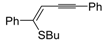
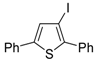
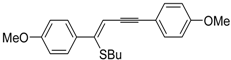
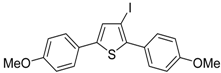
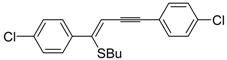
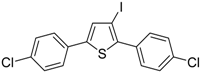
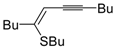
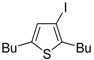
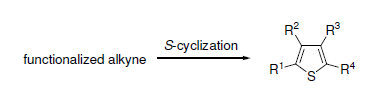
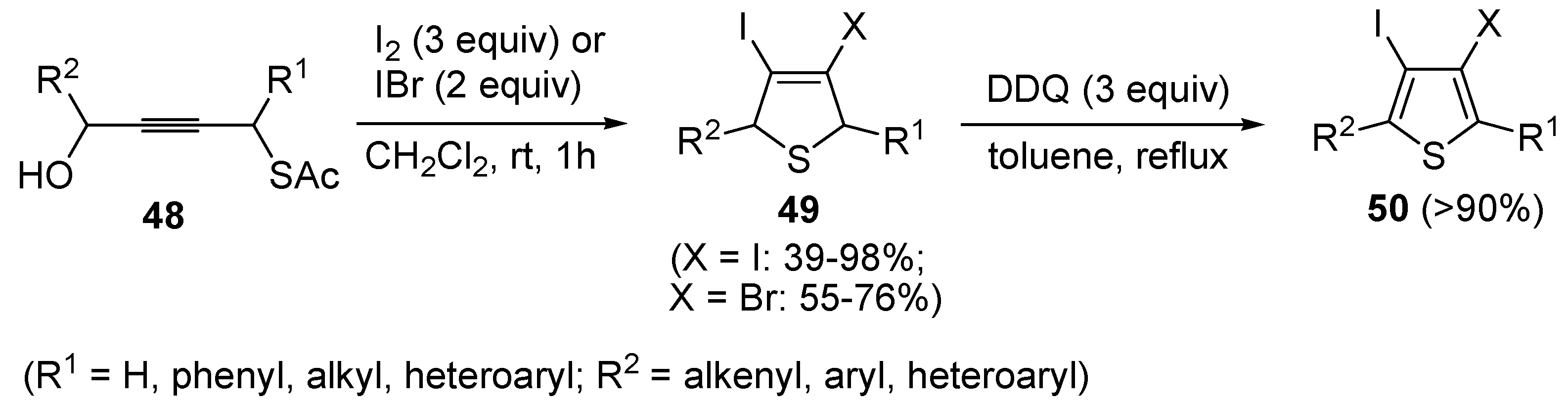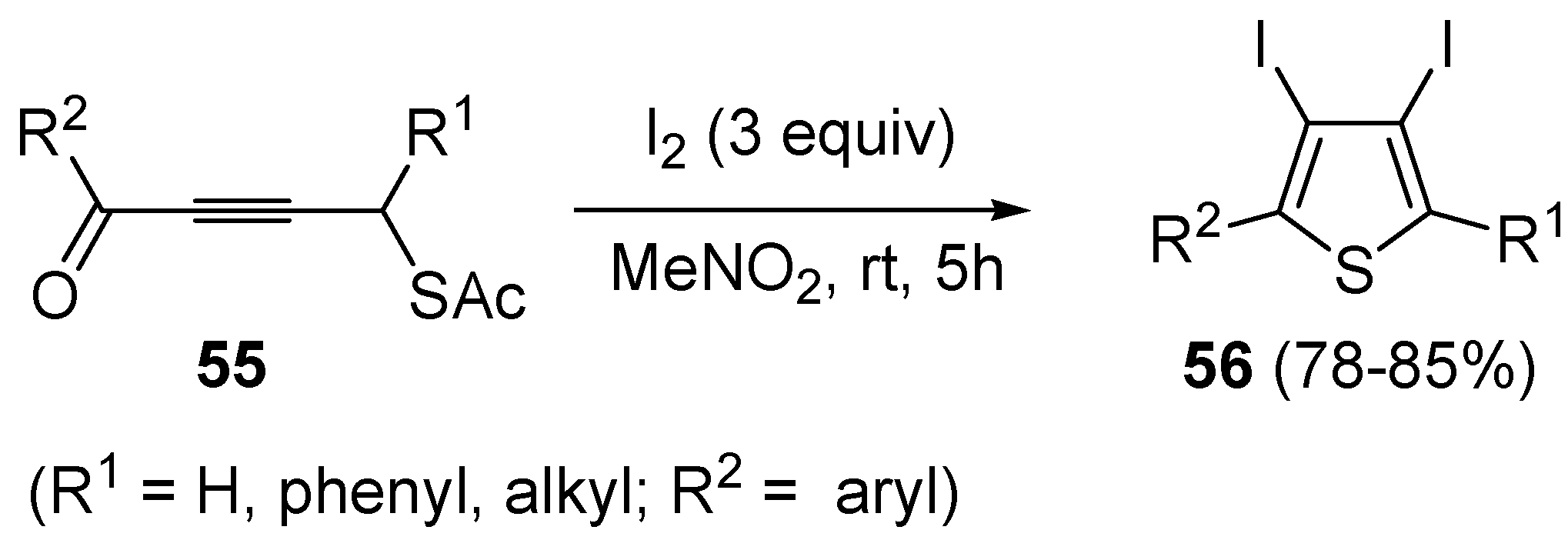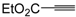1. Introduction
Substituted thiophenes are among the most important aromatic heterocyclic derivatives. Many molecules incorporating the thiophene nucleus have, in fact, shown important pharmacological activities [
1,
2,
3,
4]. Moreover, thiophene derivatives find large application in material science [
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15] and in coordination chemistry [
16,
17], and as intermediate in organic synthesis [
18,
19].
The classical approaches to substituted thiophenes are mainly based on condensation-like reactions or on subsequent functionalization of the thiophene ring [
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30]. However, during the last years, innovative approaches to the regioselective synthesis of substituted thiophenes starting from acyclic precursors have been developed, mainly based on heterocyclization of functionalized alkynes [
31].
In this review, we will highlight some recently developed efficient and selective syntheses of thiophene derivatives by cyclization of readily available S-containing alkyne substrates, which have allowed a significant step forward toward a direct and atom-economical entry to this very important class of aromatic heterocycles. As a matter of fact, these processes may allow the construction of the thiophene ring with the desired substitution pattern in a regiospecific manner and in only one step, usually with high atom economy (particularly in the case of cycloisomerization reactions), and starting from readily available starting materials (as the acetylenic S-contaning precursors can be easily prepared in a few step from commercially available compounds through simple synthetic steps).
As will be seen, many of these cyclization reactions leading to thiophenes have been performed under mild conditions (even at rt, in particular with iodocyclizations) in classical organic solvents, either dipolar aprotic (such as N,N-dimethylacetamide (DMA), dimethylsulfoxide (DMSO), or MeCN), apolar or slightly polar (such as toluene, THF, or CH2Cl2), or protic ones (such as MeOH). However, particularly during the last years, the possibility to carry out these processes in unconventional solvents, such as ionic liquids (ILs) has been successfully verified. This has allowed the easy and convenient recycling of the reaction medium and/or of the catalyst (in the case of metal-catalyzed heterocyclizations).
We have structured the review into different Sections.
Section 2 will deal with metal-catalyzed or base-promoted heterocyclizations, while in
Section 3 iodocyclization reactions will be discussed. Carbocyclization of
S-containing alkyne substrates is the topic of
Section 4. In
Section 5, some miscellaneous methods that cannot be classified into the previous categories are treated. We would like to point out here that most of the mechanisms shown in this review are based on mechanistic pathways proposed by the authors, on the basis of the existing knowledge and, in some cases, of some additional experimental evidences (product stereochemistry, reactivity pattern of the substrates, and so on). Only in a few cases these hypotheses have been corroborated by computation calculations (one example is the iodocyclization of 1-mercapto-3-yn-2-ols
23 in ionic liquids,
Section 3, while, to the best of our knowledge, no kinetic studies have been reported so far. Another aspect worth mentioning concerns the reaction conditions reported in the review: they refer to the optimized conditions, usually established after a careful study on the influence of the reaction parameters (such as the catalyst loading, reagents molar ratios, solvent, temperature and so on) on substrate reactivity and product yield.
2. Synthesis of Thiophene Derivatives by Metal-Catalyzed or Base-Promoted Heterocyclization of S-Containing Alkyne Substrates
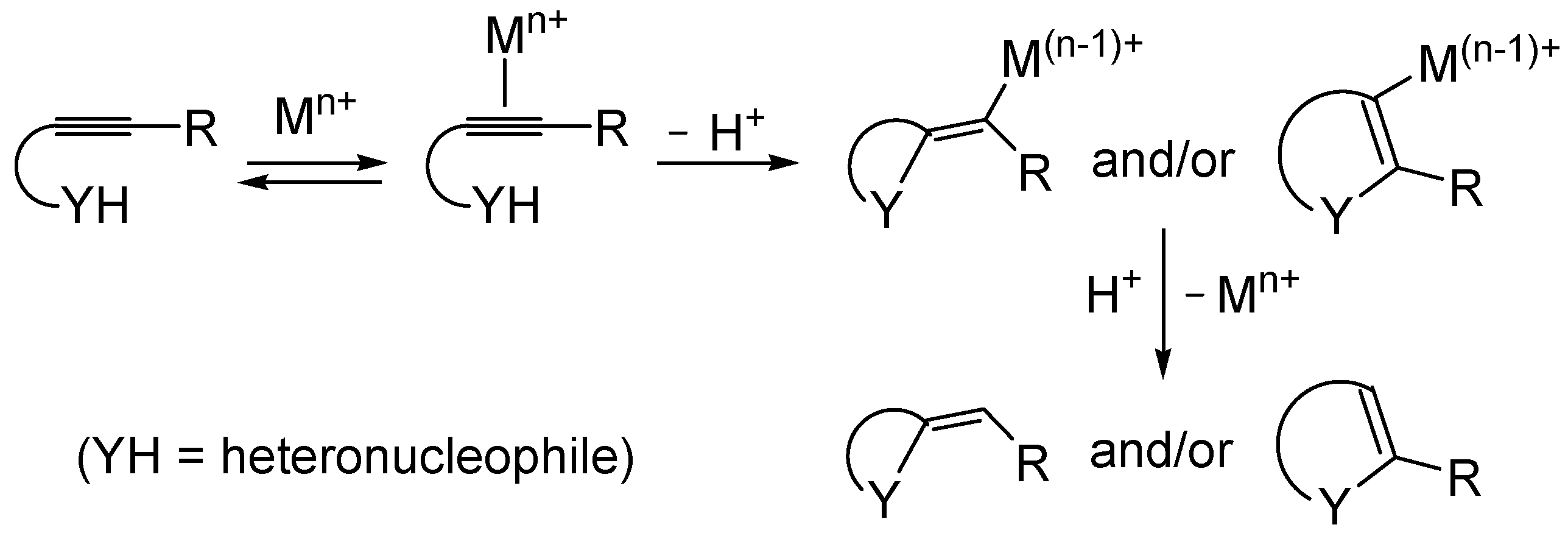
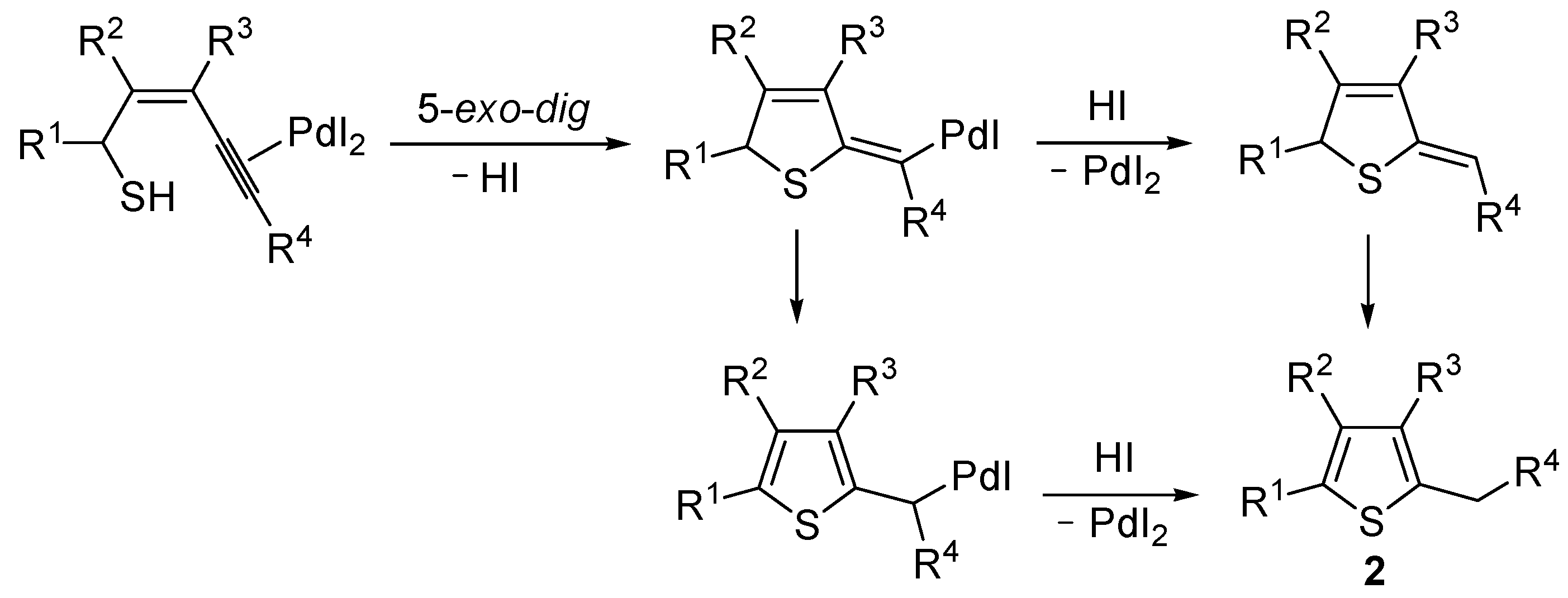
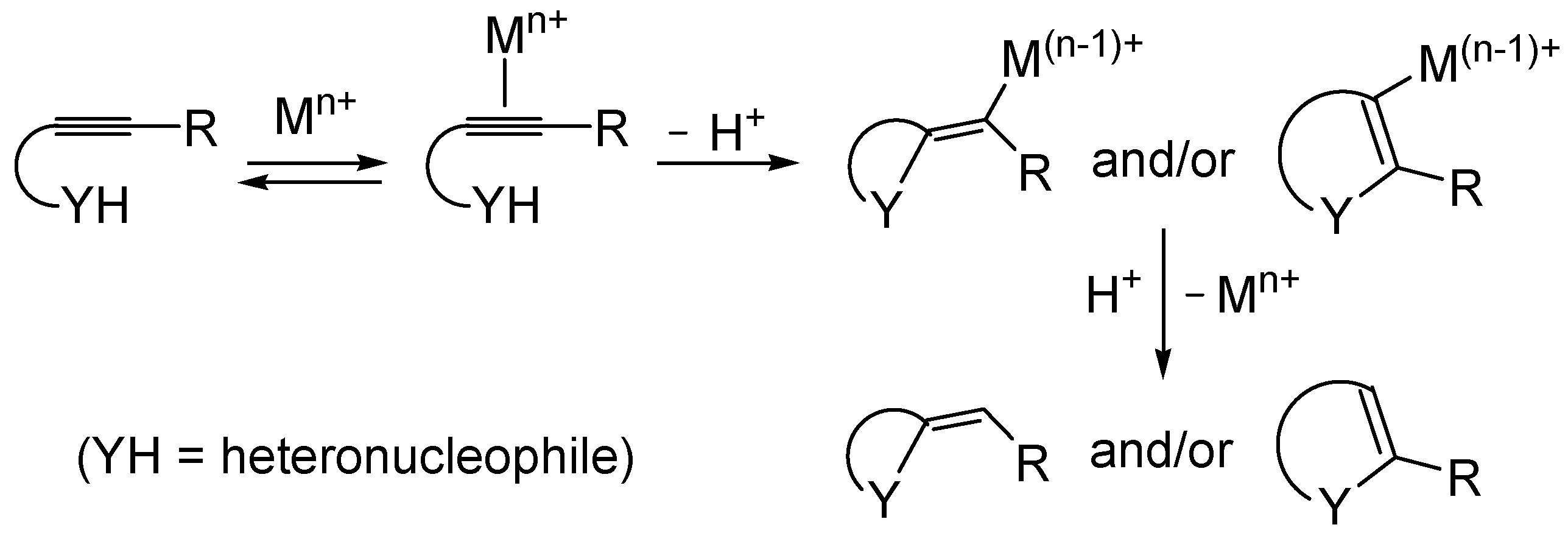
Metal-catalyzed heterocyclization of functionalized alkynes bearing a suitably placed heteronucleophilic group is a powerful methodology for the regioselective and atom-economical synthesis of substituted heterocycles starting from readily available acyclic substrates (
Scheme 1, Y = heteroatom) [
31,
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49]. The generally accepted mechanism for this important transformation involves the electrophilic activation of the triple bond by coordination to the metal center, followed by either
exo or
endo cyclization (ensuing from intramolecular nucleophilic attack by the –YH group to the coordinated triple bond) and protonolysis (
Scheme 1).
Compared to the considerable number of examples reported in the literature of the synthesis of
O- or
N-heterocycles (
Scheme 1, Y = O or N), there are still relatively few examples of metal-catalyzed heterocyclizations of
S-containing alkyne substrates leading to sulfur heterocycles (
Scheme 1, Y = S).
Scheme 1.
Metal-catalyzed heterocyclization of functionalized alkynes bearing a suitably placed nucleophilic group leading to heterocycles through activation of the triple bond by the metal species followed by intramolecular nucleophilic attack by the heteronucleophile and protonolysis.
Scheme 1.
Metal-catalyzed heterocyclization of functionalized alkynes bearing a suitably placed nucleophilic group leading to heterocycles through activation of the triple bond by the metal species followed by intramolecular nucleophilic attack by the heteronucleophile and protonolysis.
This is probably connected with the “poisoning” effect exerted by the sulfur atom on the metal catalyst, owing to its strong coordinating and adsorptive properties [
50,
51]. Nevertheless, progress in organometallic catalysis has recently permitted to develop several important processes involving the metal-catalyzed carbon-sulfur bond formation [
52,
53,
54,
55]. Although most of these processes concern the formation of sulfurated acyclic molecules, during the last years several important
S-cyclization reactions, involving the formation of the C-S bond and leading to S-heterocycles, have been developed.
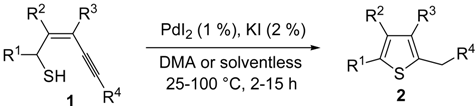
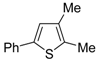
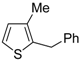
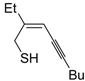
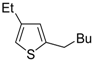
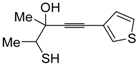
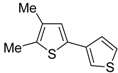
The first example of the formation of thiophenes by a metal-catalyzed cycloisomerization approach of alkynylthiol derivatives was reported in 2000 by our research group [
56]. It concerned the reaction of (
Z)-2-en-4-yne-1-thiols
1 (readily obtainable from the corresponding (
Z)-2-en-4-yn-1-ols [
57]) in
N,
N-dimethylacetamide (DMA) as the solvent or under solventless conditions at 25–100 °C, carried out in the presence of a particularly simple catalytic system, consisting of PdI
2 (1 mol %) in conjunction with KI (2 mol %) (
Table 1). The use of KI was necessary in order to make PdI
2 soluble and to stabilize the formation of the catalytically active species PdI
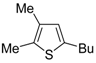
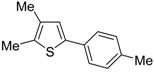
42−. With low-boiling substrates, solventless conditions were used to facilitate product recovery. In other cases, several polar solvent were tested (a polar solvent was necessary to ensure the dissolution of the ionic catalyst), and, between them, DMA gave the best results in terms of substrate reactivity and product yield. Substrates bearing a terminal as well as an internal triple bond could be employed, while alkyl as well aryl substitution was tolerated on the double bond and at C-1 (
Table 1) [
56]. One indubitable advantage of this new protocol consisted in the practically neutral conditions employed for realizing the thiocyclization, as compared with the strongly basic conditions previously used (
t-BuOK in
t-BuOH in the presence of 18-crown-6), which were not compatible with base-sensitive substrates such as those bearing a terminal triple bond [
58].
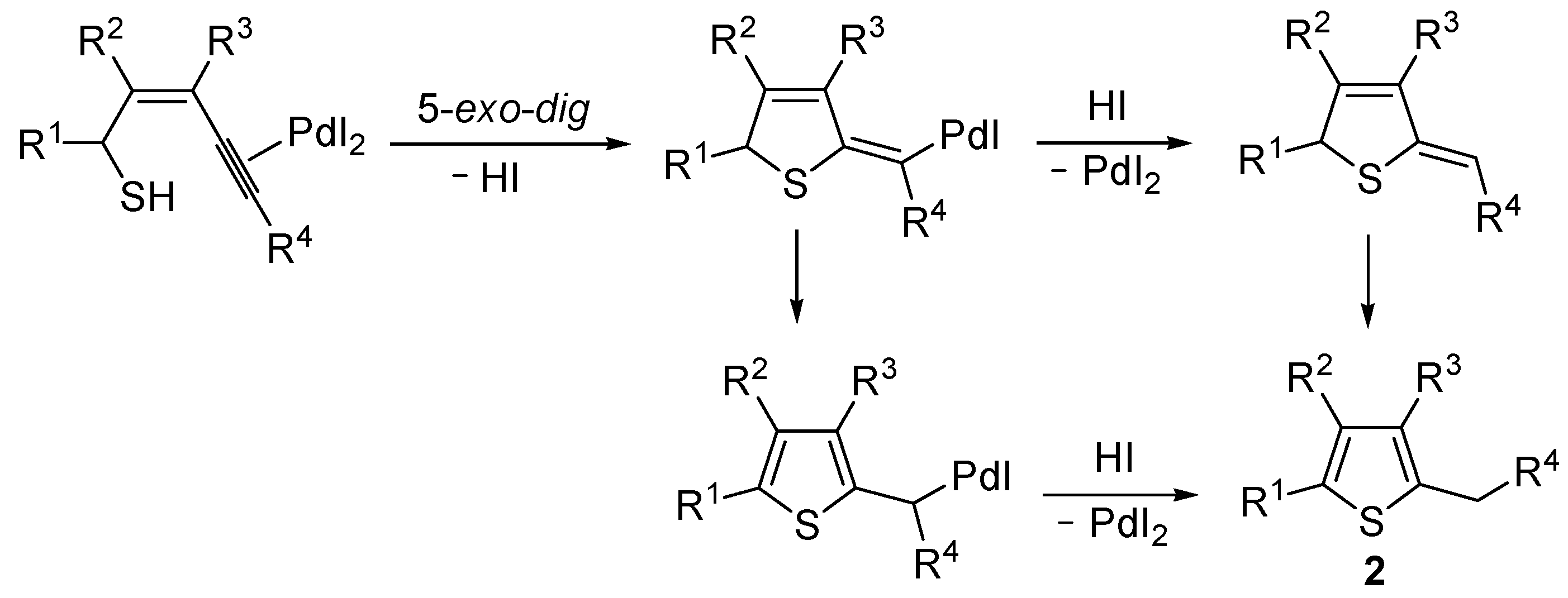
Mechanistically, the reaction is believed to proceed through
anti 5-
exo-
dig intramolecular nucleophilic attack by the thiolic group to the triple bond coordinated to Pd(II), with formal elimination of HI, followed by protonolysis and aromatization or vice versa (
Scheme 2; anionic iodide ligand are omitted for clarity). This mechanistic hypothesis was in agreement with the experimental observation that substrates bearing a terminal triple bond were more reactive with respect to those bearing an internal triple bond. With an internal triple bond, in fact, Pd(II) coordination is less favored for steric reasons. A nucleophilic attack by the –SH group on the triple bond, with Pd(II) being coordinated from the opposite site (
anti attack), was also in agreement with the significantly higher reactivity observed with substrates unsubstituted at C-3 with respect to enynethiols substituted at C-3 (
Table 1, compare Entries 5 and 6). This is clearly related to the fact that an
anti-coordination of the triple bond to Pd(II) may be less efficient, for steric reasons, in the presence of a substituent at C-3 [
56].
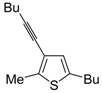
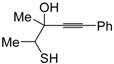
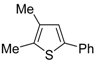
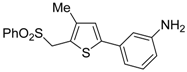
Table 1.
PdI
2/KI-catalyzed cycloisomerization of (
Z)-2-en-4-yne-1-thiols
1 to substituted thiophenes
2 a [
56].
![Molecules 19 15687 i001]()
Scheme 2.
Proposed mechanistic pathways for the PdI
2-catalyzed cycloisomerization of (
Z)-2-en-4-yne-1-thiols
1 leading to thiophenes
2 [
56].
Scheme 2.
Proposed mechanistic pathways for the PdI
2-catalyzed cycloisomerization of (
Z)-2-en-4-yne-1-thiols
1 leading to thiophenes
2 [
56].
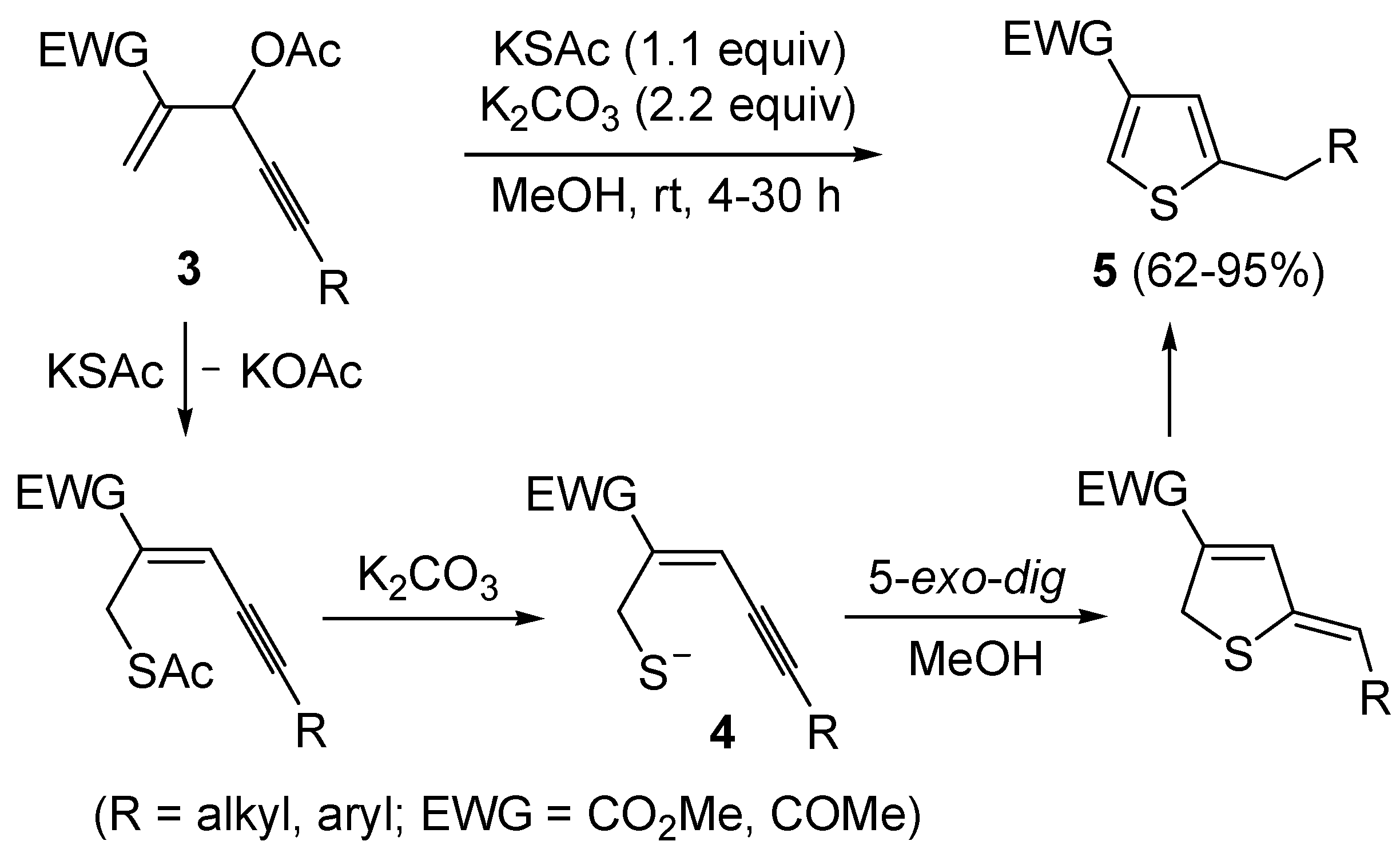
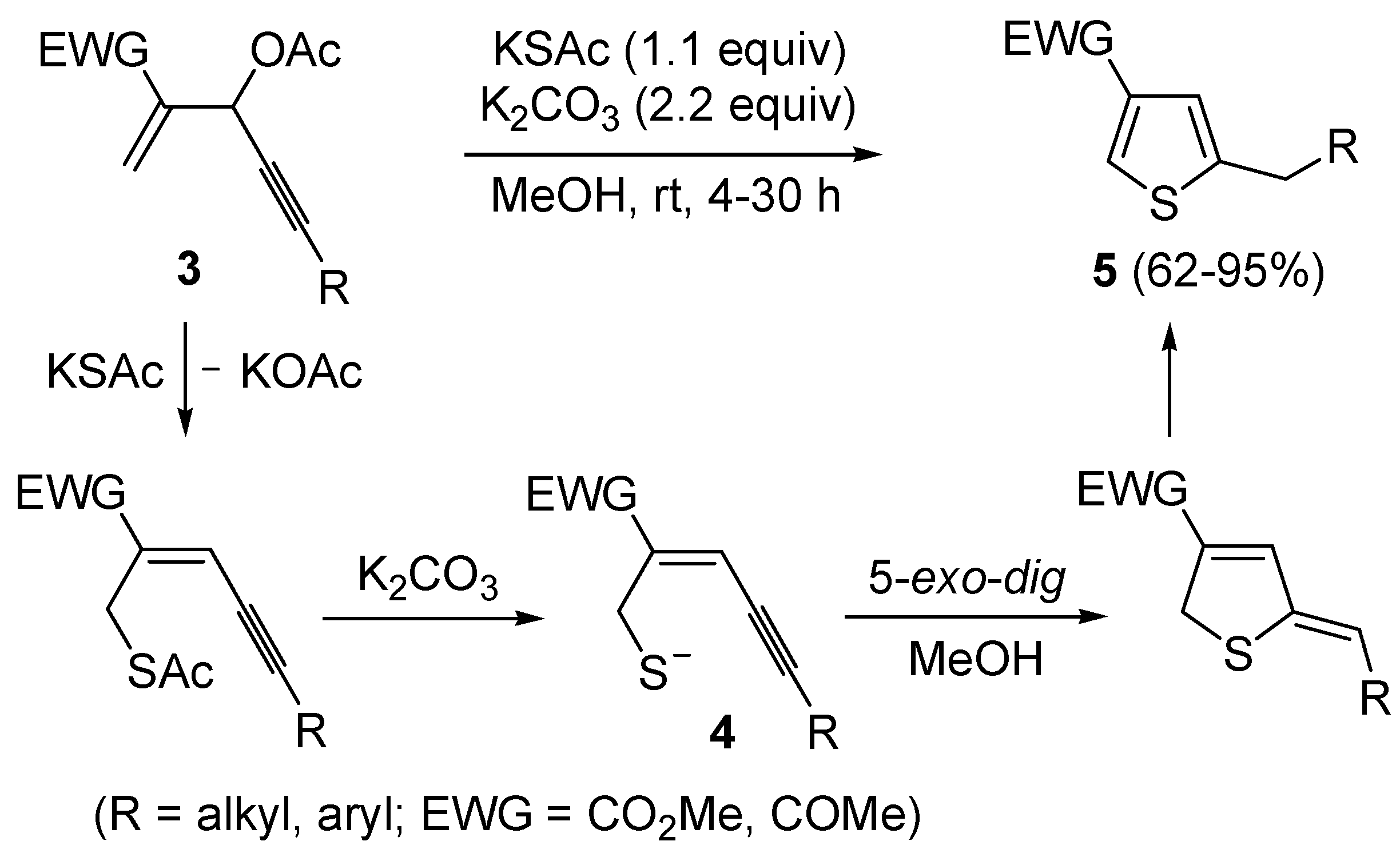
More recently, a strictly related method has been published, regarding the metal-free cyclization of 4-en-1-yn-3-yl acetates
3 to give 2,4-disubstituted thiophenes
5 through the intermediate formation of (
Z)-2-en-4-yne-1-thiolate derivatives
4, formed
in situ by allylic nucleophilic substitution with KSAc followed by base-promoted deacylation (
Scheme 3) [
59]. Intermediates
4 were then converted into thiophenes
5 by 5-
exo-
dig cyclization and aromatization. The reaction has been applied to the synthesis of several 2,4-disubstitued thiophenes, but presented limitations due to the strong basic conditions employed (for example, the reaction could not be applied to substrates bearing a terminal triple bond) and to the need for the presence of an electron-withdrawing group (EWG) at the C-4 of the starting material (
Scheme 3) [
59].
Scheme 3.
Synthesis of 2,4-disubstitued thiophenes
5 from 4-en-1-yn-3-yl acetates
3 by sequential allylic nucleophilic substitution with KSAc followed by base-promoted deacylation, to give (
Z)-2-en-4-yne-1-thiolate derivatives
4, and base-promoted thiocyclization [
59].
Scheme 3.
Synthesis of 2,4-disubstitued thiophenes
5 from 4-en-1-yn-3-yl acetates
3 by sequential allylic nucleophilic substitution with KSAc followed by base-promoted deacylation, to give (
Z)-2-en-4-yne-1-thiolate derivatives
4, and base-promoted thiocyclization [
59].
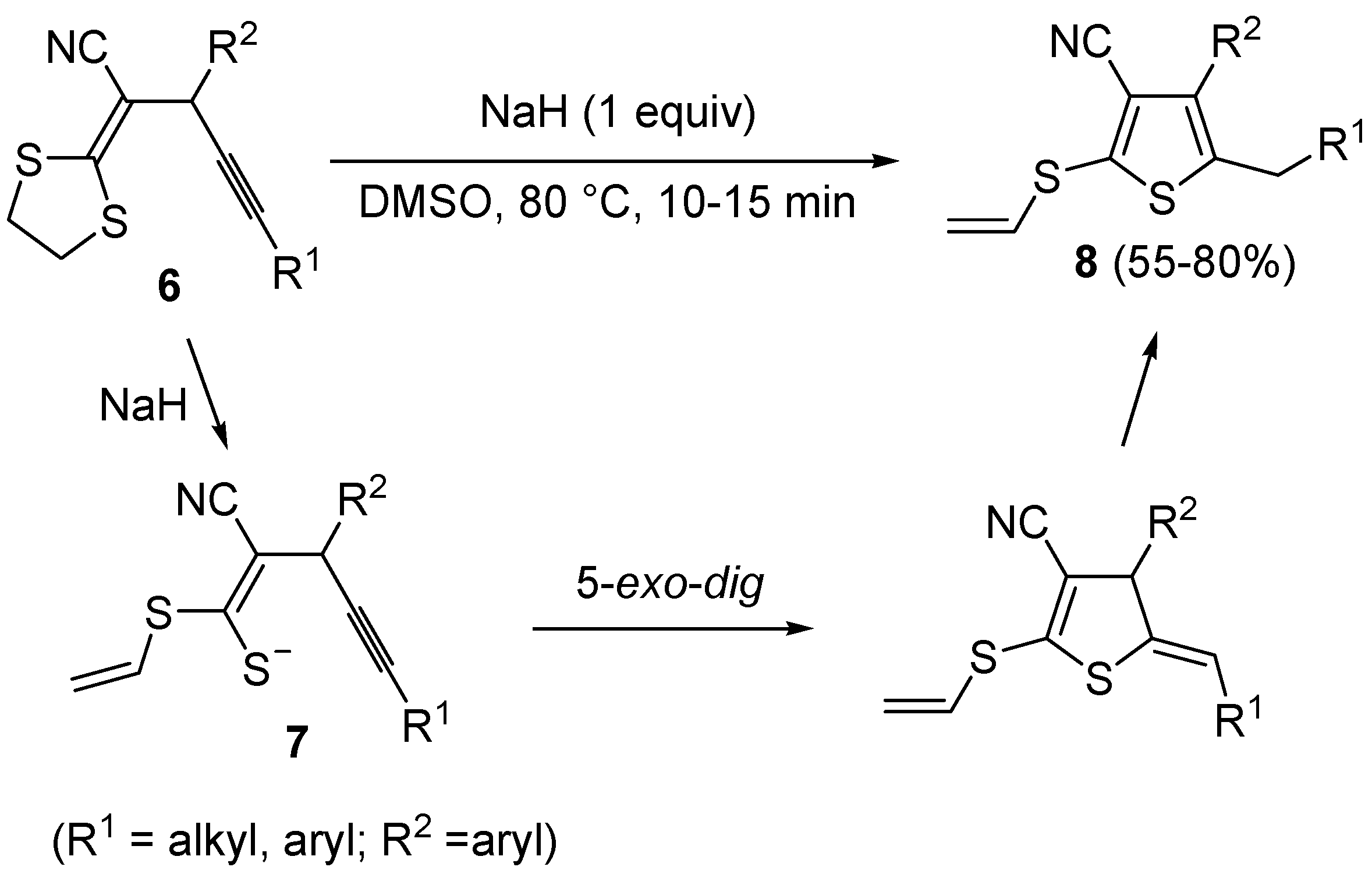
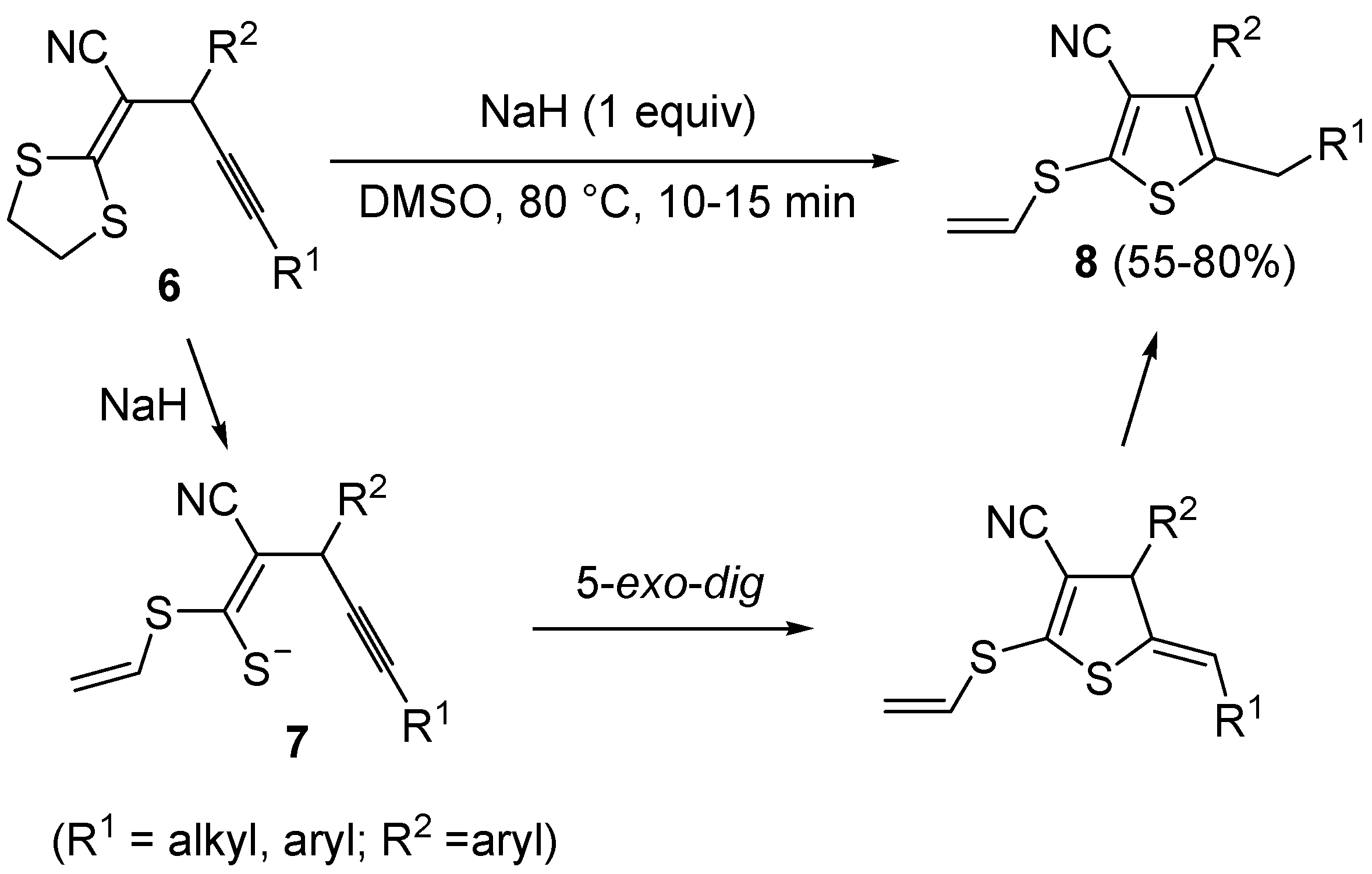
Scheme 4.
Synthesis of 3-cyano-2-(vinylthio)thiophenes
8 from 2-(1,3-dithiolan-2-ylidene)-4-ynenitriles
6 by NaH-induced ring opening, to give (
Z)-1-en-4-yne-1-thiolates
7, followed by 5-
exo-
dig cyclization and aromatization [
60].
Scheme 4.
Synthesis of 3-cyano-2-(vinylthio)thiophenes
8 from 2-(1,3-dithiolan-2-ylidene)-4-ynenitriles
6 by NaH-induced ring opening, to give (
Z)-1-en-4-yne-1-thiolates
7, followed by 5-
exo-
dig cyclization and aromatization [
60].
In a similar way, 3-cyano-2-(vinylthio)thiophenes
8 were obtained from 2-(1,3-dithiolan-2-ylidene)-4-ynenitriles
6 by NaH-induced ring opening of the 1,3-dithiolan-2-ylidene group (ensuing from deprotonation of the methylene moiety bonded to sulfur), leading to the corresponding (
Z)-1-en-4-yne-1-thiolates
7, followed by 5-
exo-
dig cyclization and aromatization (
Scheme 4) [
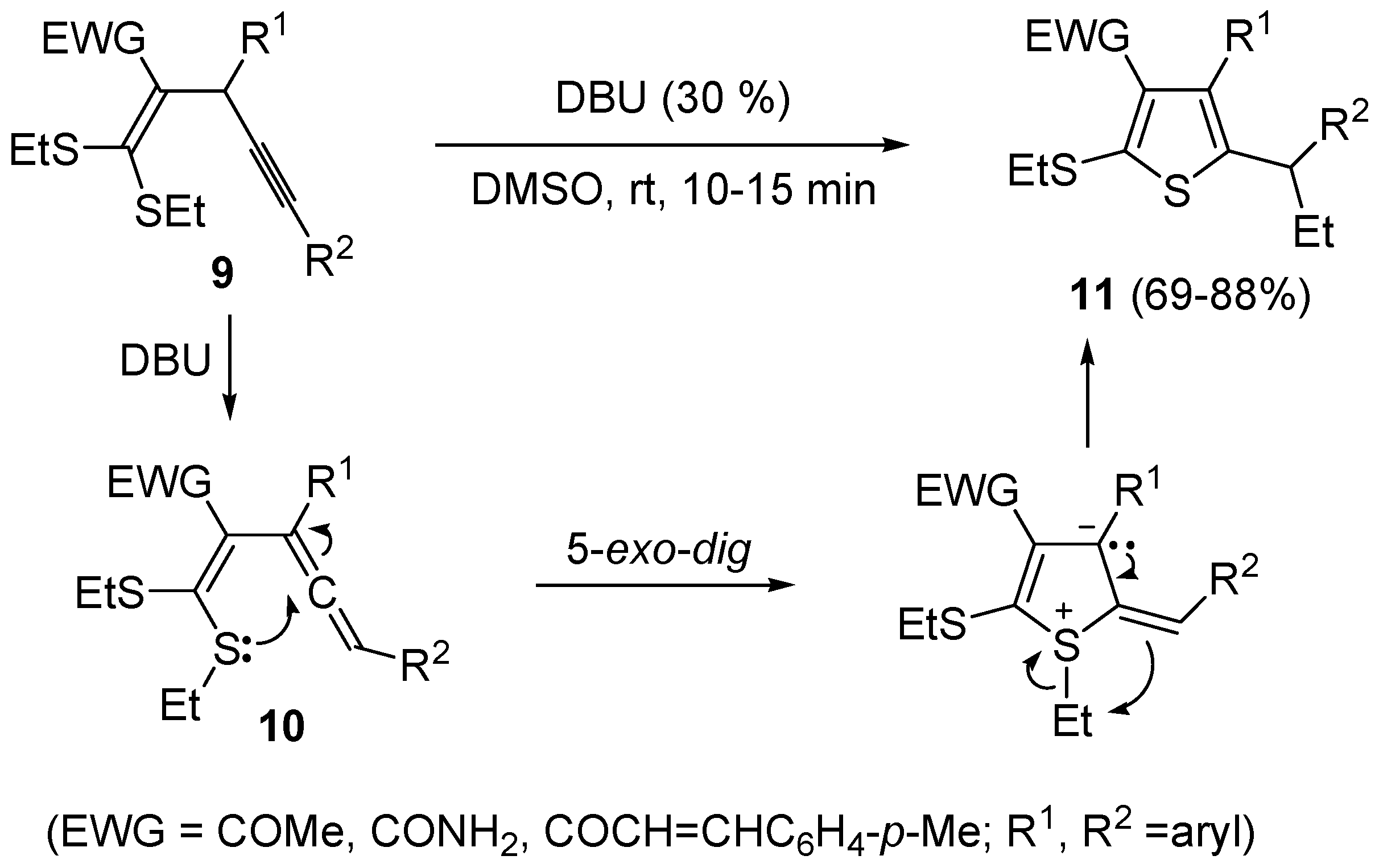
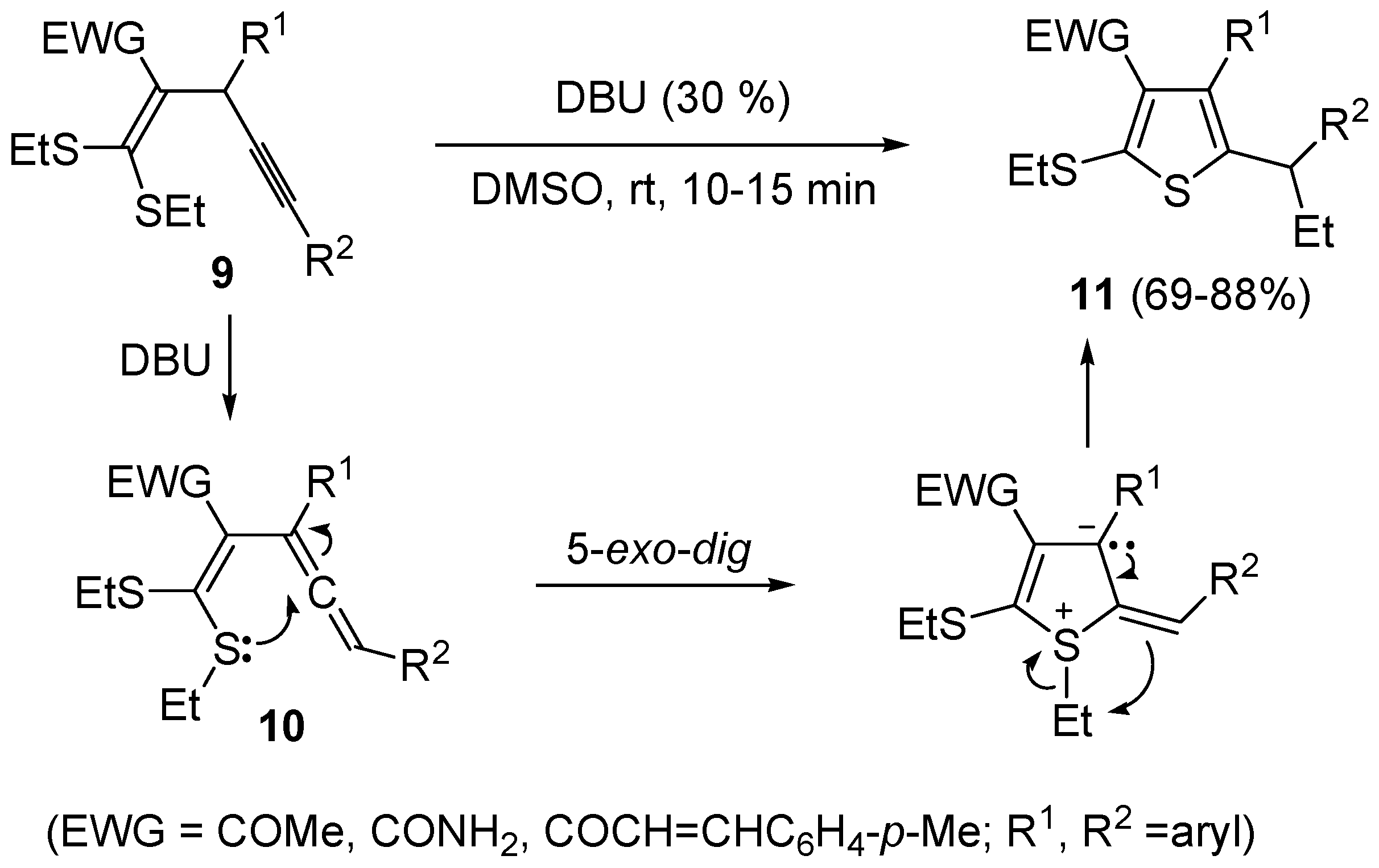
60]. Interestingly, the similar substrates 1,1-
bis(ethylthio)-1-en-4-ynes
9, bearing an electron withdrawing group, such as the carbonyl, at the 2 position, reacted in a rather different way when treated with a base such as DBU. In this case, in fact, the intermediate formation of
gem-dialkylthiovinylallenes
10 took place, followed by 5-
exo-
dig S-cyclization and 1,3-migration of the ethyl group from sulfur to the benzylic carbon, eventually leading to substituted thiophenes
11 in good to high yields (
Scheme 5) [
61].
Scheme 5.
Synthesis of thiophenes
11 from 1,1-
bis(ethylthio)-1-en-4-ynes
9 by DBU-induced isomerization to
gem-dialkylthiovinylallenes
10 followed by 5-
exo-
dig S-cyclization and 1,3-migration of the ethyl group [
61].
Scheme 5.
Synthesis of thiophenes
11 from 1,1-
bis(ethylthio)-1-en-4-ynes
9 by DBU-induced isomerization to
gem-dialkylthiovinylallenes
10 followed by 5-
exo-
dig S-cyclization and 1,3-migration of the ethyl group [
61].
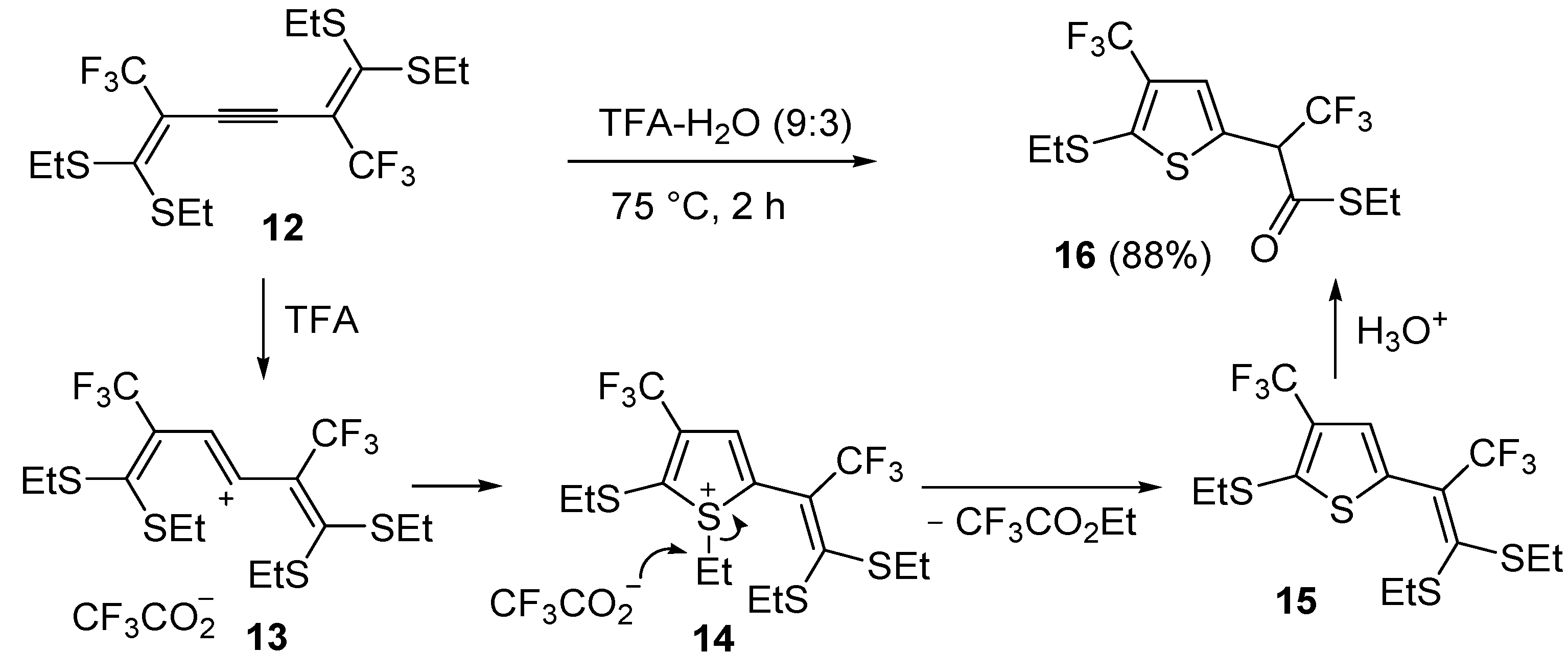
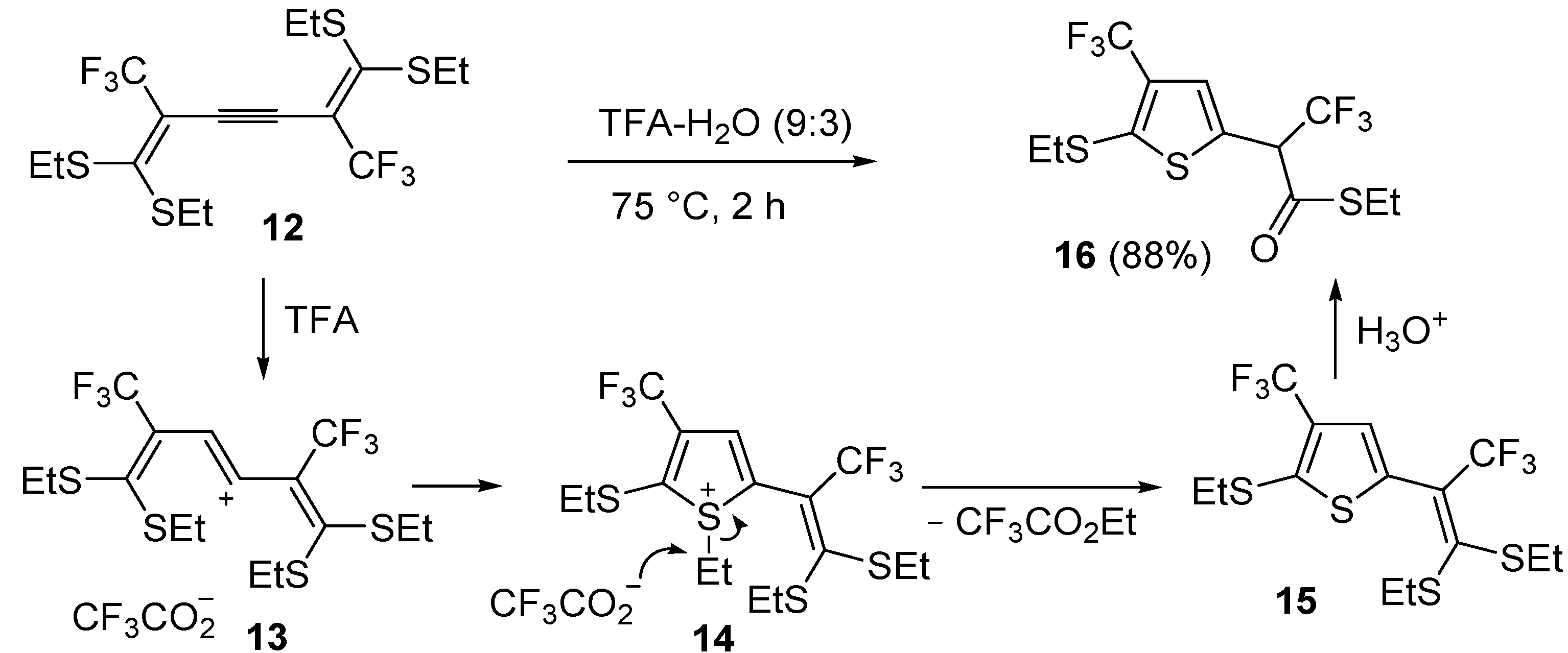
An interesting approach to multifunctionalized thiophene
16 from 1,1,6,6-
tetrakis(ethylthio)-2,5-
bis(trifluoromethyl)hexa-1,5-dien-3-yne (
12) has been reported, based on treatment of
12 with a mixture of trifluoroacetic acid and water (TFA-H
2O 9:3) at 75 °C for 2 h (
Scheme 6) [
62].
Scheme 6.
Synthesis of
S-ethyl 2-(5-(ethylthio)-4-(trifluoromethyl)thiophen-2-yl)-3,3,3-trifluoropropanethioate (
16) from 1,1,6,6-
tetrakis(ethylthio)-2,5-
bis(trifluoromethyl)hexa-1,5-dien-3-yne (
12) through the intermediate formation of 5-(3,3-
bis(ethylthio)-1,1,1-trifluoroprop-2-en-2-yl)-2-(ethylthio)-3-(trifluoromethyl)thiophene (
15) [
62].
Scheme 6.
Synthesis of
S-ethyl 2-(5-(ethylthio)-4-(trifluoromethyl)thiophen-2-yl)-3,3,3-trifluoropropanethioate (
16) from 1,1,6,6-
tetrakis(ethylthio)-2,5-
bis(trifluoromethyl)hexa-1,5-dien-3-yne (
12) through the intermediate formation of 5-(3,3-
bis(ethylthio)-1,1,1-trifluoroprop-2-en-2-yl)-2-(ethylthio)-3-(trifluoromethyl)thiophene (
15) [
62].
Formation of
S-ethyl 2-(5-(ethylthio)-4-(trifluoromethyl)thiophen-2-yl)-3,3,3-trifluoropropane-thioate (
16) is explained to occur by triple bond protonation to give stabilized carbocation
13 followed by sulfur attack to the carbocation and nucleophilic attack of the trifluoroacetate anion to the resulting sufonium cation
14. This leads to the formation of 5-(3,3-
bis(ethylthio)-1,1,1-trifluoroprop-2-en-2-yl)-2-(ethylthio)-3-(trifluoromethyl)thiophene intermediate (
15), which has been isolated under appropriate conditions, and which upon hydrolysis leads to the final product
16 (
Scheme 6) [
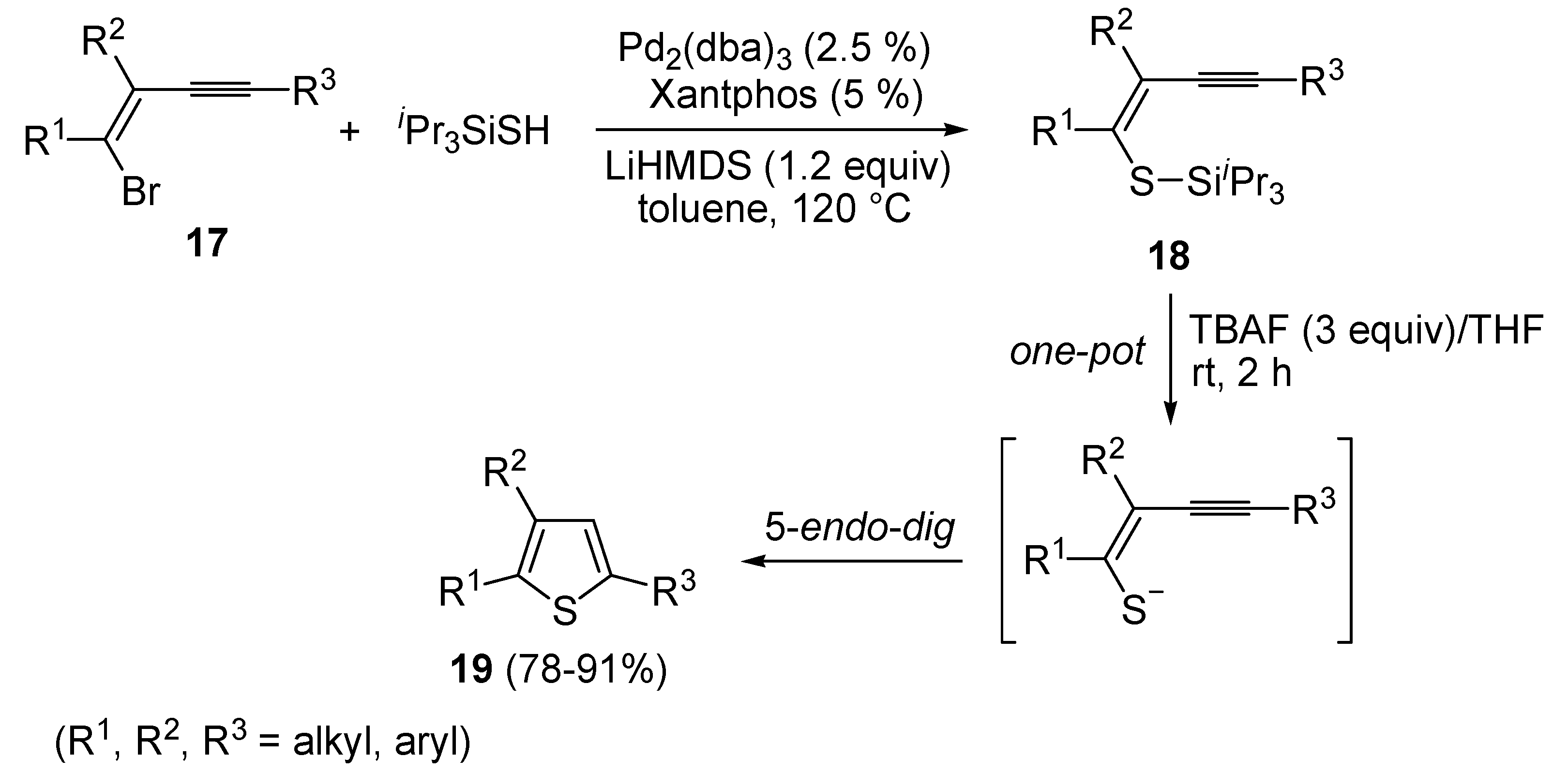
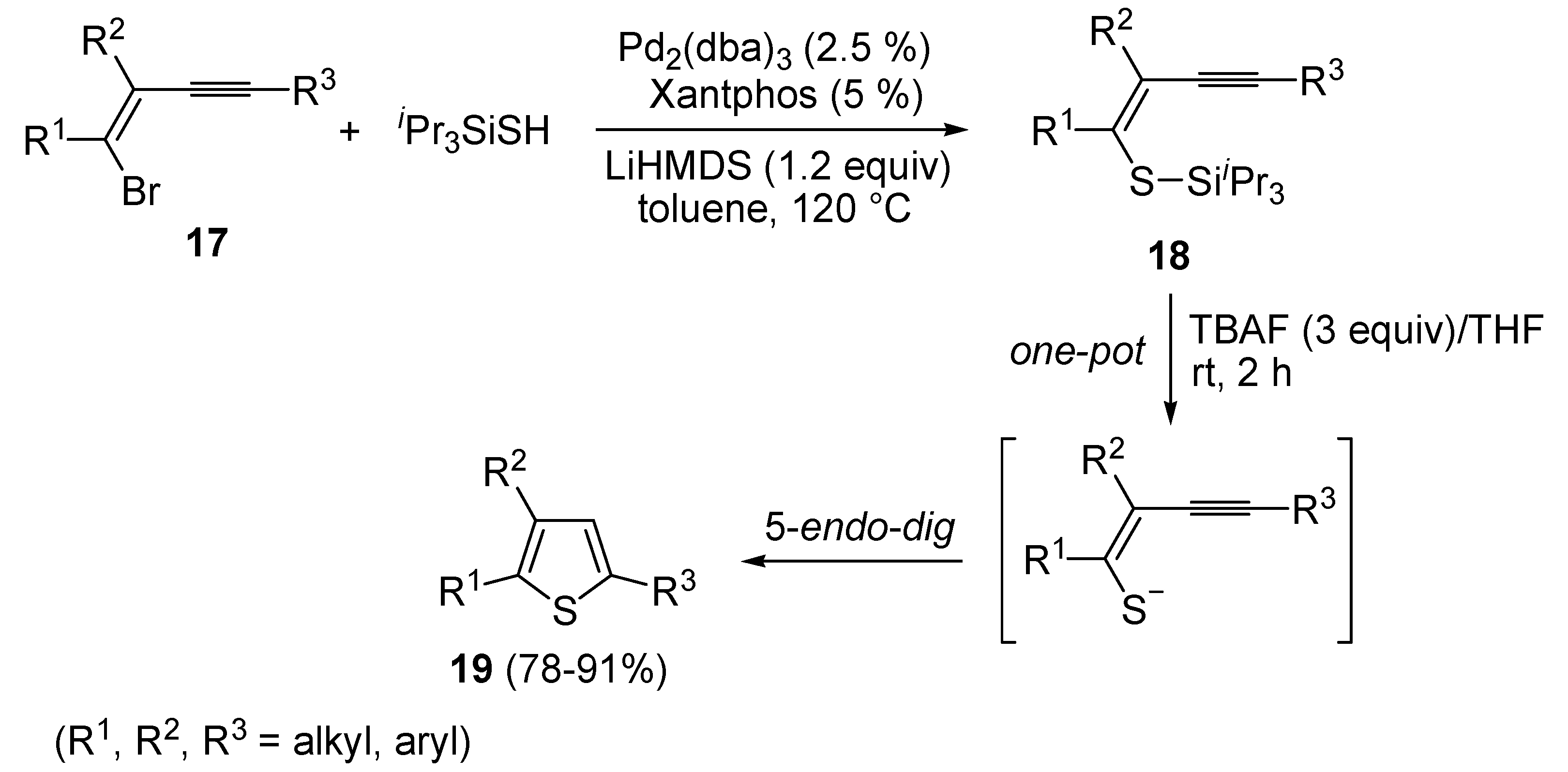
62]. A one-pot C-S coupling/heterocyclization approach to substituted thiophenes
19 has been recently reported [
63]. It involves the Pd-catalyzed reaction of (
Z)-1-bromo-1-en-3-ynes
17 with triisopropylsilanethiol (1.2 equiv), carried out in the presence of Xantphos as ligand and lithium hexamethyldisilazane (LiHMDS) as the base, to give (
Z)-(1-en-3-ynylthio)triisopropylsilanes
18, followed by 5-
endo-
dig cyclization, induced by desilylation with tetrabutylammonium fluoride (TBAF) (
Scheme 7) [
63].
Scheme 7.
Synthesis of thiophenes
19 from (
Z)-1-bromo-1-en-3-ynes
17 by Pd-catalyzed coupling with triisopropylsilanethiol, to give (
Z)-(1-en-3-ynylthio)triisopropylsilanes
18, followed by one-pot 5-
endo-
dig cyclization, induced by desilylation with tetrabutylammonium fluoride (TBAF) [
63].
Scheme 7.
Synthesis of thiophenes
19 from (
Z)-1-bromo-1-en-3-ynes
17 by Pd-catalyzed coupling with triisopropylsilanethiol, to give (
Z)-(1-en-3-ynylthio)triisopropylsilanes
18, followed by one-pot 5-
endo-
dig cyclization, induced by desilylation with tetrabutylammonium fluoride (TBAF) [
63].
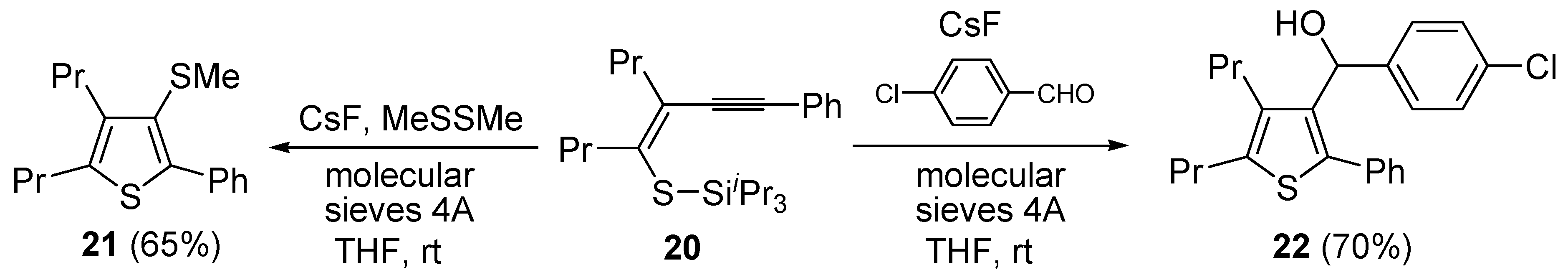
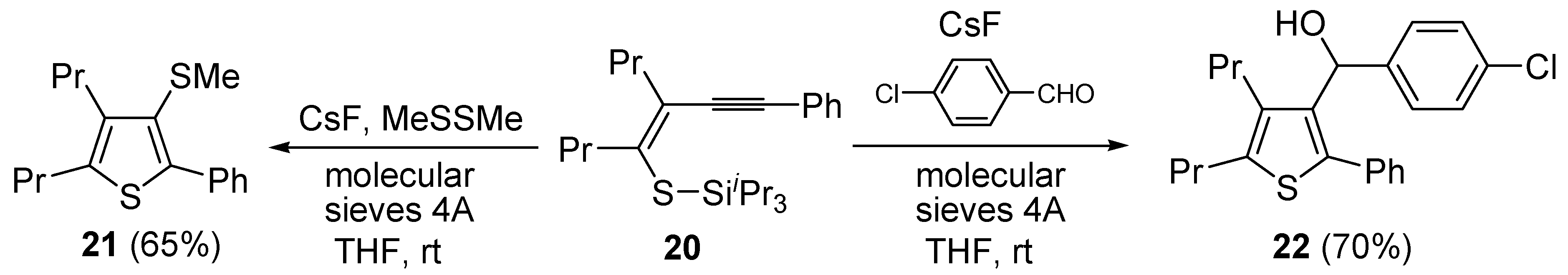
Further functionalization of the final product, to give thiophenes
21 and
22, could be introduced by reacting purified (
Z)-triisopropyl-(5-(2-phenylethynyl)oct-4-en-4-ylthio)silane (
20) with CsF in the presence of a suitable electrophile, such as dimethyldisulfide or
p-chlorobenzaldehyde, and molecular sieves 4A (
Scheme 8) [
63].
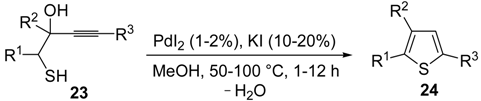
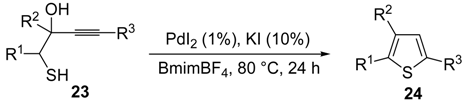
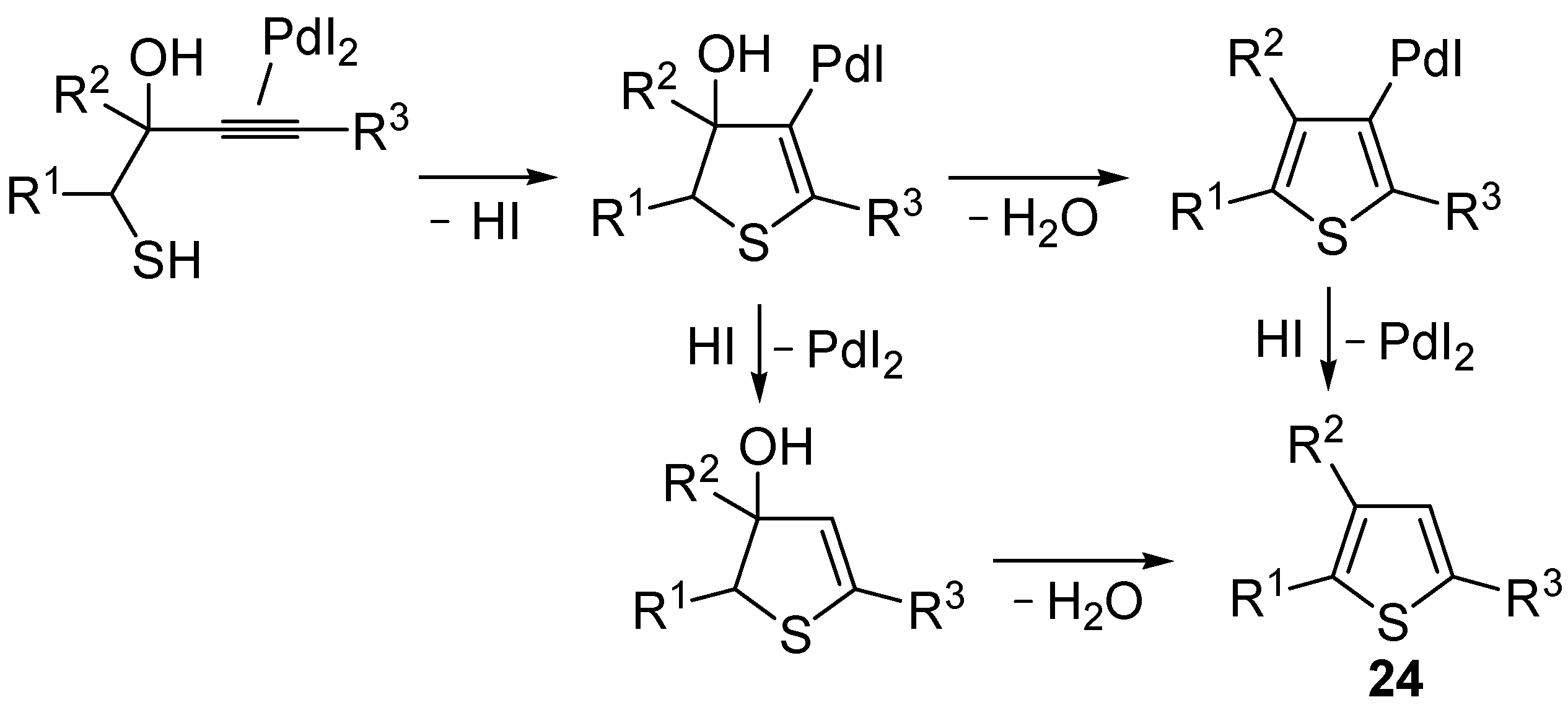
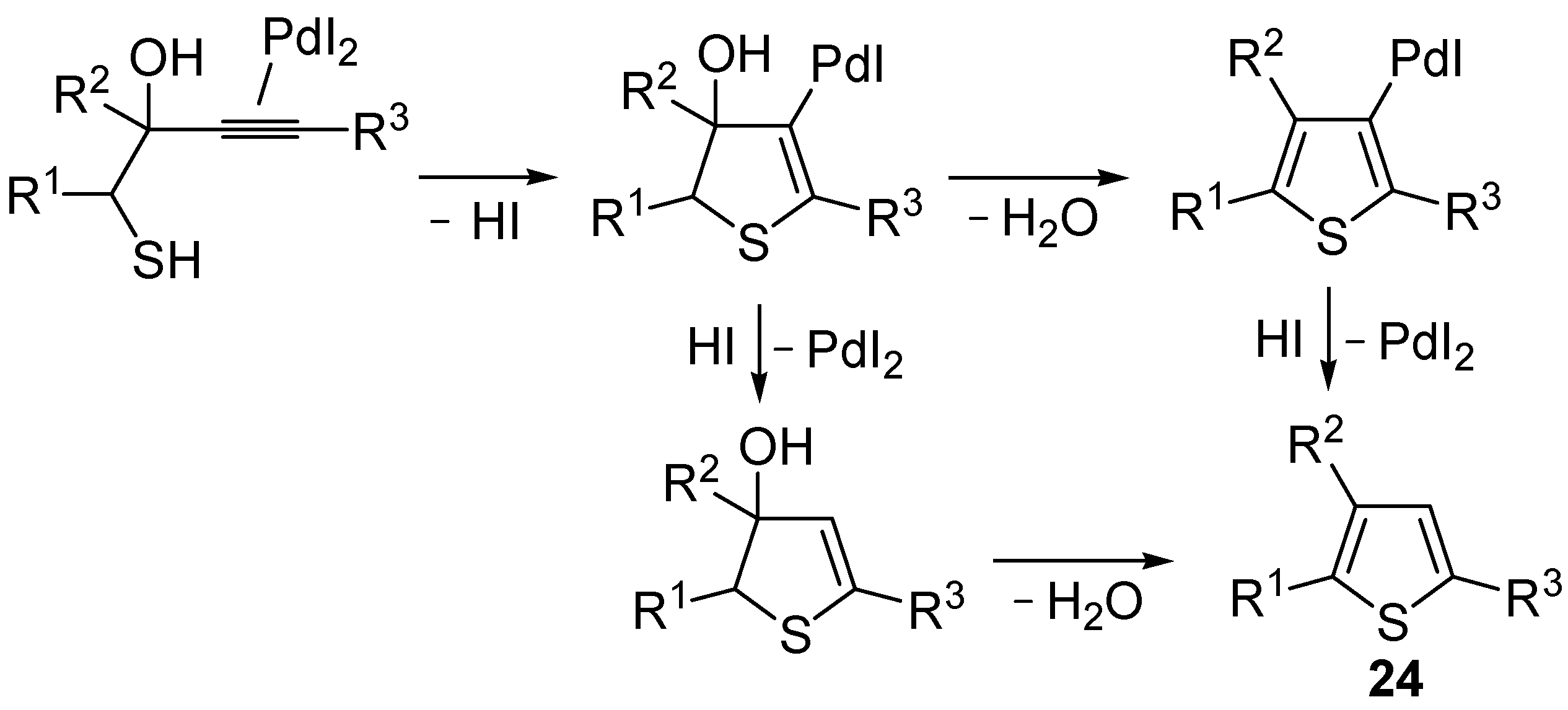
A 5-
endo-
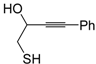
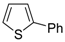
dig S-cyclization was also involved in the synthesis of substituted thiophenes
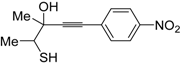
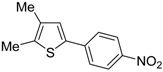
24 by Pd-catalyzed heterocyclodehydration of readily available 1-mercapto-3-yn-2-ols
23, recently reported by our research group [
64]. The cyclization reaction is catalyzed by PdI
2 in conjunction with an excess (10:1 molar ratio) of KI, and takes place either in MeOH at 50–100 °C (
Table 2) or in an ionic liquid, such as BmimBF
4, at 80 °C. These are optimized conditions, after a careful study on the influence of the reaction parameters (such as the KI:PdI
2 molar ratio, the catalyst loading, and so on) on substrate reactivity and product yield. In the case of the reactions carried out in BmimBF
4, the catalyst-solvent system could be recycled several times without appreciable loss of activity (
Table 3) [
64]. This protocol generalized the previous finding by Aponick and coworkers, who reported the Au/Ag-catalyzed transformation of 1-mercapto-4-phenylbut-3-yn-2-ol into 2-phenylthiophene, carried out using 5 mol % of Au[P(
t-Bu)
2(
o-biphenyl)]Cl and 5 mol % of AgOTf in THF at 40 °C in the presence of molecular sieves 4A [
65]. The heterocyclodehydration process takes place by 5-
endo-
dig intramolecular nucleophilic attack of the thiol group to the triple bond coordinated to the metal center, with elimination of HI, followed by dehydration and protonolysis or
vice versa (
Scheme 9; anionic iodide ligands are omitted for clarity) [
64].
Scheme 8.
Synthesis of 3-(methylthio)-2-phenyl-4,5-dipropylthiophene (
21) and (4-chlorophenyl)(2-phenyl-4,5-dipropylthiophen-3-yl)methanol (
22) from (
Z)-triisopropyl(5-(2-phenylethynyl)oct-4-en-4-ylthio)silane (
20) by CsF-induced 5-
exo-
dig S-cyclization in the presence of a suitable electrophile (dimethyldisulfide or
p-chlorobenzaldehyde respectively) [
63].
Scheme 8.
Synthesis of 3-(methylthio)-2-phenyl-4,5-dipropylthiophene (
21) and (4-chlorophenyl)(2-phenyl-4,5-dipropylthiophen-3-yl)methanol (
22) from (
Z)-triisopropyl(5-(2-phenylethynyl)oct-4-en-4-ylthio)silane (
20) by CsF-induced 5-
exo-
dig S-cyclization in the presence of a suitable electrophile (dimethyldisulfide or
p-chlorobenzaldehyde respectively) [
63].
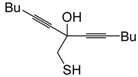
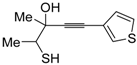
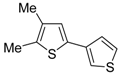
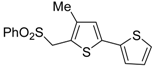
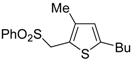
Table 2.
PdI
2/KI-catalyzed heterocyclodehydration of 1-mercapto-3-yn-2-ols
23 to substituted thiophenes
24 a [
64].
![Molecules 19 15687 i014]()
Table 3.
Recyclable catalytic synthesis of substituted thiophenes
24 by PdI
2/KI-catalyzed heterocyclodehydration of 1-mercapto-3-yn-2-ols
23 in BmimBF
4 a [
64].
![Molecules 19 15687 i033]()
Scheme 9.
Proposed mechanistic pathways for the PdI
2-catalyzed heterocyclodehydration of 1-mercapto-3-yn-2-ols
23 leading to thiophenes
24 [
64].
Scheme 9.
Proposed mechanistic pathways for the PdI
2-catalyzed heterocyclodehydration of 1-mercapto-3-yn-2-ols
23 leading to thiophenes
24 [
64].
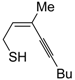
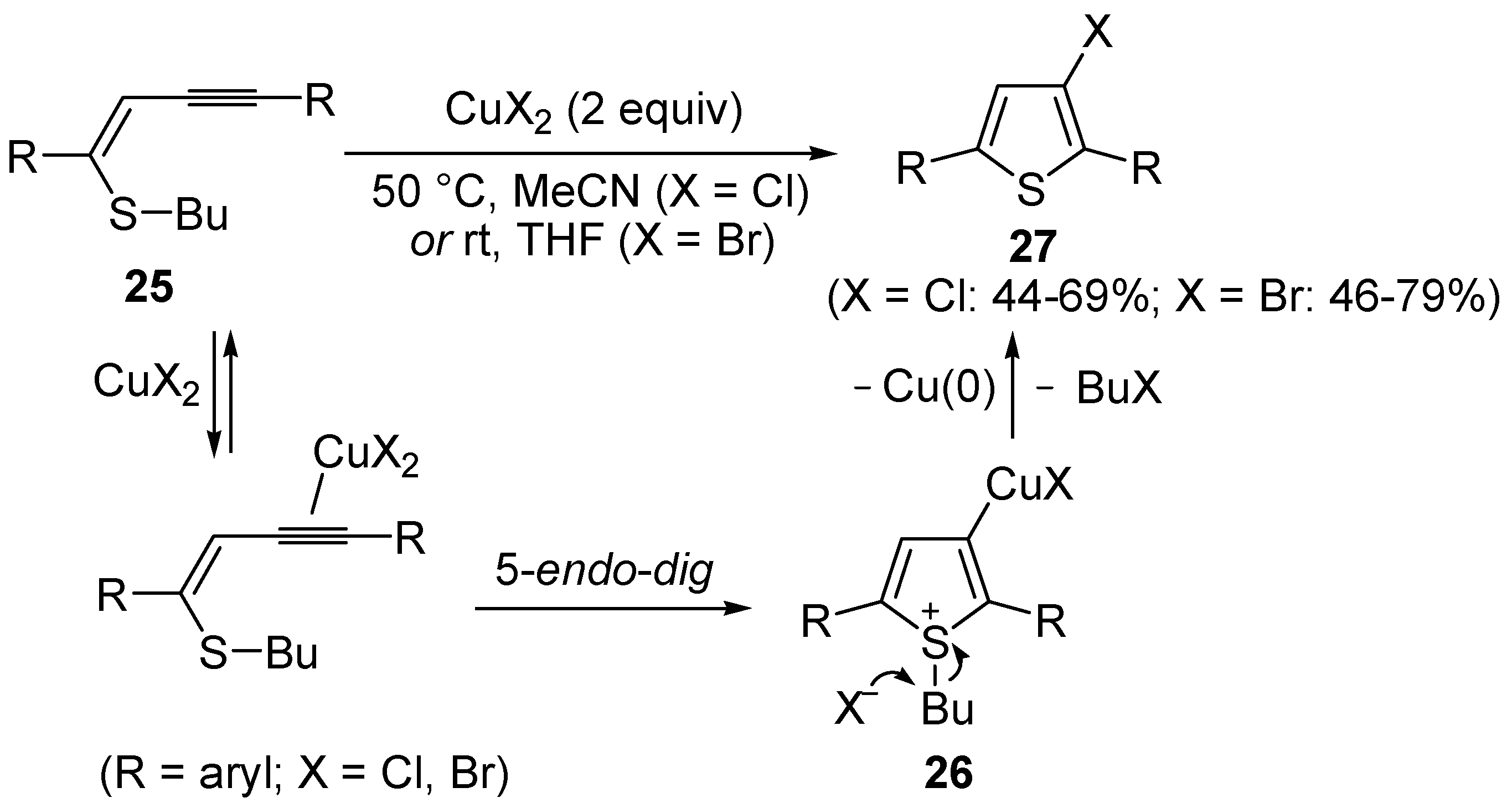
Copper-promoted or –catalyzed cyclization of
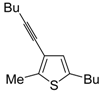
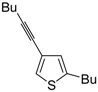
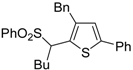
S-containing alkyne derivatives to give thiophenes has also been reported. Thus, (
Z)-1-en-3-ynyl(butyl)sulfanes
25 were converted into the corresponding substituted 3-halothiophenes
27 (X = Cl or Br) when treated with 2 equiv of CuX
2 in MeCN (X = Cl) or THF (X = Br) as the solvent (
Scheme 10) [
66]. The process is believed to proceed through CuX
2-promoted 5-
endo-
dig S-cyclization, to give the sulfonium salt
26, followed by reductive elimination with simultaneous nucleophilic attack by the X
− anion to the butyl group bonded to the sulfur atom of
26 (
Scheme 10) [
66].
Scheme 10.
Synthesis of thiophenes
27 from (
Z)-1-en-3-ynyl(butyl)sulfanes
25 by CuX
2-promoted 5-
endo-
dig S-cyclization, to give the sulfonium salt
26, followed by elimination of BuX and Cu(0) [
66].
Scheme 10.
Synthesis of thiophenes
27 from (
Z)-1-en-3-ynyl(butyl)sulfanes
25 by CuX
2-promoted 5-
endo-
dig S-cyclization, to give the sulfonium salt
26, followed by elimination of BuX and Cu(0) [
66].
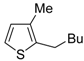
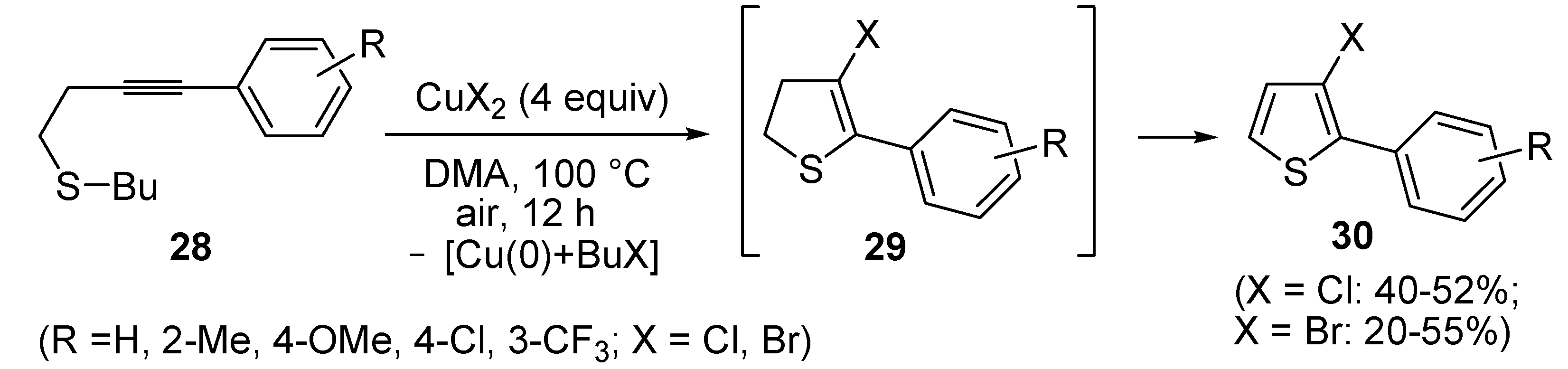
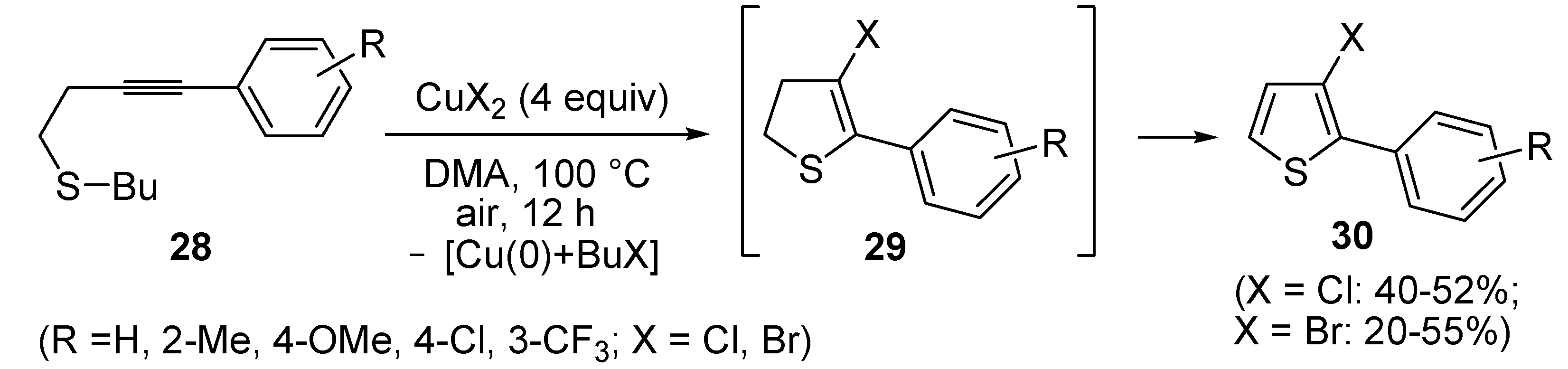
In a strictly related process, 2-aryl-3-halothiophenes
30 were obtained in moderate yields from but-3-ynyl(butyl)sulfanes
28, when working in the presence of 4 equiv of CuX
2 (X = Cl, Br) in DMA under air at 100 °C for 12 h, through the intermediate formation of dihydrothiophenes
29 (
Scheme 11) [
67].
Scheme 11.
Synthesis of 2-aryl-3-halothiophenes
30 from but-3-ynyl(butyl)sulfanes
28 by CuX
2-mediated 5-
endo-
dig S-cyclization, elimination of BuX and Cu(0), and
in situ oxidation of dihydrothiophene intermediates
29 [
67].
Scheme 11.
Synthesis of 2-aryl-3-halothiophenes
30 from but-3-ynyl(butyl)sulfanes
28 by CuX
2-mediated 5-
endo-
dig S-cyclization, elimination of BuX and Cu(0), and
in situ oxidation of dihydrothiophene intermediates
29 [
67].
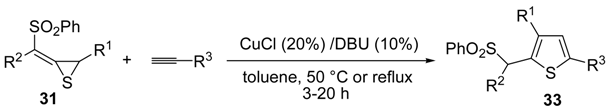
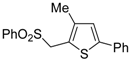
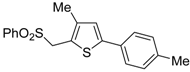
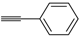
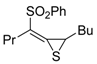
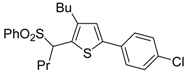
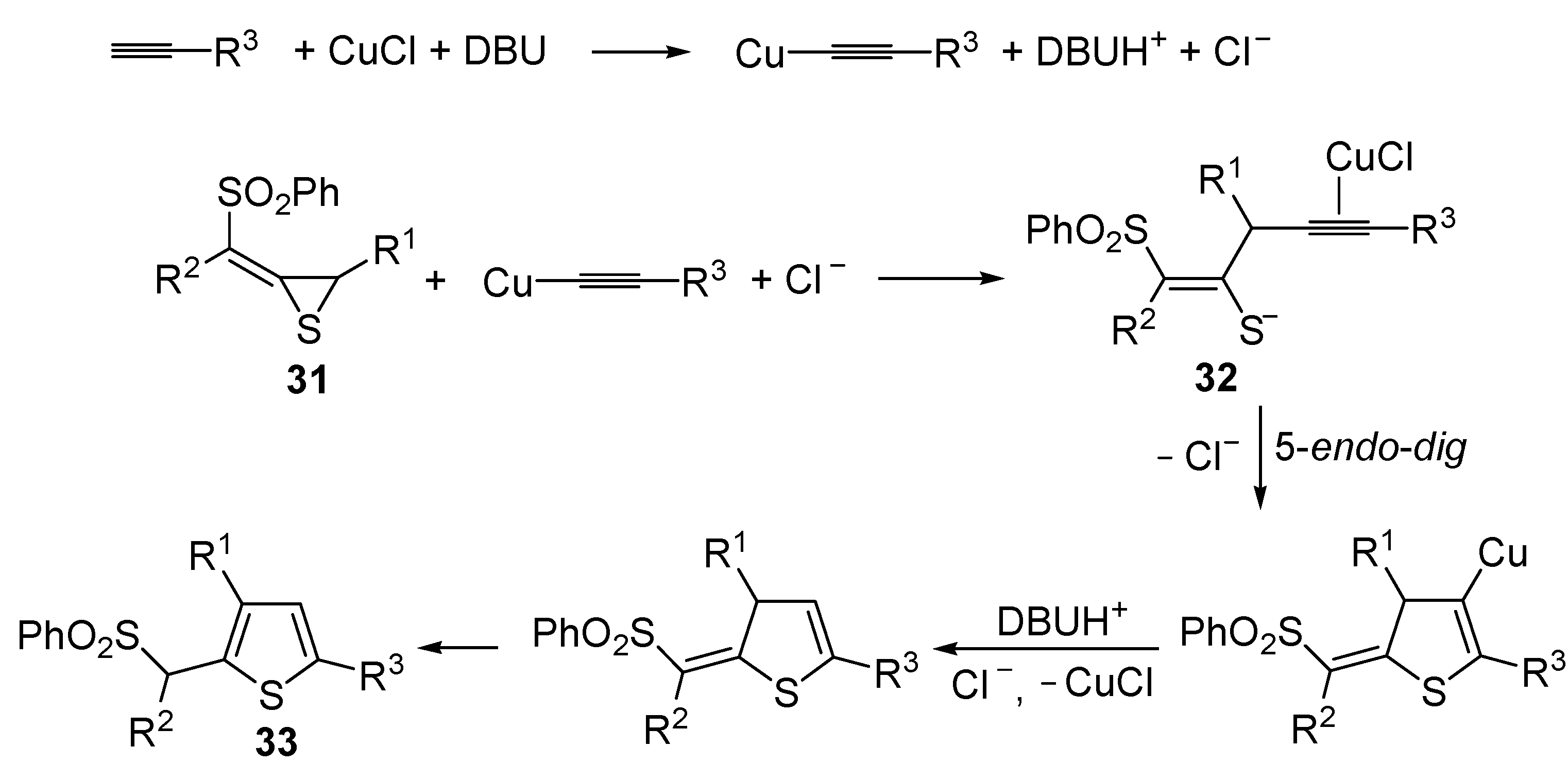
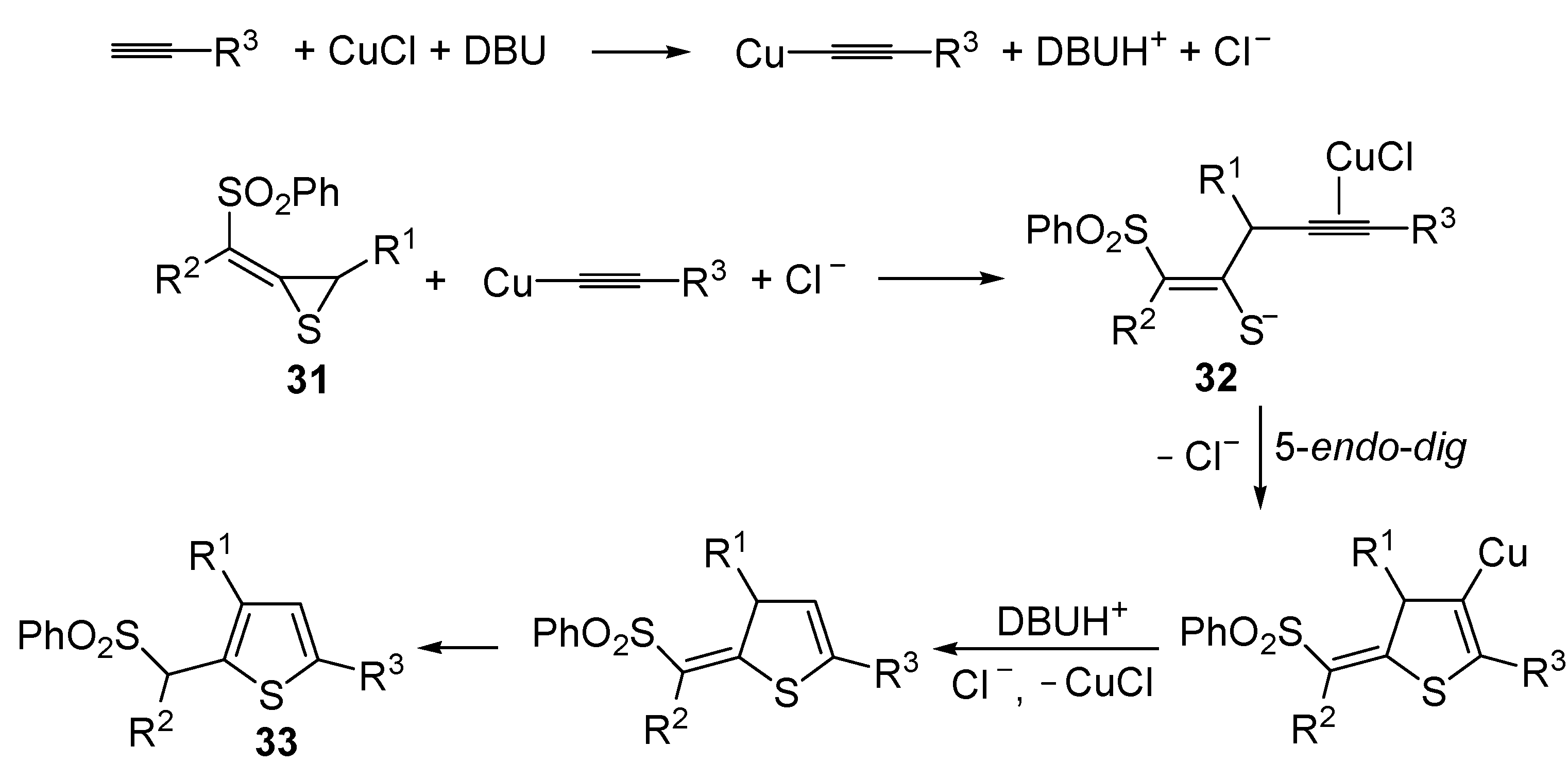
The Cu(I)-catalyzed tandem addition of terminal alkynes to 1-phenylsulfonylalkylidenethiiranes
31/cycloisomerization has allowed a convenient synthesis of functionalized thiophenes
33 (
Table 4) [
68].
Table 4.
CuCl/DBU-catalyzed tandem addition/cycloisomerization of 1-phenylsulfonylalkylidenethiiranes
31 with terminal alkynes leading to thiophenes
33 a [
68].
![Molecules 19 15687 i042]()
Reactions were carried out in toluene at 50 °C or under reflux with a molar ratio of
31: alkyne:CuCl of 1:1.5:0.2, in the presence of DBU (10 mol % with respect to
31). The reaction is believed to proceed through base-promoted formation of an alkynylcopper intermediate, whose regiospecific attack to the C-2 of the thiirane ring affords (
Z)-1-phenylsulfonyl-1-en-4-yne-2-thiolate intermediate
32. 5-
Endo-
dig cyclization of the latter, ensuing from intramolecular nucleophilic attack of the thiolate group to the triple bond coordinated to CuCl, followed by protonolysis and isomerization, eventually leads to the final thiophene derivative
33 (
Scheme 12) [
68].
Scheme 12.
Formation of thiophenes
33 by CuCl/DBU-catalyzed tandem addition/cycloisomerization of 1-phenylsulfonylalkylidenethiiranes
31 with terminal alkynes through the intermediate formation of (
Z)-1-phenylsulfonyl-1-en-4-yne-2-thiolate
32 [
68].
Scheme 12.
Formation of thiophenes
33 by CuCl/DBU-catalyzed tandem addition/cycloisomerization of 1-phenylsulfonylalkylidenethiiranes
31 with terminal alkynes through the intermediate formation of (
Z)-1-phenylsulfonyl-1-en-4-yne-2-thiolate
32 [
68].
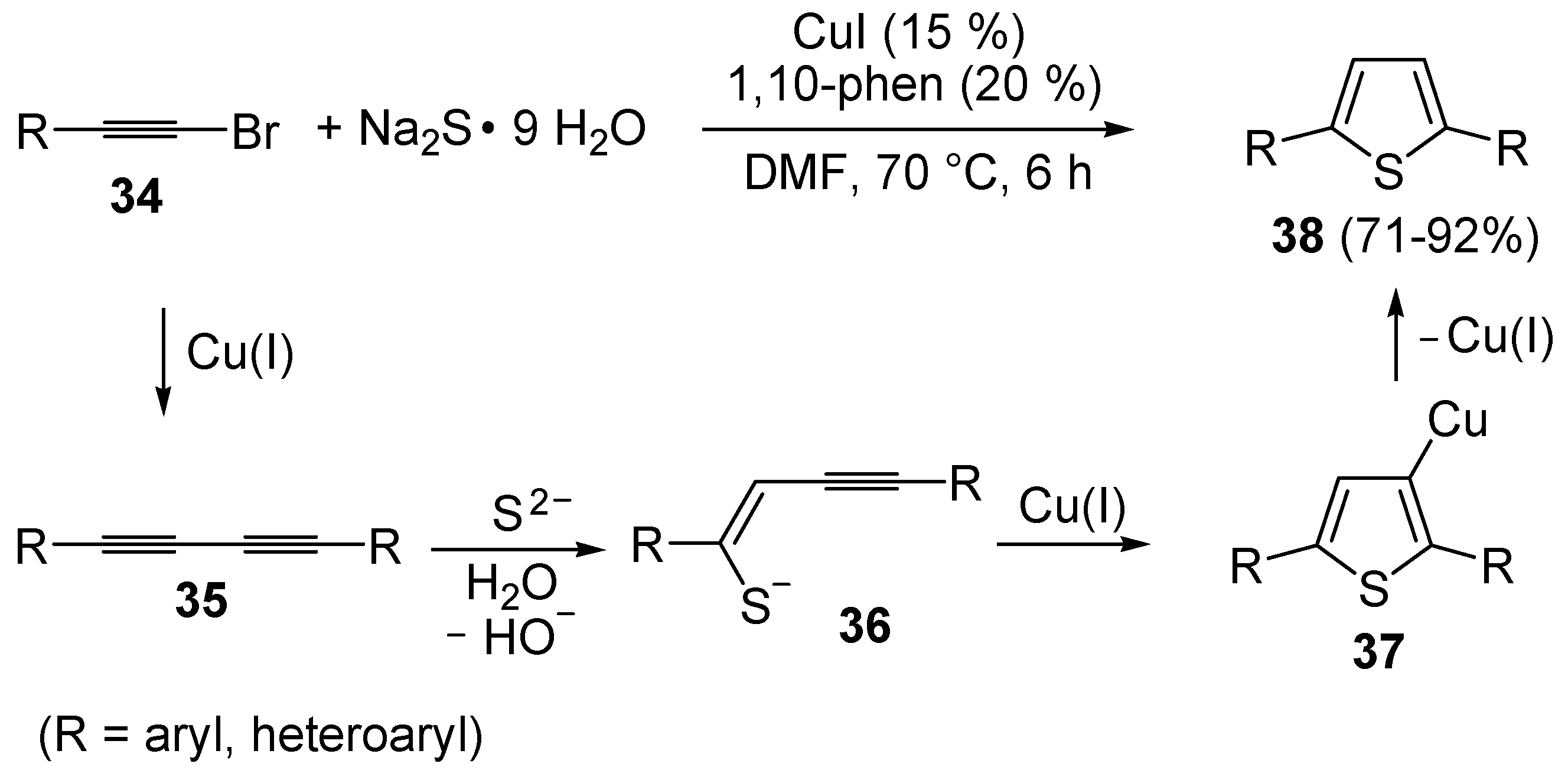
An interesting approach to 2,5-disubstituted thiophenes
38, starting from 1-bromoalkynes
34 and Na
2S (5 equiv) in the presence of CuI (15 mol %) and 1,10-phenanthroline (20 mol %), in DMF at 70 °C, has been recently developed (
Scheme 13) [
69]. The proposed mechanism starts with the Cu(I)-catalyzed formation of 1,3-diynes
35, followed by sulfide attack to the triple bond to give enynethiolate intermediate
36. Cu-promoted 5-
endo-
dig cyclization of the latter, ensuing from intramolecular attack by the sulfur atom to the triple bond coordinated to CuI, leads to 3-thienylcopper complex
37, from which the final product is formed by protonolysis (
Scheme 13) [
69].
Scheme 13.
Synthesis of thiophenes
38 from 1-bromoalkynes
34 and Na
2S by CuI-induced 5-
endo-
dig S-cyclization of enynethiolate intermediate
36, formed by sulfide addition to 1,3-diynes
35, deriving in their turn from CuI-catalyzed homocoupling of
34 [
69].
Scheme 13.
Synthesis of thiophenes
38 from 1-bromoalkynes
34 and Na
2S by CuI-induced 5-
endo-
dig S-cyclization of enynethiolate intermediate
36, formed by sulfide addition to 1,3-diynes
35, deriving in their turn from CuI-catalyzed homocoupling of
34 [
69].
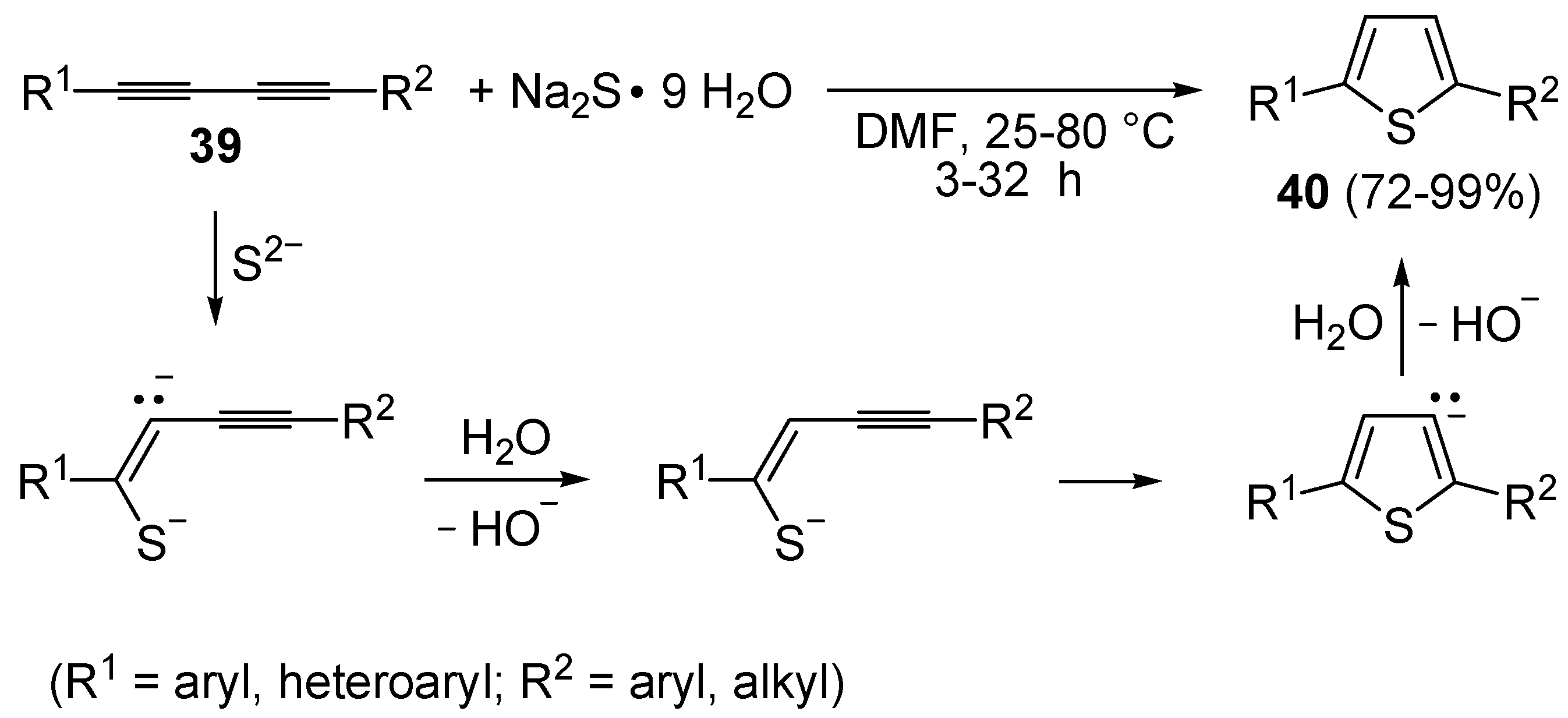
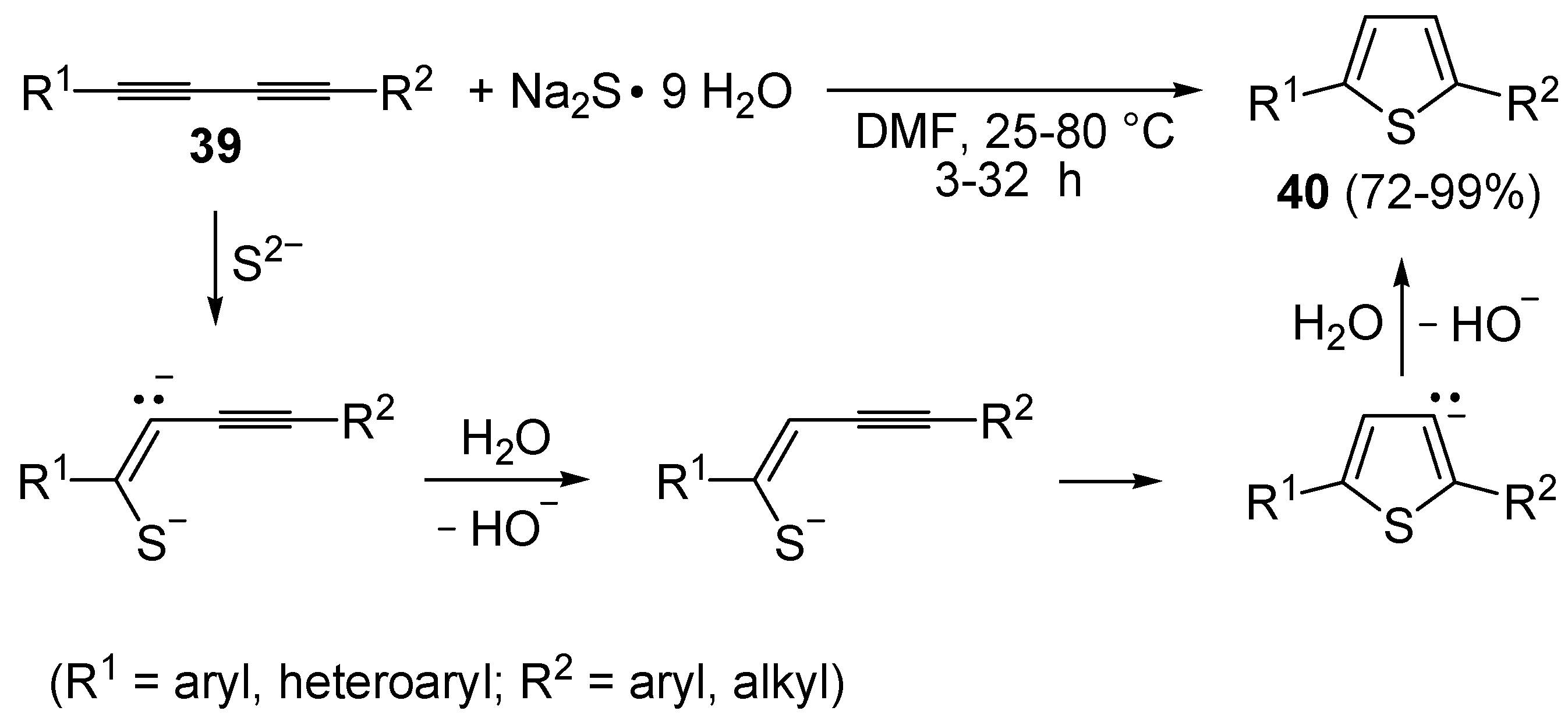
The direct, metal-free conversion of 1,3-diynes
39 to thiophenes
40, by reaction with a 3-fold excess of NaSH or Na
2S•9H
2O in DMF at 25–80 °C for 1–48 h, has also been reported, as exemplified in
Scheme 14 [
70].
Scheme 14.
Metal-free synthesis of thiophenes
40 from 1,3-diynes
39 and Na
2S [
70].
Scheme 14.
Metal-free synthesis of thiophenes
40 from 1,3-diynes
39 and Na
2S [
70].
3. Synthesis of Thiophene Derivatives by Iodocyclization of S-Containing Alkyne Substrates
The iodocyclization of suitably functionalized alkynes is a very important synthetic tool for the direct preparation of iodine-containing carbo- and heterocycles starting from readily available starting materials [
71,
72,
73,
74,
75,
76,
77,
78,
79]. The utility of the method is further demonstrated by the possibility to elaborate the final products through various cross-coupling reactions (such as Heck, Suzuki-Miyaura, and Sonogashira reactions). The process is usually carried out under mild conditions, and takes place trough intramolecular nucleophilic attack of the nucleophilic group of the substrate to the iodonium ion formed by the reaction between the triple bond and the electrophilic iodine species (indicated with I
+); both
exo and
endo cyclization modes are possible, as shown in
Scheme 15. The process is usually carried out in the presence of a base, to buffer the acid generated during the process.
Scheme 15.
Iodocyclization of alkynes bearing a suitably placed nucleophilic group leading to iodinated carbo- or heterocycles [
71,
72,
73,
74,
75,
76,
77,
78,
79].
Scheme 15.
Iodocyclization of alkynes bearing a suitably placed nucleophilic group leading to iodinated carbo- or heterocycles [
71,
72,
73,
74,
75,
76,
77,
78,
79].
Recently, several novel approaches to iodinated thiophenes have been reported, starting from readily available sulfur-containing alkynes. As an extension of the previously reported syntheses of 3-iodofuran and 3-iodopyrrole derivatives by the iodocyclization of 3-yne-1,2 diols [
80,
81,
82,
83,
84] and
N-protected 1-amino-3-yn-1-ols [
80,
85], respectively, our research group has reported a particularly facile and convenient synthesis of 3-iodothiopenes
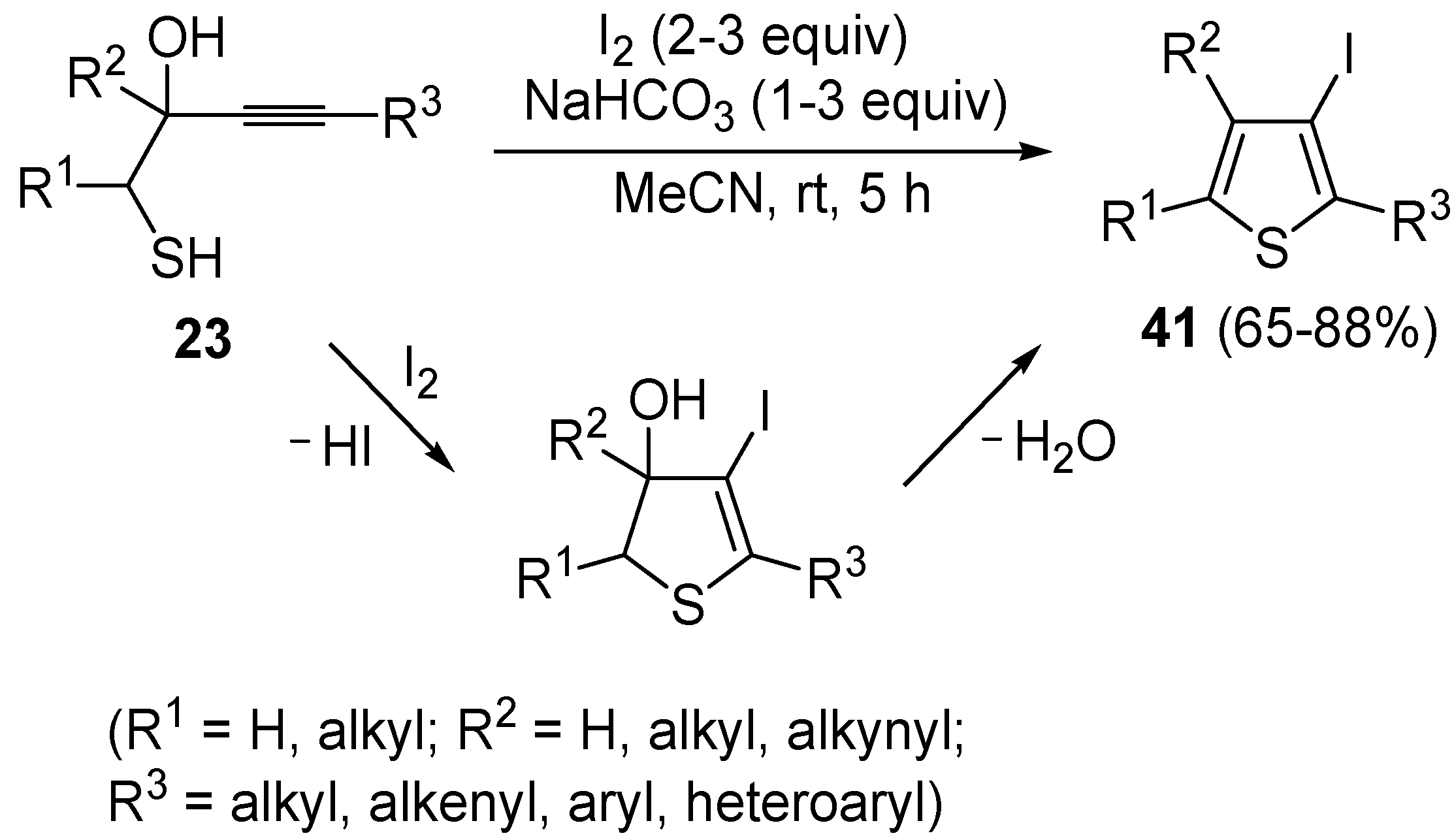
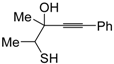
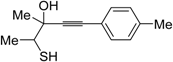
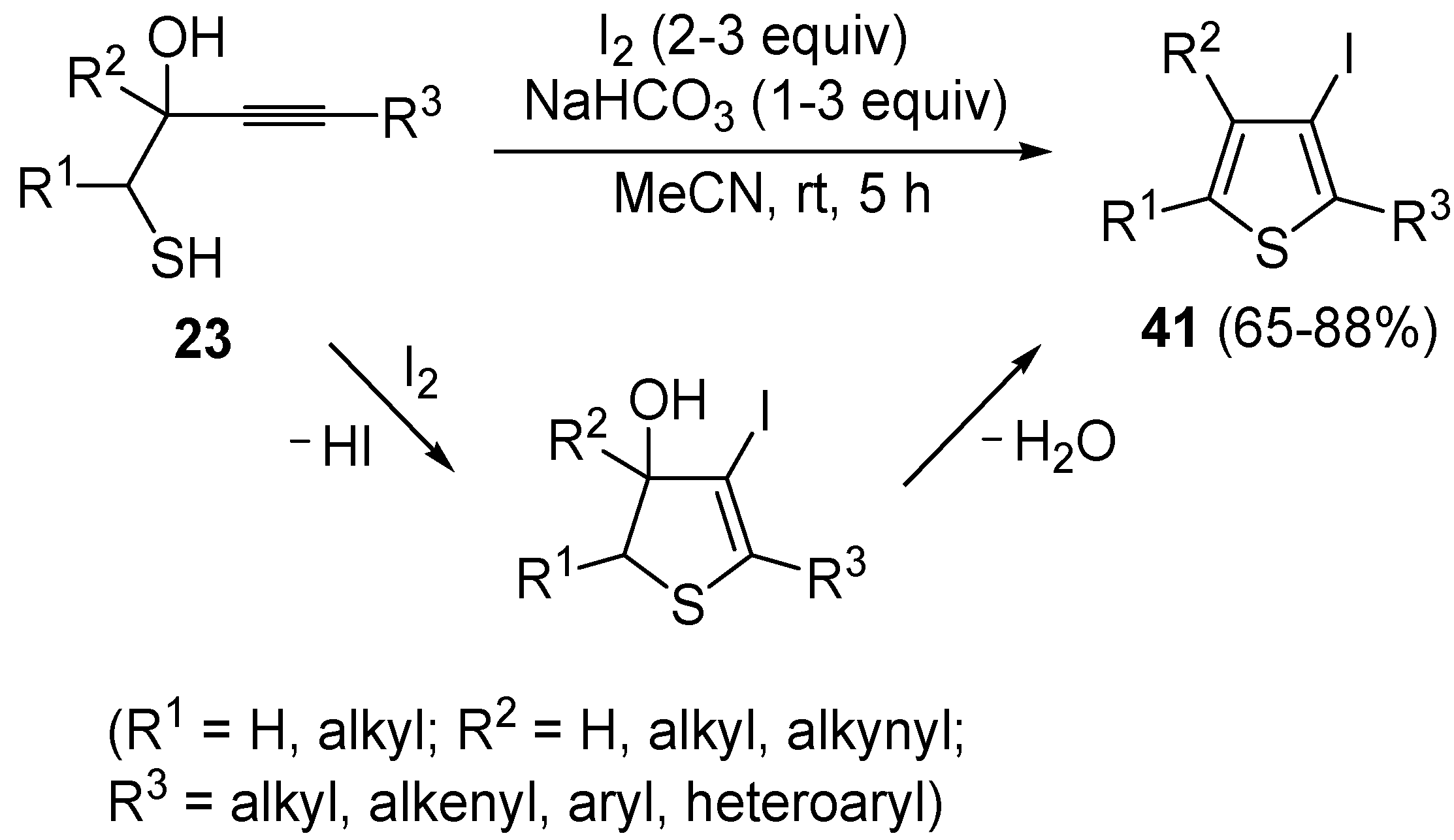
41 by dehydrative iodocyclization of 1-mercapto-3-yn-2-ols
23, according to
Scheme 16 [
86]. Reactions were carried out in MeCN at room temperature for 5 h, using molecular iodine as the electrophilic iodine species (2–3 equiv) and NaHCO
3 as the base (1–3 equiv). Iodine-induced 5-
endo-
dig cyclization was followed by dehydration with aromatization to give
41 in fair to high yields (65%–88%,
Scheme 16) [
86].
Scheme 16.
Synthesis of 3-iodothiophenes
41 by 5-
endo-
dig iodocyclization/dehydration of 1-mercapto-3-yn-2-ols
23 [
86].
Scheme 16.
Synthesis of 3-iodothiophenes
41 by 5-
endo-
dig iodocyclization/dehydration of 1-mercapto-3-yn-2-ols
23 [
86].
Interestingly, the process also took place in an ionic liquid bearing a basic anionic moiety, such as 1-ethyl-3-methylimidazolium ethylsulfate (EmimEtSO
4), as the solvent, in the absence of external bases [
87]. As reported in
Table 5, the reaction medium could be recycled several times without significantly affecting the reaction outcome. Theoretical calculations have confirmed the role of the ethylsulfate anion in the deprotonation of the thiolic group of the substrate [
87].
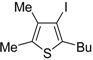
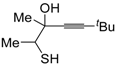
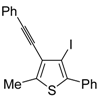
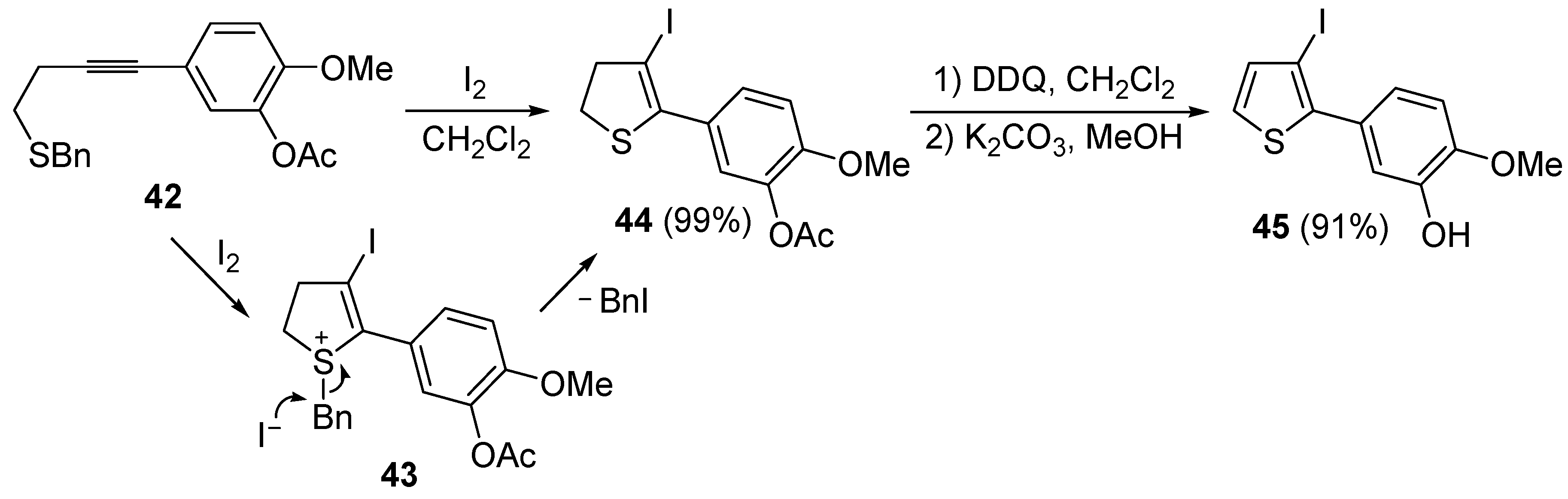
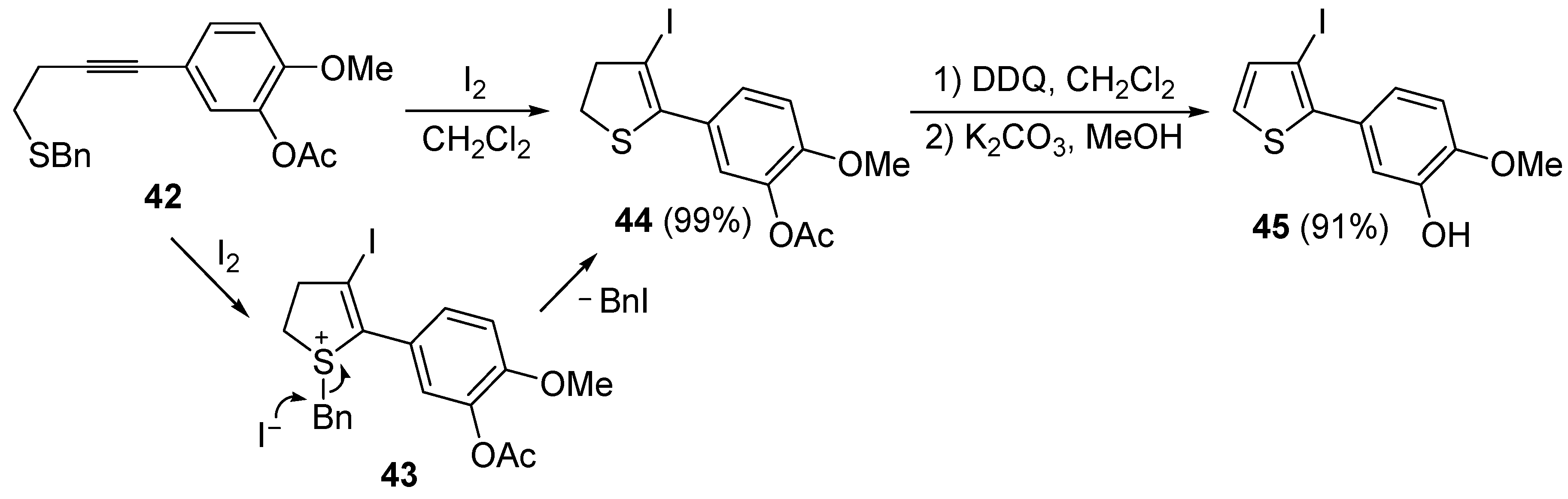
A thioether or thioester group can also act as intramolecular nucleophile in an iodocyclization process, eventually leading to a thiophene derivative. Thus, 5-(4-(benzylthio)but-1-ynyl)-2-methoxyphenyl acetate
42 was easily transformed into 5-(3-iodothiophen-2-yl)-2-methoxyphenol
45 in almost quantitative yield by a three-step procedure, involving iodocyclization (carried out with I
2 in CH
2Cl
2 at room temperature), to give dihydrothiophene
44, followed by oxidation with DDQ and deacylation with K
2CO
3 in MeOH (
Scheme 17) [
88].
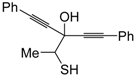
Table 5.
Recyclable and base-free synthesis of 3-iodothiophenes
41 by iodoheterocyclization of 1-mercapto-3-yn-2-ols
23 in EmimEtSO
4 a [
87].
![Molecules 19 15687 i066]()
Scheme 17.
Synthesis of 5-(3-iodothiophen-2-yl)-2-methoxyphenol
45 by 5-
endo-
dig iodocyclization of 5-(4-(benzylthio)but-1-ynyl)-2-methoxyphenyl acetate
42 to give 5-(3-iodo-4,5-dihydrothiophen-2-yl)-2-methoxyphenyl acetate
44 (through the intermediate formation of sulfonium ion salt
43) followed by oxidation and deacylation [
88].
Scheme 17.
Synthesis of 5-(3-iodothiophen-2-yl)-2-methoxyphenol
45 by 5-
endo-
dig iodocyclization of 5-(4-(benzylthio)but-1-ynyl)-2-methoxyphenyl acetate
42 to give 5-(3-iodo-4,5-dihydrothiophen-2-yl)-2-methoxyphenyl acetate
44 (through the intermediate formation of sulfonium ion salt
43) followed by oxidation and deacylation [
88].
As concerns the mechanism leading to
44, as shown in
Scheme 17, the initial iodocyclization is followed by nucleophilic attack by the iodide anion on the benzyl group of the sulfonium intermediate
43. In a similar way, (
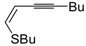
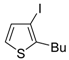
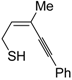
Z)-1-en-3-ynyl(butyl)sulfanes
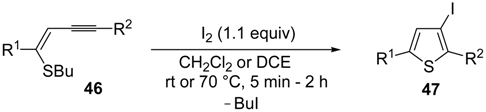
46 were smoothly converted into 3-iodothiophenes
47 when treated with 1.1 equiv of I
2 in CH
2Cl
2 or 1,2-dichloroethane (DCE) at rt or 70 °C for 5 min–2 h, as shown in
Table 6 [
89,
90].
Table 6.
Synthesis of 3-iodothiophenes
47 by iodocyclization of (
Z)-1-en-3-ynyl(butyl)sulfanes
46 a [
89,
90].
![Molecules 19 15687 i077]()
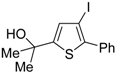
3,4-Dihalodihydrothiophenes
49 have been obtained in moderate to excellent yields (39%–98%) by the iodocyclization of
S-4-hydroxybut-2-ynyl ethanethioate
48, carried out with an excess of I
2 or IBr (2–3 equiv) in CH
2Cl
2 at rt for 1 h, according to
Scheme 18 [
91,
92]. These products could be conveniently converted into the corresponding 3,4-dihalothiophenes
50 through oxidation with DDQ and then further elaborated by the Heck or Sonogashira reactions [
91].
Scheme 18.
Synthesis of 3,4-dihalodihydrothiophenes
49 by iodocyclization of
S-4-hydroxybut-2-ynyl ethanethioate
48 and their oxidation into 3,4-dihalothiophenes
50 [
91,
92].
Scheme 18.
Synthesis of 3,4-dihalodihydrothiophenes
49 by iodocyclization of
S-4-hydroxybut-2-ynyl ethanethioate
48 and their oxidation into 3,4-dihalothiophenes
50 [
91,
92].
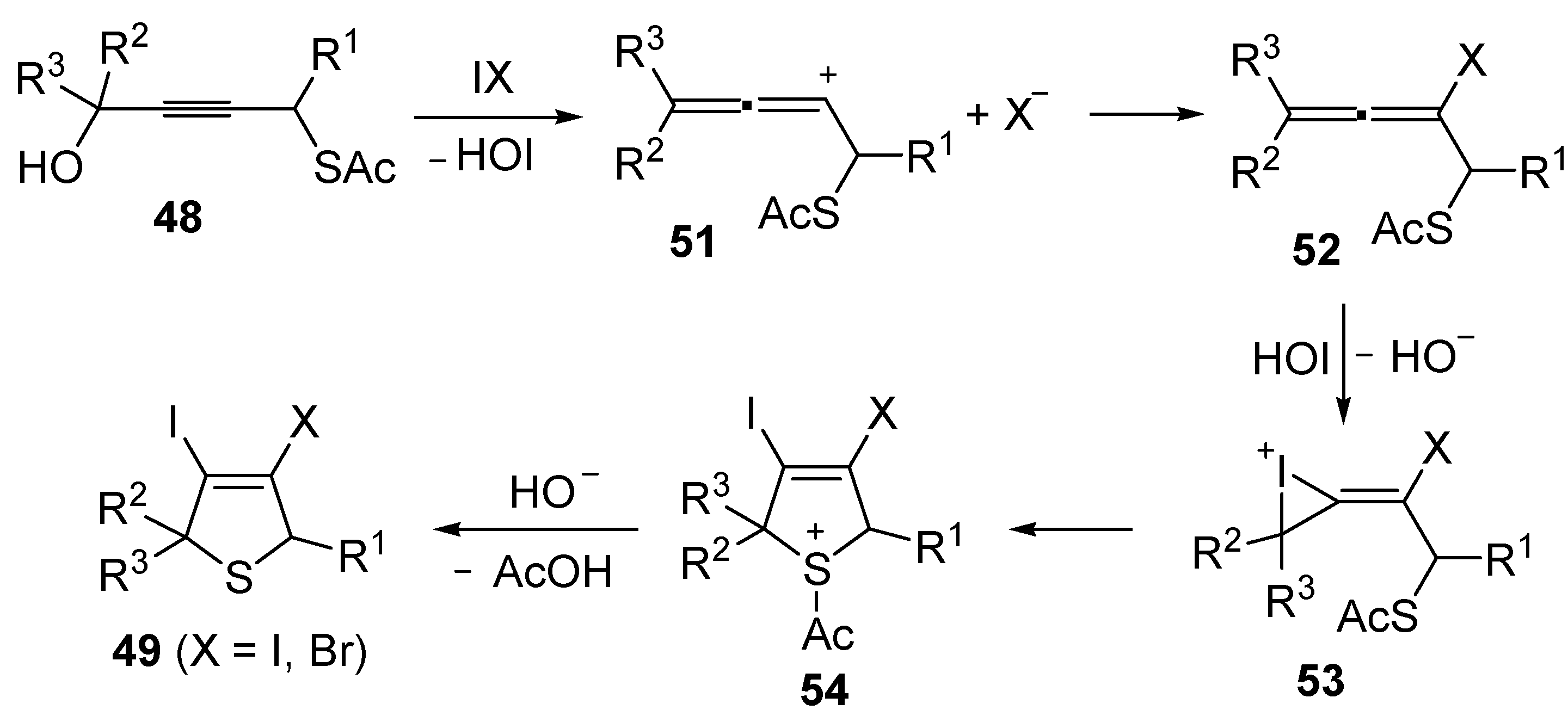
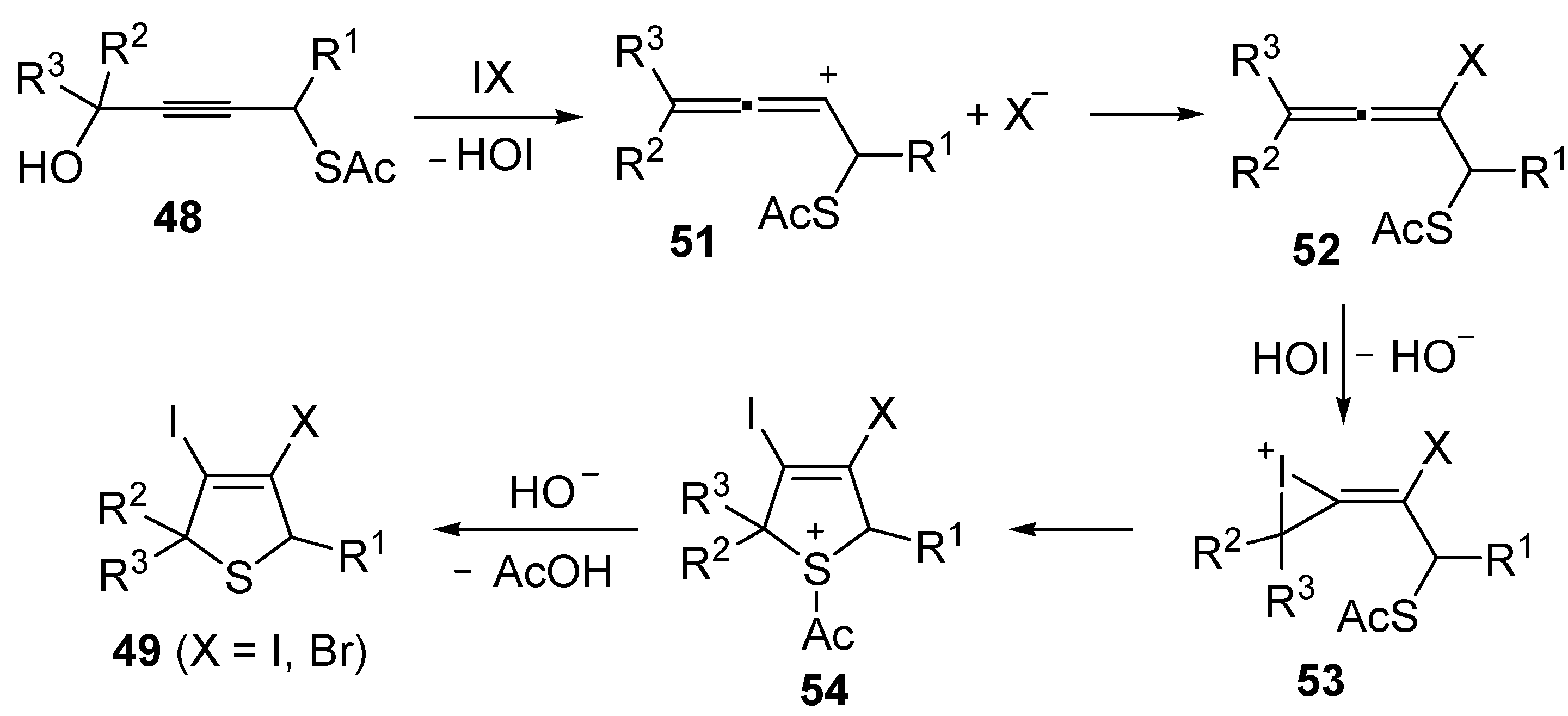
Mechanistically, the idocyclization process is believed to occur through I
2- or IBr-induced formation of an allenic carbocation
51 (with simultaneous formation of HOI), followed by iodide (or bromide) attack to give an iodoallene (or bromoallene) intermediate
52 (
Scheme 19). Reaction of the latter with HOI affords the iodonium intermediate
53, which then undergoes intramolecular nucleophilic attack by the sulfur atom to give sulfonium cation
54. Deacylation of the latter by the previously generated hydroxide anion eventually affords 3,4-dihalodihydrothiophenes
49 (
Scheme 19) [
91,
92].
Scheme 19.
Proposed mechanism for the formation of 3,4-dihalodihydrothiophenes
49 by iodocyclization of
S-4-hydroxybut-2-ynyl ethanethioate
48 [
91,
92].
Scheme 19.
Proposed mechanism for the formation of 3,4-dihalodihydrothiophenes
49 by iodocyclization of
S-4-hydroxybut-2-ynyl ethanethioate
48 [
91,
92].
Starting from
S-4-oxobut-2-ynylethanethioates
55, the formation of 3,4-diodothiophenes
56 could be obtained directly, using 3 equiv of I
2 in nitromethane at rt for 5 h (
Scheme 20) [
92].
Scheme 20.
Synthesis of 3,4-diiodothiophenes
56 by iodocyclization of
S-4-oxobut-2-ynylethanethioates
55 [
92].
Scheme 20.
Synthesis of 3,4-diiodothiophenes
56 by iodocyclization of
S-4-oxobut-2-ynylethanethioates
55 [
92].
4. Synthesis of Thiophene Derivatives by Carbocyclization of S-Containing Alkyne Substrates
Only a few methods have been reported so far in the literature for the synthesis of thiophenes through carbocyclization of
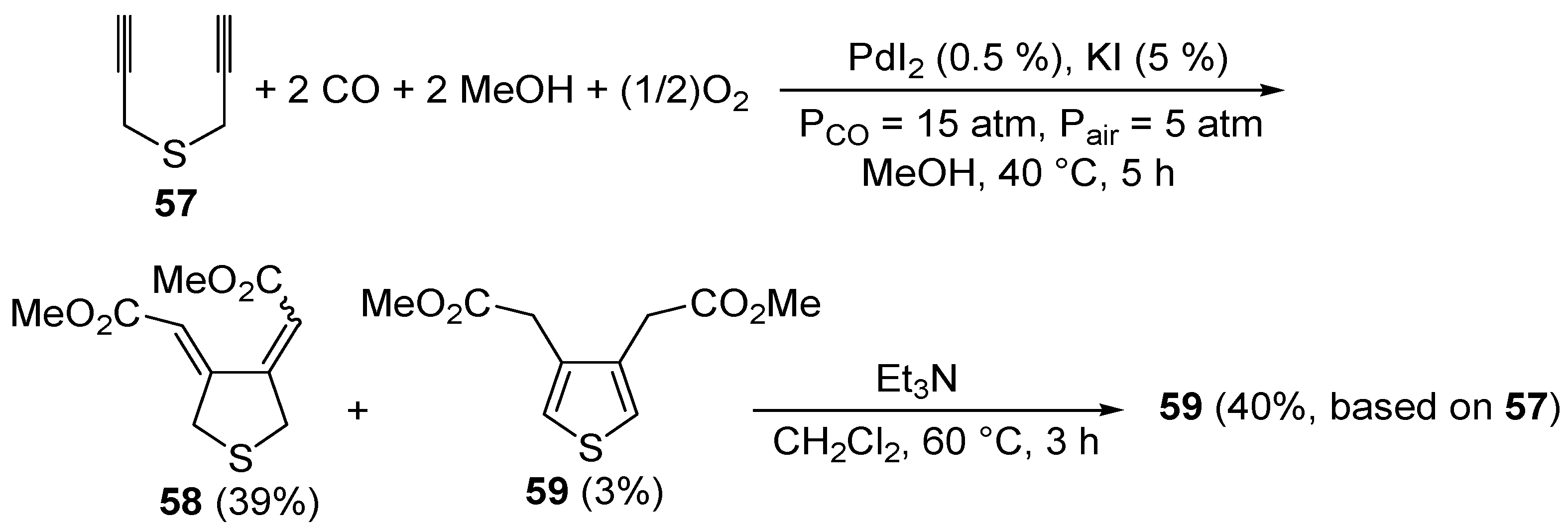
S-containing acetylenes. To the best of our knowledge, the first catalytic example of such an approach was reported by our research group in 1999 [
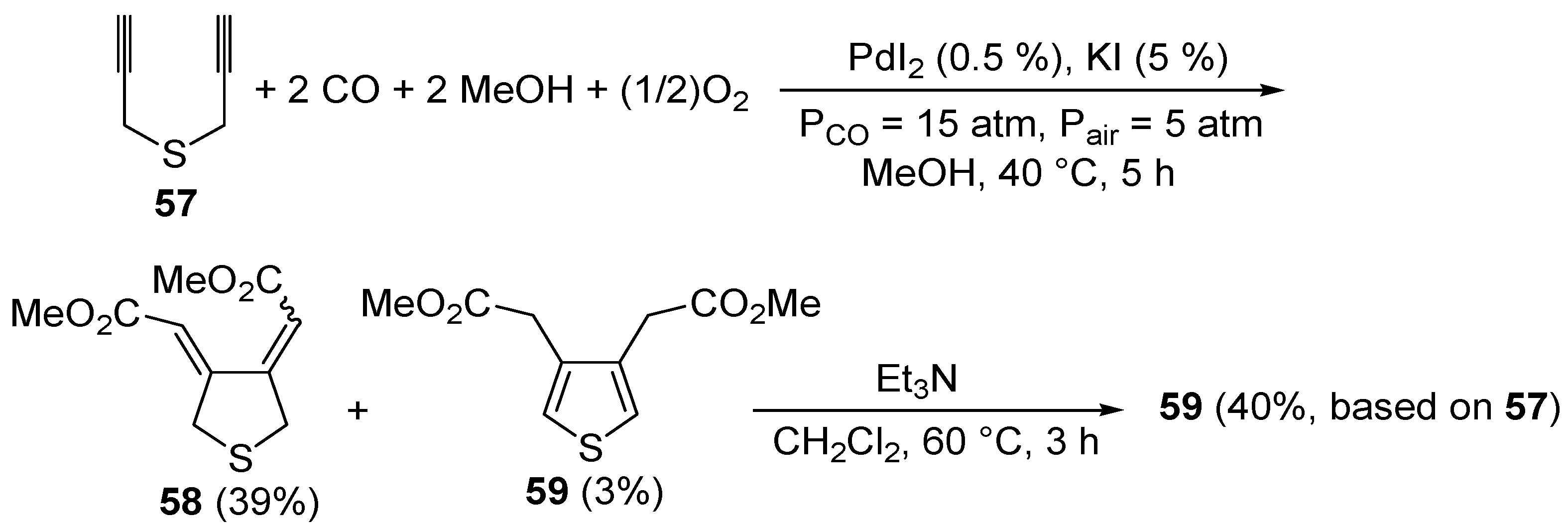
93]. It involved the PdI
2/KI-catalyzed carbonylative carbocyclization of dipropargyl sulfide (
57) to afford a mixture of 3,4-
bis(methoxycarbonylmethylene)tetrahydrothiophene (
58, 39%,
Z,
Z:
E,
E ca. 1:1) and 3,4-
bis- (methoxycarbonylmethyl)thiophene (
59, 3%), which could be treated directly, without further purification, with Et
3N in CH
2Cl
2 at 60 °C for 3 h to selectively give the novel thiophene derivatative
59 in 40% isolated yield based on starting
57 (
Scheme 21). The carbonylation reaction was carried out in MeOH as the solvent at 40 °C and under 20 atm of a 3:1 mixture of CO-air, in the presence of 0.5 mol % of PdI
2 and 5 mol % of KI for 5 h [
93].
Scheme 21.
Synthesis of 3,4-
bis(methoxycarbonylmethyl)thiophene (
59) by PdI
2-catalyzed oxidative carbonylative carbocyclization of dipropargyl sulfide
57 followed by base-promoted isomerization [
93].
Scheme 21.
Synthesis of 3,4-
bis(methoxycarbonylmethyl)thiophene (
59) by PdI
2-catalyzed oxidative carbonylative carbocyclization of dipropargyl sulfide
57 followed by base-promoted isomerization [
93].
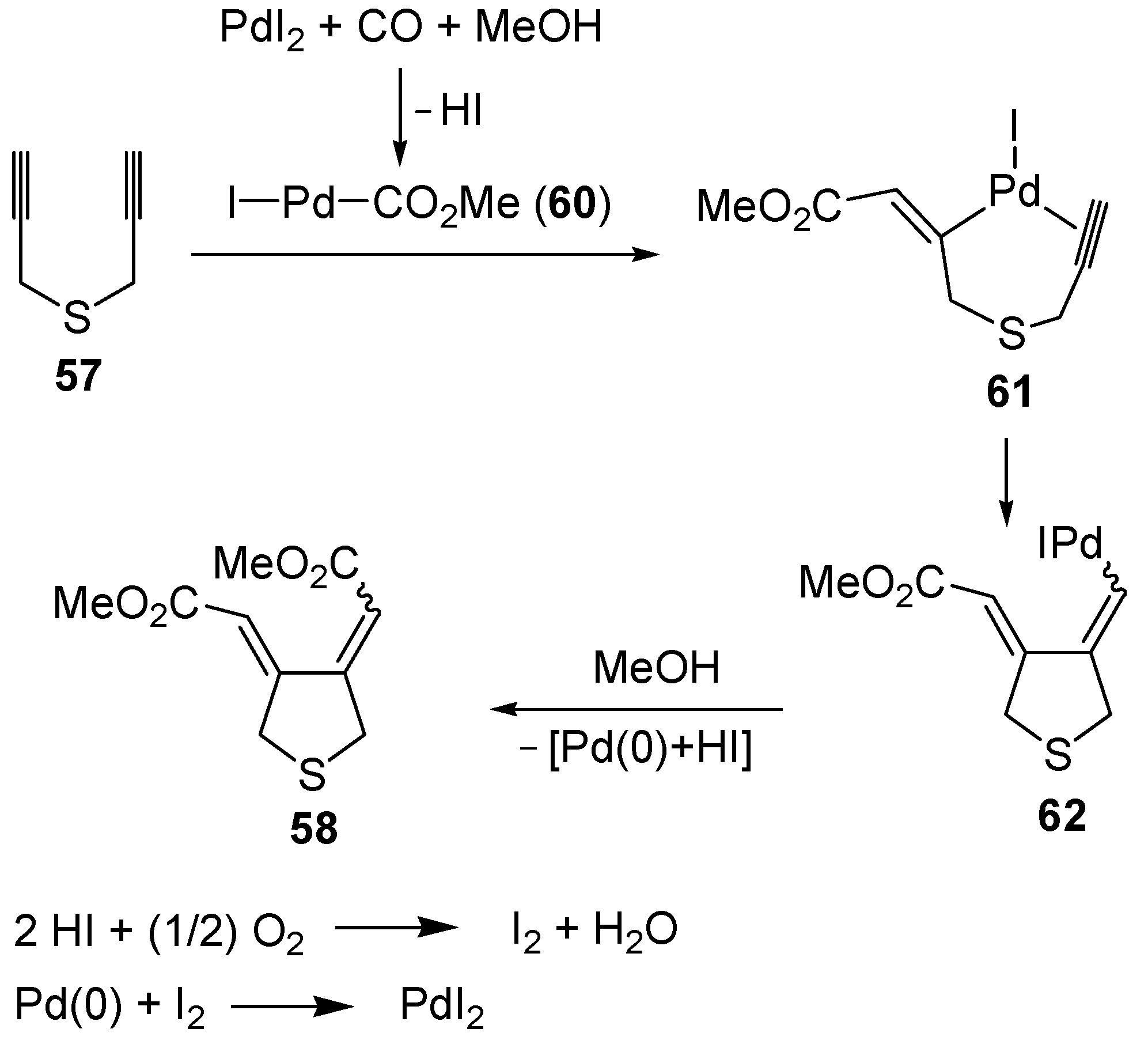
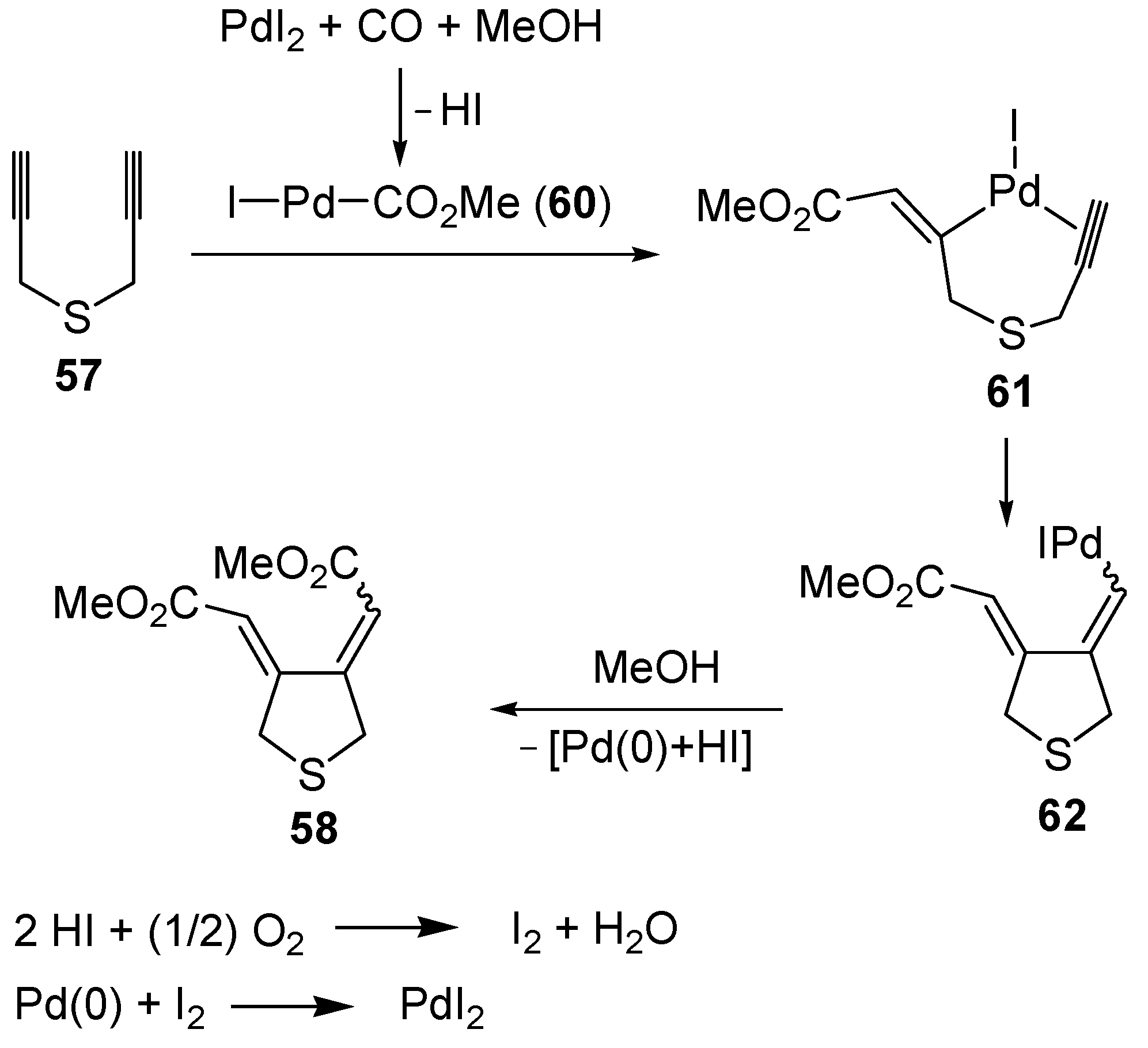
The carbonylative carbocyclization process started with the formation of a methoxycarbonylpalladium iodide intermediate
60 from the reaction between PdI
2, CO, and MeOH [
49,
94,
95,
96,
97,
98,
99,
100], followed by the insertion of the triple bond of
57 into the palladium-carbon bond to give complex
61, stabilized by the chelation from the second triple bond (
Scheme 22; anionic iodide ligands are omitted for clarity). Further insertion of the triple bond leads to the carbocyclized vinylpalladium intermediate
62, from which the final product
58 is obtained from nucleophilic displacement by MeOH. In the last step, Pd(0) was generated, which is then reoxidized back to PdI
2 according to a mechanism involving initial oxidation of 2 mol of HI (also ensuing from the carbonylation process) to give I
2, followed by oxidative addition of I
2 to Pd(0) [
49,
94,
95,
96,
97,
98,
99,
100] (
Scheme 22).
Later on, the anionic carbocyclization of some dipropargylic sulfides was studied, using
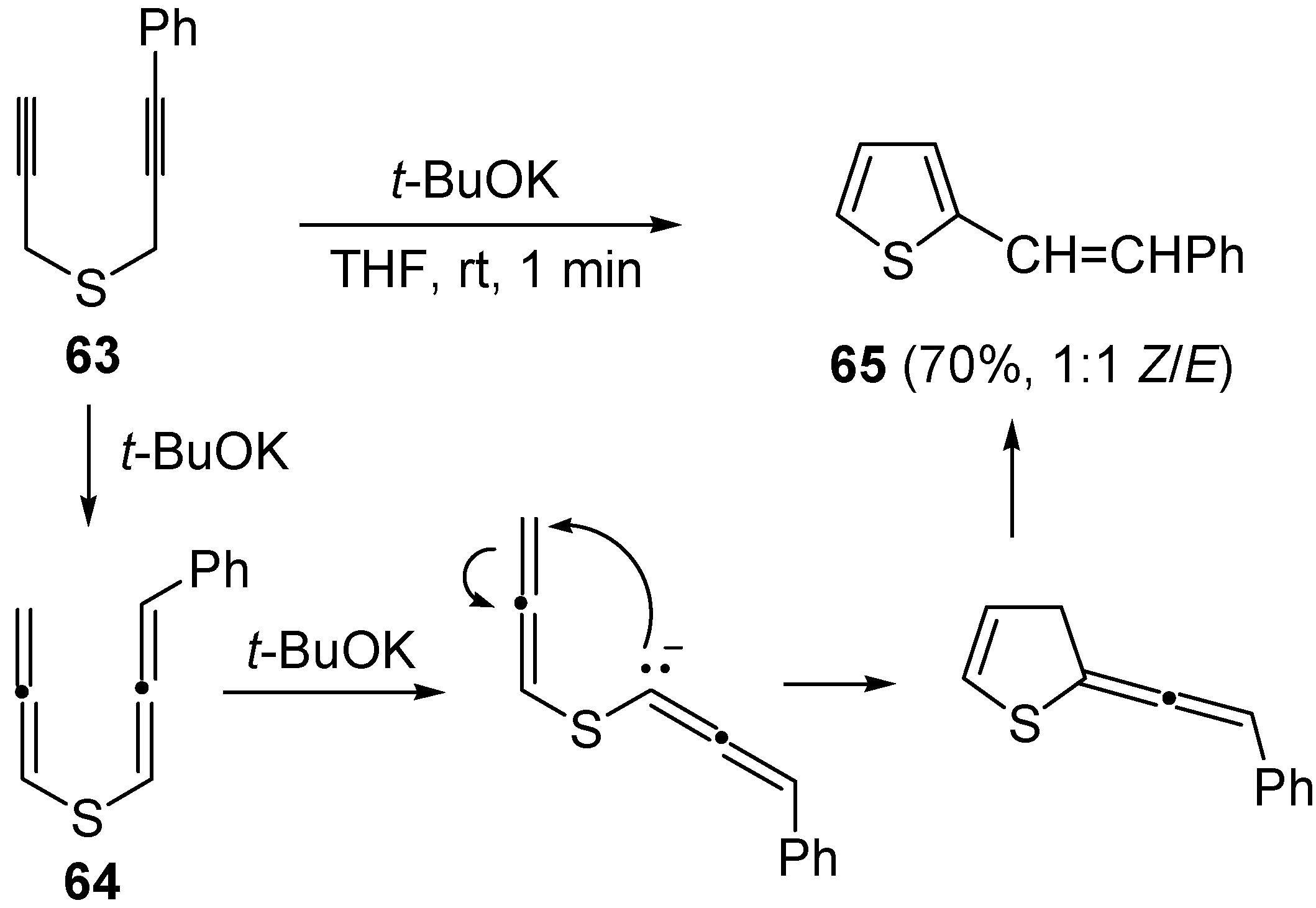
t-BuOK in THF at rt for 1 min [
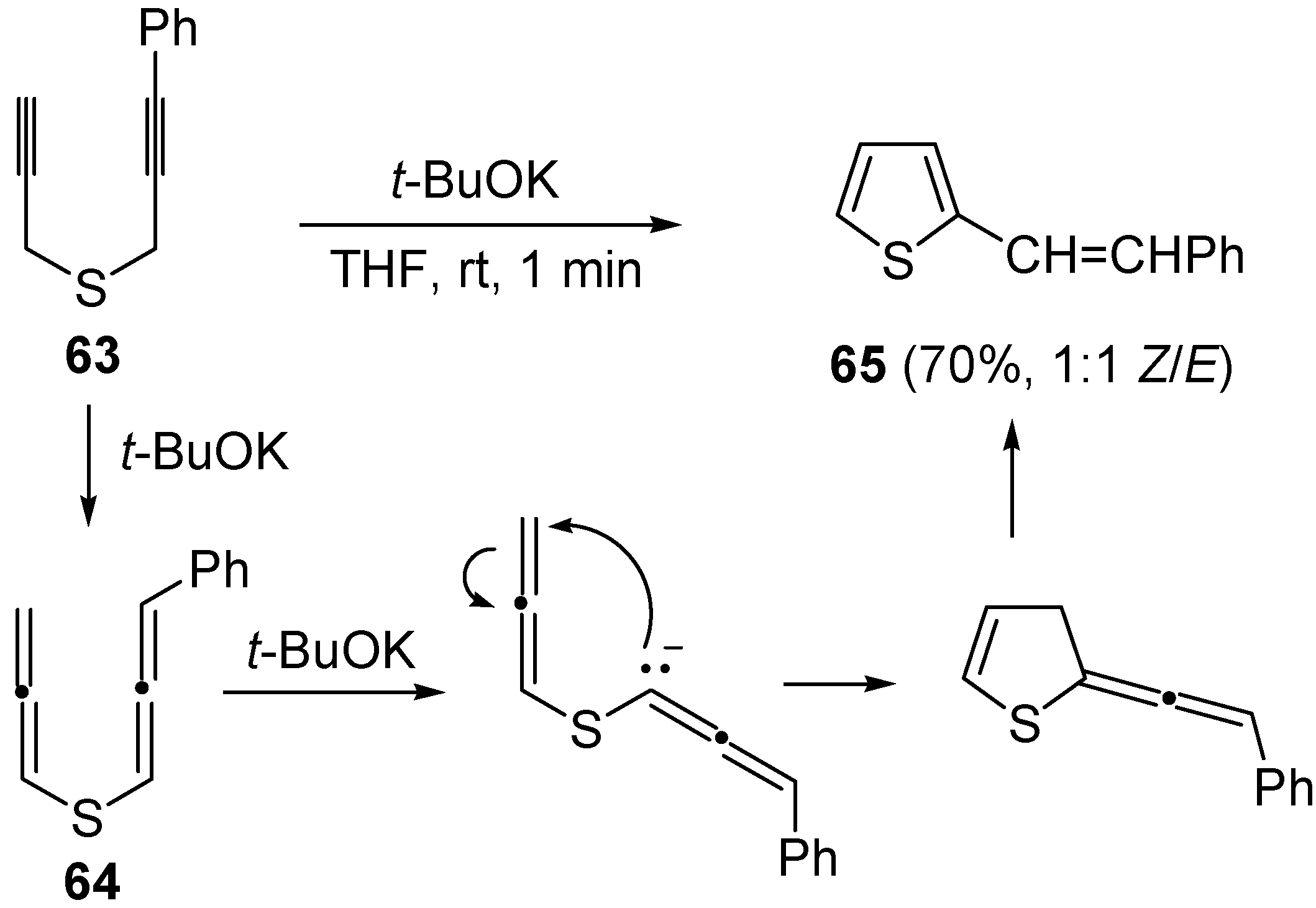
101]. The reaction of (3-phenylprop-2-ynyl)(prop-2-ynyl)sulfane (
63) led to a 1:1 diastereoisomeric mixture of 2-styrylthiophene (
65) in 70% yield, through a mechanism involving the formation of diallenyl sulfide
64 as intermediate (
Scheme 23). Similar results were obtained with (4,4-dimethylpent-2-ynyl)(3-phenylprop-2-ynyl)sulfane [
101].
Scheme 22.
Proposed mechanism for the formation of
bis(methoxycarbonylmethylene)-tetrahydrothiophene (
58) by PdI
2-catalyzed oxidative carbonylative carbocyclization of dipropargyl sulfide (
57) [
93].
Scheme 22.
Proposed mechanism for the formation of
bis(methoxycarbonylmethylene)-tetrahydrothiophene (
58) by PdI
2-catalyzed oxidative carbonylative carbocyclization of dipropargyl sulfide (
57) [
93].
Scheme 23.
Formation of 2-styrylthiophene (
65) by base-promoted carbocyclization of (3-phenylprop-2-ynyl)(prop-2-ynyl)sulfane (
65) [
101].
Scheme 23.
Formation of 2-styrylthiophene (
65) by base-promoted carbocyclization of (3-phenylprop-2-ynyl)(prop-2-ynyl)sulfane (
65) [
101].
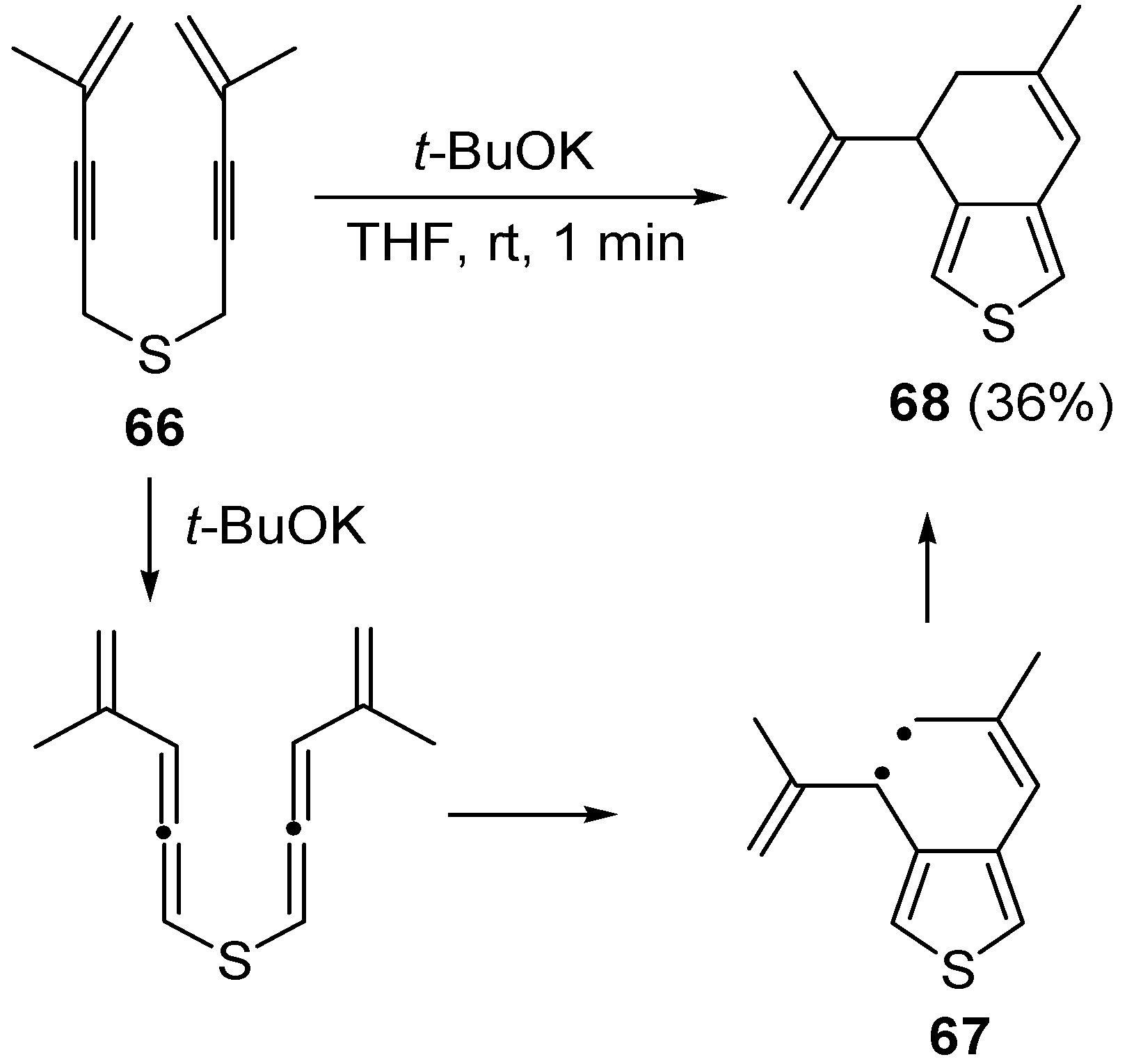
A more complicated reaction mixture was observed from the reaction of
bis(3-phenylprop-2-ynyl)sulfane, with formation of products deriving from radical cycloaromatization besides the expected vinylthiophene. A radical cycloaromatization mechanism was also at work in the case of
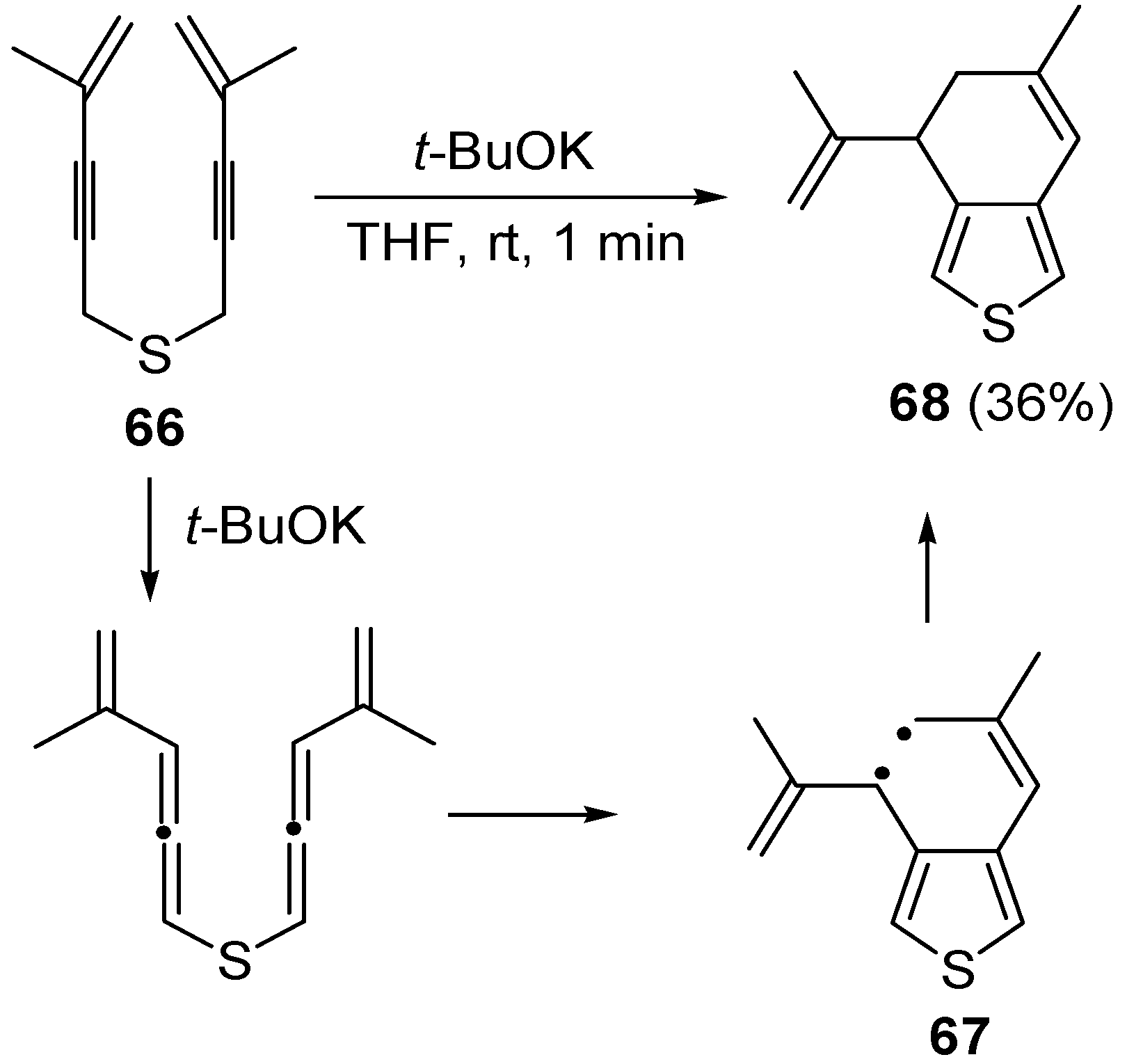
bis(4-methylpent-4-en-2-ynyl)sulfane (
66) with formation of 6-methyl-4-(prop-1-en-2-yl)-4,5-dihydrobenzo[
c]thiophene
68 in 36% yield, through the formation of the diradical intermediate
67 [
101] (
Scheme 24).
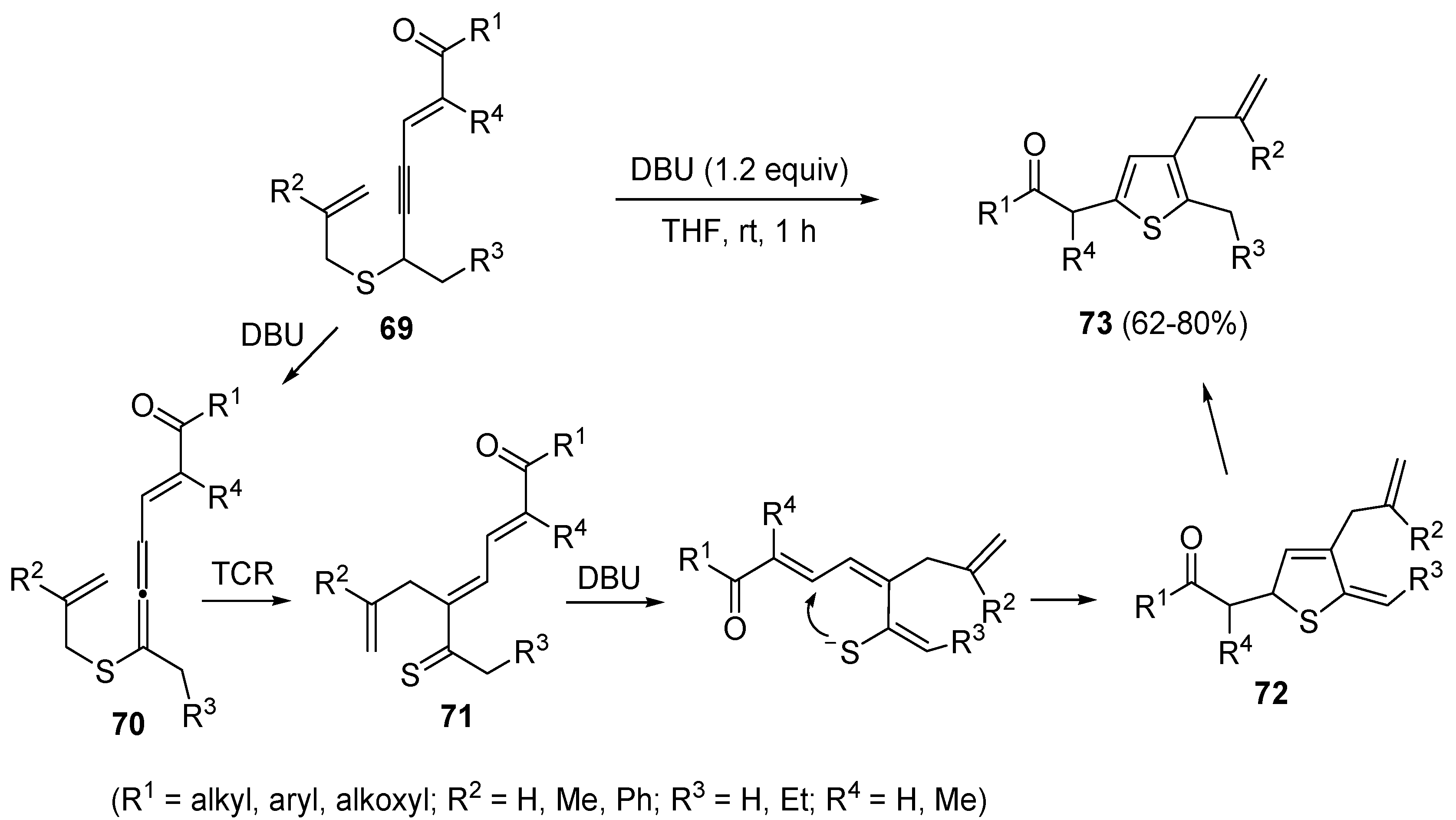
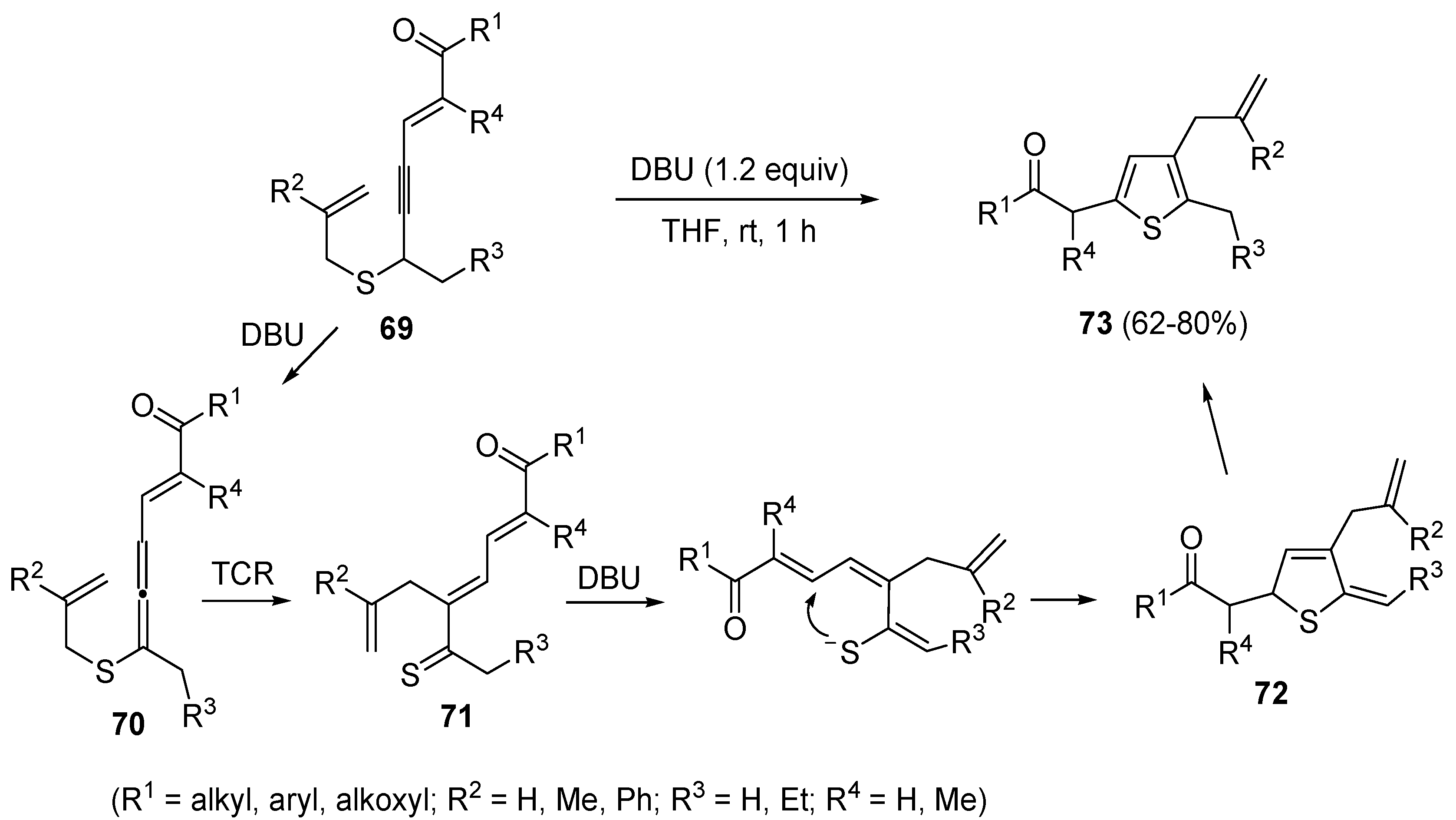
A propargyl-allenyl isomerization was also the first step in the formation of β-allyl thiophene derivatives
73 starting from functionalized allyl(4-en-2-ynyl)sulfanes
69, using DBU as the base in THF at rt [
102]. As shown in
Scheme 25, the initially formed eneallynyl intermediate
70 underwent thio-Claisen rearrangement (TCR) to give trienethione
71. Deprotonation of the latter followed by intramolecular conjugate addition afforded 5-methylene-2,5-dihydrothiophene
72, whose aromatization led to the final thiophene derivative
73 [
102].
Scheme 24.
Formation of 6-methyl-4-(prop-1-en-2-yl)-4,5-dihydrobenzo[
c]thiophene (
68) by base-promoted carbocyclization of
bis(4-methylpent-4-en-2-ynyl)sulfane (
66) [
101].
Scheme 24.
Formation of 6-methyl-4-(prop-1-en-2-yl)-4,5-dihydrobenzo[
c]thiophene (
68) by base-promoted carbocyclization of
bis(4-methylpent-4-en-2-ynyl)sulfane (
66) [
101].
Scheme 25.
Synthesis of β-allyl thiophene derivatives
73 by base-promoted isomerization/thio-Claisen rearrangement/conjugate addition/aromatization of allyl(4-en-2-ynyl)sulfanes
69 [
102].
Scheme 25.
Synthesis of β-allyl thiophene derivatives
73 by base-promoted isomerization/thio-Claisen rearrangement/conjugate addition/aromatization of allyl(4-en-2-ynyl)sulfanes
69 [
102].
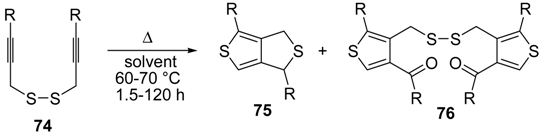
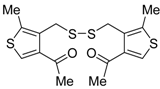
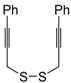
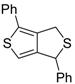
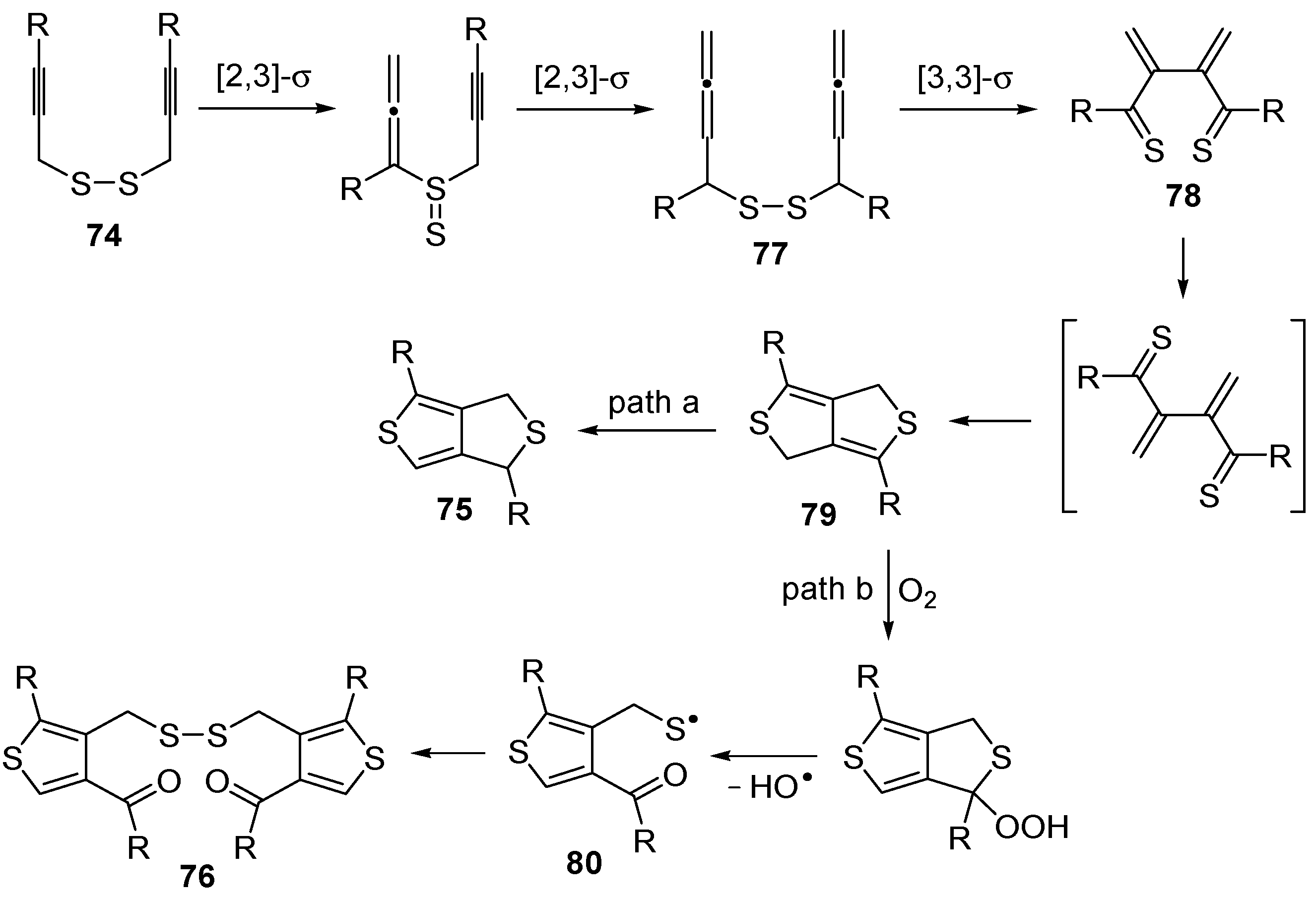
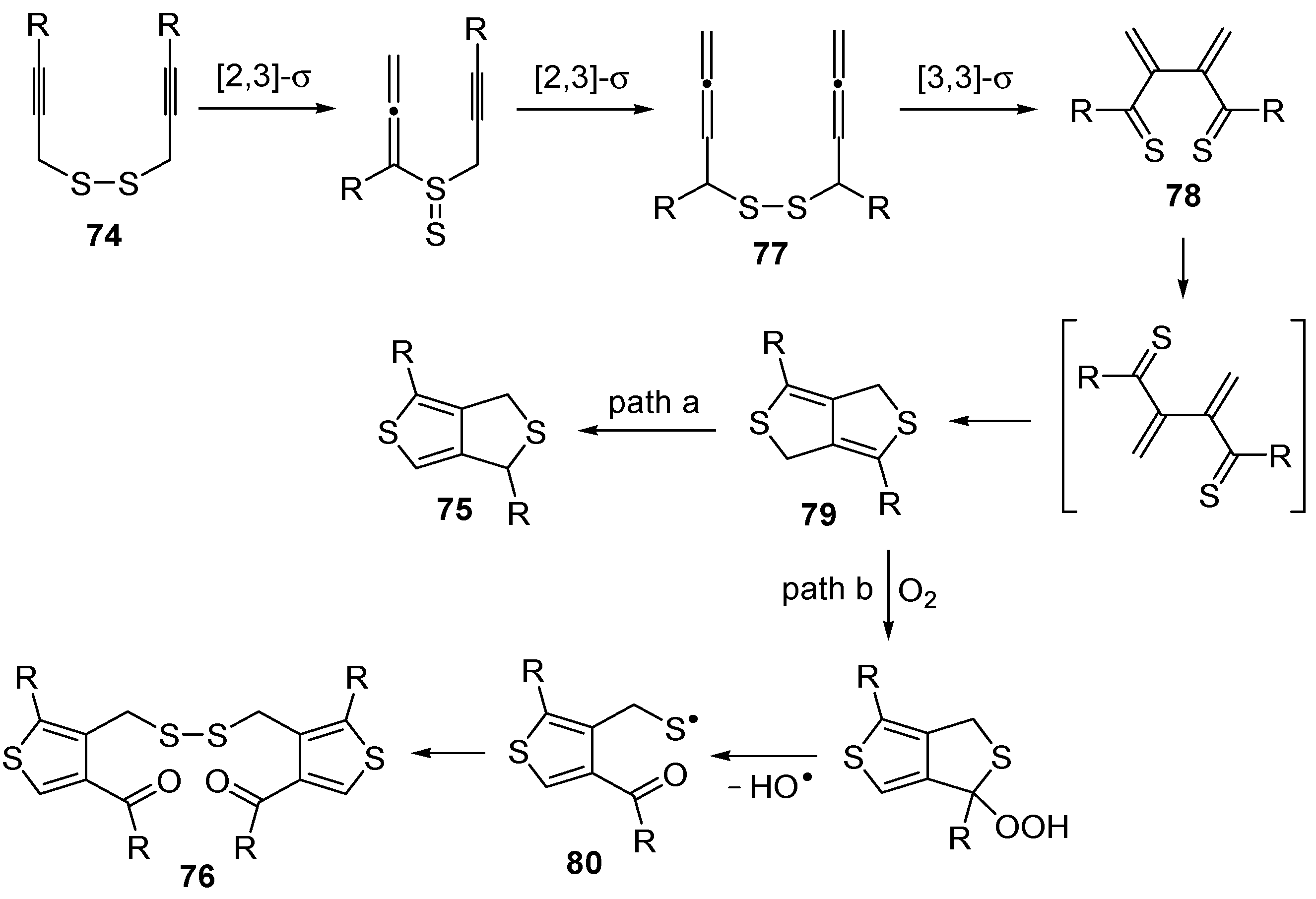
An interesting tandem thermal rearrangement/carbocyclization process of dipropargylic disulfides
74, leading to a mixture of 1,3-dihydrothieno[3,4,-
c]thiophenes
75 and thienyl disulfides
76, has been reported recently [
103]. As shown in
Table 7, the reaction takes place in CHCl
3, MeCN, or DMSO as the solvent at 60–70 °C for 1.5–160 h [
103].
Mechanistically, the reaction leading to
75 is believed to occur via an initial double [2,3]-sigmatropic rearrangement to give diallenyl disulfides
77, which may then undergo a [3,3]-sigmatropic rearrangement to give 2,3-dimethylene-1,4-dithione
78, followed by a double conjugate addition of the sulfur atoms to the double bonds of
78, to give 1,4-dihydrothieno[3,4-
c]thiophene
79, and isomerization (
Scheme 26, path a). On the other hand,
76 may be formed by dimerization of the thiyl radical intermediate
80, formed in its turn from
79 by the action of O
2 (
Scheme 26, path b). Accordingly, the formation of
76 could be minimized working in the absence of air under argon atmosphere (entry 2,
Table 8) [
103].
Table 7.
Synthesis of 1,3-dihydrothieno[3,4-
c]thiophenes
75 and thienyl disulfides
76 by tandem thermal rearrangement/carbocyclization process of dipropargylic disulfides
74 a [
103].
![Molecules 19 15687 i092]()
Scheme 26.
Proposed mechanistic pathway for the formation of 1,3-dihydrothieno[3,4-c]-thiophenes
75 and thienyl disulfides
76, by tandem thermal rearrangement/carbocyclization of dipropargylic disulfides
74 [
103].
Scheme 26.
Proposed mechanistic pathway for the formation of 1,3-dihydrothieno[3,4-c]-thiophenes
75 and thienyl disulfides
76, by tandem thermal rearrangement/carbocyclization of dipropargylic disulfides
74 [
103].
Table 8.
Multicomponent synthesis of ethyl 2-(2-(dimethylamino)thiophen-3-yl)-2-oxoacetate derivatives
84 starting from acetylenic esters
81, tetramethylthiourea
82, and ethyl bromopyruvate
83 a [
104].
![Molecules 19 15687 i102]()
5. Synthesis of Thiophene Derivatives by Miscellaneous Methods Starting from Functionalized Alkyne Substrates
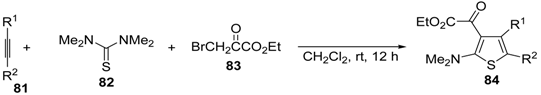
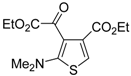
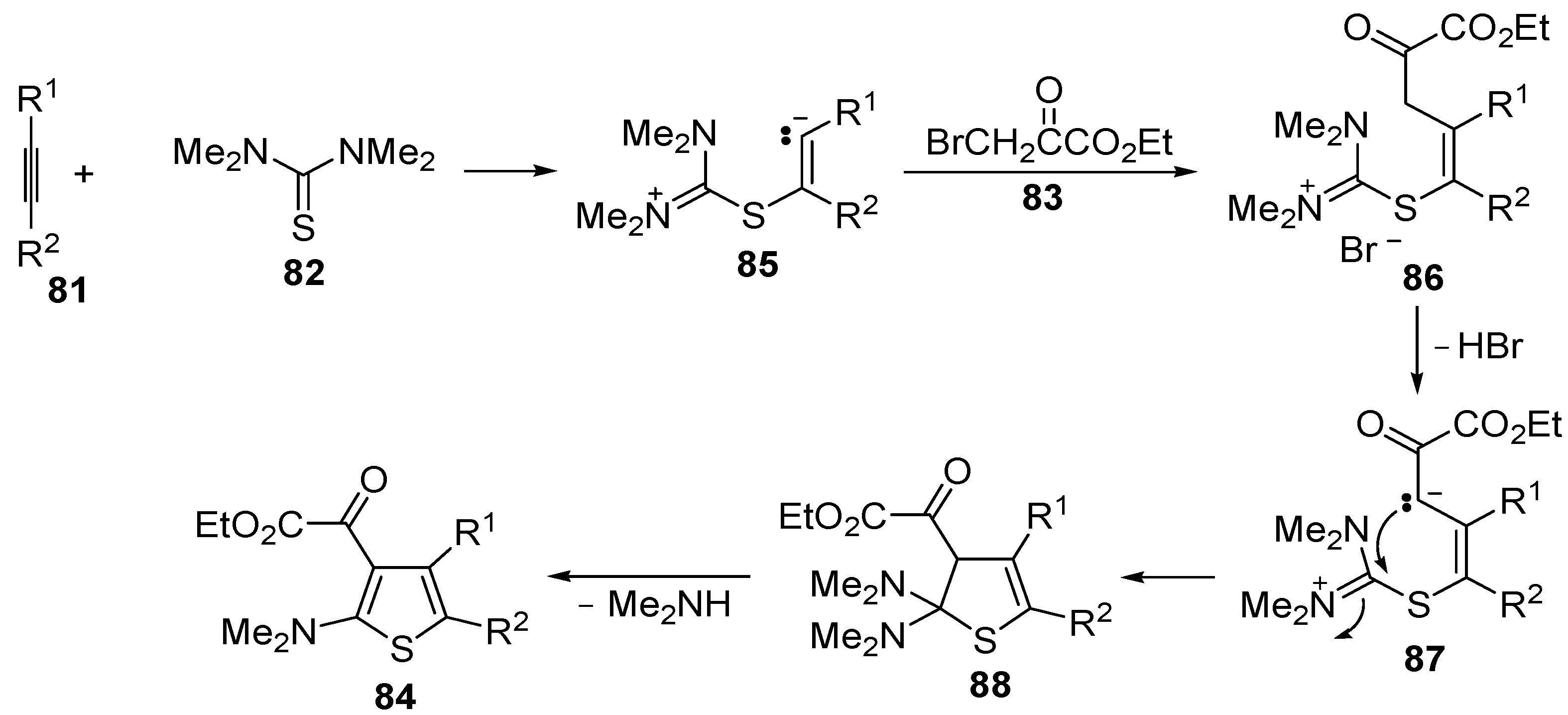
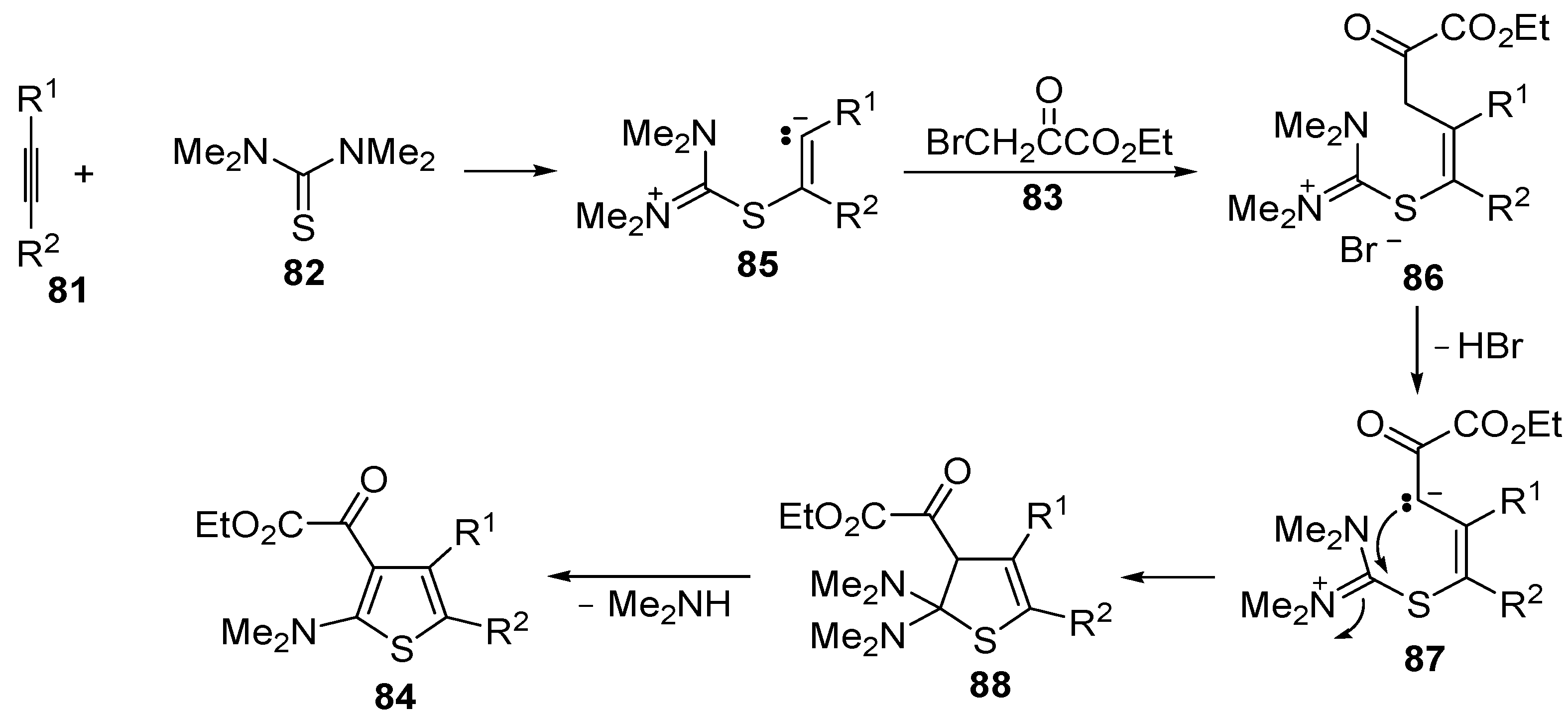
Miscellaneous methods that cannot be classified into the previous categories are reviewed here. Acetylenic esters can be useful precursors for the construction of the thiophene ring. Thus, ethyl 2-(2-(dimethylamino)thiophen-3-yl)-2-oxoacetate derivatives
84 have been conveniently synthesized through a multicomponent approach, employing acetylenic esters
81, tetramethylthiourea
82, and ethyl bromopyruvate
83 as starting materials [
104]. Reactions were carried out in CH
2Cl
2 at rt using equimolar amounts of
81,
82, and
83, to afford the corresponding thiophenes
84 in good to high yields (
Table 8) [
104].
The proposed reaction mechanism involves the formation of a 1,5-dipolar intermediate
85 from the reaction between
81 and
82, followed by nucleophilic attack by the carbanion to
83 to give the organic salt
86. The final thiophene derivative
84 is then formed from
86 by elimination of HBr to give the dipolar intermediate
87, followed by intramolecular nucleophilic attack, affording dihydrothiophene
88, and elimination of dimethylamine (
Scheme 27) [
104].
Scheme 27.
Proposed mechanism for the formation of ethyl 2-(2-(dimethylamino)thiophen-3-yl)-2-oxoacetate derivatives
84 starting from acetylenic esters
81, tetramethylthiourea
82, and ethyl bromopyruvate
83 [
104].
Scheme 27.
Proposed mechanism for the formation of ethyl 2-(2-(dimethylamino)thiophen-3-yl)-2-oxoacetate derivatives
84 starting from acetylenic esters
81, tetramethylthiourea
82, and ethyl bromopyruvate
83 [
104].
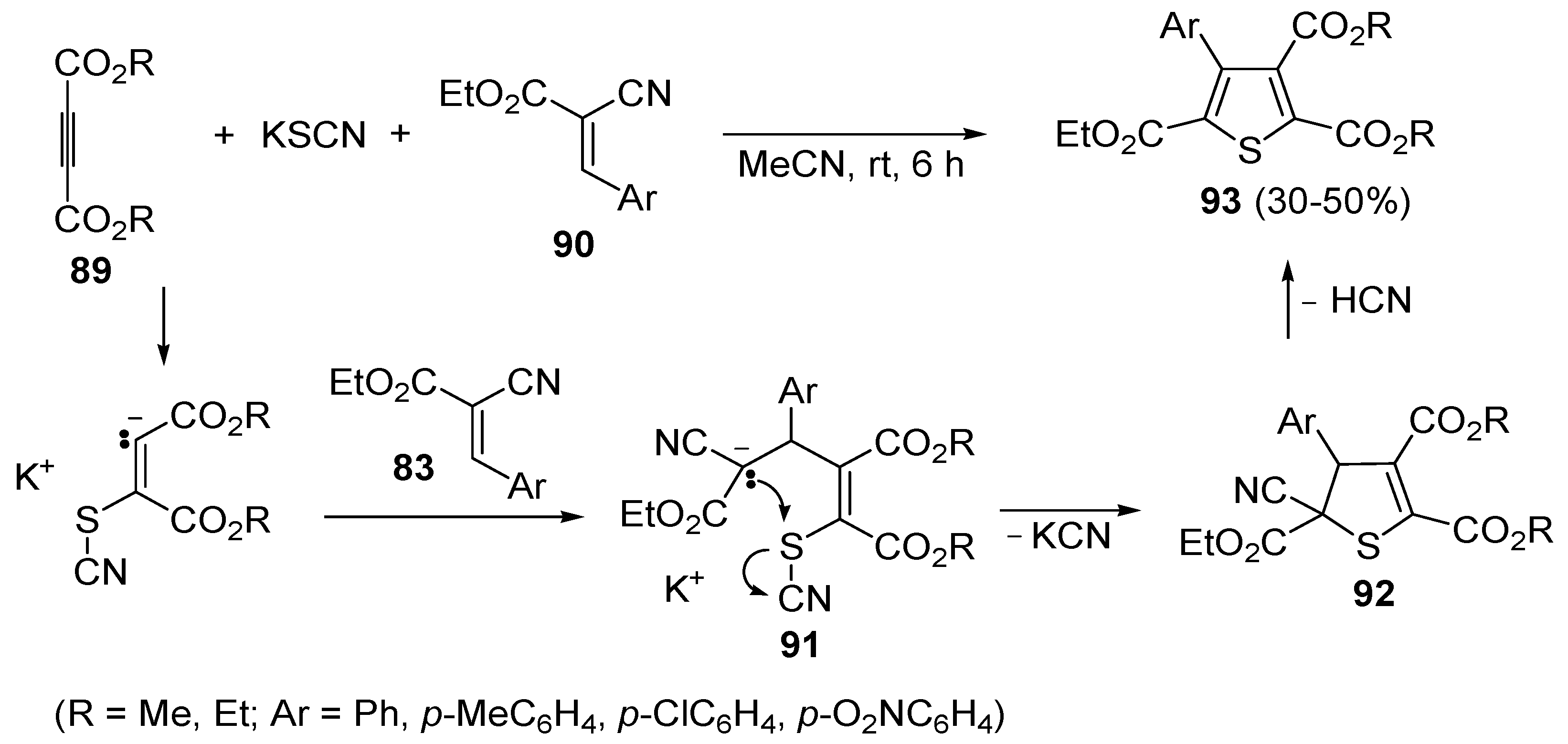
In a similar way, trialkyl 4-arylthiophene-2,3,5-tricarboxylates
93 were obtained in moderate yields (30%–50%) from the reaction between dialkyl acetylenedicarboxylates
89, KSCN, and 3-aryl-2-cyanoacrylates
90, carried out in MeCN at rt for 6 h, through the intermediate formation of the organic salt
91, which undergoes cyclization with loss of KCN (to give dihydrothiophene derivative
92) followed by elimination of HCN (
Scheme 28) [
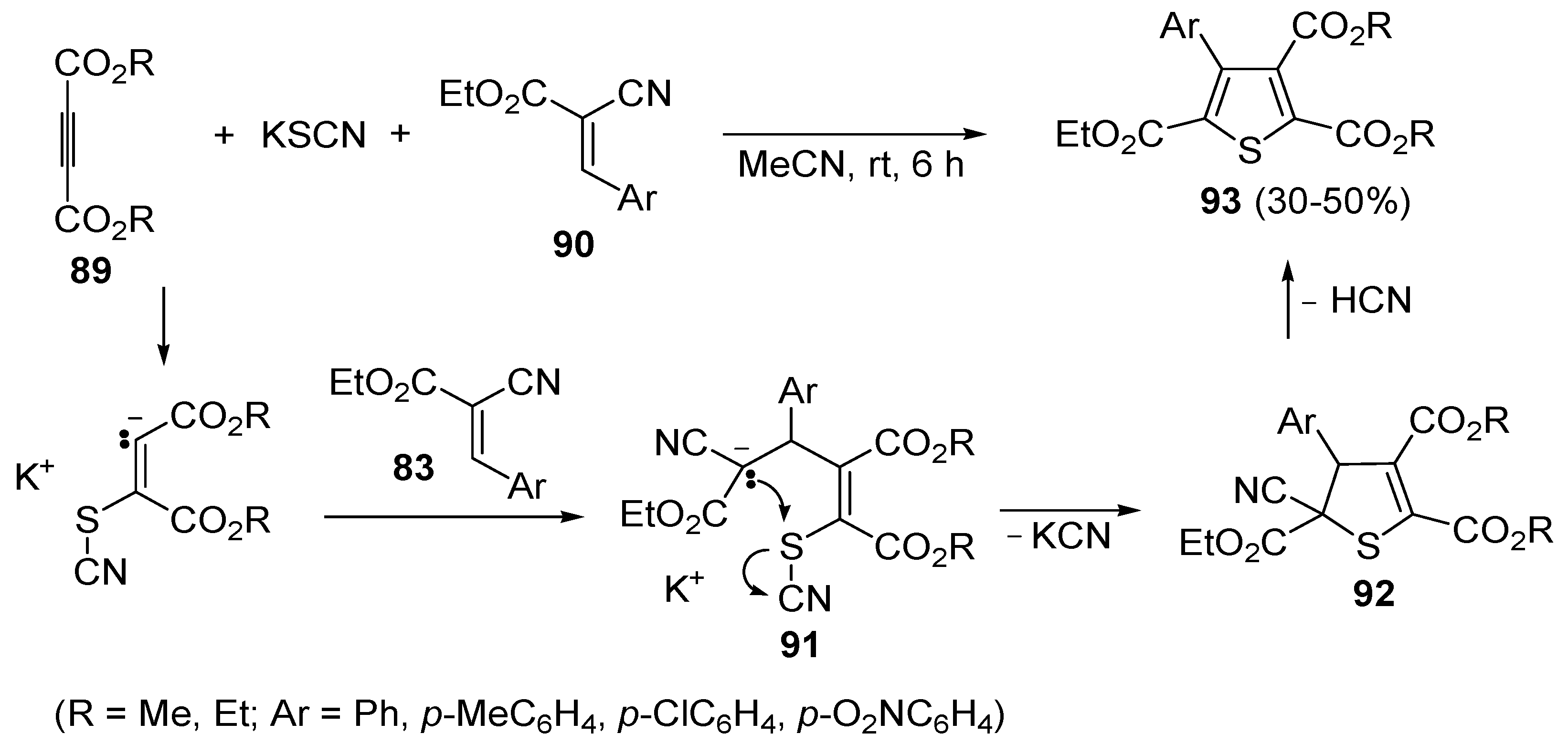
105].
Another useful utilization of acetylenic diesters for the thiophene synthesis has been reported recently. It involves the reaction between β-oxodithioesters
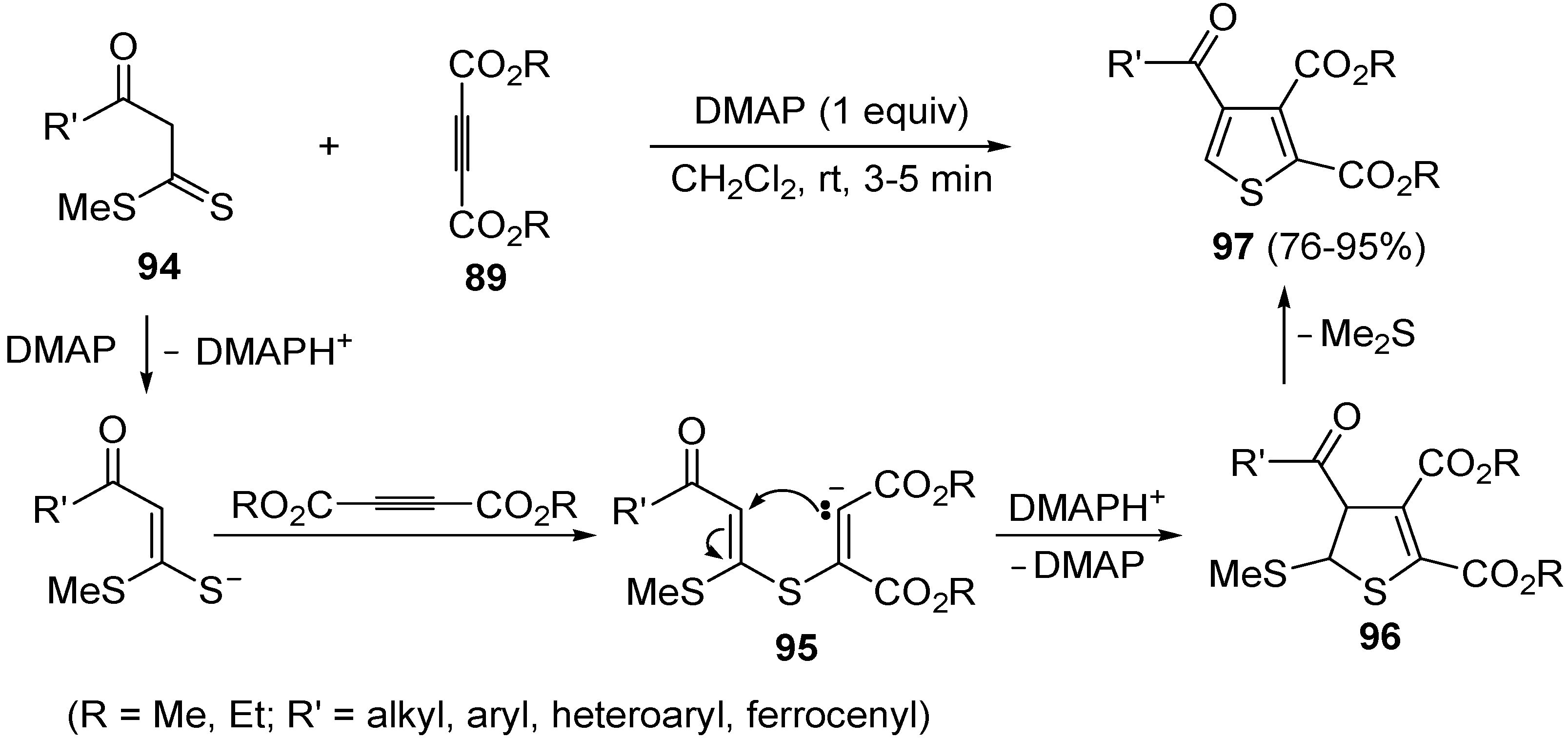
94 and dialkyl acetylenedicarboxylates
89 (1:1 molar ratio) carried out in the presence of an equimolar amount of dimethylaminopyridine (DMAP) in CH
2Cl
2 at rt for 3–5 min (
Scheme 29) [
106]. The process takes place through α-deprotonation of
94 by DMAP and intermolecular conjugate addition (from nucleophilic attack by the sulfur atom to the triple bond of
89, to give anionic intermediate
95), followed by intramolecular conjugate addition to give dihydrothiophene
96, and elimination of Me
2S, eventually leading to dialkyl thiophene-2,3-dicarboxylate derivatives
97 (
Scheme 29) [
106].
Scheme 28.
Synthesis of trialkyl 4-arylthiophene-2,3,5-tricarboxylates
93 starting from dialkyl acetylenedicarboxylates
89, KSCN, and 3-aryl-2-cyanoacrylates
90 [
105].
Scheme 28.
Synthesis of trialkyl 4-arylthiophene-2,3,5-tricarboxylates
93 starting from dialkyl acetylenedicarboxylates
89, KSCN, and 3-aryl-2-cyanoacrylates
90 [
105].
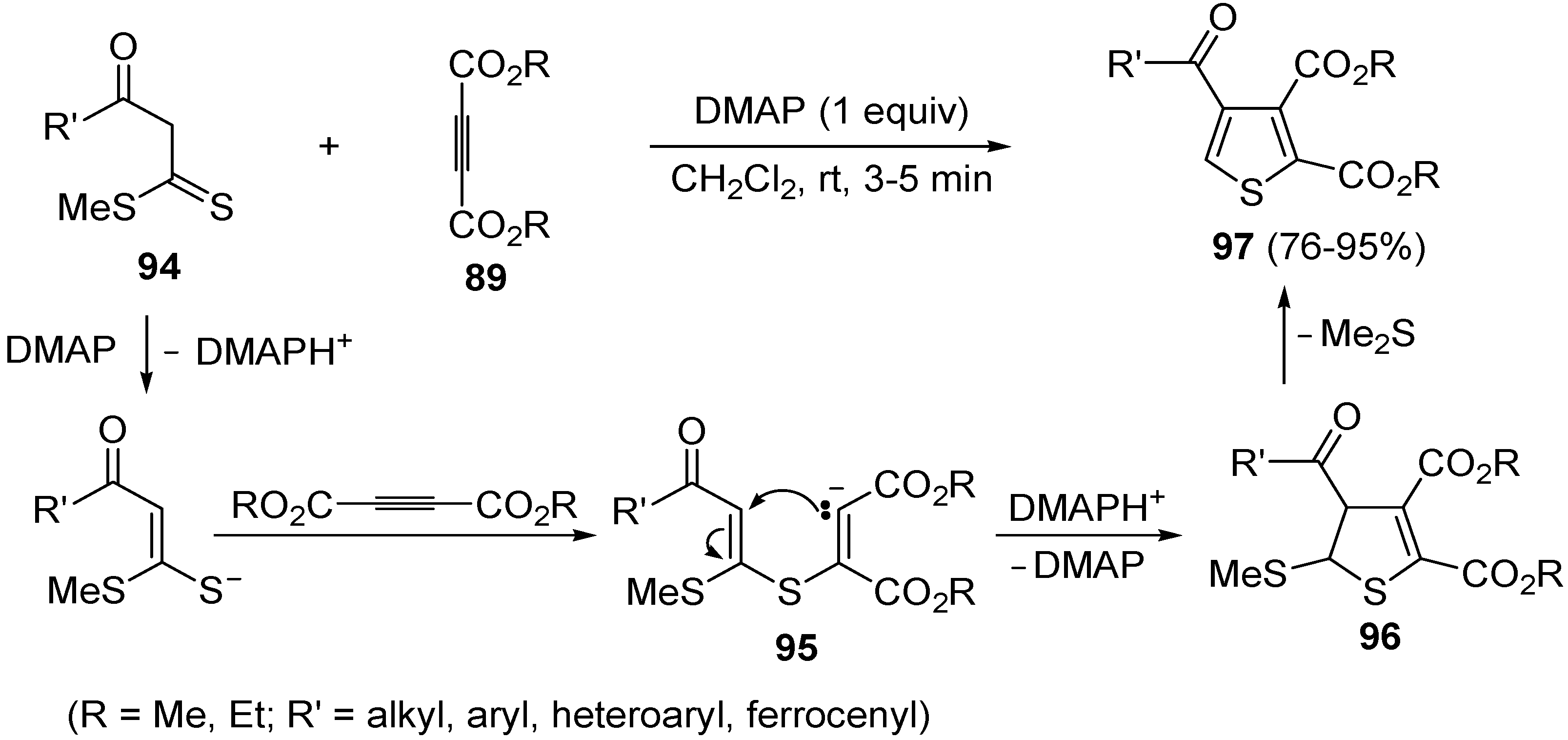
Scheme 29.
Synthesis of dialkyl thiophene-2,3-dicarboxylate derivatives
97 starting from β-oxodithioesters
94 and dialkyl acetylenedicarboxylates
89 in the presence of DMAP [
106].
Scheme 29.
Synthesis of dialkyl thiophene-2,3-dicarboxylate derivatives
97 starting from β-oxodithioesters
94 and dialkyl acetylenedicarboxylates
89 in the presence of DMAP [
106].
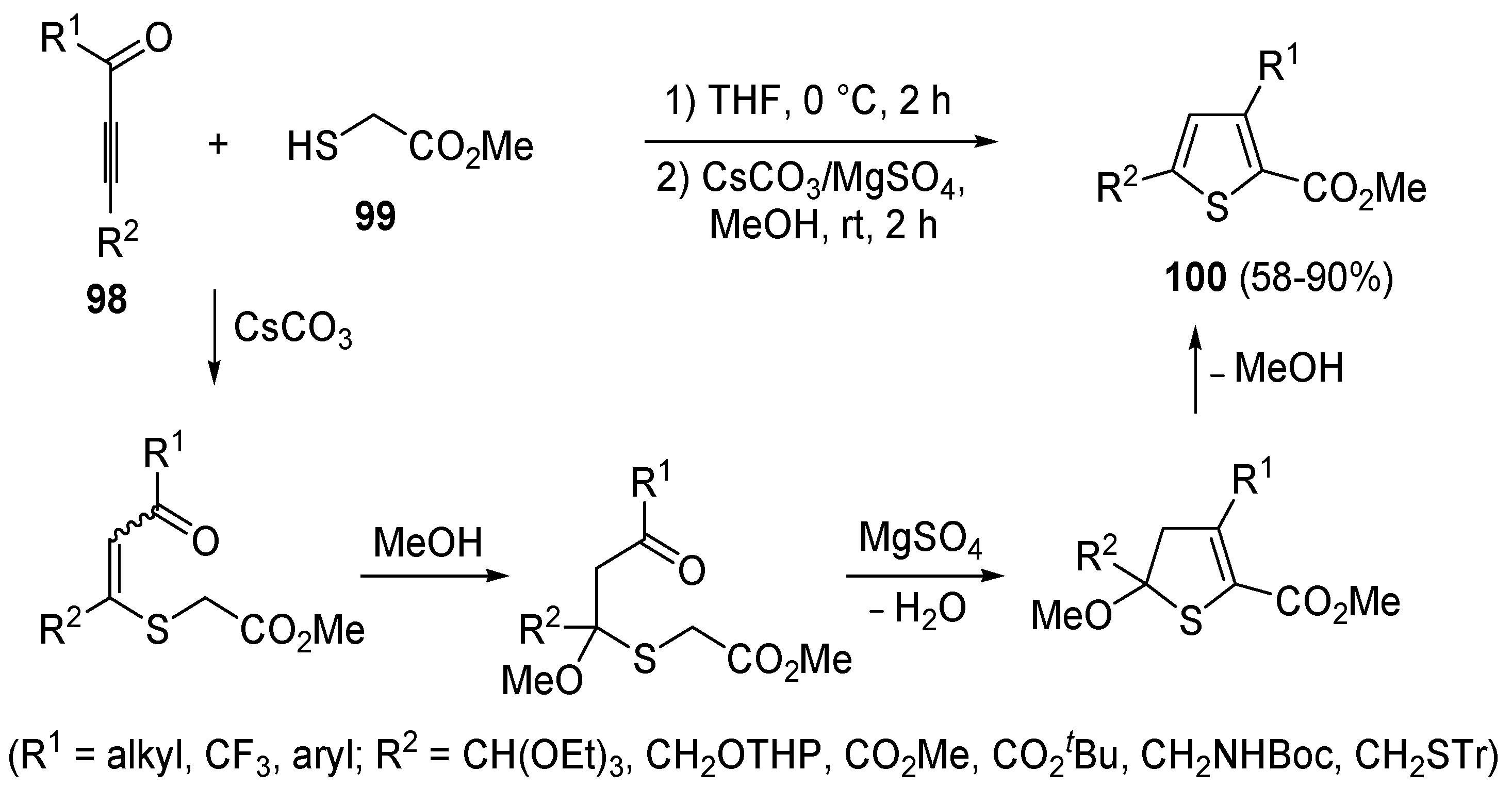
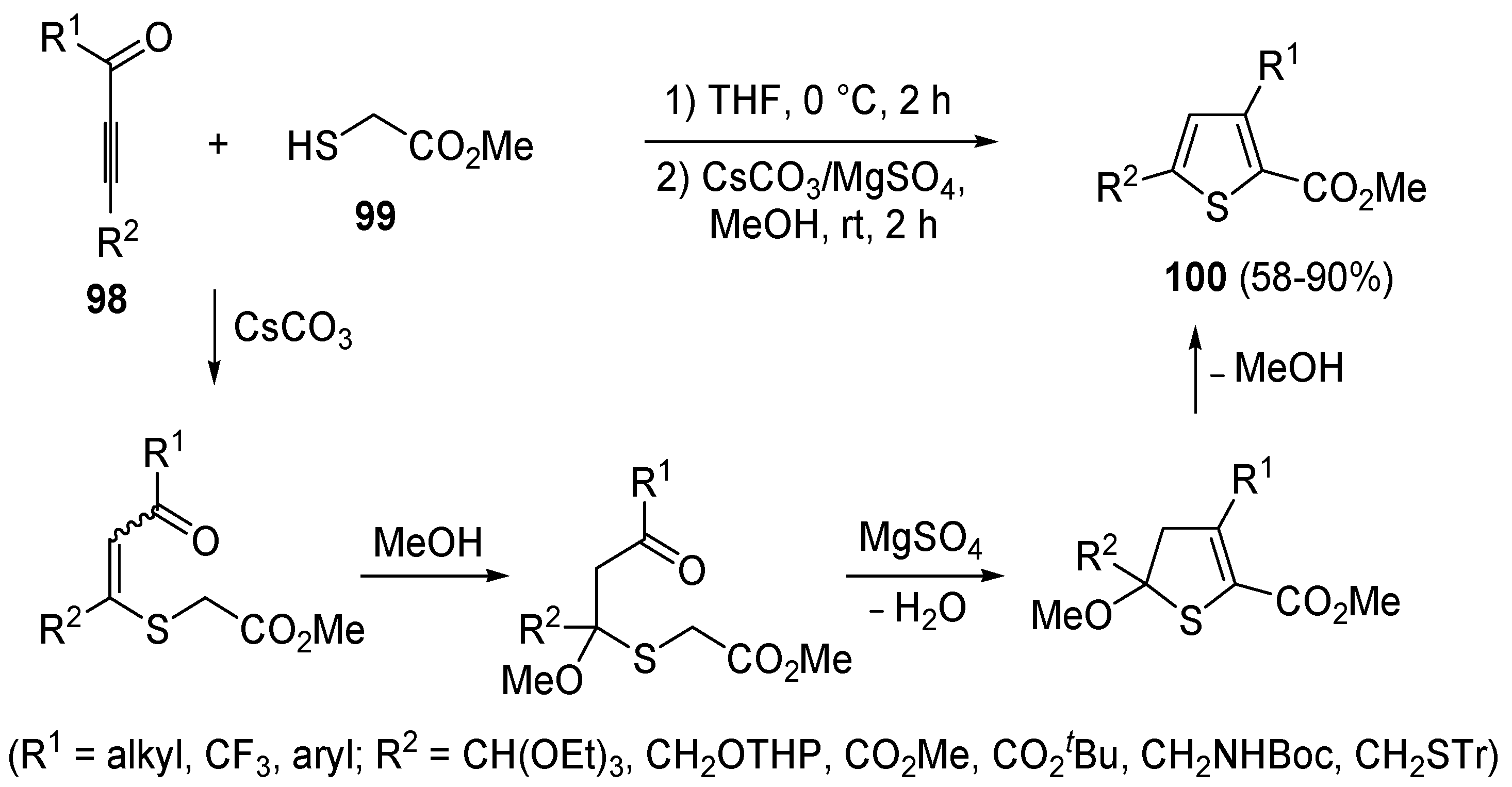
A mechanism involving a conjugate addition by a thiolate anion to an activate triple bond was also at work in a modification of the classical Fiesselmann thiophene synthesis [
107], involving the reaction between acetylenic ketones
98 and methyl thioglycolate
99 to give methyl thiophene-2-carboxylates
100 (
Scheme 30) [
108]. Reactions were carried out by dissolving an equimolar amount of
98 and
99 in THF at 0 °C followed, after 2 h, by the addition of a 1:2 mixture of CsCO
3/MgSO
4 in MeOH and stirring at rt for 2 h [
108].
Scheme 30.
Synthesis of methyl thiophene-2-carboxylates
100 from acetylenic ketones
98 and methyl thioglycolate (
99) in the presence of CsCO
3, MgSO
4, and MeOH [
108].
Scheme 30.
Synthesis of methyl thiophene-2-carboxylates
100 from acetylenic ketones
98 and methyl thioglycolate (
99) in the presence of CsCO
3, MgSO
4, and MeOH [
108].
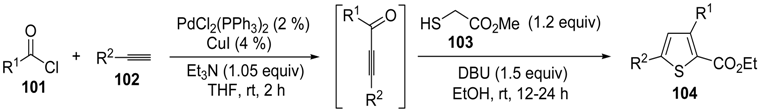
A particularly convenient approach to 2,4-disubstituted thiophenes
104, based on a sequential three-component Sonogashira coupling/Fiesselmann-type cyclocondensation, has been reported recently [
109]. Thus, the reaction between (hetero)aroyl chlorides
101 and terminal alkynes
102 (1.1 equiv) (carried out at rt in THF for 2 h, in the presence of 2 mol % of PdCl
2(PPh
3)
2, 4 mol % of CuI, and 1.05 equiv of Et
3N) was followed by the addition of EtOH, ethyl thioglycolate
103 (1.2 equiv) and DBU (1.5 equiv), to give, after stirring for 12–24 h at rt, thiophene derivatives
104 in moderate to excellent yields (32%–97%,
Table 9). The method has also been successfully applied to the synthesis of luminescent terthiophenes and pentathiophenes [
109].
Table 9.
Synthesis of ethyl thiophene-2-carboxylates
104 starting from sequential Sonogashira coupling between (hetero)aroyl chlorides
101 and terminal alkynes
102/Fiesselmann-type cyclocondensation
a [
109].
![Molecules 19 15687 i113]()
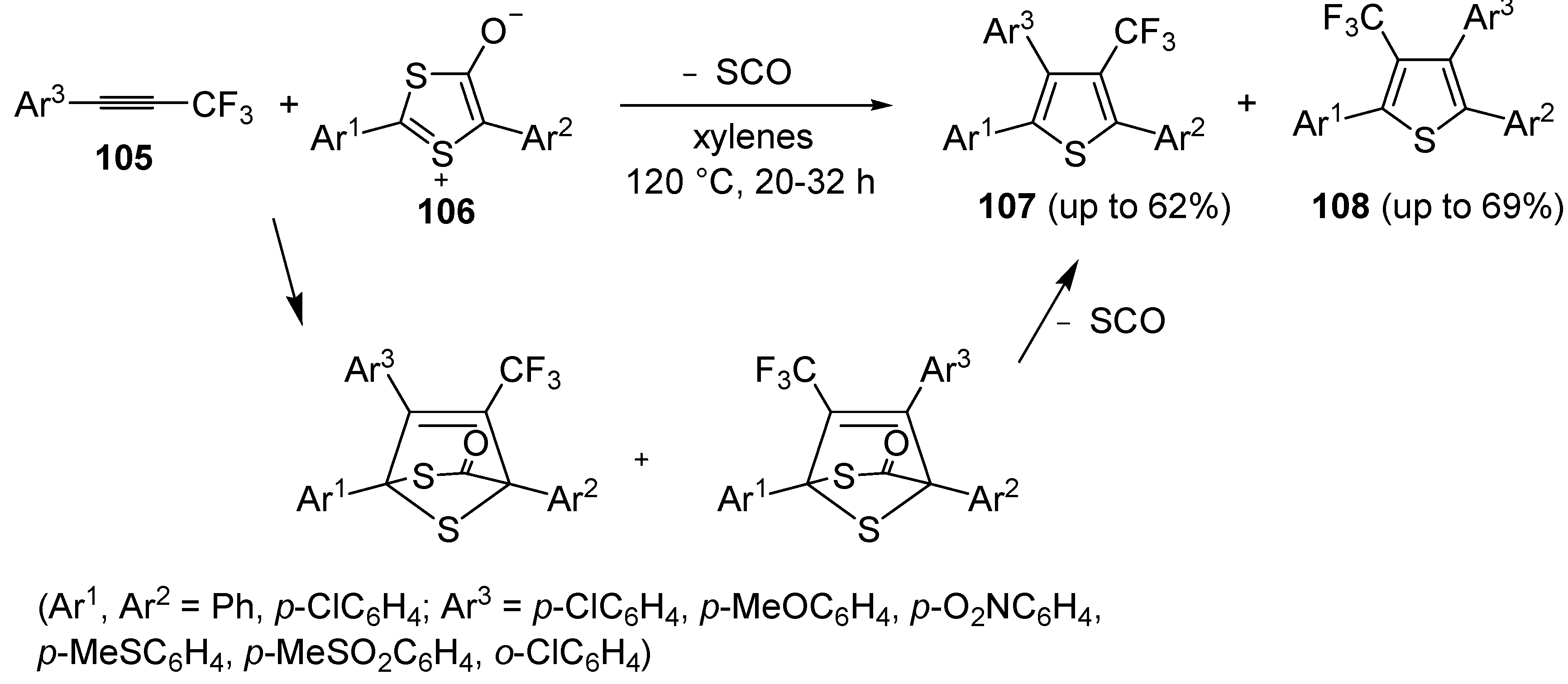
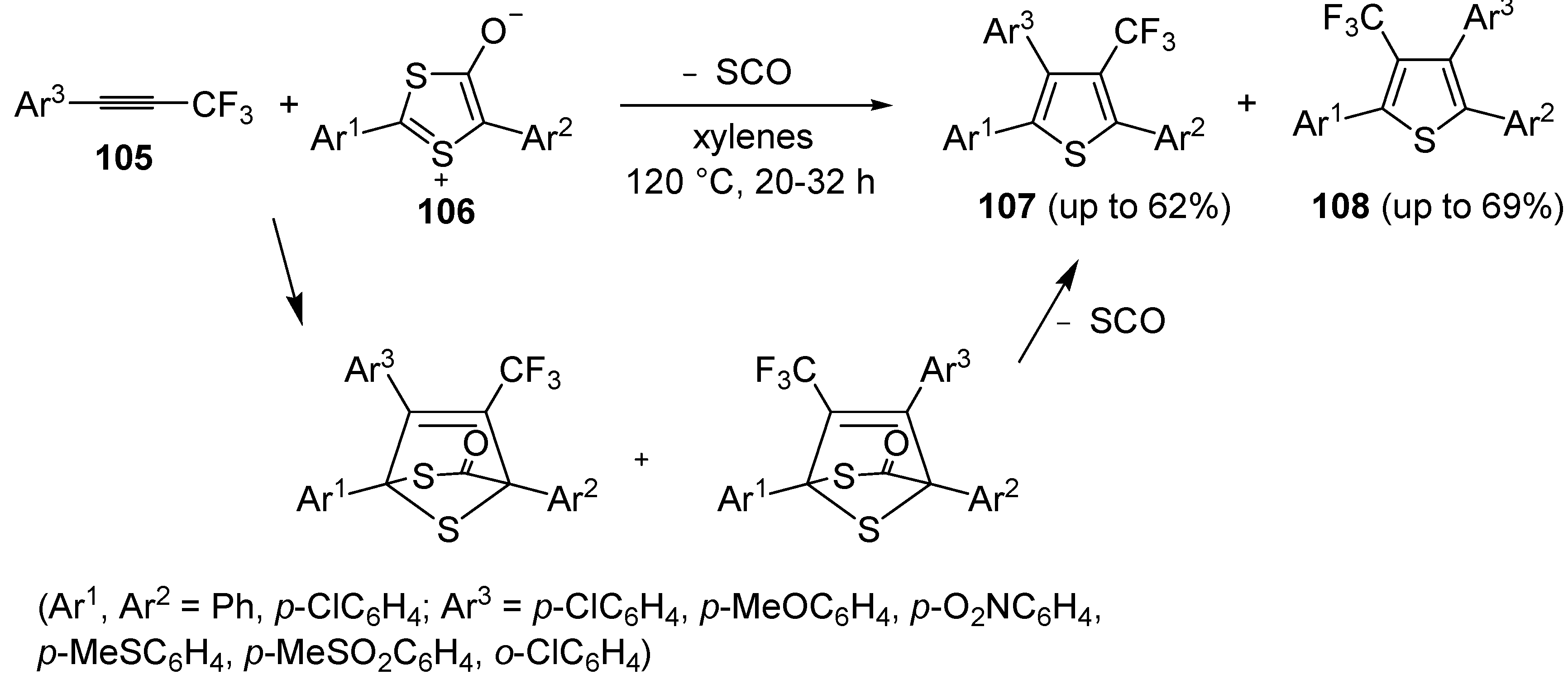
An interesting approach to regioisomeric 2,3,5-triaryl-4-trifluoromethylthiphenes
107 and
108, based on 1,3-dipolar cycloaddition between 1-aryl-3,3,3-trifluoro-1-propynes
105 and 1,3-dithiolium-4-olates
106 (1:1 molar ratio), was developed some years ago (
Scheme 31) [
109]. Reactions were carried out in xylenes at 120 °C for 20–32 h [
110].
Scheme 31.
Synthesis of 2,3,5-triaryl-4-trifluoromethylthiophenes
107 and
108 from 1-aryl-3,3,3-trifluoro-1-propynes
105 and 1,3-dithiolium-4-olates
106 [
110].
Scheme 31.
Synthesis of 2,3,5-triaryl-4-trifluoromethylthiophenes
107 and
108 from 1-aryl-3,3,3-trifluoro-1-propynes
105 and 1,3-dithiolium-4-olates
106 [
110].





















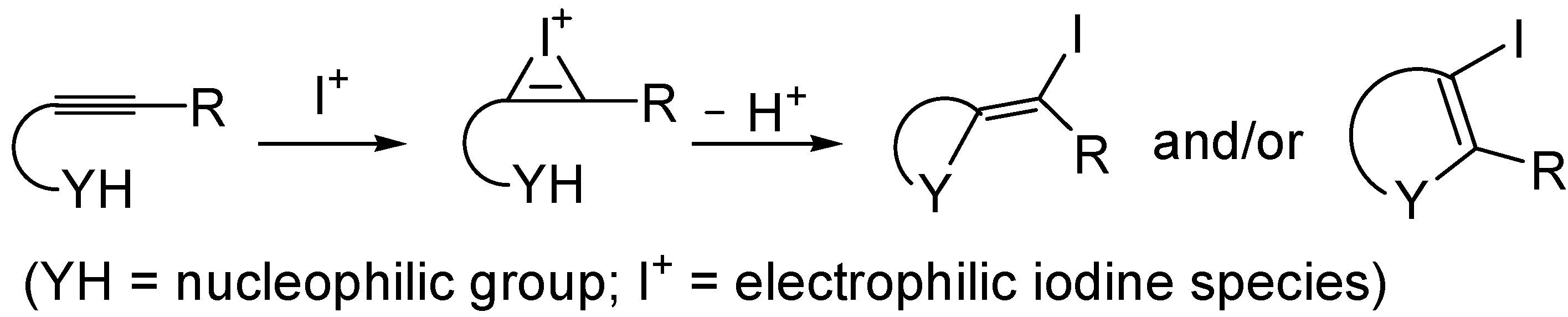





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
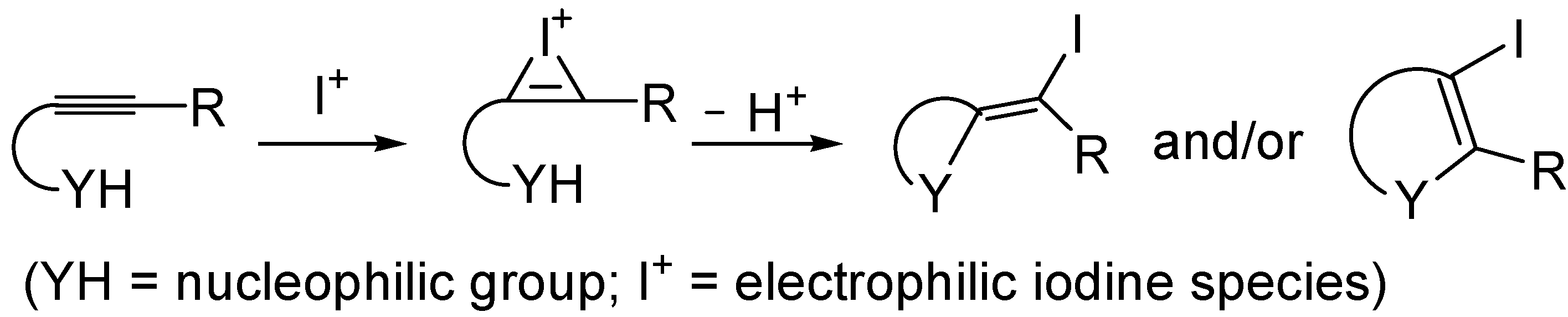{kind=link}
{kind=link}
{kind=link}
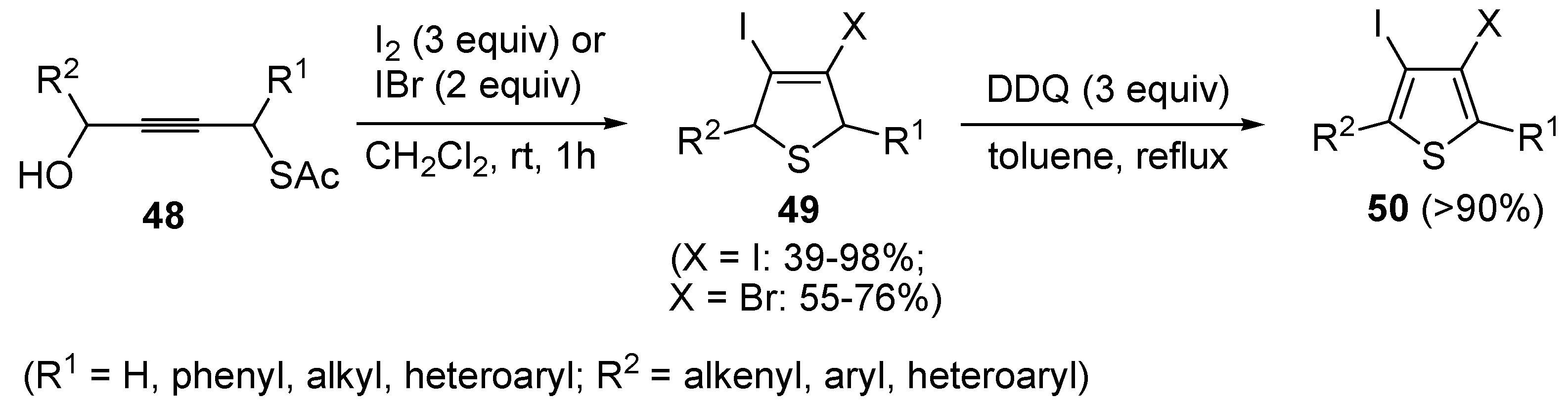{kind=link}
{kind=link}
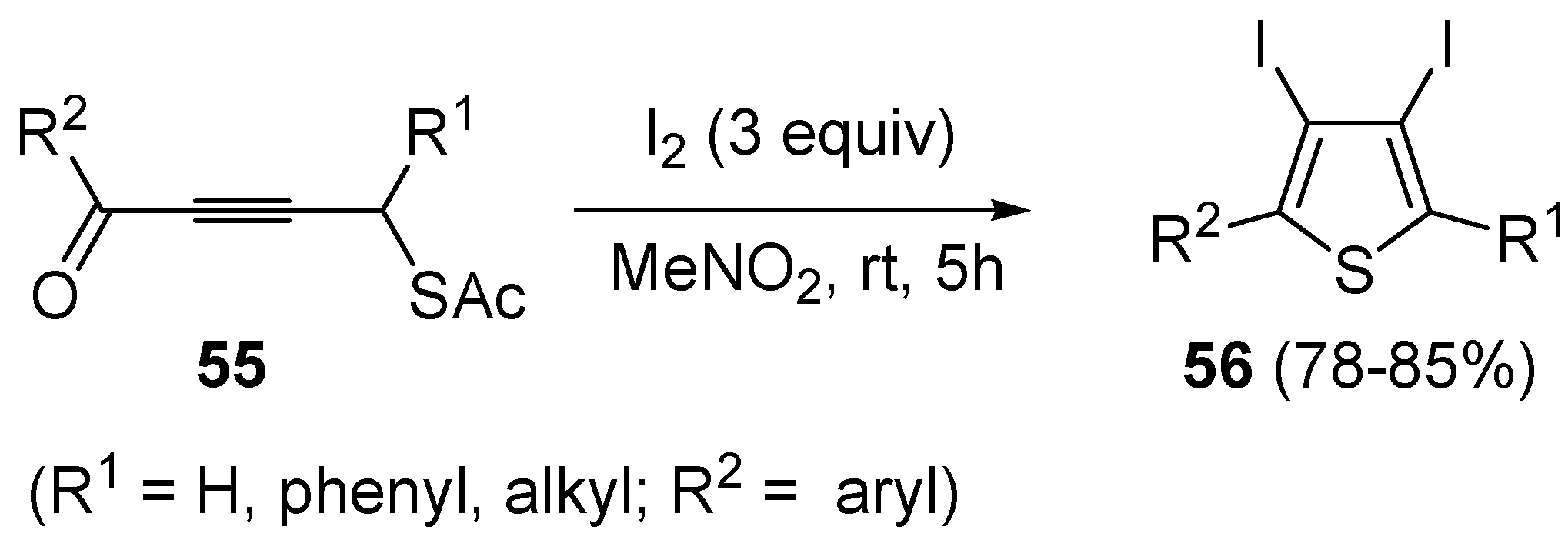{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}