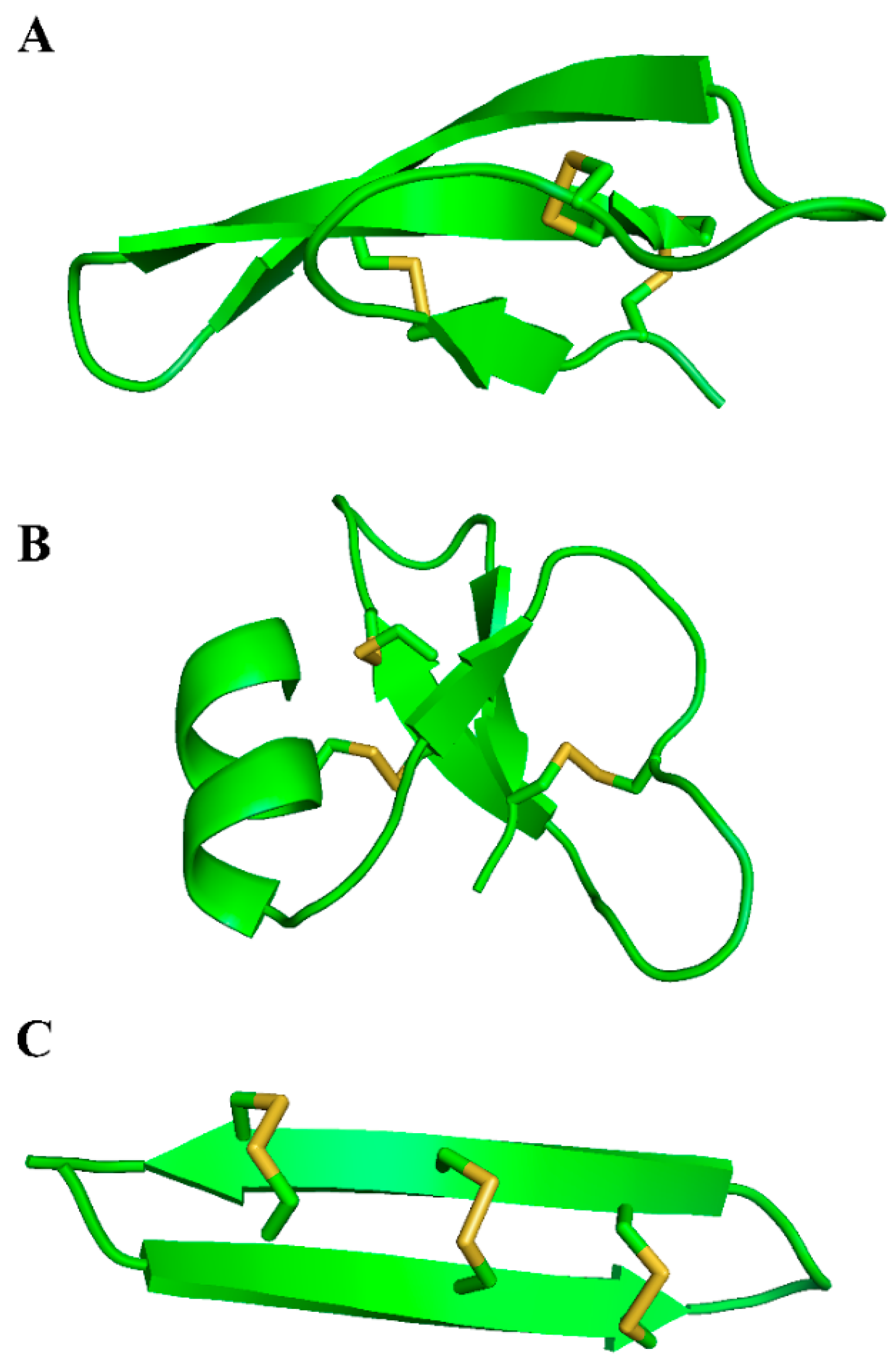
4.2. Human β-Defensins
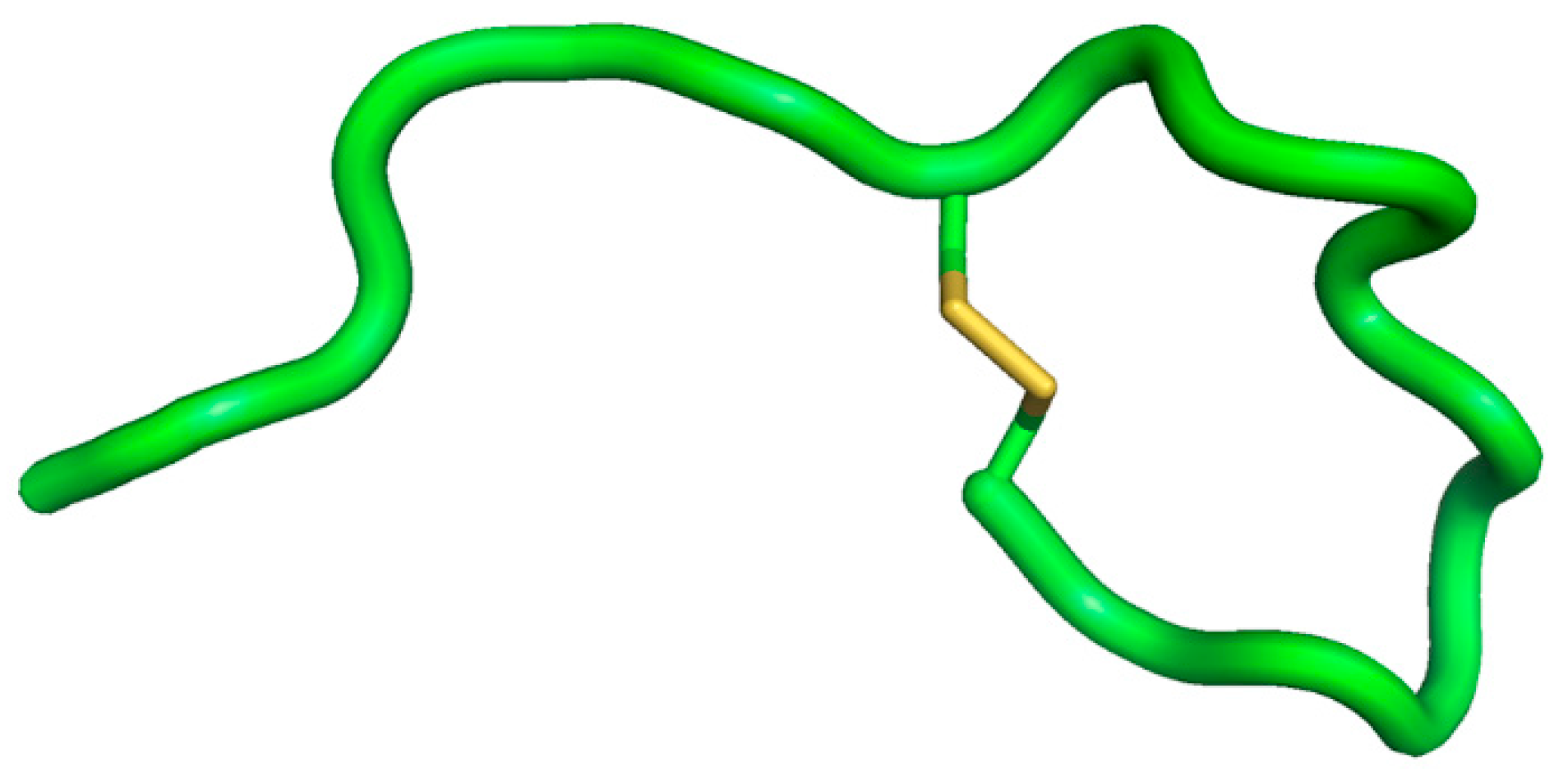
4.3. θ-Defensin
4.4. De Novo Design of Cyclic Peptides Starting from Defensins
5. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Falanga, A.; Berisio, R.; Grieco, P.; Morelli, G.; Galdiero, M. Antimicrobial peptides as an opportunity against bacterial diseases. Curr. Med. Chem. 2015, 22, 1665–1677. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.P.; Hancock, R.E. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Shai, Y. Can we predict biological activity of antimicrobial peptides from their interactions with model phospholipid membranes? Peptides 2003, 24, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Pept. Sci. 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Wieprecht, T. Structural features of helical antimicrobial peptides: Their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1999, 1462, 71–87. [Google Scholar] [CrossRef]
- Dennison, S.R.; Wallace, J.; Harris, F.; Phoenix, D.A. Amphiphilic α-helical antimicrobial peptides and their structure/function relationships. Prot. Pept. Lett. 2005, 12, 31–39. [Google Scholar] [CrossRef]
- Bradshaw, J. Cationic antimicrobial peptides: Issues for potential clinical use. BioDrugs 2003, 17, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Hahm, K.S. Novel short AMP: Design and activity study. Prot. Pept. Lett. 2012, 19, 652–656. [Google Scholar] [CrossRef]
- Danial, M.; van Dulmen, T.H.; Aleksandrowicz, J.; Potgens, A.J.; Klok, H.A. Site-specific PEGylation of HR2 peptides: Effects of PEG conjugation position and chain length on HIV-1 membrane fusion inhibition and proteolytic degradation. Bioconjug. Chem. 2012, 23, 1648–1660. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.P.; Bhonsle, J.B.; Venugopal, D.; Koser, B.W.; Magill, A.J. De novo design of selective antibiotic peptides by incorporation of unnatural amino acids. J. Med. Chem. 2007, 50, 3026–3036. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Oren, Z.; Pag, U.; Sahl, H.G.; Shai, Y. The consequence of sequence alteration of an amphipathic α-helical antimicrobial peptide and its diastereomers. J. Biol. Chem. 2002, 277, 33913–33921. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, O.; Galdiero, S.; Cantisani, M.; Di Noto, R.; Vitiello, M.; Galdiero, M.; Naclerio, G.; Cassiman, J.J.; Pedone, C.; Castaldo, G.; et al. Novel synthetic, salt-resistant analogs of human β-defensins 1 and 3 endowed with enhanced antimicrobial activity. Antimicrob. Agents Chemother. 