Chemical Synthesis, Backbone Cyclization and Oxidative Folding of Cystine-knot Peptides — Promising Scaffolds for Applications in Drug Design

Abstract
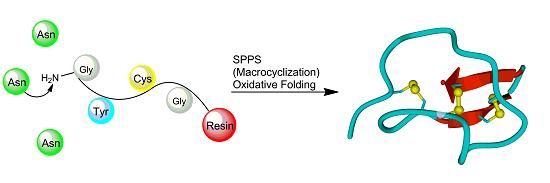
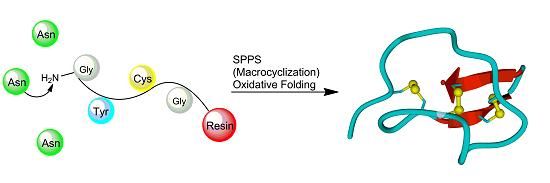
:
1. Introduction
2. Structure
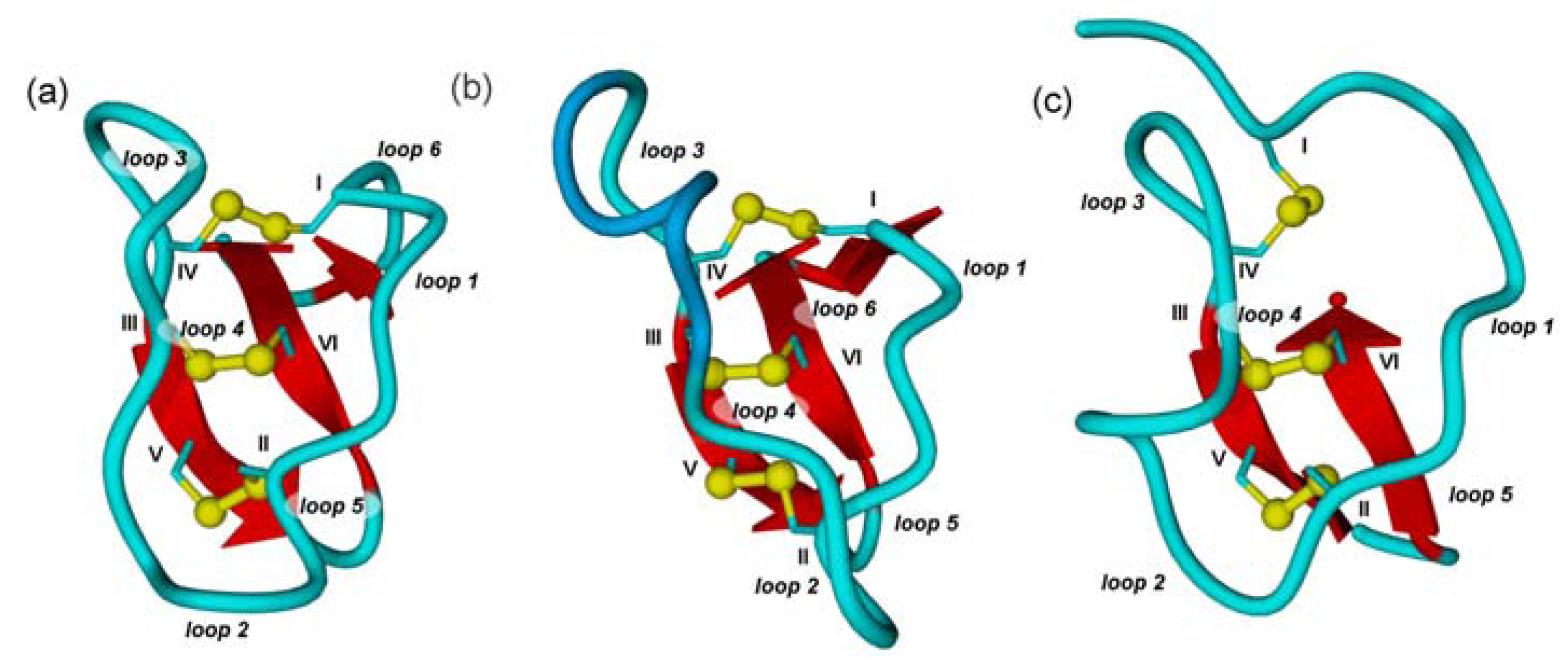
2.1. The Cystine-Knot Motif
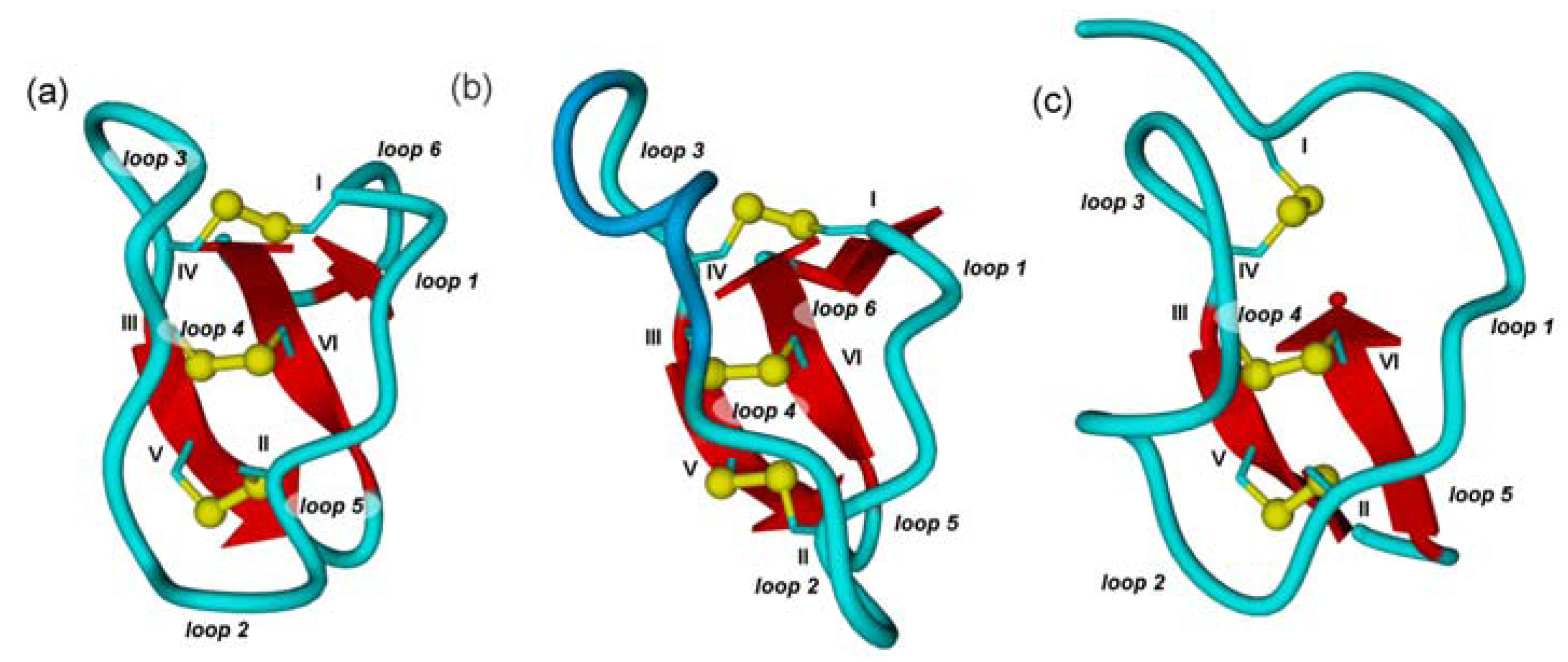
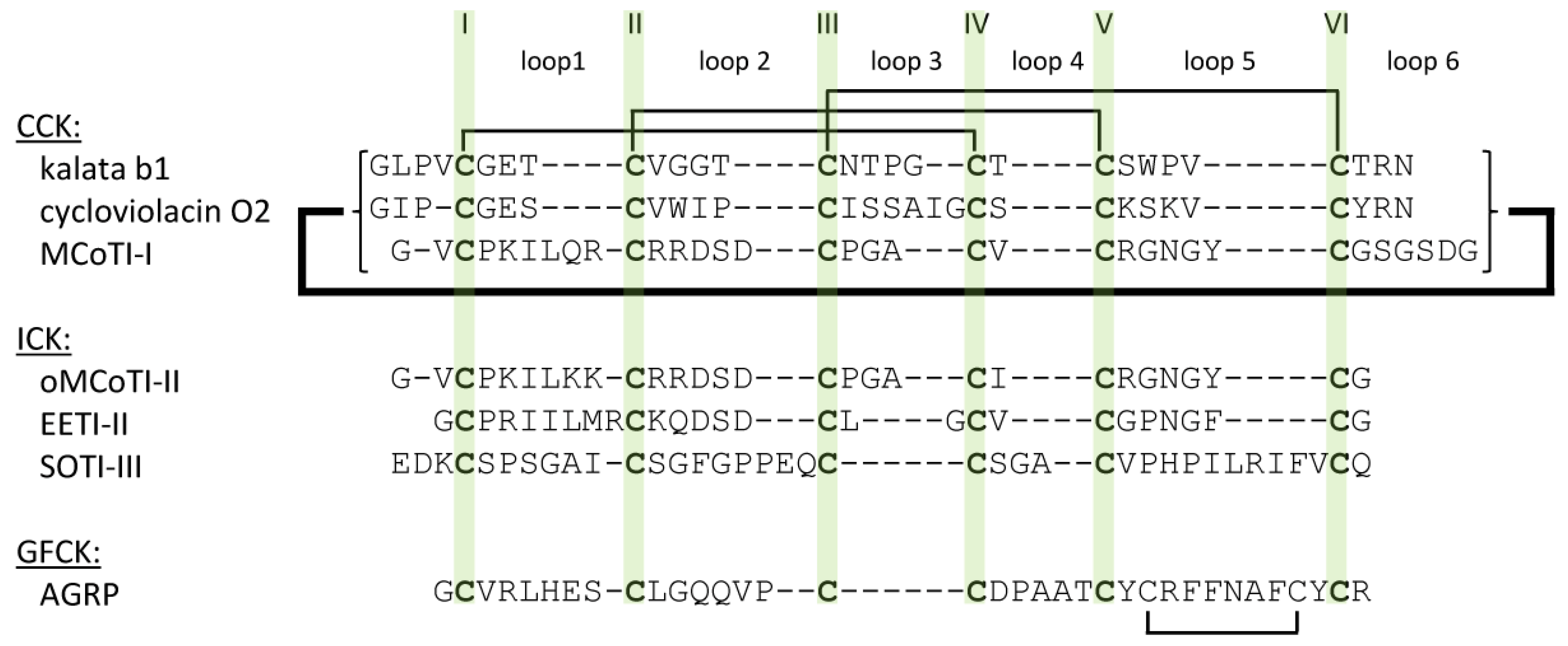
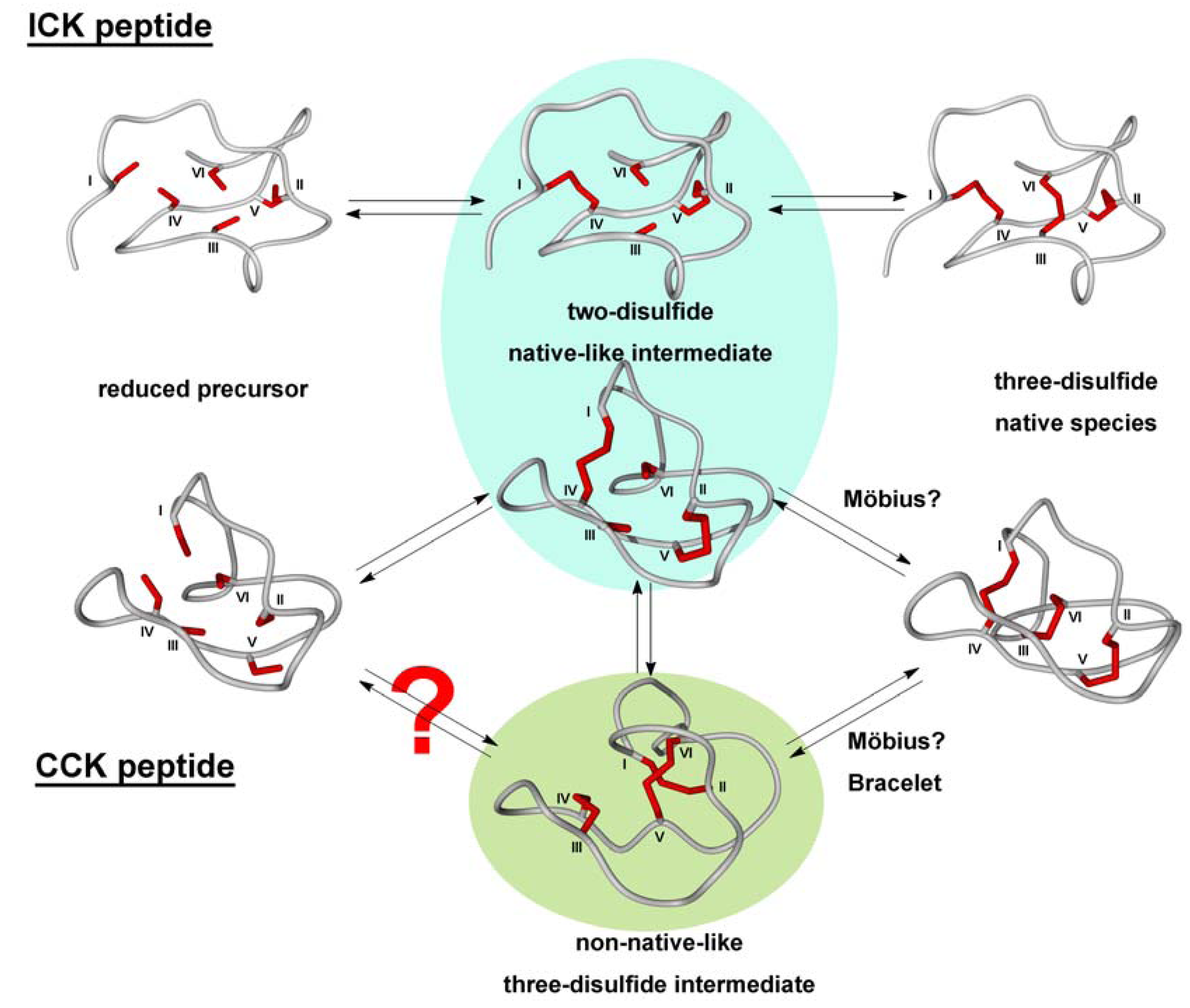
2.2. Cyclic Cystine Knots
2.3. Inhibitor Cystine Knots
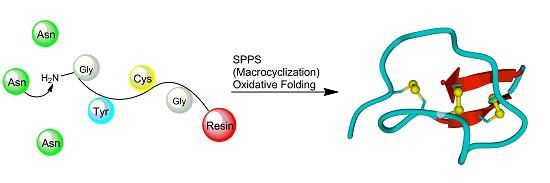
3. Synthesis of Cystine-Knot Peptides
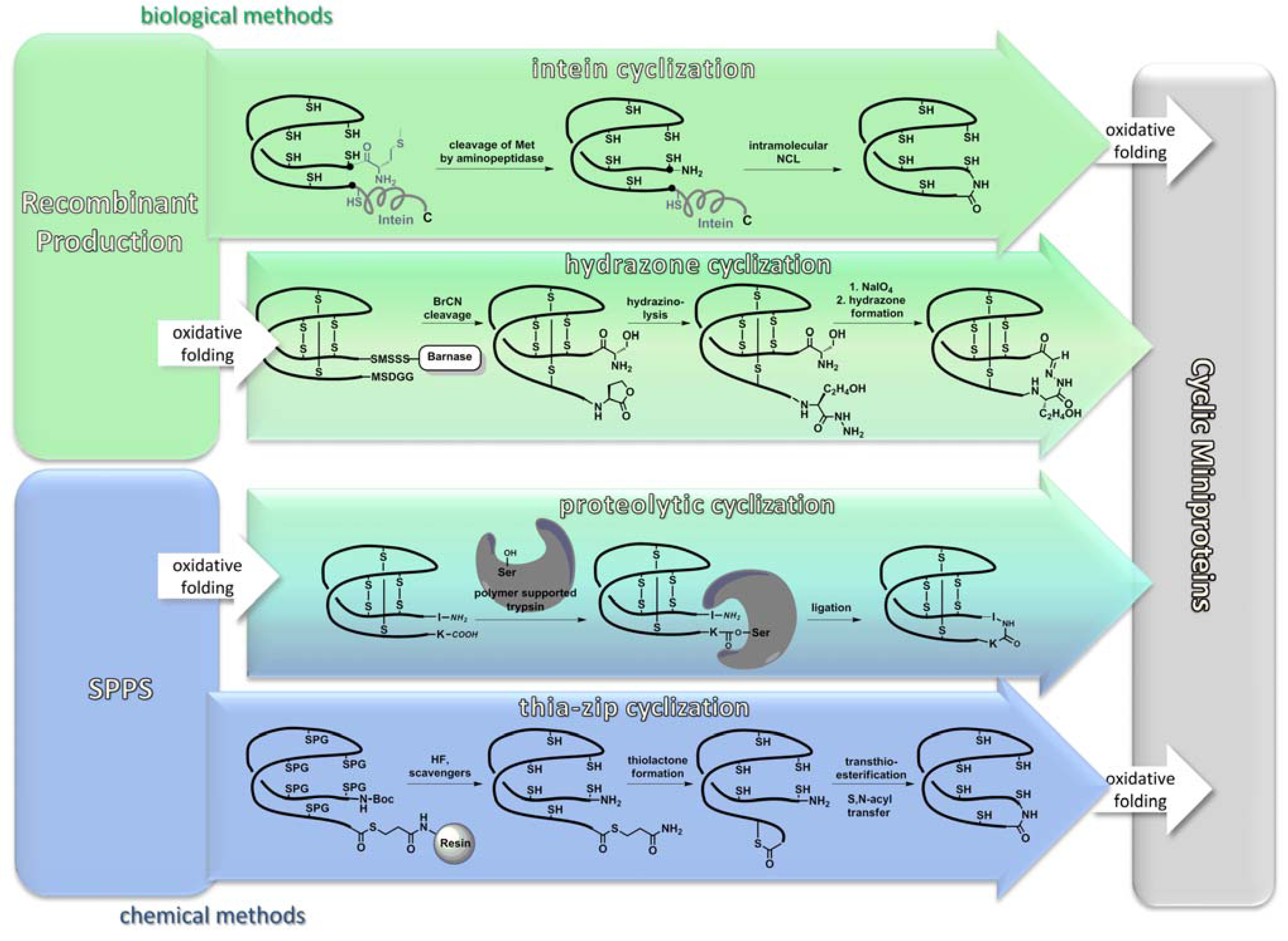
3.1. Recombinant Production


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Type | Folding conditions | Yield/Conversion | Reference |
|---|---|---|---|---|
| ocMCoTI | ICK | 0.5 mM HCl, 200 mM NaHCO3, pH = 9.1, 1–1.5 mg/L peptide. | 16% a | [48] |
| 29% b | ||||
| cMCoTI | ICK | 100 mM NH4OAc, pH = 8.5, GSH (1‑5 mM), 0.1 mg/L peptide | 90% c | [49] |
| Variants of ocMCoTI | ICK | 50% MeCN in 100 mM (NH4)2CO3, GSH (4 eq.) | 1.8–7.7% a | [45] |
| 36–72% b | ||||
| ICK toxins | ICK | GSSG/GSH (0.3 mM/0.15 mM) in 2 M urea, 100 mM Tris-HCl | 3–6% a | [50] |
| EETI-II | ICK | 100 mM NH4OAc, pH = 9.1 | >80% c | [51] |
| Gurmarin | ICK | 1. Orthogonal cysteine protecting groups 2. GSH/cystamine in 100 mM Tris-HCl, pH = 7.8 | 1.: 0.55% a | [52] |
| 2.: 14.1% a | ||||
| GVIA and analogues | Cono-toxin | Cysteine-selenocysteine exchange, GSSG/GSH (1 mM/2 mM) | 60–78% c | [53] |
| Cycloviolacin O2 | CCK | 35% DMSO, 6% Brij 35 (an oil dispersant), addition of GSH/cystamine after 24 h (2 mM/2 mM) in 100 mM Tris‑HCl, pH = 8.5 | 52% c | [54] |
| Kalata B1 | CCK | 35% DMSO, 6%, Brij 35 (an oil dispersant), GSH/cystamine (2 mM/2 mM) in 100 mM NH4HCO3, pH = 8.5 | >95% c | [31] |
| Kalata B2 | CCK | 50% i-PrOH, GSSG/GSH (2 mM/2 mM) in 100 mM NH4HCO3, pH = 8.5 | >95% c | [31] |
| Kalata B8 | CCK | 50% i-PrOH, GSSG/GSH (2 mM/2 mM) in 100 mM NH4HCO3, pH = 8.5 | >80% c | [31] |
| Cyclic hedyotide B1 | CCK | 70–80% i-PrOH, pH = 8.5 0.033 mg/L peptide | 48% c | [55] |
| ASIP | GFCK | 100 mM Tris-buffer, pH = 7.7–7.9, 1 mM EDTA, 1 M GuHCl, GSSG/GSH (1:10) | 10% b | [56] |
3.2. Chemical Synthesis
3.2.1. Chain Assembly
3.2.2. Oxidative Folding
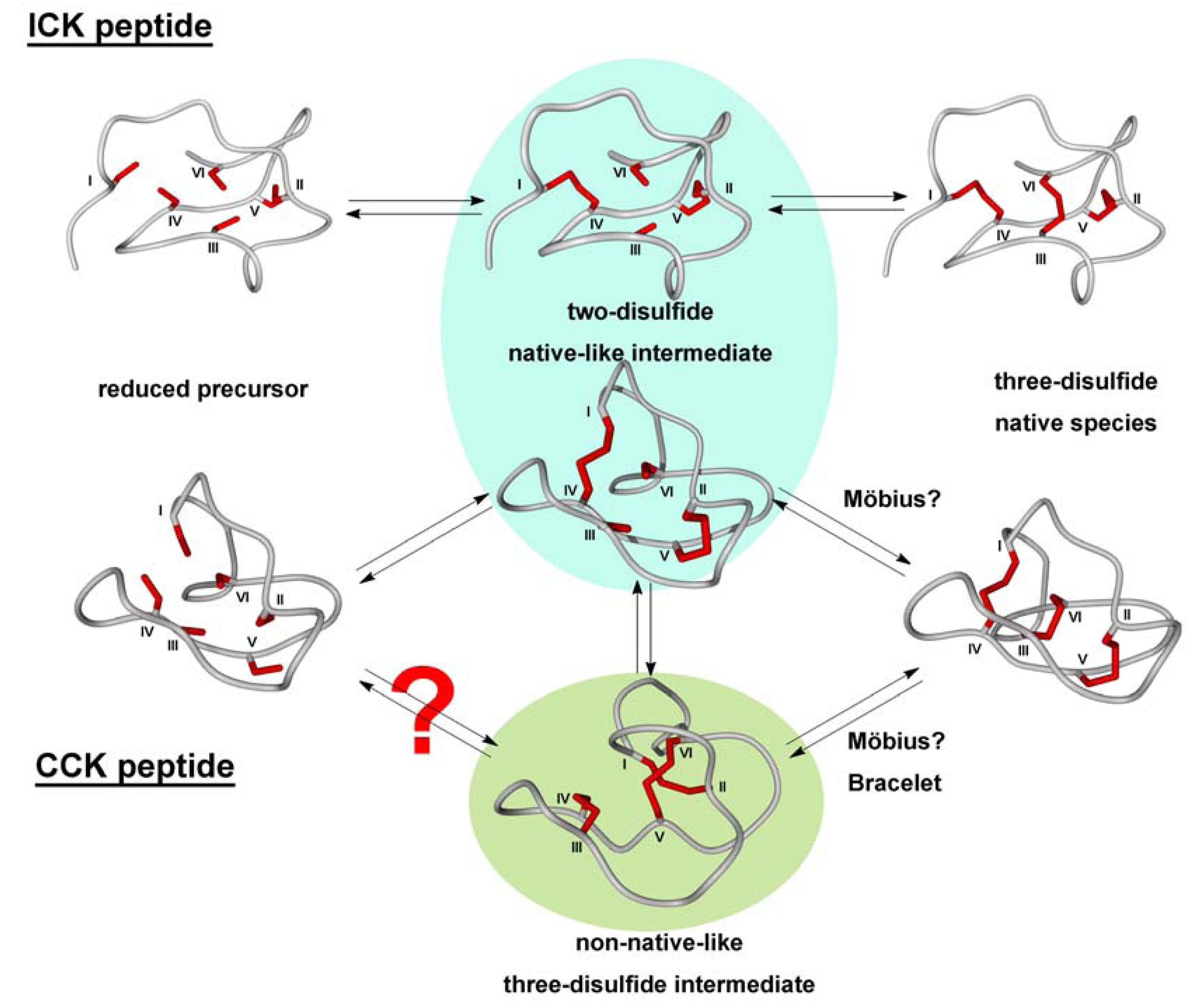
3.2.3. Backbone Macrocyclization
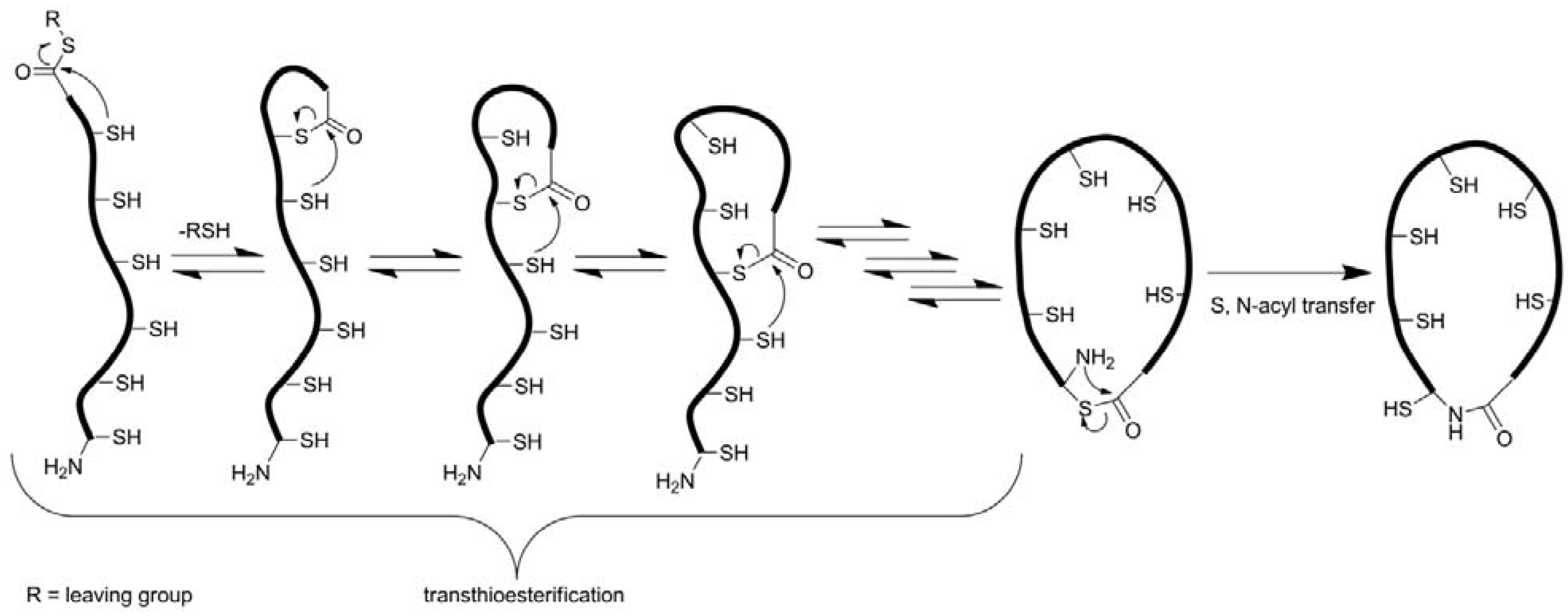
| Peptide | Type | Cyclization reaction; conditions | Yield/Conversion | Reference |
|---|---|---|---|---|
| Variants of cMCoTI | ICK | Immobilized trypsin; 100 mM phosphate buffer, pH = 7.4 | 90–94% a | [44,45] |
| Variants of cMCoTI | ICK | Folding and NCL as one-pot reaction; 50% GSSG (1 mM) in 100 mM carbonate buffer, 50% peptide (3 mM) in acetonitrile | 63–72% b | [45] |
| Variants of cMCoTI | ICK | Hydrazone linkage; multiple reactions from recombinantly produced barnase fusion | 0.5–1 mg/L c | [43] |
| Kalata B1 | CCK | Amide bond; HBTU or BOP, respectively (1–10 eq.), DIEA in DMF | ~25% a | [69] |
| Kalata B1 | CCK | NCL; 100 mM NaH2PO4, TCEP (6 eq.), pH = 7.4, 1 mg/mL peptide | 100% a | [69] |
| hB1 | CCK | NCL; 100 mM NaH2PO4, 6 M GuHCl, thiophenol (100 eq.), pH = 7.5 | 100% a | [55] |
| Cyclic MrIA | Cyclic cono-toxin | NCL; 100 mM Tris-HCl, pH = 7.8, 6 M GuHCl, sodium 2‑sulfonylethane sulfonate (1 mg/mL), anaerobic | 100% a | [98] |
3.2.4. Analysis of Cystine Knots
4. Applications to Drug Design
5. Conclusions and Outlook
Acknowledgments
References
- Kolmar, H. Natural and engineered cystine knot miniproteins for diagnostic and therapeutic applications. Curr. Pharm. Des. 2011, 17, 4329–4336. [Google Scholar] [CrossRef]
- Gelly, J.C.; Gracy, J.; Kaas, Q.; Le-Nguyen, D.; Heitz, A.; Chiche, L. The KNOTTIN website and database: A new information system dedicated to the knottin scaffold. Nucleic Acids Res. 2004, 32, D156–D159. [Google Scholar] [CrossRef]
- Gracy, J.; Le-Nguyen, D.; Gelly, J.C.; Kaas, Q.; Heitz, A.; Chiche, L. KNOTTIN: The knottin or inhibitor cystine knot scaffold in 2007. Nucleic Acids Res. 2008, 36, D314–D319. [Google Scholar]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Daly, N.L.; Craik, D.J. Bioactive cystine knot proteins. Curr. Opin. Chem. Biol. 2011, 15, 362–368. [Google Scholar] [CrossRef]
- Gran, L. On the effect of a polypeptide isolated from “Kalata-Kalata” (Oldenlandia affinis DC) on the oestrogen dominated uterus. Acta Pharmacol. Toxicol. (Copenh.) 1973, 33, 400–408. [Google Scholar]
- McDonald, N.Q.; Lapatto, R.; Murray-Rust, J.; Gunning, J.; Wlodawer, A.; Blundell, T.L. New protein fold revealed by a 2.3-A resolution crystal structure of nerve growth factor. Nature 1991, 354, 411–414. [Google Scholar]
- Felizmenio-Quimio, M.E.; Daly, N.L.; Craik, D.J. Circular proteins in plants: Solution structure of a novel macrocyclic trypsin inhibitor from Momordica cochinchinensis. J. Biol. Chem. 2001, 276, 22875–22882. [Google Scholar]
- Chiche, L.; Heitz, A.; Gelly, J.C.; Gracy, J.; Chau, P.T.; Ha, P.T.; Hernandez, J.F.; Le-Nguyen, D. Squash inhibitors: From structural motifs to macrocyclic knottins. Curr. Protein Pept. Sci. 2004, 5, 341–349. [Google Scholar] [CrossRef]
- Kolmar, H. Biological diversity and therapeutic potential of natural and engineered cystine knot miniproteins. Curr. Opin. Pharmacol. 2009, 9, 608–614. [Google Scholar] [CrossRef]
- Craik, D.J.; Cemazar, M.; Wang, C.K.; Daly, N.L. The cyclotide family of circular miniproteins: Nature’s combinatorial peptide template. Biopolymers 2006, 84, 250–266. [Google Scholar] [CrossRef]
- Wang, C.K.; