
Synthetic Strategies for 5- and 6-Membered Ring Azaheterocycles Facilitated by Iminyl Radicals

Abstract
:
1. Introduction
2. Syntheses of Dihydropyrroles and Pyrroles
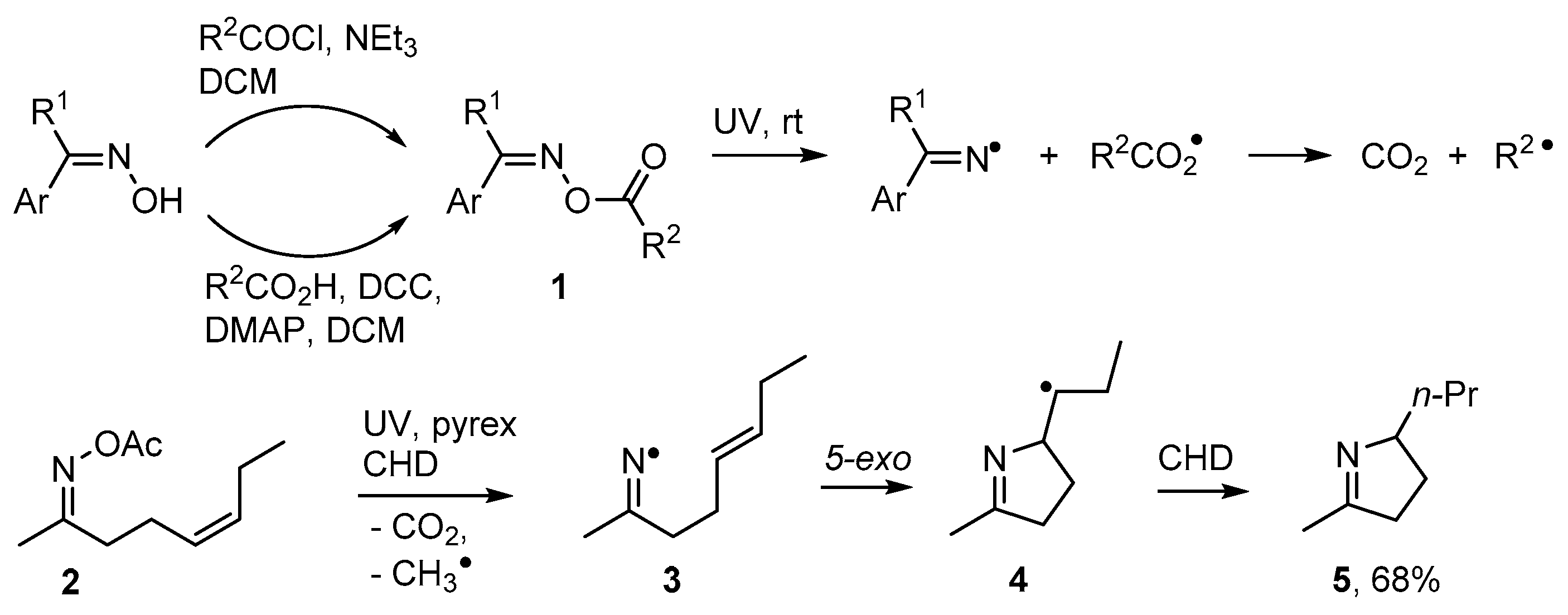
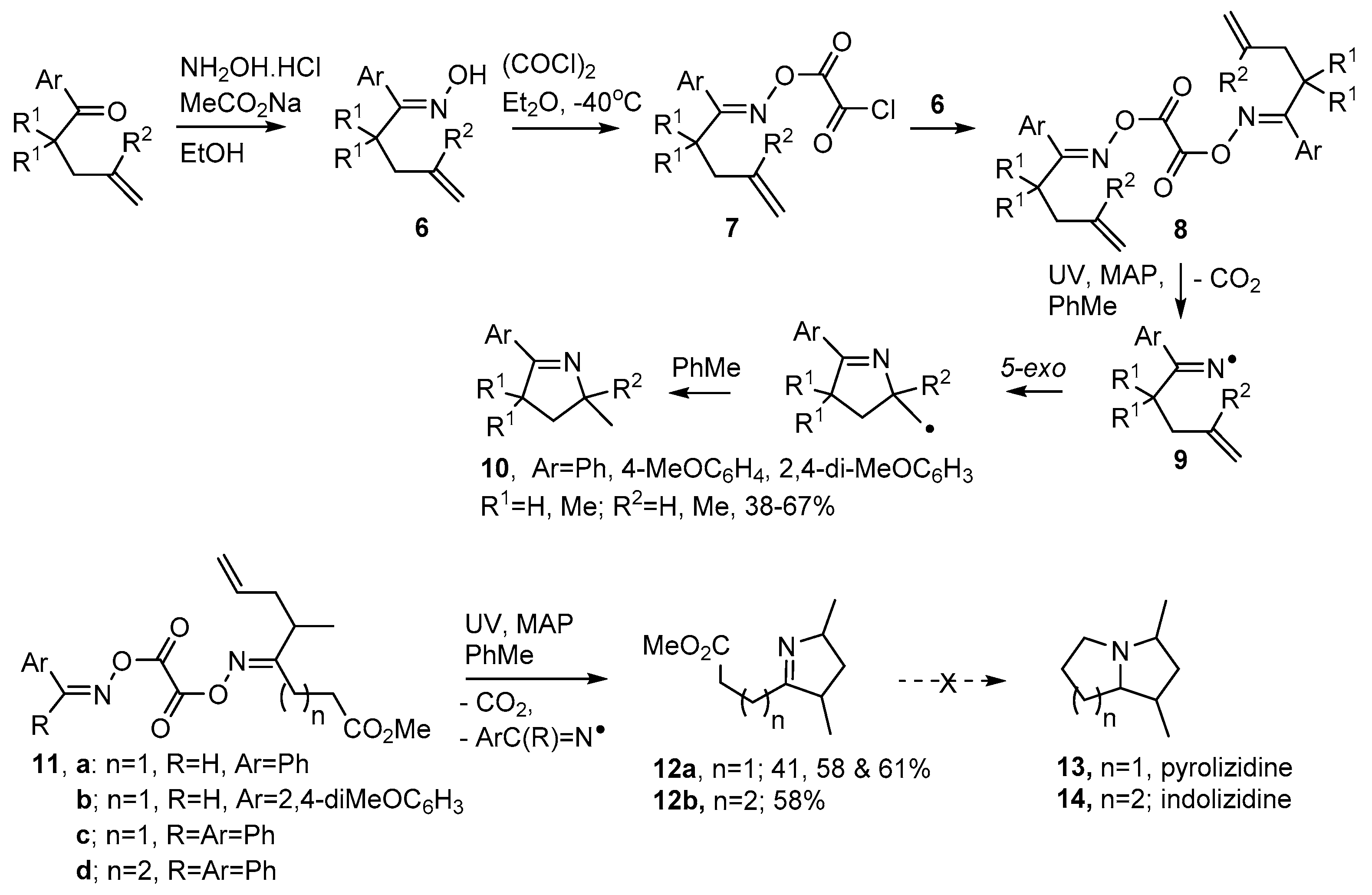
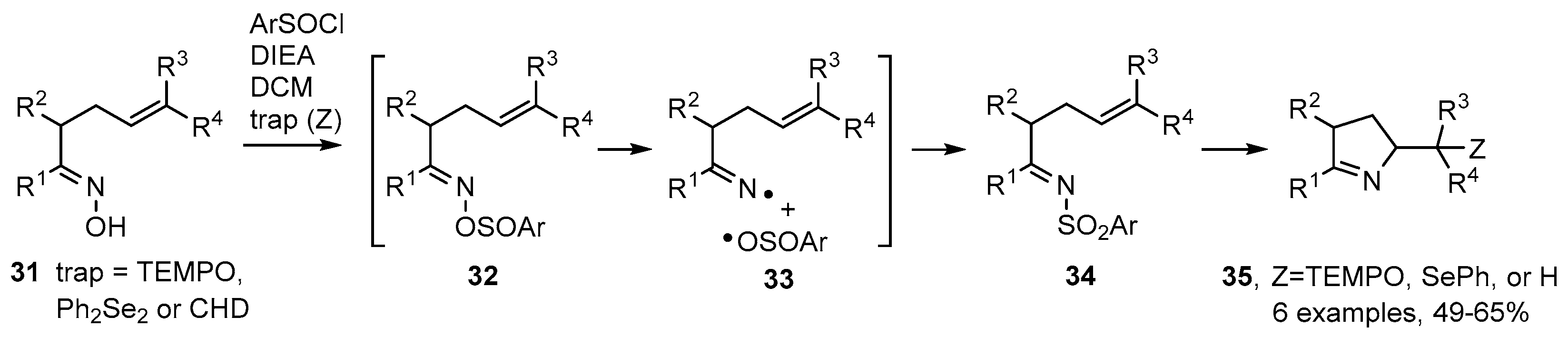
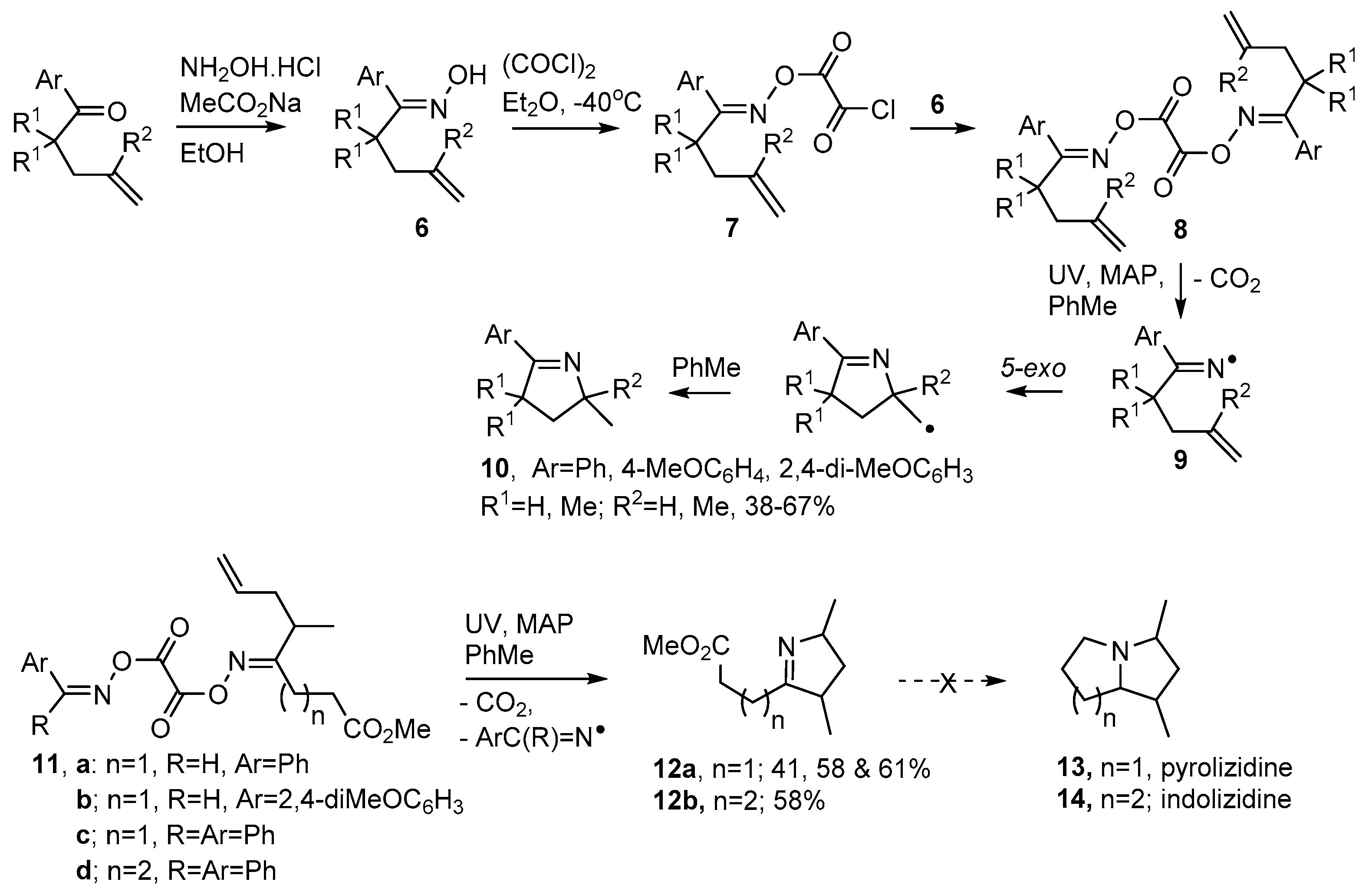
2.1. Pyrrole and Dihydropyrrole Preparations from Carbonyl Oximes
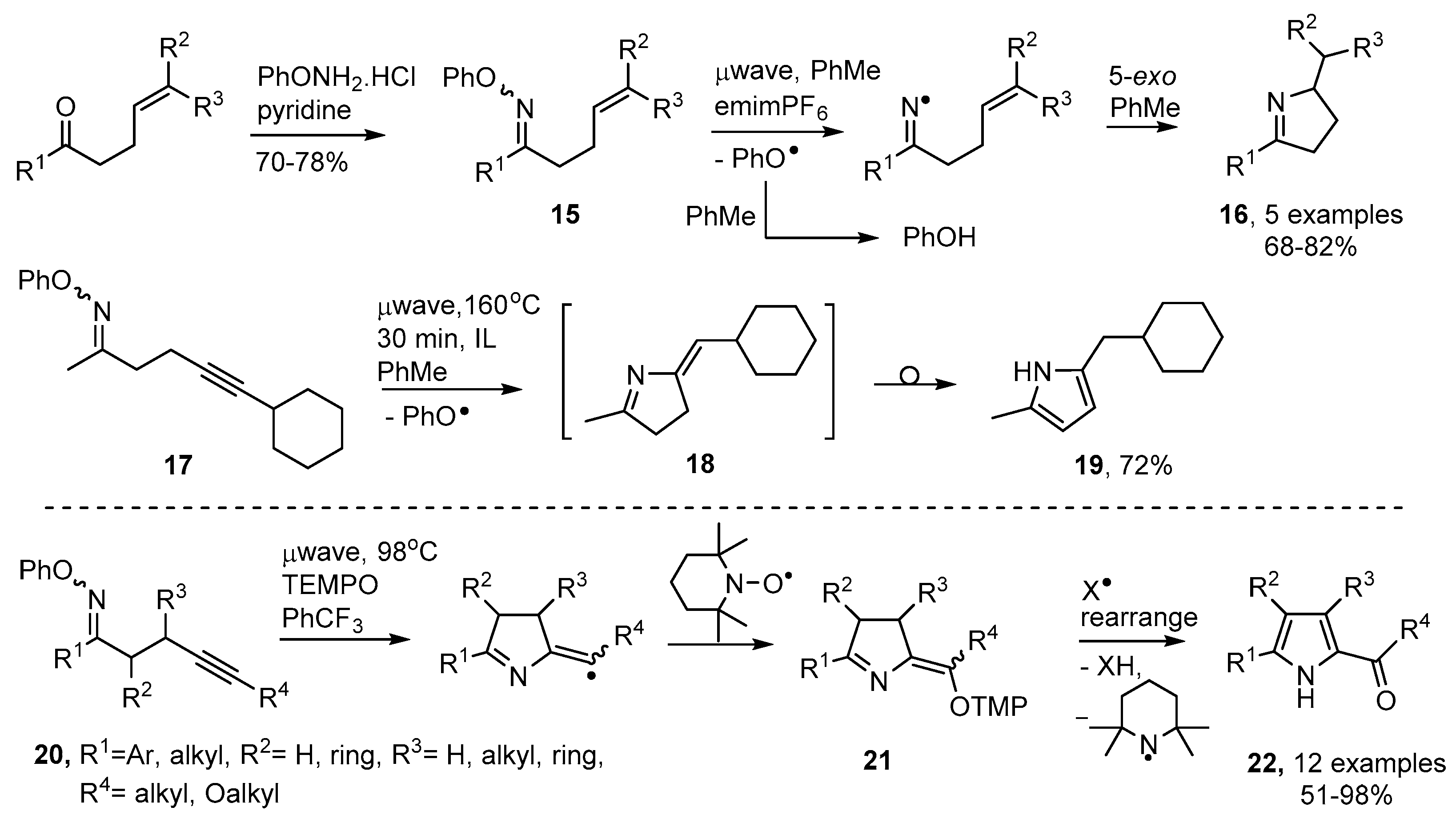
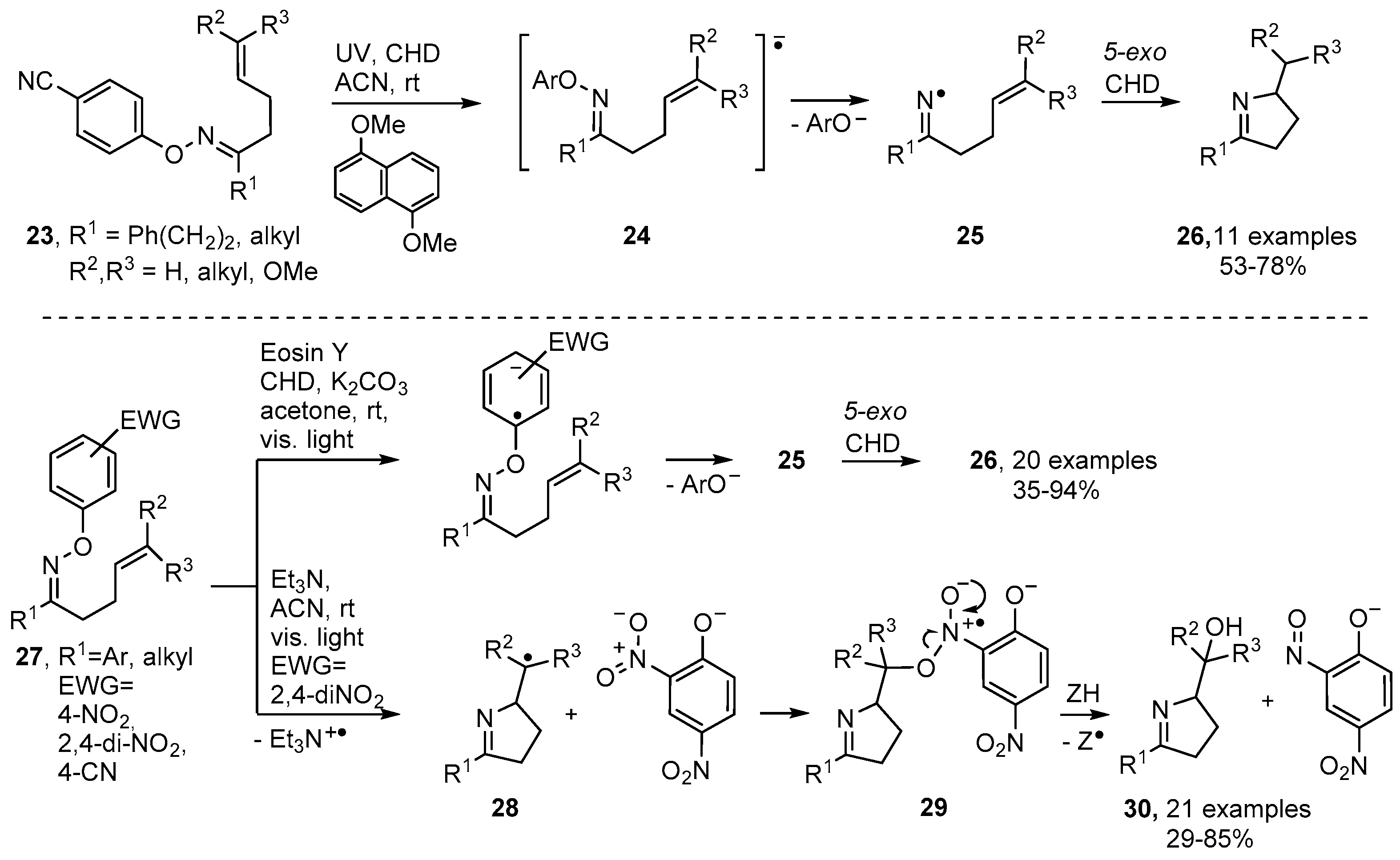
2.2. Pyrrole and Dihydropyrrole Preparations from Oxime Ethers
2.3. Photoredox Catalyzed Preparations of Pyrroles and Dihydropyrroles from Oxime Derivatives
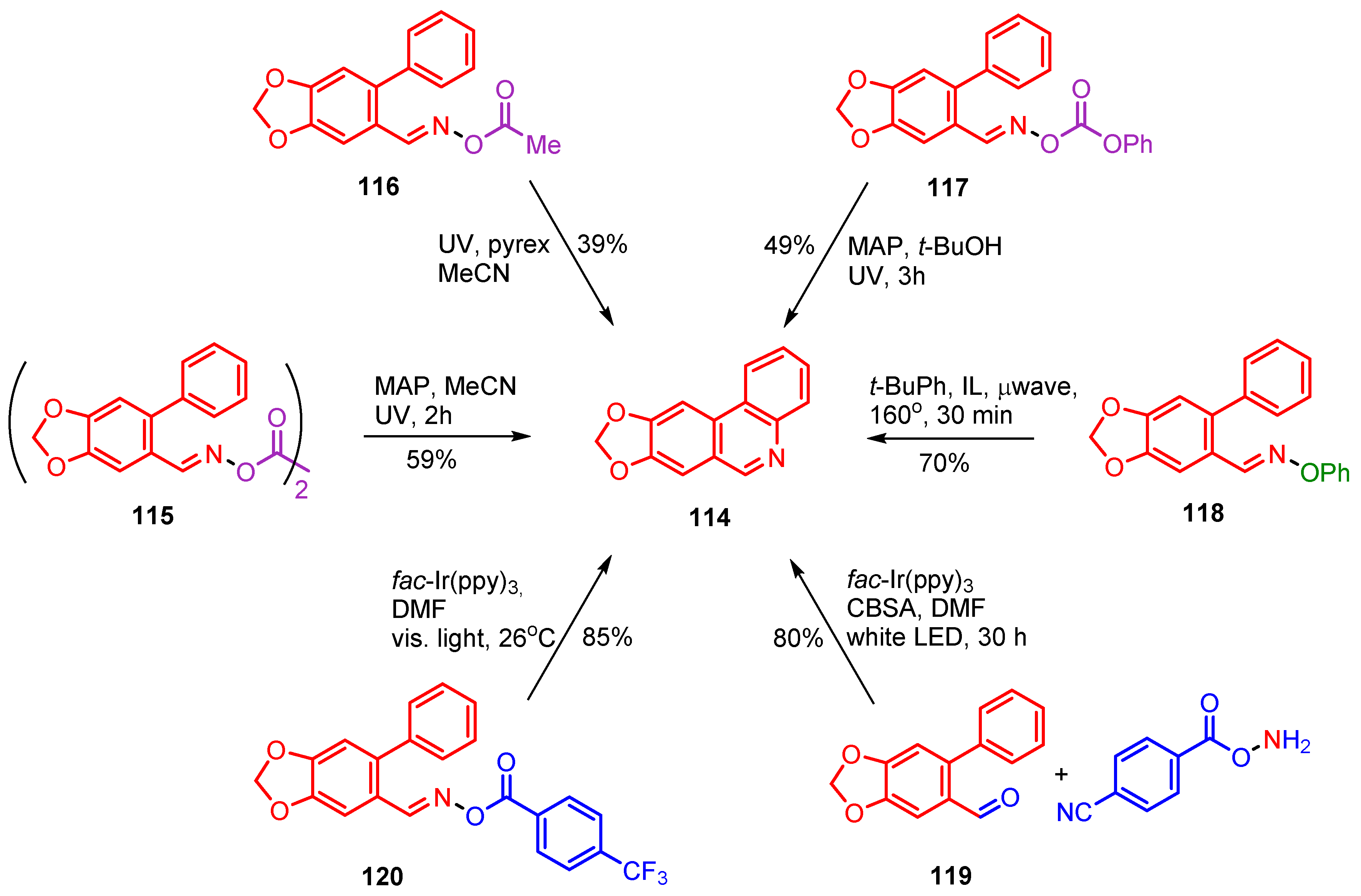
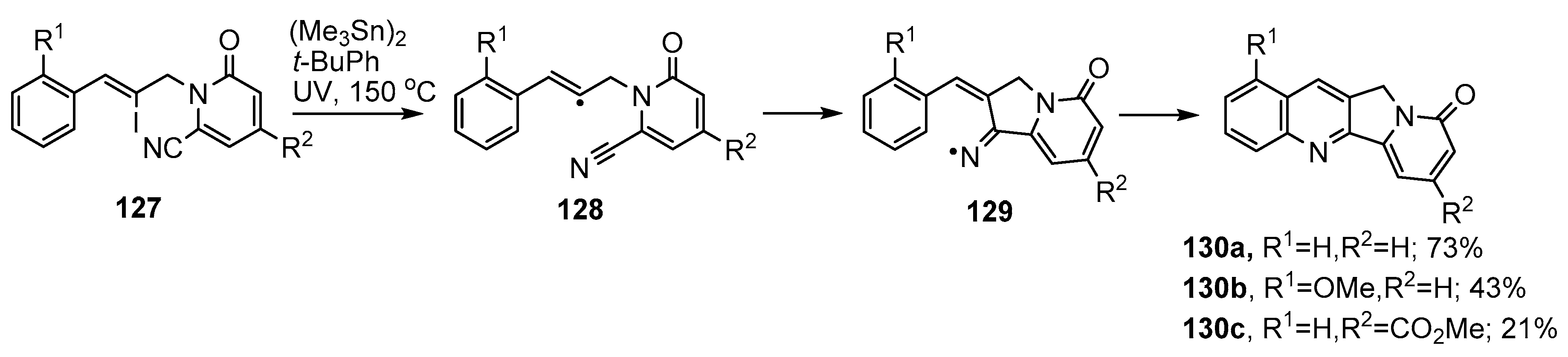
3. Preparations of Pyridine, Quinoline, Phenanthridine and Related Aza-Arenes
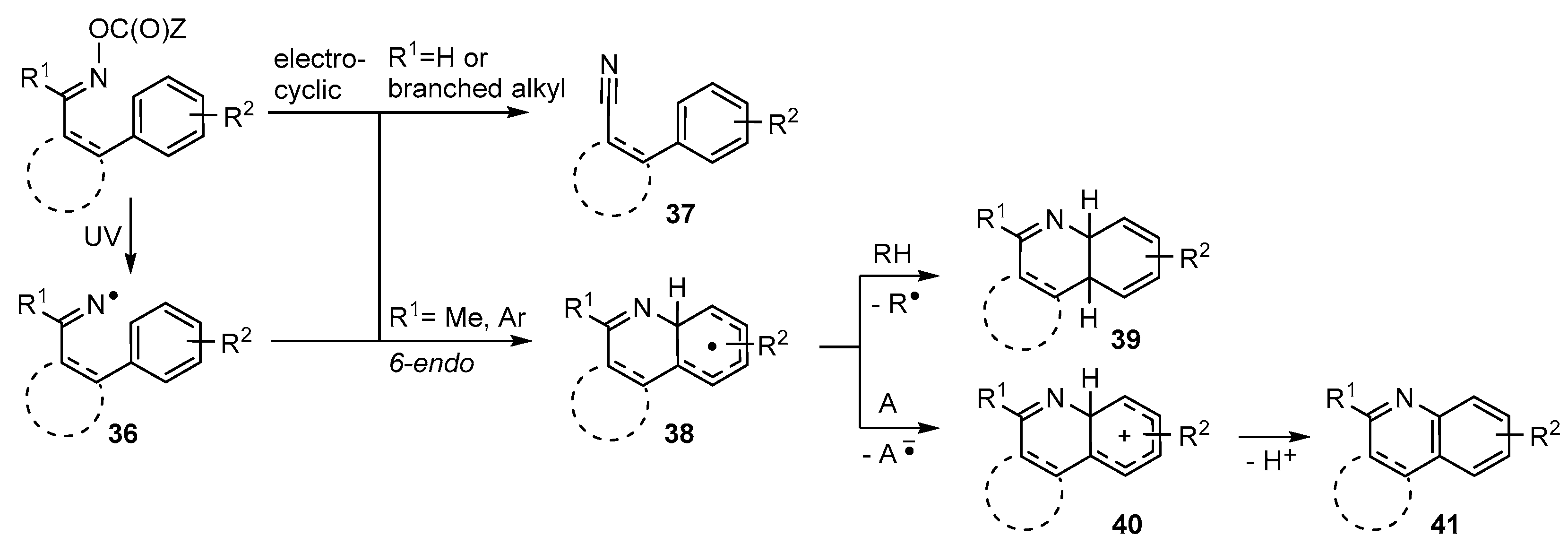
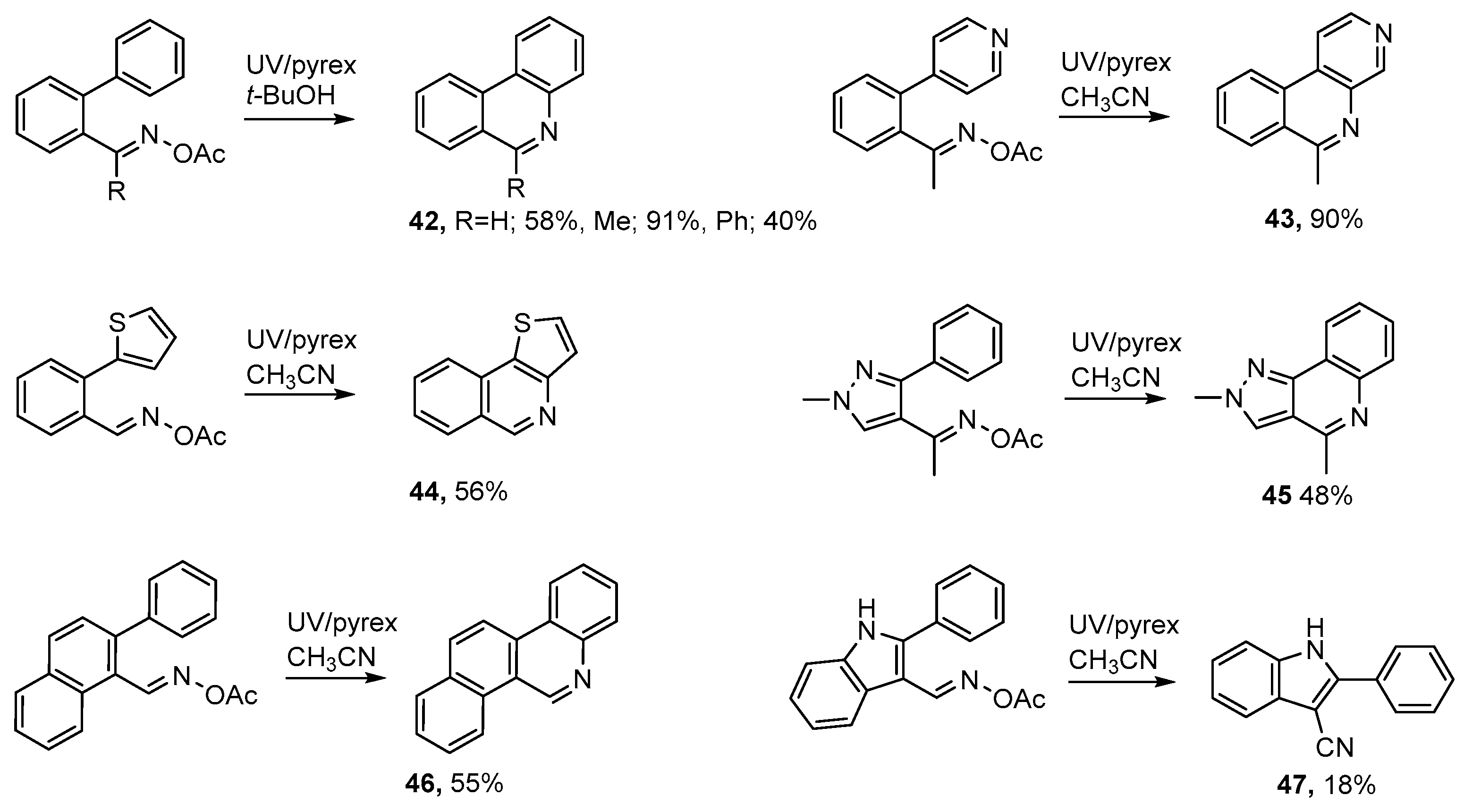
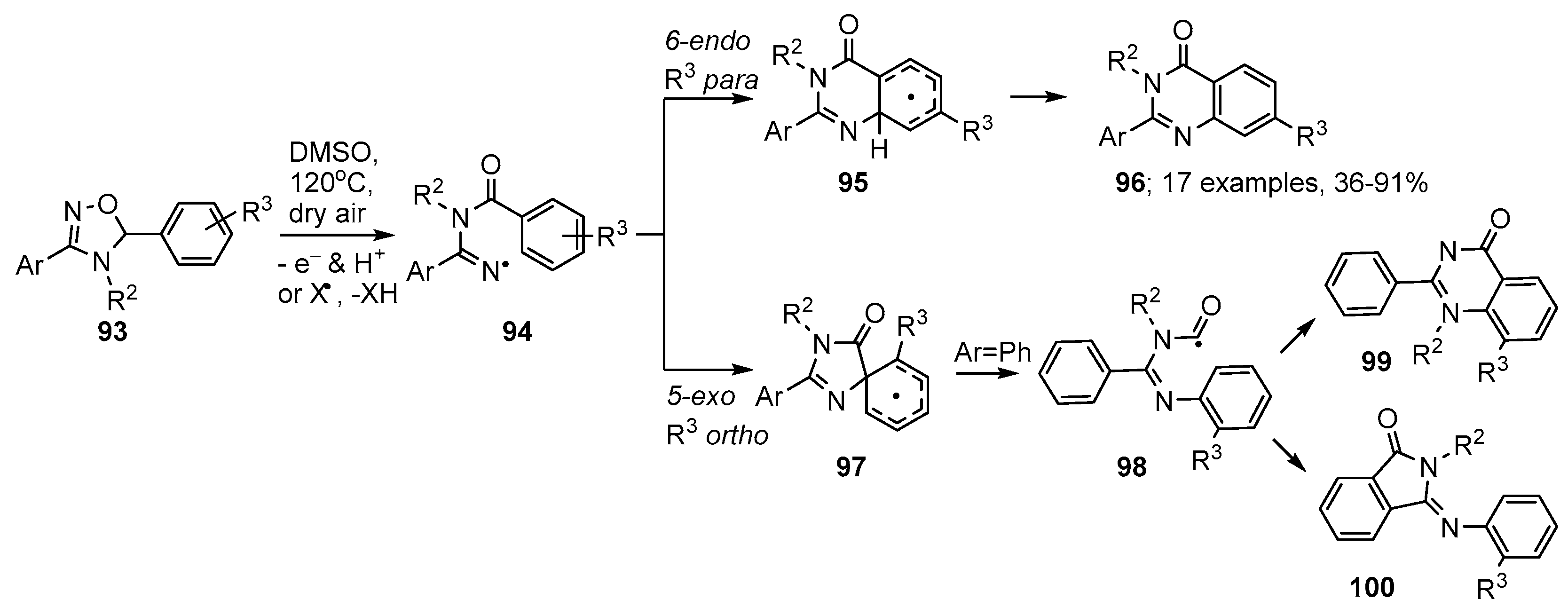
3.1. Preparations of Aza-Arenes from Carbonyl Oximes
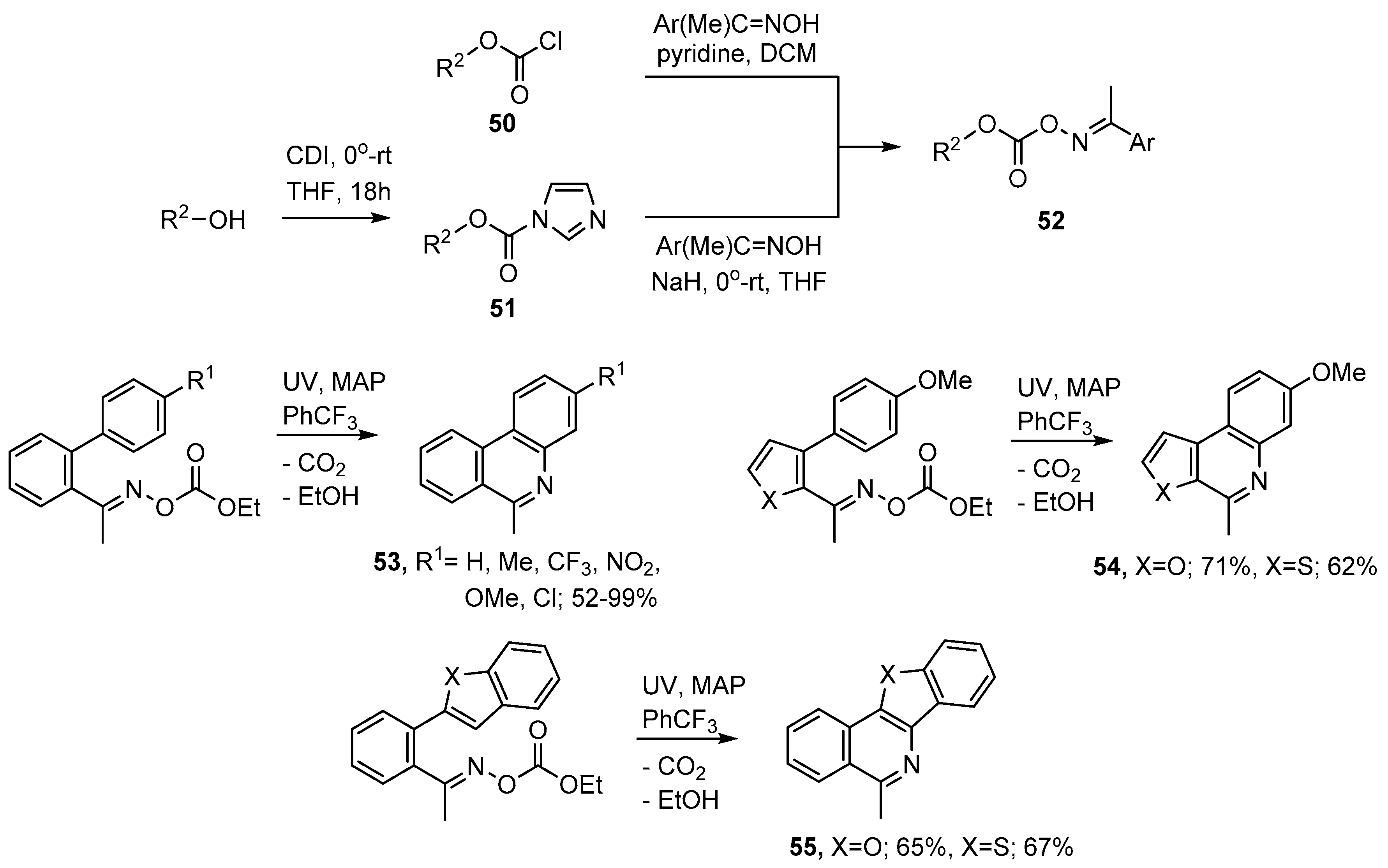
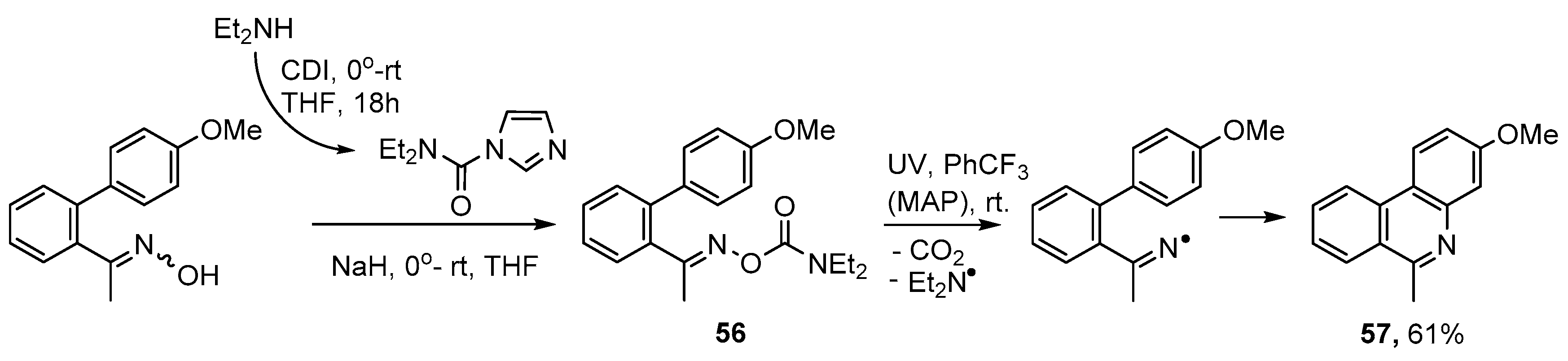
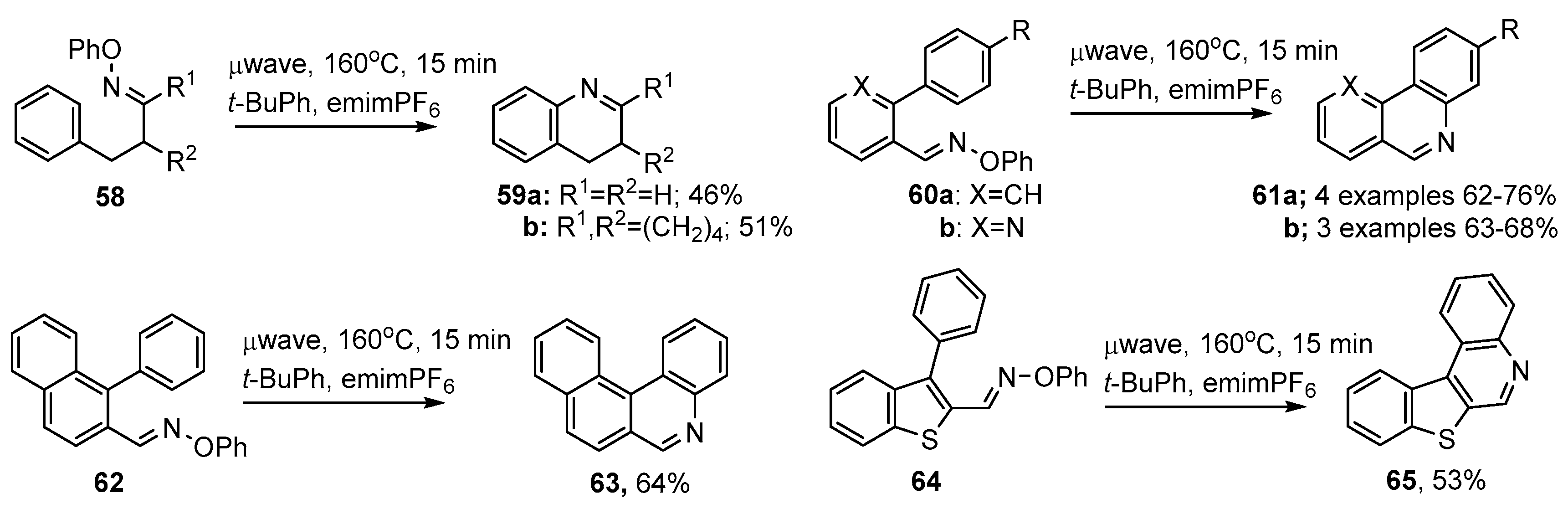
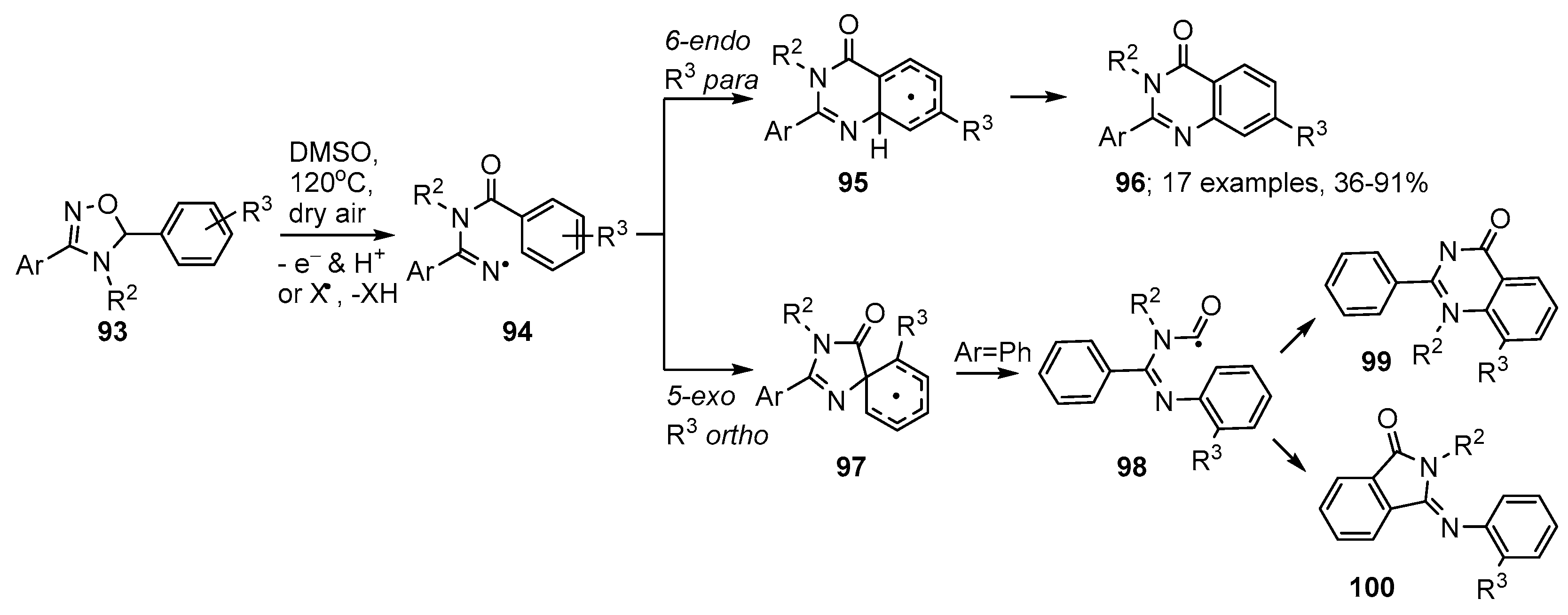
3.2. Preparations of Aza-Arenes from Oxime Ethers
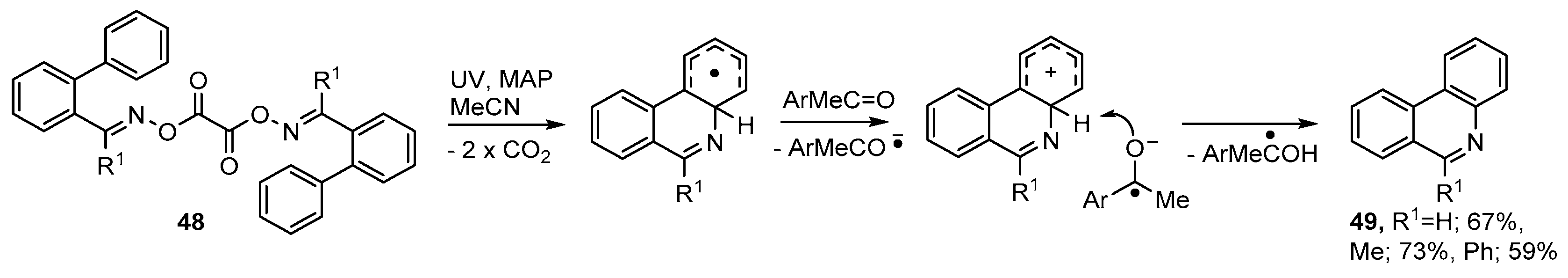
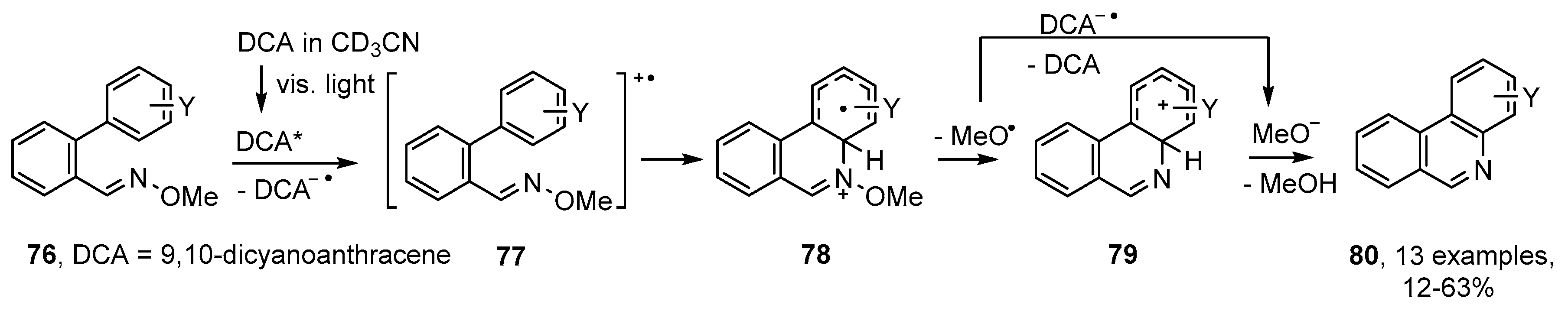
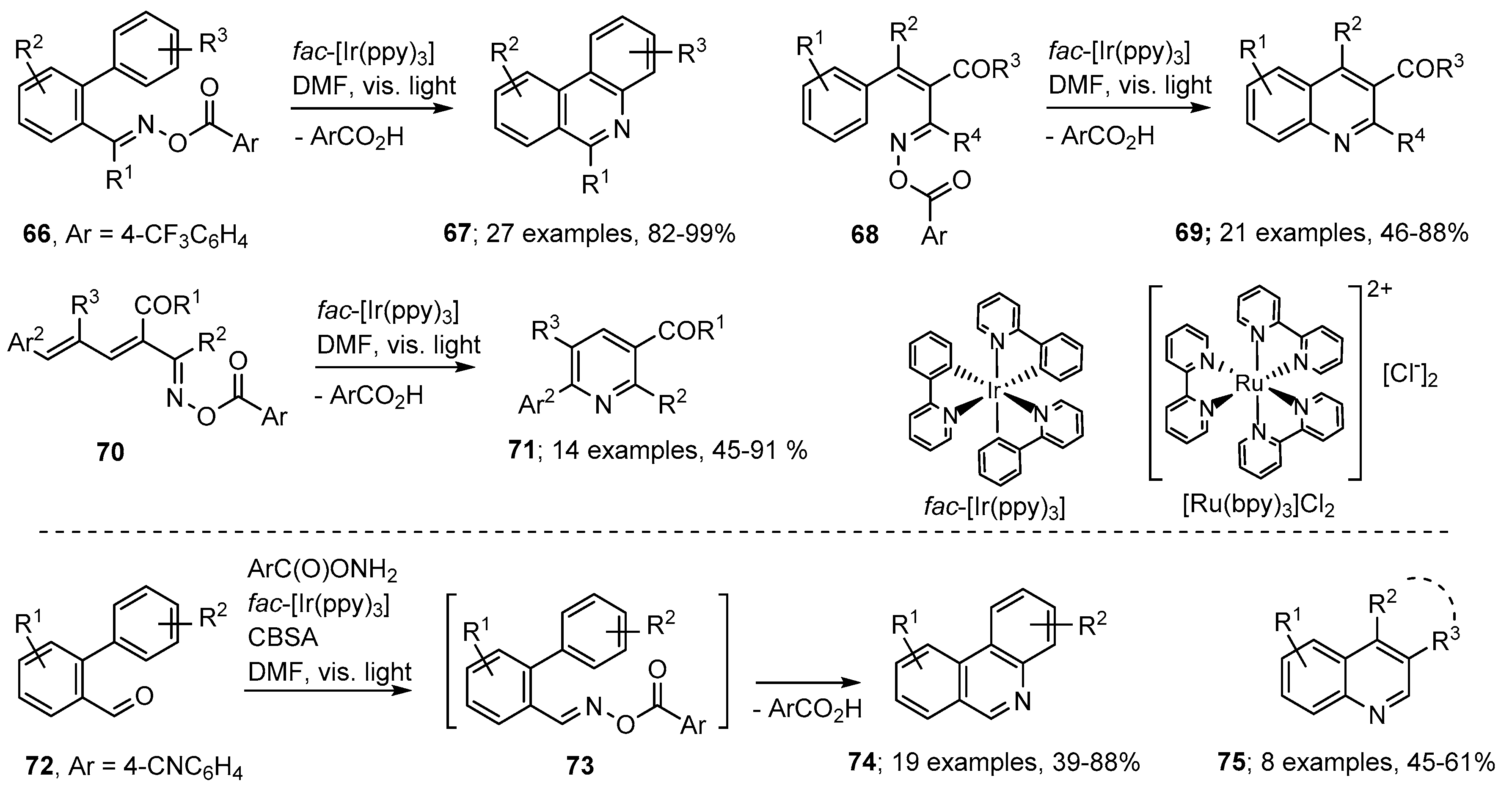
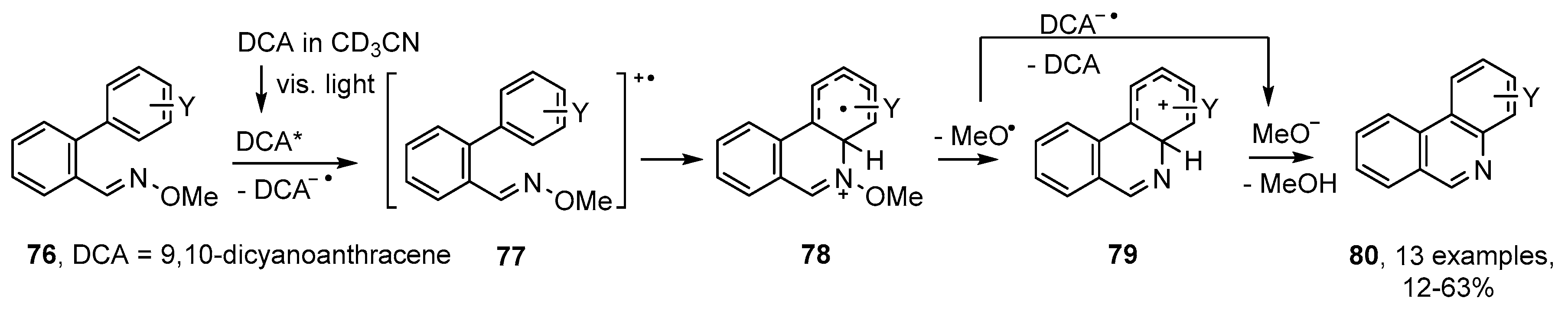
3.3. Photoredox Catalyzed Preparations of Aza-Arenes from Oxime Derivatives
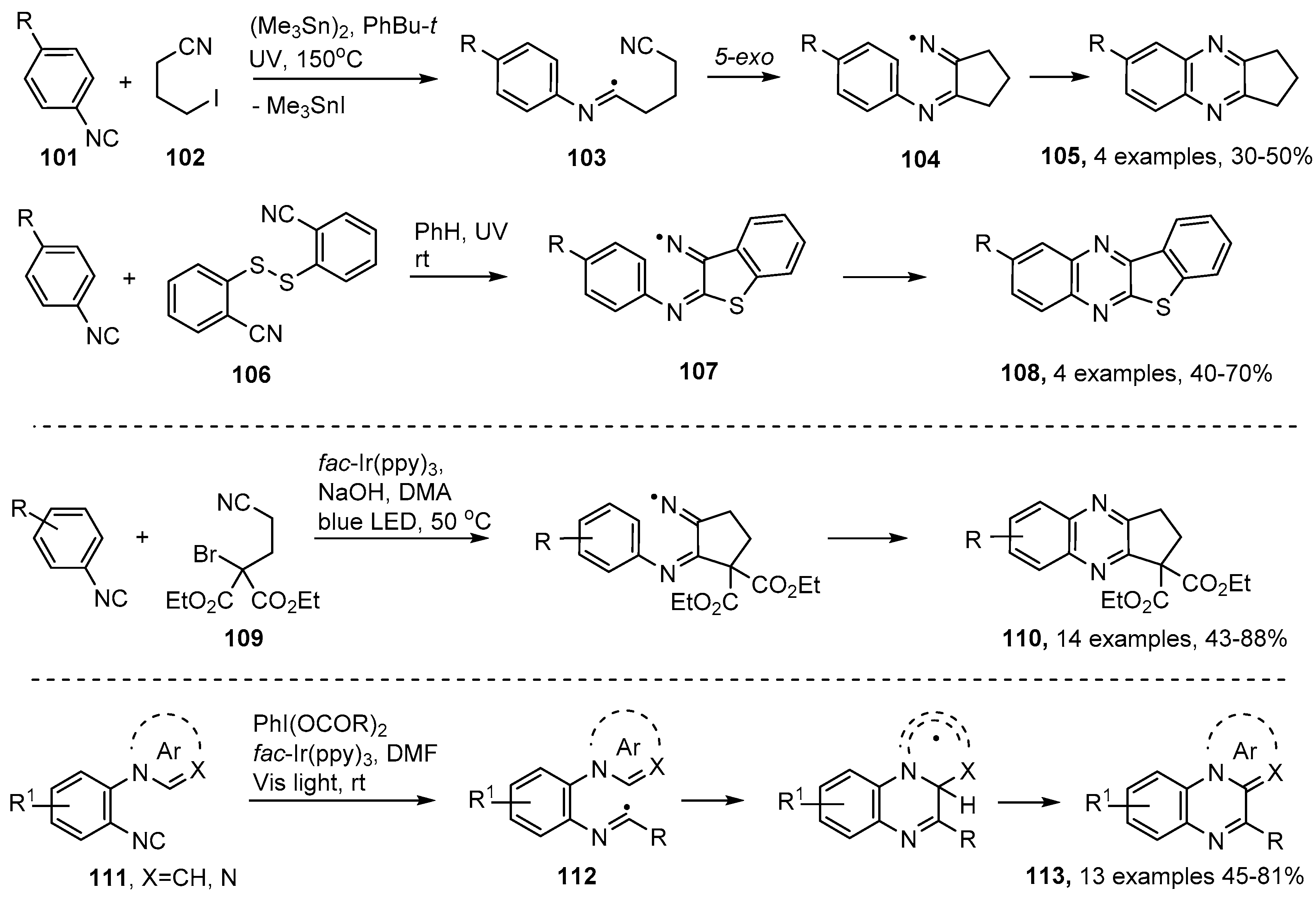
4. Preparations of Quinazoline, Quinoxaline and Related Heterocycles from Oxime Derivatives
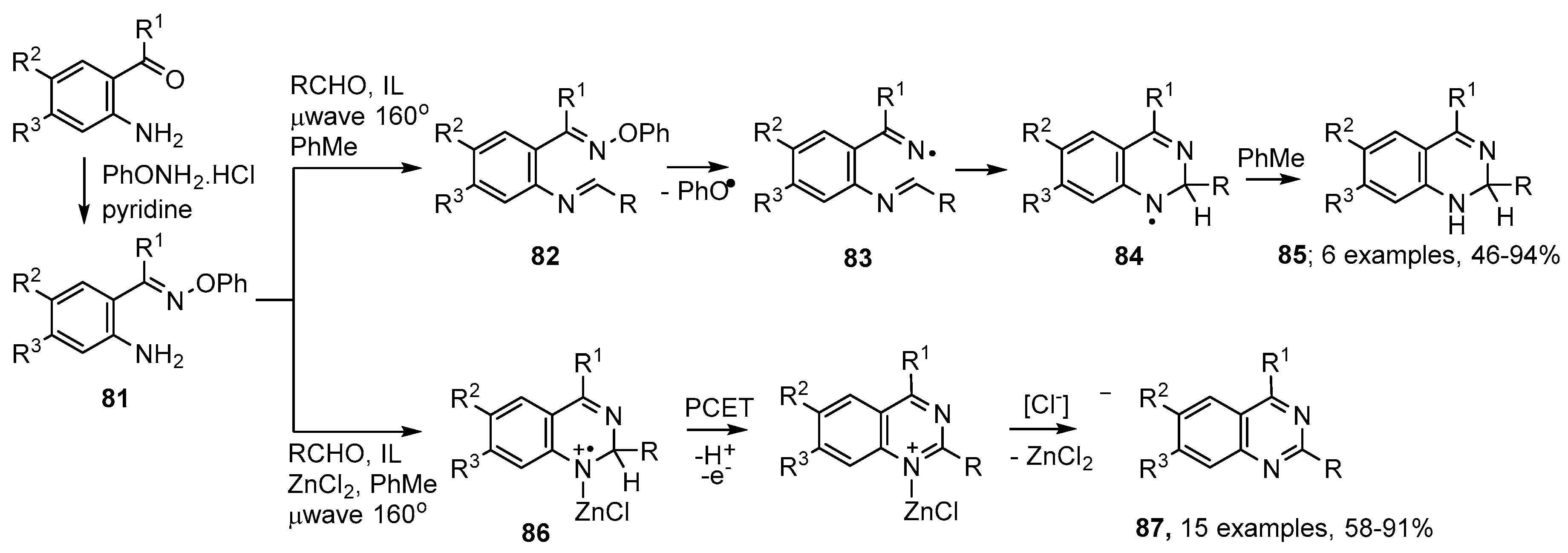
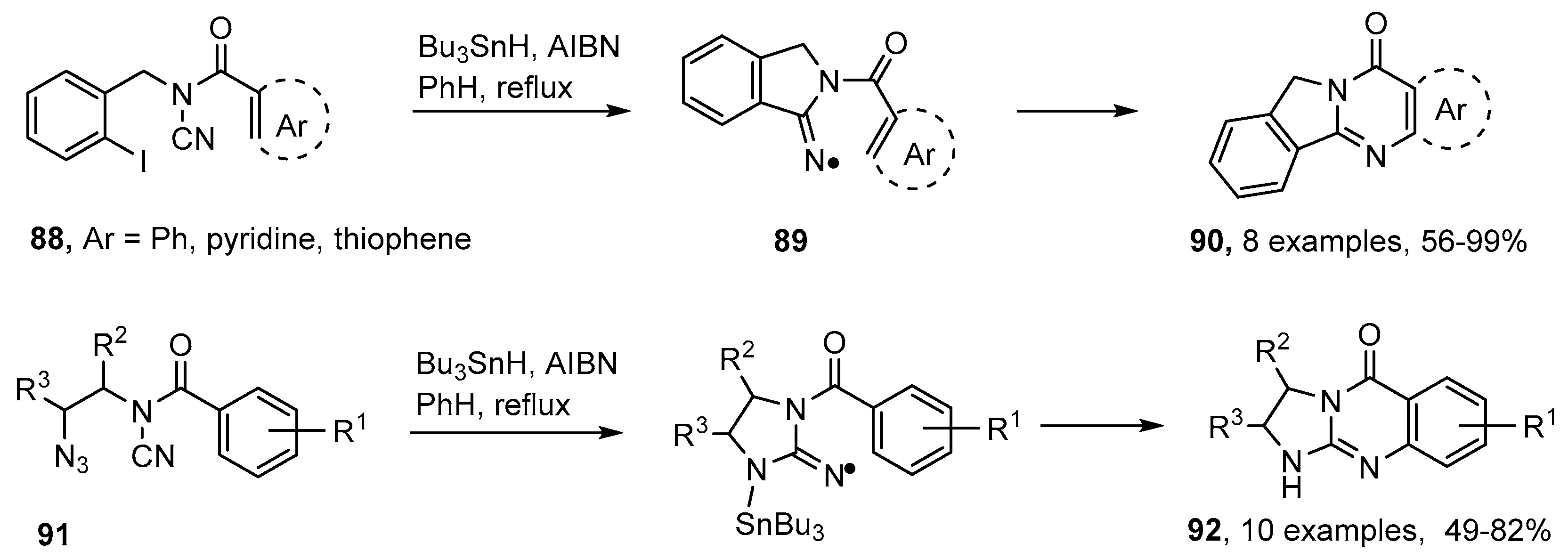
4.1. Preparations of Quinazoline Derivatives
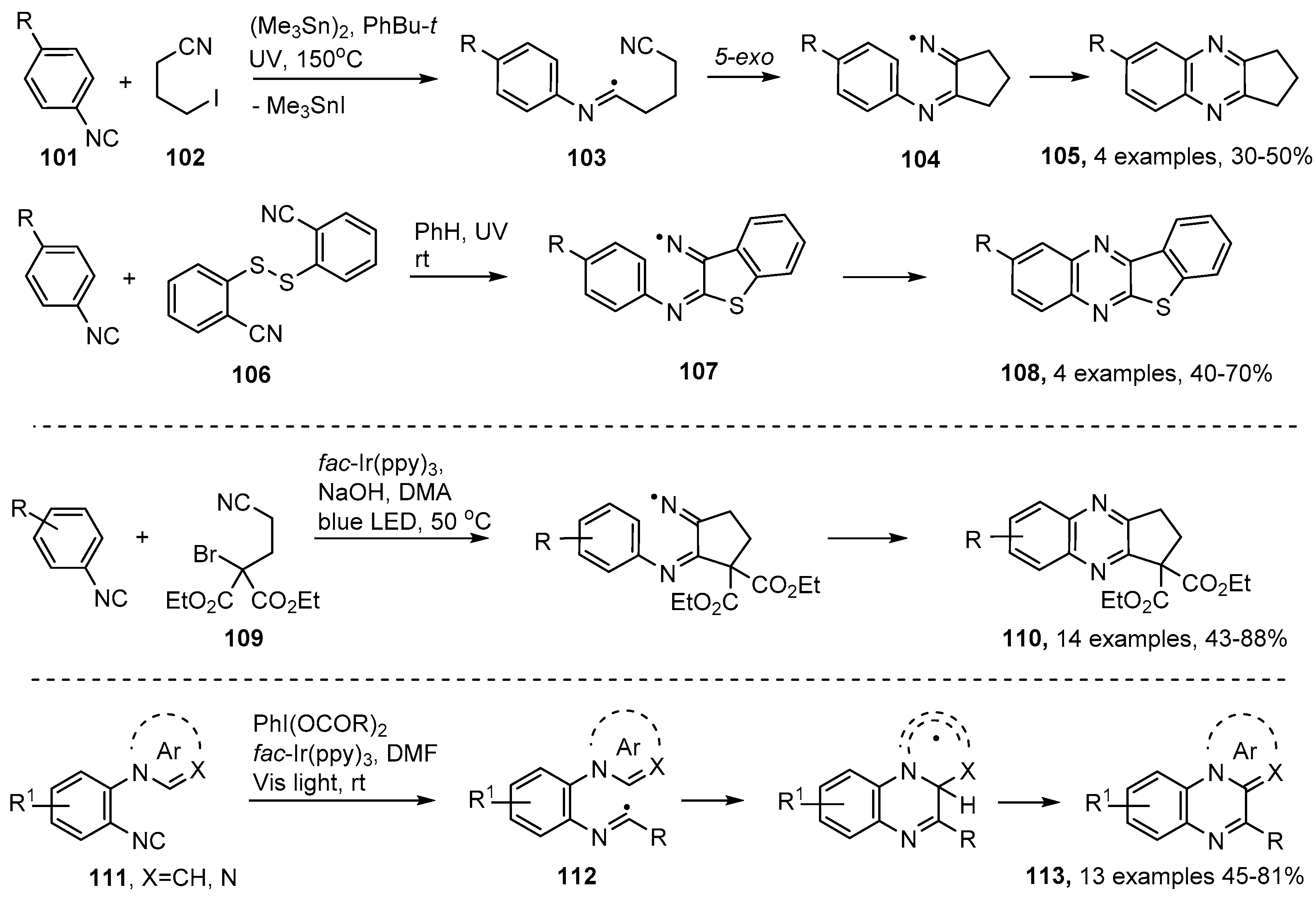
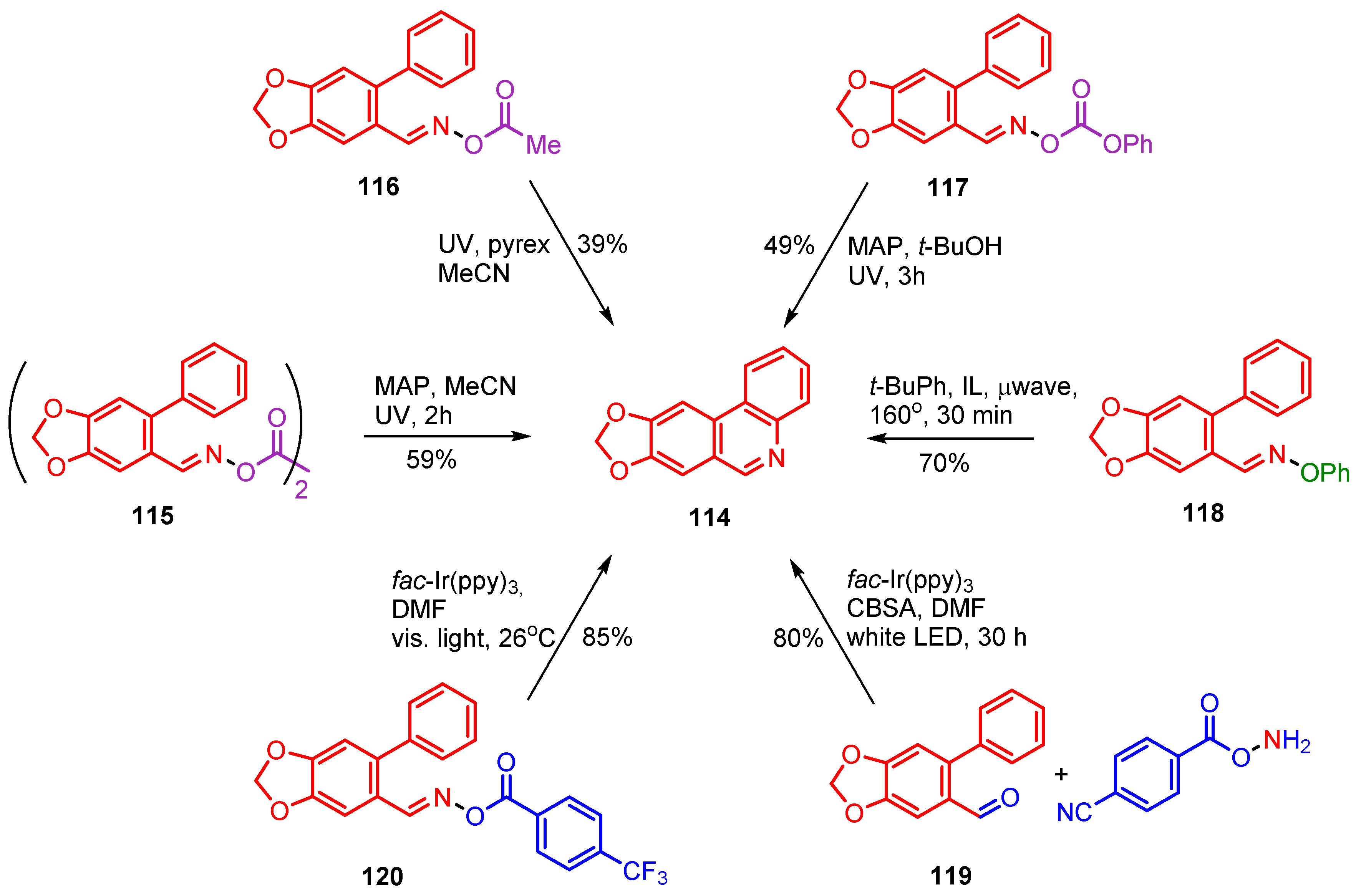
4.2. Preparations of Quinoxaline Derivatives
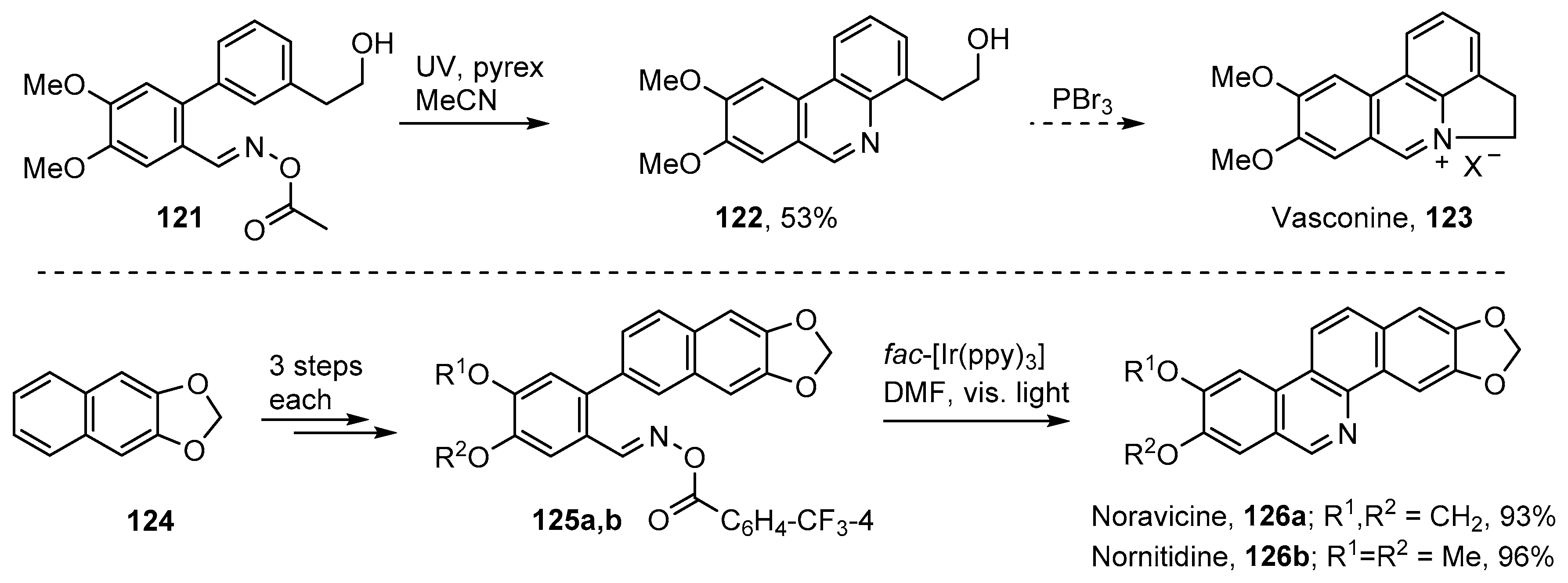
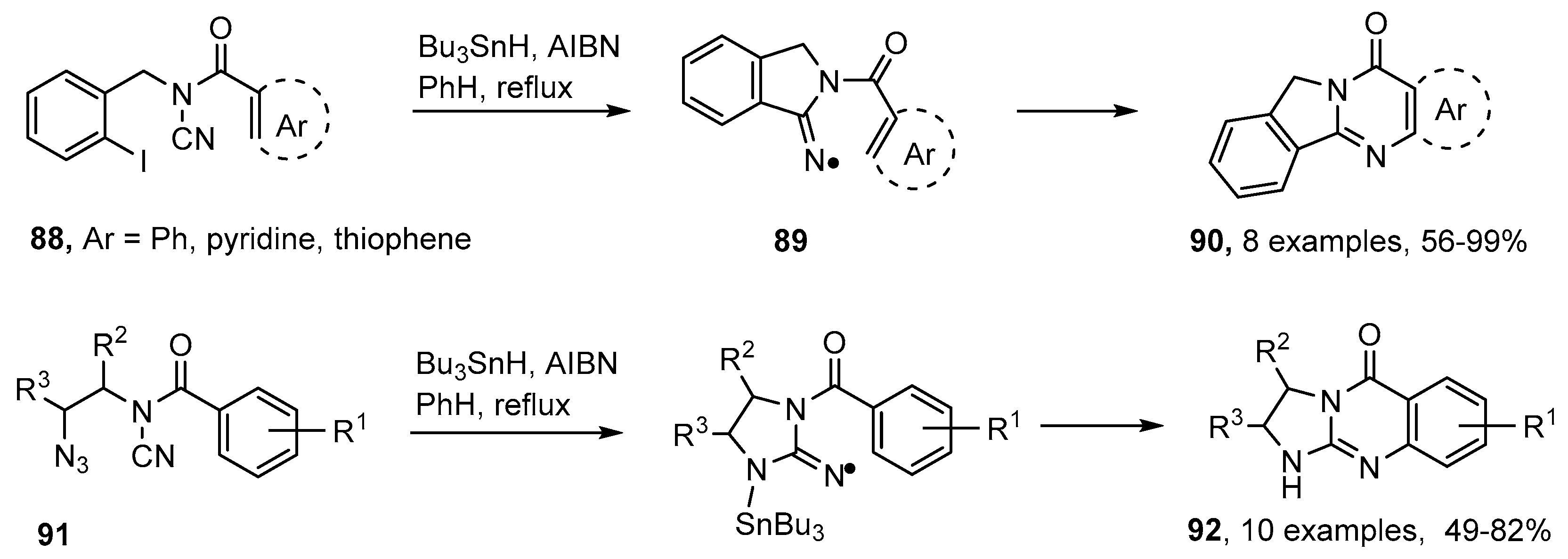
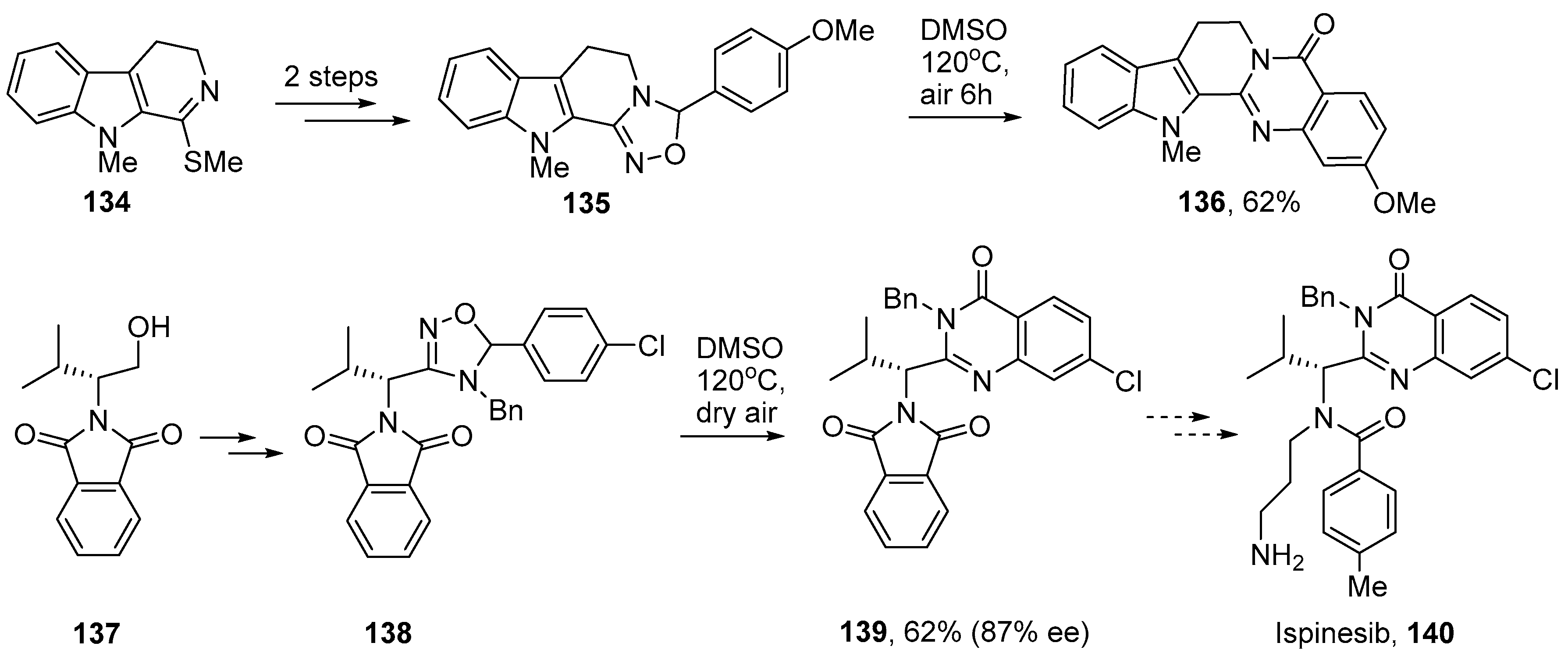
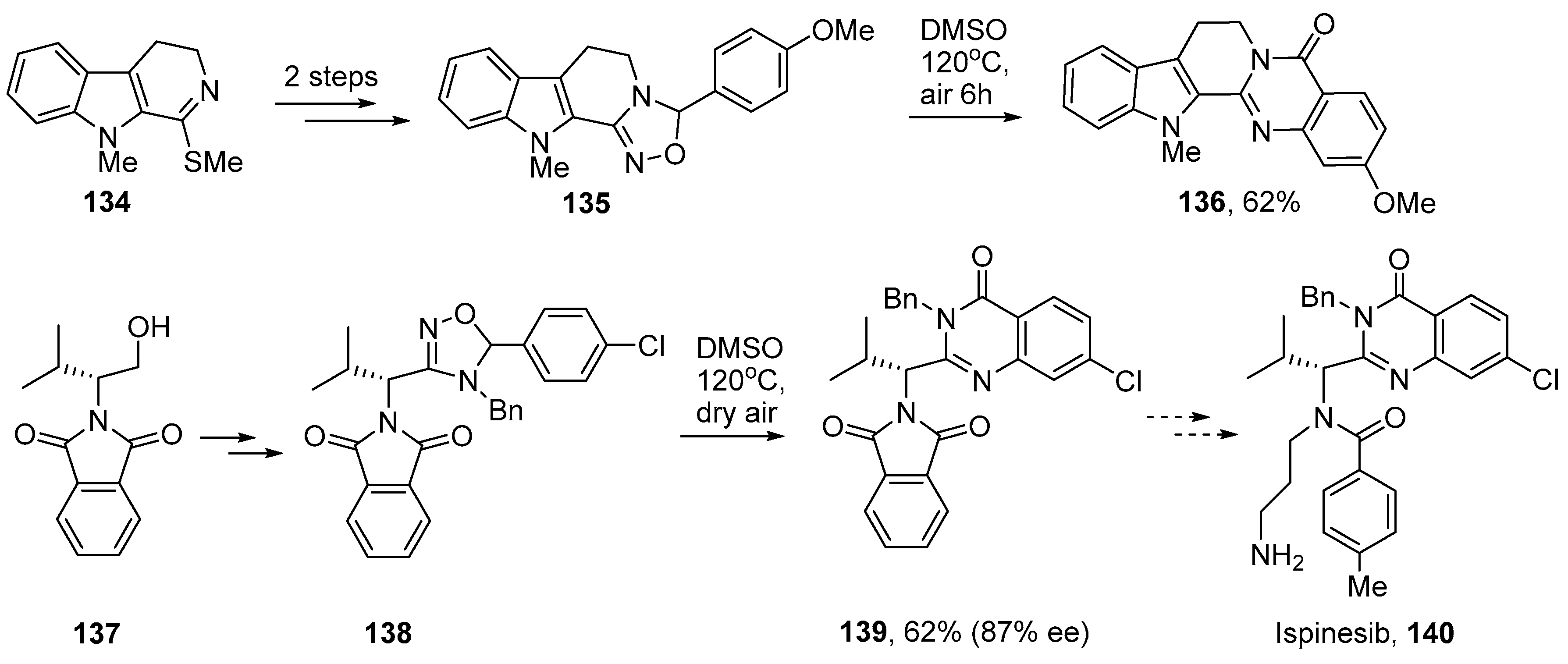
5. Iminyl Radical Mediated Preparations of Natural Products and Bioactive Compounds
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Reymond, J.-L. The chemical space project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Bon, R.S.; Waldemann, H. Bioactivity-guided navigation of chemical space. Acc. Chem. Res. 2010, 43, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P. Cheminformatics analysis of organic substituents: Identification of the most common substituents, calculation of substituent properties, and automatic identification of drug-like bioisosteric groups. J. Chem. Inf. Comput. Sci. 2003, 43, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modelling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Eigen, M. Self-organization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Jelfs, S.; Muehlbacher, J.; Schuffenhauer, A.; Selzer, P. Quest for the rings. In silico exploration of ring universe to identify novel bioactive heteroaromatic scaffolds. J. Med. Chem. 2006, 49, 4568–4573. [Google Scholar] [CrossRef] [PubMed]
- Renaud, P.; Sibi, M. (Eds.) Radicals in Organic Synthesis; Wiley: Weinheim, Germany, 2001; Volumes 1–2.
- Newcomb, M. Synthetic strategies & applications. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C., Studer, A., Eds.; Wiley: New York, NY, USA, 2012; Volume 2. [Google Scholar]
- Baguley, P.A.; Walton, J.C. Flight from the tyranny of tin: the quest for practical radical sources free from metal encumbrances. Angew. Chem. Int. Ed. 1998, 110, 3072–3082. [Google Scholar] [CrossRef]
- Studer, A.; Amrein, S. Tin hydride substitutes in reductive radical chain reactions. Synthesis 2002, 835–849. [Google Scholar] [CrossRef]
- McCarroll, A.J.; Walton, J.C. Programming organic molecules: Design and management of organic syntheses through free-radical cascade processes. Angew. Chem. Int. Ed. 2001, 40, 2224–2248. [Google Scholar] [CrossRef]
- McCarroll, A.J.; Walton, J.C. Organic syntheses through free-radical annulations and related cascade sequences. J. Chem. Soc. Perkin Trans. 1 2001, 3215–3229. [Google Scholar] [CrossRef]
- Walton, J.C.; Studer, A. Evolution of functional cyclohexadiene-based synthetic reagents: The importance of becoming aromatic. Acc. Chem. Res. 2005, 38, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Doni, E.; Mondal, B.; O’Sullivan, S.; Tuttle, T.; Murphy, J.A. Overturning established chemoselectivities: Selective reduction of arenes over malonates and cyanoacetates by photoactivated organic electron donors. J. Am. Chem. Soc. 2013, 135, 10934–10937. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.A. Discovery and development of organic super-electron-donors. J. Org. Chem. 2014, 79, 3731–3746. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.S.; Doni, E.; Traboulsee, K.T.; Coulthard, G.; Murphy, J.A.; Dyker, C.A. Pushing the limits of neutral organic electron donors: A tetra(iminophosphorano)-substituted bispyridinylidene. Angew. Chem. Int. Ed. 2015, 54, 11236–11239. [Google Scholar] [CrossRef] [PubMed]
- Darmency, V.; Renaud, P. Tin-free radical reactions mediated by organoboron compounds. Top. Curr. Chem. 2006, 263, 71–106. [Google Scholar]
- Luethy, M.; Darmency, V.; Renaud, P. Modified B-alkylcatecholboranes as radical precursors. Eur. J. Org. Chem. 2011, 2011, 547–552. [Google Scholar] [CrossRef]
- Ueng, S.-H.; Solovyev, A.; Yuan, X.; Geib, S.J.; Fensterbank, L.; Lacote, E.; Malacria, M.; Newcomb, M.; Walton, J.C.; Curran, D.P. N-Heterocyclic carbene boryl radicals: A new class of boron-centered radical. J. Am. Chem. Soc. 2009, 131, 11256–11262. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.C. Linking borane with N-heterocyclic carbenes: Effective hydrogen-atom donors for radical reactions. Angew. Chem. Int. Ed. 2009, 48, 1726–1728. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.C.; Brahmi, M.M.; Monot, J.; Fensterbank, L.; Malacria, M.; Curran, D.P.; Lacôte, E. Electron paramagnetic resonance and computational studies of radicals derived from boron-substituted N-heterocyclic carbene boranes. J. Am. Chem. Soc. 2011, 133, 10312–10321. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, T.; Geib, S.J.; Curran, D.P. Radical reactions of N-heterocyclic carbene boranes with organic nitriles: Cyanation of NHC-boranes and reductive decyanation of malononitriles. J. Am. Chem. Soc. 2015, 137, 8617–8622. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.P.; Ischay, M.A.; Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2010, 2, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Narayanam, J.M.R.; Stephenson, C.R.J. Visible light photoredox catalysis: Applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.-Q.; Chen, J.-R.; Xiao, W.-J. Homogeneous visible-light photoredox catalysis. Angew. Chem. Int. Ed. 2013, 52, 11701–11703. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Booth, S.G.; Essafi, S.; Dryfe, R.A.W.; Leonori, D. Visible-light-mediated generation of nitrogen-centered radicals: Metal-free hydroimination and iminohydroxylation cyclization reactions. Angew. Chem. Int. Ed. 2015, 54, 14017–14021. [Google Scholar] [CrossRef] [PubMed]
- Hofstra, J.L.; Grassbaugh, B.R.; Tran, Q.M.; Armada, N.R.; de Lijser, H.J.P. Catalytic oxidative cyclization of 2′-arylbenzaldehyde oxime ethers under photoinduced electron transfer conditions. J. Org. Chem. 2015, 80, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Manley, D.W.; McBurney, R.T.; Miller, P.; Howe, R.F.; Rhydderch, S.; Walton, J.C. Unconventional titania photocatalysis: Direct deployment of carboxylic acids in alkylations and annulations. J. Am. Chem. Soc. 2012, 134, 13580–13583. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, N. Photocatalysis with TiO2 applied to organic synthesis. Aust. J. Chem. 2015, 68, 1621–1639. [Google Scholar] [CrossRef]
- Manley, D.W.; Walton, J.C. Preparative semiconductor photoredox catalysis: An emerging theme in organic synthesis. Beilstein J. Org. Chem. 2015, 11, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
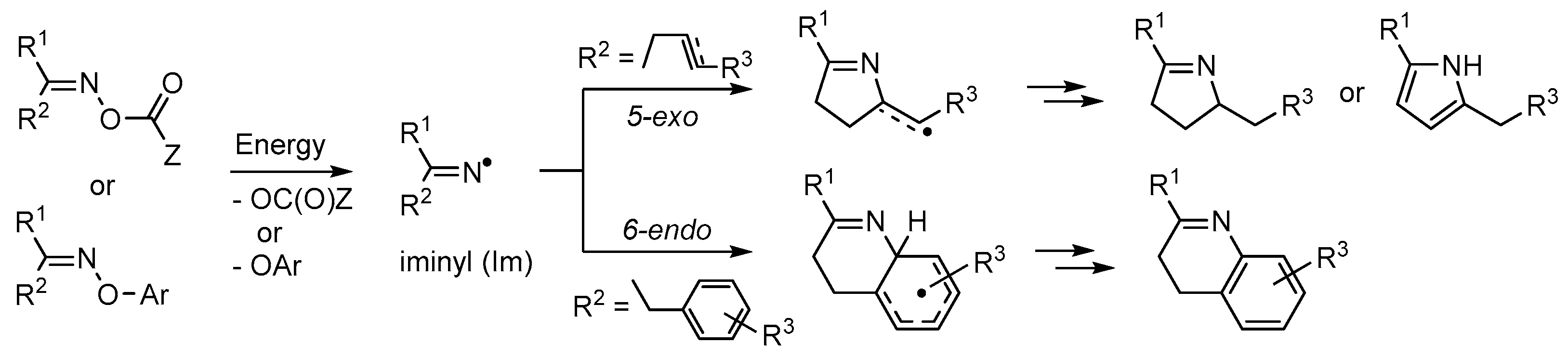
- Walton, J.C. Functionalised oximes: Emergent precursors for carbon-, nitrogen- and oxygen-centred radicals. Molecules 2016, 21, 63. [Google Scholar] [CrossRef] [PubMed]
- Zard, S.Z. Recent progress in the generation and use of nitrogen-centred radicals. Chem. Soc. Rev. 2008, 37, 1603–1618. [Google Scholar] [CrossRef] [PubMed]
- Gagosz, F.; Zard, S. Generation and capture of iminyl radicals from ketoxime xanthates. Synlett 1999, 1978–1980. [Google Scholar] [CrossRef]
- Nanni, D.; Calestani, G.; Leardini, R.; Zanardi, G. On the regioselectivity of imidoyl radical cyclisations. Eur. J. Org. Chem. 2000, 2000, 707–711. [Google Scholar] [CrossRef]
- Leardini, R.; McNab, H.; Minozzi, M.; Nanni, D.; Reed, D.; Wright, A.G. Reactions of 1-(2-alkoxyphenyl)alkaniminyl radicals. J. Chem. Soc. Perkin Trans. 1 2001, 2704–2710. [Google Scholar] [CrossRef]
- Minozzi, M.; Nanni, D.; Spagnolo, P. From azides to nitrogen-centered radicals: Applications of azide radical chemistry to organic synthesis. Chem. Eur. J. 2009, 15, 7830–7840. [Google Scholar] [CrossRef] [PubMed]
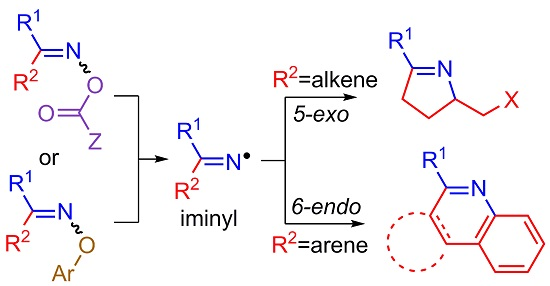
- Aldabbagh, F.; Bowman, W.R. Synthesis of heterocycles by radical cyclization. Contemp. Org. Synth. 1997, 4, 261–280. [Google Scholar] [CrossRef]
- Aldabbagh, F.; Bowman, W.R.; Mann, E.; Slawin, A.M.Z. Bu3SnH mediated oxidative radical cyclization onto imidazoles and pyrroles. Tetrahedron 1999, 55, 8111–8128. [Google Scholar] [CrossRef]
- Bowman, W.R.; Bridge, C.F.; Cloonan, M.O.; Leach, D.C. Synthesis of heteroarenes via radical cyclisation onto nitriles. Synlett 2001, 765–768. [Google Scholar] [CrossRef]
- Fallis, A.G.; Brinza, I.M. Free radical cyclizations involving nitrogen. Tetrahedron 1997, 53, 17543–17594. [Google Scholar] [CrossRef]
- Zhang, B.; Studer, A. Recent advances in the synthesis of nitrogen heterocycles via radical cascade reactions using isonitriles as radical acceptors. Chem. Soc. Rev. 2015, 44, 3505–3521. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Barlocco, D. Privileged structures as leads in medicinal chemistry. Curr. Med. Chem. 2006, 13, 65–85. [Google Scholar] [CrossRef] [PubMed]
- Bellina, F.; Rossi, R. Synthesis and biological activity of pyrrole, pyrroline and pyrrolidine derivatives with two aryl groups on adjacent positions. Tetrahedron 2006, 62, 7213–7256. [Google Scholar] [CrossRef]
- Domagala, A.; Jarosz, T.; Lapkowski, M. Living on pyrrolic foundations - advances in natural and artificial bioactive pyrrole derivatives. Eur. J. Med. Chem. 2015, 100, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Cullen, K.E.; Sharp, J.T. Reactions of diene-conjugated 1,3-dipolar intermediates: A versatile and efficient route to dibenz[c,e]azepines via benzonitrile o-arylbenzyl ylides. J. Chem. Soc. Perkin Trans. 1 1993, 2961–2967. [Google Scholar] [CrossRef]
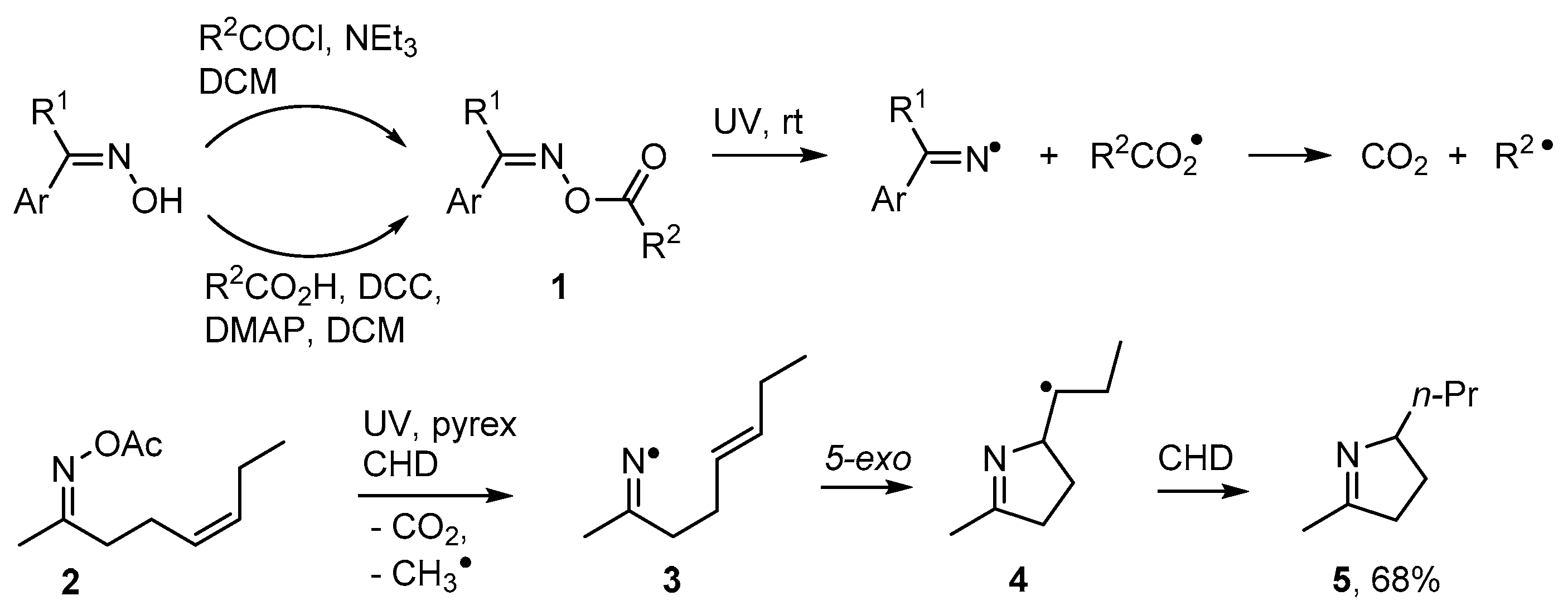
- McCarroll, A.J.; Walton, J.C. Exploitation of aldoxime esters as radical precursors in preparative and EPR spectroscopic roles. J. Chem. Soc. Perkin Trans. 2 2000, 2399–2409. [Google Scholar] [CrossRef]
- McCarroll, A.J.; Walton, J.C. Enhanced radical delivery from aldoxime esters for EPR and ring closure applications. Chem. Commun. 2000, 351–352. [Google Scholar] [CrossRef]
- Alonso, R.; Campos, P.J.; Garcıa, B.; Rodrıguez, M.A. New light-induced iminyl radical cyclization reactions of acyloximes to isoquinolines. Org. Lett. 2006, 8, 3521–3423. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Campos, P.J.; Rodrıguez, M.A.; Sampedro, D. Photocyclization of iminyl radicals: Theoretical study and photochemical aspects. J. Org. Chem. 2008, 73, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Caballero, A.; Campos, P.J.; Rodrıguez, M.A. Photochemistry of acyloximes: Synthesis of heterocycles and natural products. Tetrahedron 2010, 66, 8828–8831. [Google Scholar] [CrossRef]
- Jochims, J.C.; Hehl, S.; Herzberger, S. Preparation and Beckmann rearrangement of O-(chlorooxalyl)oximes. Synthesis 1990, 1128–1133. [Google Scholar] [CrossRef]
- Portela-Cubillo, F.; Lymer, J.; Scanlan, E.M.; Scott, J.S.; Walton, J.C. Dioxime oxalates; new iminyl radical precursors for syntheses of N-heterocycles. Tetrahedron 2008, 64, 11908–11916. [Google Scholar] [CrossRef]
- Blake, J.A.; Pratt, D.A.; Lin, S.; Walton, J.C.; Mulder, P.; Ingold, K.U. Thermolyses of O-phenyl oxime ethers. A new source of iminyl radicals and a new source of aryloxyl radicals. J. Org. Chem. 2004, 69, 3112–3120. [Google Scholar] [CrossRef] [PubMed]
- Portela-Cubillo, F.; Scott, J.S.; Walton, J.C. Microwave-assisted preparations of dihydropyrroles from alkenone O-phenyl oximes. Chem. Commun. 2007, 4041–4043. [Google Scholar] [CrossRef] [PubMed]
- Portela-Cubillo, F.; Scott, J.S.; Walton, J.C. Microwave-assisted syntheses of N-heterocycles using alkenone-, alkynone- and aryl-carbonyl O-phenyl oximes: Formal synthesis of neocryptolepine. J. Org. Chem. 2008, 73, 5558–5565. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Jalan, A.; Kubosumi, A.R.; Castle, S.L. Microwave-promoted tin-free iminyl radical cyclization with TEMPO trapping: A practical synthesis of 2‑acylpyrroles. Org. Lett. 2015, 17, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Visible light photoredox-controlled reactions of N-radicals and radical ions. Chem. Soc. Rev. 2016, 45, 2044–2056. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Narasaka, K. Photochemical transformation of γ,δ-unsaturated ketone O-(p-cyanophenyl)oximes to 3,4-dihydro-2H-pyrrole derivatives. Chem. Lett. 2000, 29, 338–339. [Google Scholar] [CrossRef]
- Lin, X.; Artman, G.D.; Stien, D.; Weinreb, S.M. Development of efficient new methodology for generation, cyclization and functional trapping of iminyl and amidyl radicals. Tetrahedron 2001, 57, 8779–8791. [Google Scholar] [CrossRef]
- Madapa, S.; Tusi, Z.; Batra, S. Advances in the syntheses of quinoline and quinoline-annulated ring systems. Curr. Org. Chem. 2008, 12, 1116–1183. [Google Scholar] [CrossRef]
- Chung, P.-Y.; Bian, Z.-X.; Pun, H.-Y.; Chan, D.; Chan, A.S.-C.; Chui, C.-H.; Tang, J.C.-O.; Lam, K.-H. Recent advances in research of natural and synthetic bioactive quinolines. Future Med. Chem. 2015, 7, 947–967. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Ding, K.-M.; Zhang, L.; Cheng, X.-M.; Wang, C.-H.; Wang, Z.-T. Acetylcholinesterase and butyrylcholinesterase inhibitory activities of β-carboline and quinoline alkaloids derivatives from the plants of genus Peganum. J. Chem. 2013, 2013. [Google Scholar] [CrossRef]
- Cheng, P.; Gu, Q.; Liu, W.; Zou, J.-F.; Ou, Y.-Y.; Luo, Z.-Y.; Zeng, J.-G. Synthesis of quinolin-2-one alkaloid derivatives and their inhibitory activities against HIV-1 reverse transcriptase. Molecules 2011, 16, 7649–7661. [Google Scholar] [CrossRef] [PubMed]
- Byler, K.G.; Wang, C.; Setzer, W.N. Quinoline alkaloids as intercalative topoisomerase inhibitors. J. Mol. Model. 2009, 15, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Fujioka, H.; Kita, Y. Synthesis of the marine pyrroloiminoquinone alkaloids, discorhabdins. Mar. Drugs 2010, 8, 1394–1416. [Google Scholar] [CrossRef] [PubMed]
- Truchado, P.; Martos, I.; Bortolotti, L.; Sabatini, A.G.; Ferreres, F.; Tomas-Barberan, F.A. Use of quinoline alkaloids as markers of the floral origin of chestnut honey. J. Agric. Food Chem. 2009, 57, 5680–5686. [Google Scholar] [CrossRef] [PubMed]
- Tumir, L.-M.; Stojkovic, M.R.; Piantanida, I. Come-back of phenanthridine and phenanthridinium derivatives in the 21st century. Beilstein J. Org. Chem. 2014, 10, 2930–2954. [Google Scholar] [CrossRef] [PubMed]
- Dostal, J.; Slavik, J. Some aspects of the chemistry of quaternary benzo[c]phenanthridine alkaloids. Stud. Nat. Prod. Chem. 2002, 27, 155–184. [Google Scholar]
- Dvorak, Z.; Kuban, V.; Klejdus, B.; Hlavac, J.; Vicar, J.; Ulrichova, J.; Simanek, V. Quaternary benzo[c]phenanthridines sanguinarine and chelerythrine: A review of investigations from chemical and biological studies. Heterocycles 2006, 68, 2403–2422. [Google Scholar] [CrossRef]
- Hammerova, J.; Uldrijan, S.; Taborska, E.; Slaninova, I. Benzo[c]phenanthridine alkaloids exhibit strong anti-proliferative activity in malignant melanoma cells regardless of their p53 status. J. Dermatol. Sci. 2011, 62, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Slaninova, I.; Pencikova, K.; Urbanova, J.; Slanina, J.; Taborska, E. Antitumour activities of sanguinarine and related alkaloids. Phytochem. Rev. 2014, 13, 51–68. [Google Scholar] [CrossRef]
- Cao, F.-J.; Yang, R.; Lv, C.; Ma, Q.; Lei, M.; Geng, H.-L.; Zhou, L. Pseudocyanides of sanguinarine and chelerythrine and their series of structurally simple analogues as new anticancer lead compounds: Cytotoxic activity, structure-activity relationship and apoptosis induction. Eur. J. Pharm. Sci. 2015, 67, 45–54. [Google Scholar] [CrossRef] [PubMed]
- McBurney, R.T.; Walton, J.C. Interplay of ortho- with spiro-cyclisation during iminyl radical closures onto arenes and heteroarenes. Beilstein J. Org. Chem. 2013, 9, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Cai, J.; Tang, L.; Wang, Z.; Li, F.; Deng, G.-J. Metal-free assembly of polysubstituted pyridines from oximes and acroleins. J. Org. Chem. 2016, 81, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- McBurney, R.T.; Slawin, A.M.Z.; Smart, L.A.; Yu, Y.; Walton, J.C. UV promoted phenanthridine syntheses from oxime carbonate derived iminyl radicals. Chem. Commun. 2011, 47, 7974–7976. [Google Scholar] [CrossRef] [PubMed]
- McBurney, R.T.; Walton, J.C. Dissociation or cyclization: Options for a triad of radicals released from oxime carbamates. J. Am. Chem. Soc. 2013, 135, 7349–7354. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; An, X.; Tong, K.; Zheng, T.; Zhang, Y.; Yu, S. Visible-light-promoted iminyl-radical formation from acyl oximes: A unified approach to pyridines, quinolines, and phenanthridines. Angew. Chem. Int. Ed. 2015, 54, 4055–4059. [Google Scholar] [CrossRef] [PubMed]
- An, X.-D.; Yu, S. Visible-light-promoted and one-pot synthesis of phenanthridines and quinolines from aldehydes and O‑acyl hydroxylamine. Org. Lett. 2015, 17, 2692–2695. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Kraker, A.J.; McMichael, A.; Ambroso, L.A.; Nelson, J.M.; Leopold, W.R.; Connors, R.W.; Bridges, A.J. A specific inhibitor of the epidermal growth factor receptor tyrosine kinase. Science 1994, 265, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Kunes, J.; Bazant, J.; Pour, M.; Waisser, K.; Slosarek, M.; Janota, J. Quinazoline derivatives with antitubercular activity. Farmaco 2000, 55, 725–729. [Google Scholar] [CrossRef]
- Foster, B.A.; Coffrey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [PubMed]
- Bedi, P.M.S.; Kumar, V.; Mahajan, M.P. Synthesis and biological activity of novel antibacterial quinazolines. Bioorg. Med. Chem. Lett. 2004, 14, 5211–5213. [Google Scholar] [CrossRef] [PubMed]
- Gundla, R.; Kazemi, R.; Sanam, R.; Muttineni, R.; Sarma, J.A.R.P.; Dayam, R.; Neamati, N. Discovery of novel small-molecule inhibitors of human epidermal growth factor receptor-2: Combined ligand and target-based approach. J. Med. Chem. 2008, 51, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Mendes da Silva, J.F.; Walters, M.; Al-Damluji, S.; Ganellin, C.R. Molecular features of the prazosin molecule required for activation of transport-P. Bioorg. Med. Chem. 2008, 16, 7254–7263. [Google Scholar] [CrossRef] [PubMed]
- Rewcastle, G.W.; Palmer, B.D.; Bridges, A.J.; Showalter, H.D.H.; Sun, L.; Nelson, J.; McMichael, A.; Kraker, A.J.; Fry, D.W.; Denny, W.A. Tyrosine kinase inhibitors. 9. Synthesis and evaluation of fused tricyclic quinazoline analogs as ATP site inhibitors of the tyrosine kinase activity of the epidermal growth factor receptor. J. Med. Chem. 1996, 39, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Luth, A.; Lowe, W. Syntheses of 4-(indol-3-yl)quinazolines—A new class of epidermal growth factor receptor tyrosine kinase inhibitors. Eur. J. Med. Chem. 2008, 43, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.J.; Cusack, D.; O’Sullivan, T.P.; Guiry, P.J. Synthesis of quinazolinones and quinazolines. Tetrahedron 2005, 61, 10153–10202. [Google Scholar] [CrossRef]
- Besson, T.; Chosson, E. Microwave-assisted synthesis of bioactive quinazolines and quinazolinones. Comb. Chem. High Throughput Screen. 2007, 10, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 2003, 20, 476–493. [Google Scholar] [CrossRef] [PubMed]
- Chilin, A.; Marzaro, G.; Zanatta, S.; Guiotto, A. A microwave improvement in the synthesis of the quinazoline scaffold. Tetrahedron Lett. 2007, 48, 3229–3231. [Google Scholar] [CrossRef]
- Seijas, J.A.; Vazquez-Tato, M.P.; Martinez, M.M. Microwave-enhanced synthesis of 4-aminoquinazolines. Tetrahedron Lett. 2000, 41, 2215–2217. [Google Scholar] [CrossRef]
- Yoon, D.S.; Han, Y.; Stark, T.M.; Haber, J.C.; Gregg, B.T.; Stankovich, S.B. Efficient synthesis of 4-aminoquinazoline and thieno[3,2-d]pyrimidin-4-ylamine derivatives by microwave irradiation. Org. Lett. 2004, 6, 4775–4778. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Mohan, C.; Gupta, M.; Mahajan, M.P. A catalyst- and solvent-free selective approach to biologically important quinazolines and benzo[g]quinazoline. Tetrahedron 2005, 61, 3533–3538. [Google Scholar] [CrossRef]
- Maitraie, D.; Yakaiah, T.; Srinivas, K.; Reddy, G.V.; Ravikanth, S.; Narsaiah, B.; Rao, S.P.; Ravikumar, K.; Sridhar, B. Regioselective addition of Grignard reagents to 2,6-dicyanoanilines and cyclization to new quinazoline derivatives under thermal/microwave irradiation conditions. J. Fluor. Chem. 2006, 127, 351–359. [Google Scholar] [CrossRef]
- Abdel-Jalil, R.J.; Völter, W.; Saeed, M. A novel method for the synthesis of 4(3H)-quinazolinones. Tetrahedron Lett. 2004, 45, 3475–3476. [Google Scholar] [CrossRef]
- Portela-Cubillo, F.; Scott, J.S.; Walton, J.C. 2-(Aminoaryl)alkanone O-phenyl oximes: Versatile reagents for syntheses of quinazolines. Chem. Commun. 2008, 2935–2937. [Google Scholar] [CrossRef] [PubMed]
- Portela-Cubillo, F.; Scott, J.S.; Walton, J.C. Microwave-promoted syntheses of quinazolines and dihydroquinazolines from 2-aminoarylalkanone O-phenyl oximes. J. Org. Chem. 2009, 74, 4934–4942. [Google Scholar] [CrossRef] [PubMed]
- Pouilhes, A.; Langlois, Y.; Chiaroni, A. First synthesis of marine alkaloid (±)-bengacarboline. Synlett 2003, 1488–1490. [Google Scholar] [CrossRef]
- Patil, D.A.; Patil, P.O.; Patil, G.B.; Surana, S.J. Synthesis of 2,3-disubstituted-quinazolin-4-(3H)-ones. Mini-Rev. Med. Chem. 2011, 11, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Jiang, Y.; Ma, D. Synthesis of 3-substituted and 2,3-disubstituted quinazolinones via Cu-catalyzed aryl amidation. Org. Lett. 2012, 14, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Wang, Y.; Peng, J.; Zhu, Q. Synthesis of quinazolin-4(3H)-ones via Pd(II)-catalyzed intramolecular C(sp2)-H carboxamidation of N-arylamidines. J. Org. Chem. 2011, 76, 6362–6366. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Lam, J.K.; Yu, J.-Q. Synthetic applications of Pd(II)-catalyzed C-H carboxylation and mechanistic insights: Expedient routes to anthranilic acids, oxazolinones, and quinazolinones. J. Am. Chem. Soc. 2010, 132, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Servais, A.; Azzouz, M.; Lopes, D.; Courillon, C.; Malacria, M. Radical cyclization of N-acylcyanamides: Total synthesis of luotonin A. Angew. Chem. Int. Ed. 2007, 46, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Larraufie, M.-H.; Courillon, C.; Ollivier, C.; Lacôte, E.; Malacria, M.; Fensterbank, L. Radical migration of substituents of aryl groups on quinazolinones derived from N-acyl cyanamides. J. Am. Chem. Soc. 2010, 132, 4381–4387. [Google Scholar] [CrossRef] [PubMed]
- Beaume, A.; Courillon, C.; Derat, E.; Malacria, M. Unprecedented aromatic homolytic substitutions and cyclization of amide-iminyl radicals: Experimental and theoretical study. Chem. Eur. J. 2008, 14, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Larraufie, M.-H.; Ollivier, C.; Fensterbank, L.; Malacria, M. Radical synthesis of guanidines from N-acyl cyanamides. Angew. Chem. Int. Ed. 2010, 49, 2178–2181. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, B.; Mardomingo, C.L.; Plumet, J.; Cativiela, C.; Mayoral, J.A. Orbital control in the 1,3-dipolar cycloaddition of benzonitrile oxide to benzylideneanilines. Can. J. Chem. 1987, 65, 2050–2056. [Google Scholar] [CrossRef]
- Srivastava, R.M.; Freire, M.V.S.; Chaves, A.S.S.C.; Beltrao, T.M.; Carpenter, G.B. Synthesis, spectroscopic studies and the mechanism of formation of 4,5-dihydro-1,2,4-oxadiazoles. J. Heterocycl. Chem. 1987, 24, 101–105. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Zhang, F.-L.; Chiba, S. Oxidative radical skeletal rearrangement induced by molecular oxygen: Synthesis of quinazolinones. Org. Lett. 2013, 15, 2842–2845. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.J.; Stelzer, L.S.; TenBrink, R.E.; Belonga, K.L.; Carter, D.B.; Im, H.K.; Im, W.B.; Sethy, V.H.; Tang, A.H.; Zhong, W.Z.; et al. Piperazine imidazo[1,5-a]quinoxaline ureas as high-affinity GABAA ligands of dual functionality. J. Med. Chem. 1999, 42, 1123–1144. [Google Scholar] [PubMed]
- Lawrence, D.S.; Copper, J.E.; Smith, C.D. Structure-activity studies of substituted quinoxalinones as multiple-drug-resistance antagonists. J. Med. Chem. 2001, 44, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Padvi, P.A.; Mahale, G.H.; Pawar, D.E.; Falak, C.S.; Kendre, A.V. Synthesis and biological activity of quinoxaline derivatives. World J. Pharm. Res. 2015, 4, 1892–1900. [Google Scholar]
- Ferreira, S.R.A.; Franco, M.S.F.; Diniz, E.M.L.P.; Emery, F.D.S.; Clososki, G.C. Drug likeness and selective functionalization of quinoxalines. Curr. Org. Synth. 2015, 12, 714–729. [Google Scholar] [CrossRef]
- Camaggi, C.M.; Leardini, R.; Nanni, D.; Zanardi, G. Radical annulations with nitriles: Novel cascade reactions of cyano-substituted alkyl and sulfanyl radicals with isonitriles. Tetrahedron 1998, 54, 5587–5598. [Google Scholar] [CrossRef]
- Sun, X.; Li, J.; Ni, Y.; Ren, D.; Hu, Z.; Yu, S. Synthesis of fused quinoline and quinoxaline derivatives enabled by domino radical triple bond insertions. Asian J. Org. Chem. 2014, 3, 1317–1325. [Google Scholar] [CrossRef]
- Togo, H.; Katohgi, M. Synthetic uses of organohypervalent iodine compounds through radical pathways. Synlett 2001, 565–581. [Google Scholar] [CrossRef]
- He, Z.; Bae, M.; Wu, J.; Jamison, T.F. Synthesis of highly functionalized polycyclic quinoxaline derivatives using visible-light photoredox catalysis. Angew. Chem. Int. Ed. 2014, 53, 14451–14455. [Google Scholar] [CrossRef] [PubMed]
- Viladomat, F.; Selles, M.; Codina, C.; Bastida, J. Alkaloids from Narcissus asturiensis. Planta Med. 1997, 63, 583. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Halim, O.B.; Morikawa, T.; Ando, S.; Matsuda, H.; Yoshikawa, M. New crinine-type alkaloids with inhibitory effect on induction of inducible nitric oxide synthase from Crinum yemense. J. Nat. Prod. 2004, 67, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Zupko, I.; Rethy, B.; Hohmann, J.; Molnar, J.; Ocsovszki, I.; Falkay, G. Antitumor activity of alkaloids derived from Amaryllidaceae species. In Vivo 2009, 23, 41–48. [Google Scholar] [PubMed]
- Szlavik, L.; Gyuris, A.; Minarovits, J.; Forgo, P.; Molnar, J.; Hohmann, J. Alkaloids from leucojum vernum and antiretroviral activity of amaryllidaceae alkaloids. Planta Med. 2004, 70, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Cortes, N.; Posada-Duque, R.A.; Alvarez, R.; Alzate, F.; Berkov, S.; Cardona-Gomez, G.P.; Osorio, E. Neuroprotective activity and acetylcholinesterase inhibition of five amaryllidaceae species: A comparative study. Life Sci. 2015, 122, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.M.; Lobo, A.M.; Branco, P.S.; Prabhakar, S.; Sa-da-Costa, M. New syntheses of the Amaryllidaceae alkaloids vasconine, assoanine, oxoassoanine, pratosine and ismine by radical cyclization. Tetrahedron 1997, 53, 299–306. [Google Scholar] [CrossRef]
- Parnes, J.S.; Carter, D.S.; Kurz, L.J.; Flippin, L. A. Concise synthesis of narcissus pyrrolophenanthridine alkaloids: Vasconine, Assoanine and Oxoassoanine. J. Org. Chem. 1994, 59, 3497–3499. [Google Scholar] [CrossRef]
- Nakanishi, T.; Masuda, A.; Suwa, M.; Akiyama, Y.; Hoshino-Abe, N.; Suzuki, M. Synthesis of derivatives of NK109, 7-OH benzo[c]phenanthridine alkaloid, and evaluation of their cytotoxicities and reduction-resistant properties. Bioorg. Med. Chem. Lett. 2000, 10, 2321–2323. [Google Scholar] [CrossRef]
- Nakanishi, T.; Suzuki, M. Revision of the structure of Fagaridine based on comparison of UV and NMR data of synthetic compounds. J. Nat. Prod. 1998, 61, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Potmesil, M.; Pinedo, H. Camptothecins: New Anticancer Agents; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Li, S.; Zhang, Z.; Cain, A.; Wang, B.; Long, M.; Taylor, J. Antifungal activity of camptothecin, trifolin, and hyperoside isolated from camptotheca acuminata. J. Agric. Food Chem. 2005, 53, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Tuticorin, R.G.; Kongovi, R.R.; Narayanan, V. Mappicine, a minor alkaloid from Mappia foetida miers. J. Chem. Soc. Perkin Trans. 1 1974, 1215–1217. [Google Scholar] [CrossRef]
- Zhao, X.-B.; Goto, M.; Song, Z.-L.; Morris-Natschke, S.L.; Zhao, Y.; Wu, D.; Yang, L.; Li, S.-G.; Liu, Y.-Q.; Zhu, G.-X.; et al. Design and synthesis of new 7-(N-substituted-methyl)-camptothecin derivatives as potent cytotoxic agents. Bioorg. Med. Chem. Lett. 2014, 24, 3850–3853. [Google Scholar] [CrossRef] [PubMed]
- Loza-Mejia, M.A.; Olvera-Vazquez, S.; Maldonado-Hernandez, K.; Guadarrama-Salgado, T.; Gonzalez-Sanchez, I.; Rodriguez-Hernandez, F.; Solano, J.D.; Rodriguez-Sotres, R.; Lira-Rocha, A. Synthesis, cytotoxic activity, DNA topoisomerase-II inhibition, molecular modeling and structure-activity relationship of 9-anilinothiazolo[5,4-b]quinoline derivatives. Bioorg. Med. Chem. 2009, 17, 3266–3277. [Google Scholar] [CrossRef] [PubMed]
- Liew, S.T.; Yang, L.-X. Design, synthesis and development of novel camptothecin drugs. Curr. Pharm. Des. 2008, 14, 1078–1097. [Google Scholar] [CrossRef] [PubMed]
- Gabarda, A.E.; Du, W.; Isarno, T.; Tangirala, R.S.; Curran, D.P. Asymmetric total synthesis of (20R)-homocamptothecin, substituted homocamptothecins and homosilatecans. Tetrahedron 2002, 58, 6329–6341. [Google Scholar] [CrossRef]
- Dom, B.; Curran, D.P.; Kruszewski, S.; Zimmer, S.G.; Strode, J.T.; Kohlhagen, G.; Du, W.; Chavan, A.J.; Fraley, K.A.; Bingcang, A.L.; et al. The novel silatecan 7-tert-butyldimethylsilyl-10-hydroxycamptothecin displays high lipophilicity, improved human blood stability, and potent anticancer activity. J. Med. Chem. 2000, 43, 3970–3980. [Google Scholar]
- Josien, H.; Bom, D.; Curran, D.P.; Zheng, Y.-H.; Chou, T.-C. 7-Silylcamptothecins (silatecans): A new family of camptothecin antitumor agents. Biorg. Med. Chem. Lett. 1997, 7, 3189–3194. [Google Scholar] [CrossRef]
- Curran, D.P.; Liu, H.; Josien, H.; Ko, S.-B. Tandem radical reactions of isonitriles with 2-pyridonyl and other aryl radicals: Scope and limitations, and a first generation synthesis of (±)-camptothecin. Tetrahedron 1996, 52, 11385–11404. [Google Scholar] [CrossRef]
- Curran, D.P.; Ko, S.-B.; Josien, H. Cascade radical reactions of isonitriles: A second-generation synthesis of (20S)-camptothecin, topotecan, irinotecan, and GI-147211C. Angew. Chem. Int. Ed. 1995, 34, 2683–2684. [Google Scholar] [CrossRef]
- Liu, H.; Curran, D.P. New 4 + 1 radical annulations. A formal total synthesis of (±)-camptothecin. J. Am. Chem. Soc. 1992, 114, 5863–5864. [Google Scholar]
- Zhang, W.; Luo, Z.; Chen, C.H.-T.; Curran, D.P. Solution-phase preparation of a 560-compound library of individual pure mappicine analogues by fluorous mixture synthesis. J. Am. Chem. Soc. 2002, 124, 10443–10450. [Google Scholar] [CrossRef] [PubMed]
- Bowman, W.R.; Bridge, C.F.; Brookes, P.; Cloonan, M.O.; Leach, D.C. Cascade radical synthesis of heteroarenes via iminyl radicals. J. Chem. Soc. Perkin Trans. 1 2002, 58–68. [Google Scholar] [CrossRef]
- Bowman, W.R.; Cloonan, M.O.; Fletcher, A.J.; Stein, T. Synthesis of heteroarenes using cascade radical cyclisation via iminyl radicals. Org. Biomol. Chem. 2005, 3, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-Z.; Hano, Y.; Nomura, T.; Chen, Y.-J. Two new pyrroloquinazolinoquinoline alkaloids from Peganum nigellastrum. Heterocycles 1997, 46, 541–546. [Google Scholar]
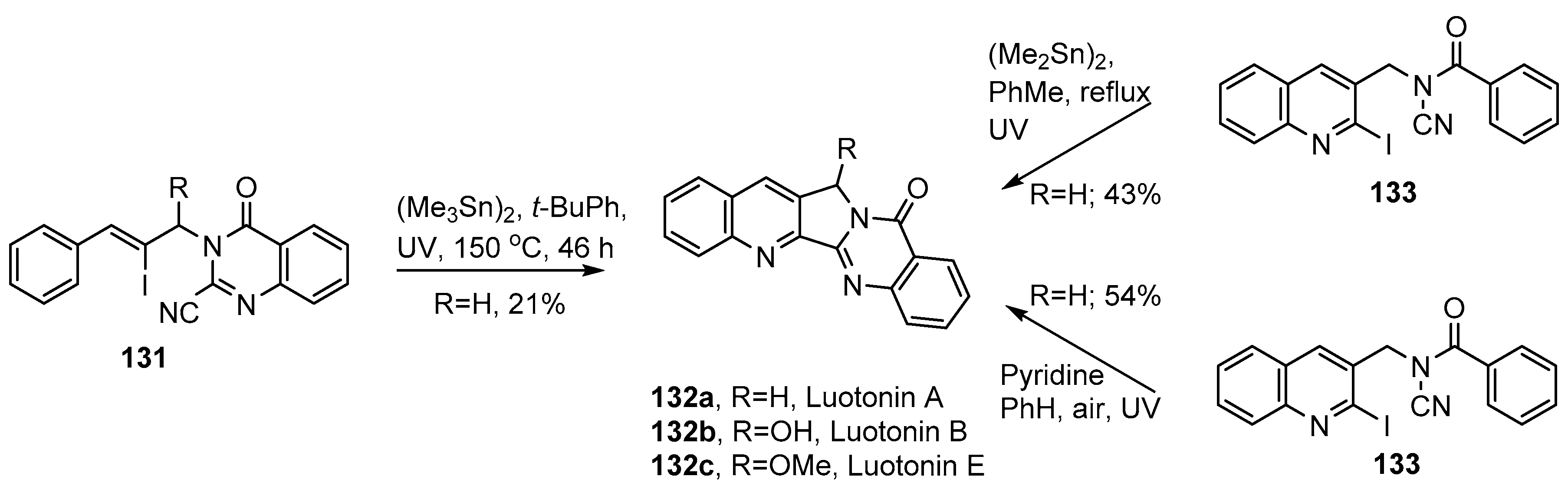
- Cagir, A.; Jones, S.H.; Gao, R.; Eisenhauer, B.M.; Hecht, S.M.; Luotonin, A. A naturally occurring human DNA topoisomerase I poison. J. Am. Chem. Soc. 2003, 125, 13628–13629. [Google Scholar] [CrossRef] [PubMed]
- Mhaske, S.B.; Argade, N.P. Regioselective quinazolinone-directed ortho lithiation of quinazolinoylquinoline: Practical synthesis of naturally occurring human DNA topoisomerase I poison Luotonin A and Luotonins B and E. J. Org. Chem. 2004, 69, 4563–4566. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Park, J.G.; Kim, S.I.; Jahng, Y. Synthesis and properties of luotonin A homologues and their aza-analogues. Heterocycles 2006, 68, 151–158. [Google Scholar]
- Ma, Z.-Z.; Hano, Y.; Nomura, T.; Chen, Y.-J. Synthesis of the cytotoxic pyrroloquinazolinoquinoline alkaloid, luotonin A. Heterocycles 1999, 51, 1593–1596. [Google Scholar]
- Toyota, M.; Komori, C.; Ihara, M. Three-step total synthesis of pyrroloquinazolinoquinoline alkaloid, luotonin A, by intramolecular hetero Diels-Alder reaction. Heterocycles 2002, 56, 101–103. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, B.V.S. Microwave-assisted rapid synthesis of the cytotoxic alkaloid luotonin A. Tetrahedron Lett. 2002, 43, 1905–1907. [Google Scholar] [CrossRef]
- Dallavalle, S.; Merlini, L. A new synthesis of the cytotoxic alkaloid Luotonine A. Tetrahedron Lett. 2002, 43, 1835–1837. [Google Scholar] [CrossRef]
- Ahn, H.; Nam, J.-W.; Seo, E.-K.; Mar, W. Induction of NAD(P)H: Quinone reductase by rutaecarpine isolated from the fruits of Evodia rutaecarpa in the murine hepatic Hepa-1c1c7 cell line. Planta Med. 2008, 74, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Christopher, E.; Bedir, E.; Dunbar, C.; Khan, I.A.; Okunji, C.O.; Schuster, B.M.; Iwu, M.M. Indoloquinazoline alkaloids from Araliopsis tabouensis. Helv. Chim. Acta 2003, 86, 2914–2918. [Google Scholar] [CrossRef]
- Sheth, P.R.; Shipps, G.W., Jr.; Seghezzi, W.; Smith, C.K.; Chuang, C.C.; Sanden, D.; Basso, A.D.; Vilenchik, L.; Gray, K.; Annis, D.A.; et al. Novel benzimidazole inhibitors bind to a unique site in the kinesin spindle protein motor domain. Biochemistry 2010, 49, 8350–8358. [Google Scholar] [CrossRef] [PubMed]
- Theoclitou, M.E.; Aquila, B.; Block, M.H.; Brassil, P.J.; Castriotta, L.; Code, E.; Collins, M.P.; Davies, A.M.; Deegan, T.; Ezhuthachan, J.; et al. Discovery of (+)-N-(3-aminopropyl)-N-[1-(5-benzyl-3-methyl-4-oxo-[1,2]thiazolo[5,4-d]pyrimidin-6-yl)-2-methylpropyl]-4-methylbenzamide (AZD4877), a kinesin spindle protein inhibitor and potential anticancer agent. J. Med. Chem. 2011, 54, 6734–6750. [Google Scholar] [CrossRef] [PubMed]
- Sorbera, L.A.; Bolós, J.; Serradell, N.; Bayés, M. Ispinesib mesilate. Drugs Future 2006, 31, 778–787. [Google Scholar] [CrossRef]

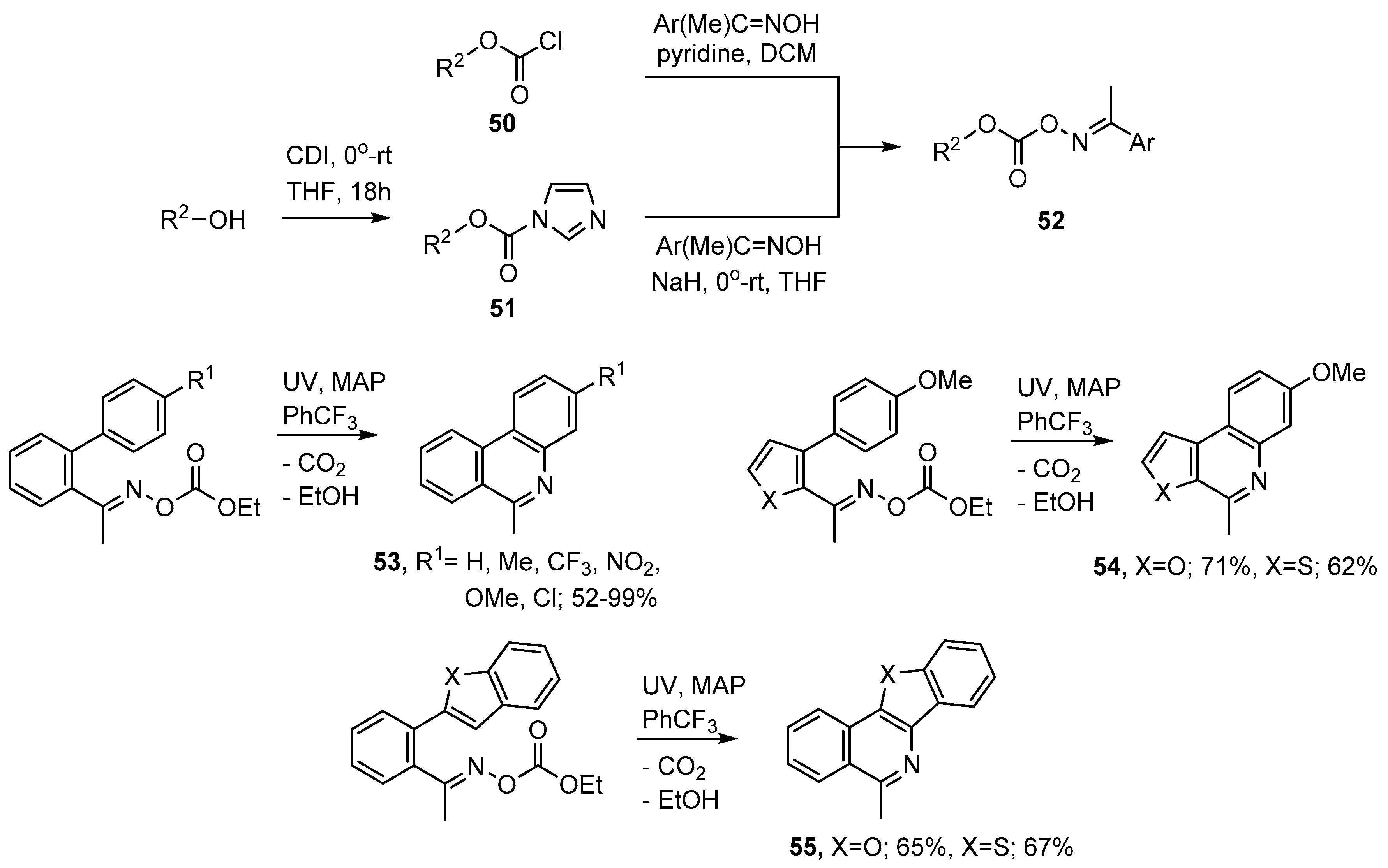
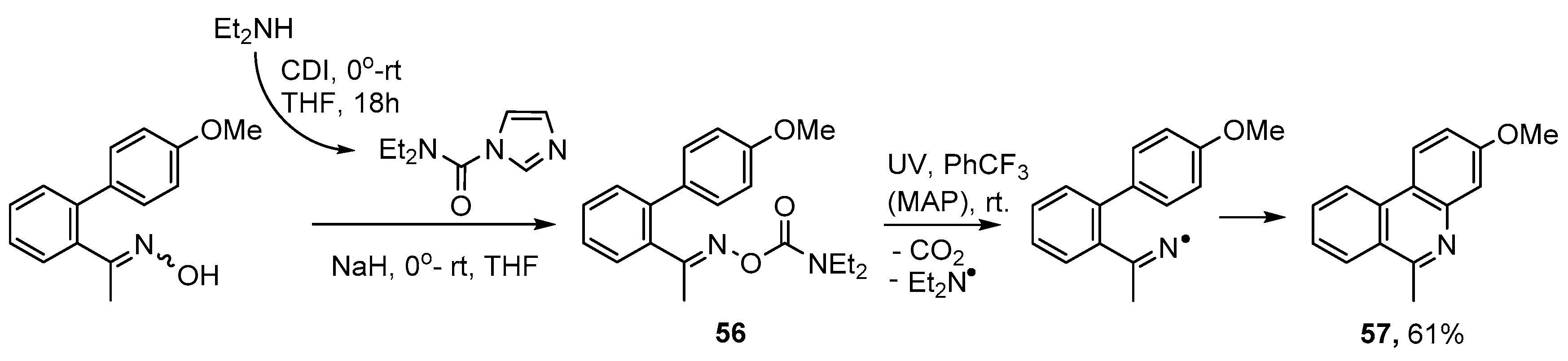
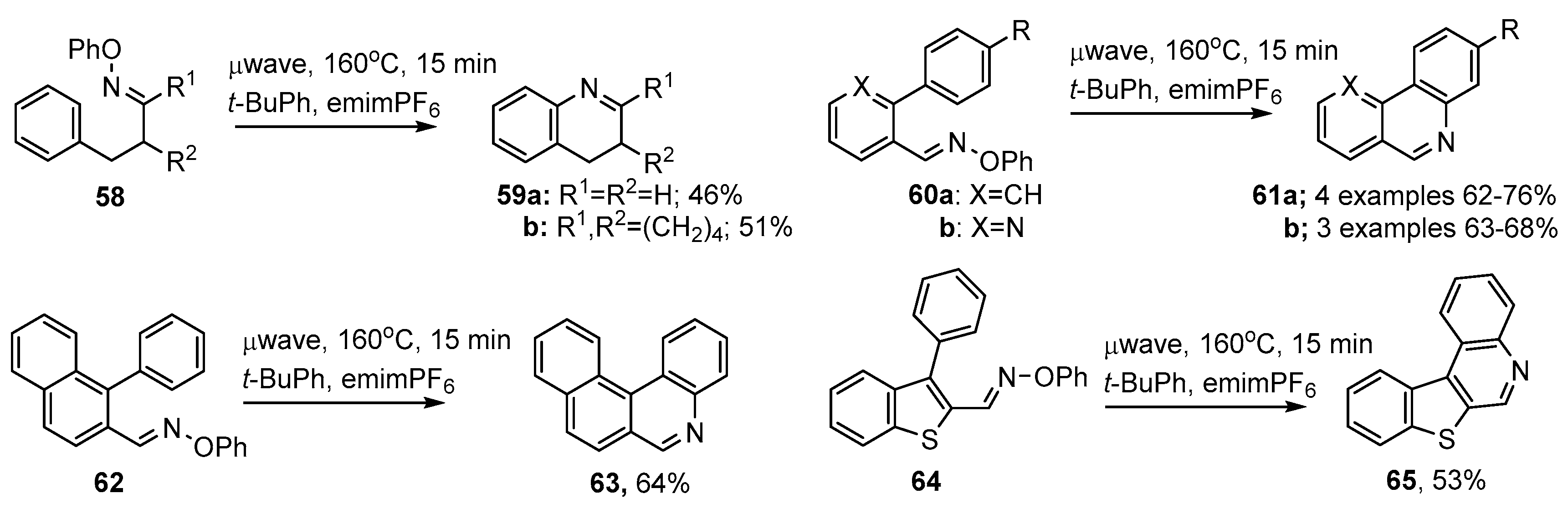
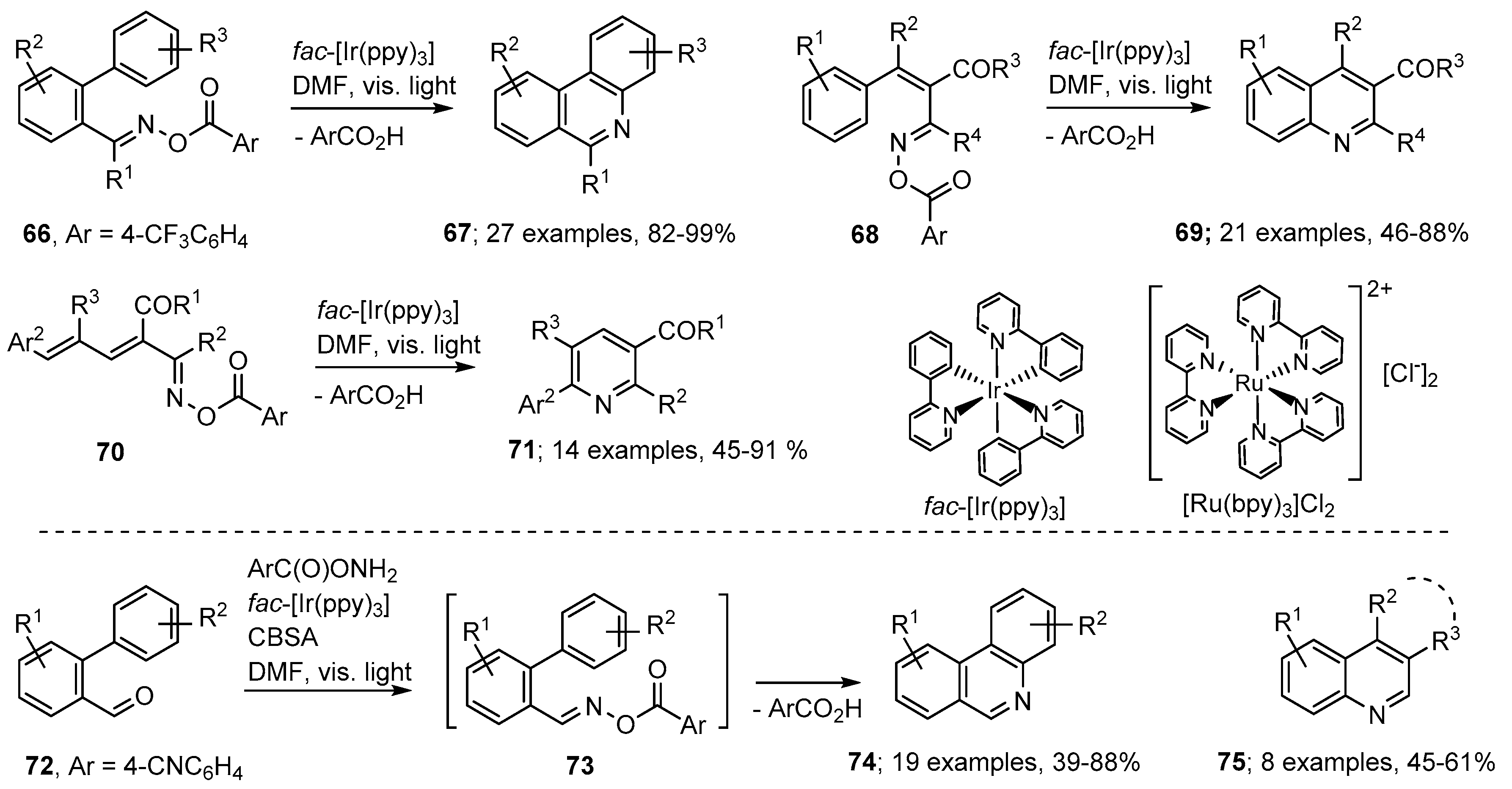
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walton, J.C. Synthetic Strategies for 5- and 6-Membered Ring Azaheterocycles Facilitated by Iminyl Radicals. Molecules 2016, 21, 660. https://doi.org/10.3390/molecules21050660
Walton JC. Synthetic Strategies for 5- and 6-Membered Ring Azaheterocycles Facilitated by Iminyl Radicals. Molecules. 2016; 21(5):660. https://doi.org/10.3390/molecules21050660
Chicago/Turabian StyleWalton, John C. 2016. "Synthetic Strategies for 5- and 6-Membered Ring Azaheterocycles Facilitated by Iminyl Radicals" Molecules 21, no. 5: 660. https://doi.org/10.3390/molecules21050660
APA StyleWalton, J. C. (2016). Synthetic Strategies for 5- and 6-Membered Ring Azaheterocycles Facilitated by Iminyl Radicals. Molecules, 21(5), 660. https://doi.org/10.3390/molecules21050660