2010, 54, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, O.; Galdiero, S.; Nigro, E.; Del Vecchio, L.; Di Noto, R.; Cantisani, M.; Colavita, I.; Galdiero, M.; Cassiman, J.J.; Daniele, A.; et al. Chimeric β-defensin analogs, including the novel 3NI analog, display salt-resistant antimicrobial activity and lack toxicity in human epithelial cell lines. Antimicrob. Agents Chemother. 2013, 57, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, O.; Nigro, E.; Cantisani, M.; Colavita, I.; Leone, M.; Mercurio, F.A.; Galdiero, M.; Pessi, A.; Daniele, A.; Salvatore, F.; et al. Design and activity of a cyclic mini-β-defensin analog: A novel antimicrobial tool. Int. J. Nanomed. 2015, 10, 6523–6539. [Google Scholar]
- Falanga, A.; Lombardi, L.; Franci, G.; Vitiello, M.; Iovene, M.R.; Morelli, G.; Galdiero, M.; Galdiero, S. Marine Antimicrobial Peptides: Nature Provides Templates for the Design of Novel Compounds against Pathogenic Bacteria. Int. J. Mol. Sci. 2016, 17, 785. [Google Scholar] [CrossRef] [PubMed]
- Shinnar, A.E.; Butler, K.L.; Park, H.J. Cathelicidin family of antimicrobial peptides: Proteolytic processing and protease resistance. Bioorg. Chem. 2003, 31, 425–436. [Google Scholar] [CrossRef]
- Cantisani, M.; Finamore, E.; Mignogna, E.; Falanga, A.; Nicoletti, G.F.; Pedone, C.; Morelli, G.; Leone, M.; Galdiero, M.; Galdiero, S. Structural insights into and activity analysis of the antimicrobial peptide myxinidin. Antimicrob. Agents Chemother. 2014, 58, 5280–5290. [Google Scholar] [CrossRef] [PubMed]
- Cantisani, M.; Leone, M.; Mignogna, E.; Kampanaraki, K.; Falanga, A.; Morelli, G.; Galdiero, M.; Galdiero, S. Structure-activity relations of myxinidin, an antibacterial peptide derived from the epidermal mucus of hagfish. Antimicrob. Agents Chemother. 2013, 57, 5665–5673. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.; Stellato, M.I.; Oliva, R.; Falanga, A.; Galdiero, M.; Petraccone, L.; D’Errico, G.; De Santis, A.; Galdiero, S.; Del Vecchio, P. Antimicrobial peptides at work: Interaction of myxinidin and its mutant WMR with lipid bilayers mimicking the P. aeruginosa and E. coli membranes. Sci. Rep. 2017, 7, 44425. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.; Carvalho, I.F.; Montelaro, R.C.; Gomes, P.; Martins, M.C. Covalent immobilization of antimicrobial peptides (AMPs) onto biomaterial surfaces. Acta Biomater. 2011, 7, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Blin, T.; Purohit, V.; Leprince, J.; Jouenne, T.; Glinel, K. Bactericidal microparticles decorated by an antimicrobial peptide for the easy disinfection of sensitive aqueous solutions. Biomacromolecules 2011, 12, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, E.; Siciliano, A.; Maselli, V.; Gesuele, R.; Guida, M.; Fulgione, D.; Galdiero, S.; Lombardi, L.; Falanga, A. An integrated study on antimicrobial activity and ecotoxicity of quantum dots and quantum dots coated with the antimicrobial peptide indolicidin. Int. J. Nanomed. 2016, 11, 4199–4211. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Pinto, S.; Evangelista, M.B.; Gil, H.; Kallip, S.; Ferreira, M.G.; Ferreira, L. High-density antimicrobial peptide coating with broad activity and low cytotoxicity against human cells. Acta Biomater. 2016, 33, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.J.; Wong, E.H.; O’Brien-Simpson, N.M.; Pantarat, N.; Blencowe, A.; Reynolds, E.C.; Qiao, G.G. Bionano Interaction Study on Antimicrobial Star-Shaped Peptide Polymer Nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 33446–33456. [Google Scholar] [CrossRef] [PubMed]
- Pal, I.; Brahmkhatri, V.P.; Bera, S.; Bhattacharyya, D.; Quirishi, Y.; Bhunia, A.; Atreya, H.S. Enhanced stability and activity of an antimicrobial peptide in conjugation with silver nanoparticle. J. Colloid Interface Sci. 2016, 483, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Yeom, J.H.; Lee, B.; Kim, D.; Lee, J.K.; Kim, S.; Bae, J.; Park, Y.; Lee, K. Gold nanoparticle-DNA aptamer conjugate-assisted delivery of antimicrobial peptide effectively eliminates intracellular Salmonella enterica serovar Typhimurium. Biomaterials 2016, 104, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Maselli, V.; Siciliano, A.; Giorgio, A.; Falanga, A.; Galdiero, S.; Guida, M.; Fulgione, D.; Galdiero, E. Multigenerational effects and DNA alterations of QDs-Indolicidin on Daphnia magna. Environ. Pollut. 2017, 224, 597–605. [Google Scholar] [CrossRef] [PubMed]
- De Alteriis, E.; Falanga, A.; Galdiero, S.; Guida, M.; Maselli, V.; Galdiero, E. Genotoxicity of gold nanoparticles functionalized with indolicidin towards Saccharomyces cerevisiae. J. Environ. Sci. 2017. [Google Scholar] [CrossRef]
- Galdiero, E.; Maselli, V.; Falanga, A.; Gesuele, R.; Galdiero, S.; Fulgione, D.; Guida, M. Integrated analysis of the ecotoxicological and genotoxic effects of the antimicrobial peptide melittin on Daphnia magna and Pseudokirchneriella subcapitata. Environ. Pollut. 2015, 203, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Ruden, S.; Hilpert, K.; Berditsch, M.; Wadhwani, P.; Ulrich, A.S. Synergistic interaction between silver nanoparticles and membrane-permeabilizing antimicrobial peptides. Antimicrob. Agents Chemother. 2009, 53, 3538–3540. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gomez, S.; Martinez-de-Tejada, G. Antimicrobial Peptides as Anti-biofilm Agents in Medical Implants. Curr. Top. Med. Chem. 2017, 17, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Yazici, H.; O’Neill, M.B.; Kacar, T.; Wilson, B.R.; Oren, E.E.; Sarikaya, M.; Tamerler, C. Engineered Chimeric Peptides as Antimicrobial Surface Coating Agents toward Infection-Free Implants. ACS Appl. Mater. Interfaces 2016, 8, 5070–5081. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Ahmed, S.; Eswari, J.S. Therapeutic cyclic lipopeptides mining from microbes: Latest strides and hurdles. World J. Microbiol. Biotechnol. 2015, 31, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Kanafani, Z.A.; Corey, G.R. Daptomycin: A rapidly bactericidal lipopeptide for the treatment of Gram-positive infections. Expert Rev. Anti-Infect. Ther. 2007, 5, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Scheinpflug, K.; Nikolenko, H.; Komarov, I.V.; Rautenbach, M.; Dathe, M. What Goes around Comes around-A Comparative Study of the Influence of Chemical Modifications on the Antimicrobial Properties of Small Cyclic Peptides. Pharmaceuticals 2013, 6, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Scheinpflug, K.; Krylova, O.; Nikolenko, H.; Thurm, C.; Dathe, M. Evidence for a novel mechanism of antimicrobial action of a cyclic R-,W-rich hexapeptide. PLoS ONE 2015, 10, e0125056. [Google Scholar] [CrossRef] [PubMed]
- Abraham, T.; Prenner, E.J.; Lewis, R.N.A.H.; Mant, C.T.; Keller, S.; Hodges, R.S.; McElhaney, R.N. Structure–activity relationships of the antimicrobial peptide gramicidin S and its analogs: Aqueous solubility, self-association, conformation, antimicrobial activity and interaction with model lipid membranes. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.L.; Hodges, R.S. Structure-activity relationships of de novo designed cyclic antimicrobial peptides based on gramicidin S. Biopolymers 2003, 71, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Klinker, K.P.; Borgert, S.J. Beyond Vancomycin: The Tail of the Lipoglycopeptides. Clin. Ther. 2015, 37, 2619–2636. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Boddy, C.N.; Brase, S.; Winssinger, N. Chemistry, Biology, and Medicine of the Glycopeptide Antibiotics. Angew. Chem. Int. Ed. Engl. 1999, 38, 2096–2152. [Google Scholar] [CrossRef]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; Van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.A.; DeMarco, S.; Gombert, F.; Moehle, K.; Obrecht, D. The design, structures and therapeutic potential of protein epitope mimetics. Drug Discov. Today 2008, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Jarczak, J.; Kosciuczuk, E.M.; Lisowski, P.; Strzalkowska, N.; Jozwik, A.; Horbanczuk, J.; Krzyzewski, J.; Zwierzchowski, L.; Bagnicka, E. Defensins: Natural component of human innate immunity. Hum. Immunol. 2013, 74, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Sankaran-Walters, S.; Hart, R.; Dills, C. Guardians of the Gut: Enteric Defensins. Front Microbiol. 2017, 8, 647. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Lu, W. α-Defensins in human innate immunity. Immunol. Rev. 2012, 245, 84–112. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Sakuragi, N.; Takakuwa, A.; Ayabe, T. Paneth cell α-defensins and enteric microbiota in health and disease. Biosci. Microbiota Food Health 2016, 35, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tolbert, W.D.; Ericksen, B.; Zhan, C.; Wu, X.; Yuan, W.; Li, X.; Pazgier, M.; Lu, W. Single, double and quadruple alanine substitutions at oligomeric interfaces identify hydrophobicity as the key determinant of human neutrophil α-defensin HNP1 function. PLoS ONE 2013, 8, e78937. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Hubert, P.; Delvenne, P.; Herfs, M. Defensins: “Simple” antimicrobial peptides or broad-spectrum molecules? Cytokine Growth Factor Rev. 2015, 26, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Mattar, E.H.; Almehdar, H.A.; Yacoub, H.A.; Uversky, V.N.; Redwan, E.M. Antimicrobial potentials and structural disorder of human and animal defensins. Cytokine Growth Factor Rev. 2016, 28, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.; Davidson, D.J.; Hancock, R.E. Immunomodulatory properties of defensins and cathelicidins. Curr. Top. Microbiol. Immunol. 2006, 306, 27–66. [Google Scholar] [PubMed]
- Fahlgren, A.; Hammarstrom, S.; Danielsson, A.; Hammarstrom, M.L. Increased expression of antimicrobial peptides and lysozyme in colonic epithelial cells of patients with ulcerative colitis. Clin. Exp. Immunol. 2003, 131, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Cobo, E.R.; Chadee, K. Antimicrobial Human β-Defensins in the Colon and Their Role in Infectious and Non-Infectious Diseases. Pathogens 2013, 2, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Suleman, H.; Otri, A.M.; Kulkarni, B.B.; Chen, P.; Hopkinson, A.; Dua, H.S. Localization and gene expression of human β-defensin 9 at the human ocular surface epithelium. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4677–4682. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Colavita, I.; Sarnataro, D.; Scudiero, O.; Zambrano, G.; Granata, V.; Daniele, A.; Carotenuto, A.; Galdiero, S.; Folliero, V.; et al. An ancestral host defence peptide within human β-defensin 3 recapitulates the antibacterial and antiviral activity of the full-length molecule. Sci. Rep. 2015, 5, 18450. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Colavita, I.; Sarnataro, D.; Scudiero, O.; Daniele, A.; Salvatore, F.; Pessi, A. Host defense peptide-derived privileged scaffolds for anti-infective drug discovery. J. Pept. Sci. 2017, 23, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Bauer, F.; Schweimer, K.; Kluver, E.; Conejo-Garcia, J.R.; Forssmann, W.G.; Rosch, P.; Adermann, K.; Sticht, H. Structure determination of human and murine β-defensins reveals structural conservation in the absence of significant sequence similarity. Protein Sci. 2001, 10, 2470–2479. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.C.; Affolter, V.K.; Bevins, C.L. Antimicrobial peptides: Agents of border protection for companion animals. Vet. Dermatol. 2012, 23, 177-e36. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Bishop, B.M.; van Hoek, L. Antimicrobial activity of human β-defensins and induction by Francisella. Biochem. Biophys. Res. Commun. 2008, 371, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Hitomi, S.; Nagase, T.; Matsui, H.; Matsuse, T.; Kimura, S.; Ouchi, Y. Effect of ions on antibacterial activity of human β-defensin 2. Microbiol. Immunol. 2000, 44, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Joly, S.; Maze, C.; McCray, P.B., Jr.; Guthmiller, J.M. Human β-defensins 2 and 3 demonstrate strain-selective activity against oral microorganisms. J. Clin. Microbiol. 2004, 42, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Wu, Z.; Nuding, S.; Groscurth, S.; Marcinowski, M.; Beisner, J.; Buchner, J.; Schaller, M.; Stange, E.F.; Wehkamp, J. Reduction of disulphide bonds unmasks potent antimicrobial activity of human β-defensin 1. Nature 2011, 469, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Raschig, J.; Mailander-Sanchez, D.; Berscheid, A.; Berger, J.; Stromstedt, A.A.; Courth, L.F.; Malek, N.P.; Brotz-Oesterhelt, H.; Wehkamp, J. Ubiquitously expressed Human β-defensin 1 (HBD1) forms bacteria-entrapping nets in a redox dependent mode of action. PLoS Pathog. 2017, 13, e1006261. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, A.; Jin, G.; Sieg, S.; McCormick, T.S. The yin and yang of human β-defensins in health and disease. Front. Immunol. 2012, 3, 294. [Google Scholar] [CrossRef] [PubMed]
- Rohrl, J.; Yang, D.; Oppenheim, J.J.; Hehlgans, T. Human β-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J. Immunol. 2010, 184, 6688–6694. [Google Scholar] [CrossRef] [PubMed]
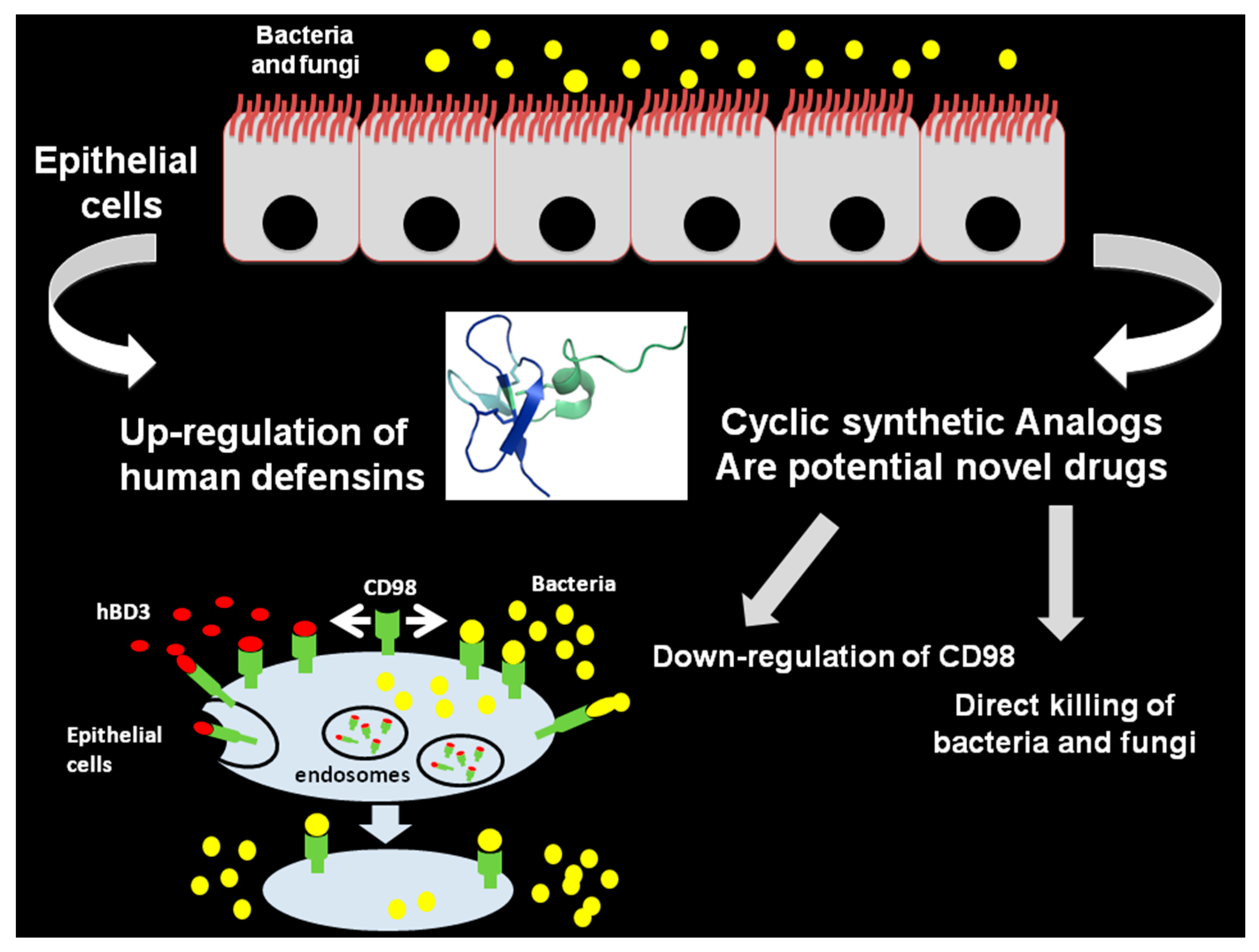
- Colavita, I.; Nigro, E.; Sarnataro, D.; Scudiero, O.; Granata, V.; Daniele, A.; Zagari, A.; Pessi, A.; Salvatore, F. Membrane protein 4F2/CD98 is a cell surface receptor involved in the internalization and trafficking of human β-Defensin 3 in epithelial cells. Chem. Biol. 2015, 22, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E. θ-Defensins: Cyclic antimicrobial peptides produced by binary ligation of truncated α-defensins. Curr. Protein Pept. Sci. 2004, 5, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.C.; Craik, D.J. The chemistry and biology of θ-defensins. Angew. Chem. Int. Ed. Engl. 2014, 53, 10612–10623. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Cole, A.M.; Selsted, M.E. θ-Defensins: Cyclic peptides with endless potential. J. Biol. Chem. 2012, 287, 27014–27019. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.A.; Wang, W.; Rawat, S.S.; Jung, G.; Waring, A.J.; Cole, A.M.; Lu, H.; Yan, X.; Daly, N.L.; Craik, D.J.; et al. θ-Defensins prevent HIV-1 Env-mediated fusion by binding gp41 and blocking 6-helix bundle formation. J. Biol. Chem. 2006, 281, 18787–18792. [Google Scholar] [CrossRef] [PubMed]
- Yasin, B.; Wang, W.; Pang, M.; Cheshenko, N.; Hong, T.; Waring, A.J.; Herold, B.C.; Wagar, E.A.; Lehrer, R.I. θ Defensins protect cells from infection by herpes simplex virus by inhibiting viral adhesion and entry. J. Virol. 2004, 78, 5147–5156. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; White, M.R.; Tecle, T.; Gantz, D.; Crouch, E.C.; Jung, G.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Hartshorn, K.L. Interactions of α-, β-, and θ-defensins with influenza A virus and surfactant protein D. J. Immunol. 2009, 182, 7878–7887. [Google Scholar] [CrossRef] [PubMed]
- Yount, N.Y.; Yeaman, M.R. Multidimensional signatures in antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 7363–7368. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.C.; Bochen, A.; Rosengren, K.J.; Stupar, P.; Wang, C.; Kessler, H.; Craik, D.J. The Cyclic Cystine Ladder of θ-defensins as a Stable, Bifunctional Scaffold: A Proof-of-Concept Study Using the Integrin-Binding RGD Motif. ChemBioChem 2014, 15, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Du, J. Cyclotides as drug design scaffolds. Curr. Opin. Chem. Biol. 2017, 38, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant Cyclotides: A Unique Family of Cyclic and Knotted Proteins that Defines the Cyclic Cystine Knot Structural Motif1. J. Mol. Biol. 1999, 294, 1327–1336. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year of Approval | Generic Name | Indication | Mode of Action | Route of Administration | Company |
|---|---|---|---|---|---|
| 2009 | Telavancin | Skin and skin structure infections, nosocomial pneumonia | Bacterial cell-wall synthesis inhibitor | IV infusion | Theravance |
| 2014 | Dalbavancin | Skin and skin structure infections | Bacterial cell-wall synthesis inhibitor | IV infusion | Durata Therapeutics/Teva |
| 2014 | Oritavancin | Skin and skin structure infections | Bacterial cell-wall synthesis inhibitor | IV infusion | The Medicines Company |
| 2006 | Anidula fungin | Fungal infections | Fungal 1,3-β-d-glucan synthase inhibitor | IV infusion | Vicuron/Pfizer |
| Phase 2 | POL7080 | P. aeruginosa infections, Gram-negative infections | LptD protein homolog inhibitor, inhibits outer-membrane biogenesis | Polyphor |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falanga, A.; Nigro, E.; De Biasi, M.G.; Daniele, A.; Morelli, G.; Galdiero, S.; Scudiero, O. Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of Human Defensin Mimetics. Molecules 2017, 22, 1217. https://doi.org/10.3390/molecules22071217
Falanga A, Nigro E, De Biasi MG, Daniele A, Morelli G, Galdiero S, Scudiero O. Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of Human Defensin Mimetics. Molecules. 2017; 22(7):1217. https://doi.org/10.3390/molecules22071217
Chicago/Turabian StyleFalanga, Annarita, Ersilia Nigro, Margherita Gabriella De Biasi, Aurora Daniele, Giancarlo Morelli, Stefania Galdiero, and Olga Scudiero. 2017. "Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of Human Defensin Mimetics" Molecules 22, no. 7: 1217. https://doi.org/10.3390/molecules22071217
APA StyleFalanga, A., Nigro, E., De Biasi, M. G., Daniele, A., Morelli, G., Galdiero, S., & Scudiero, O. (2017). Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of Human Defensin Mimetics. Molecules, 22(7), 1217. https://doi.org/10.3390/molecules22071217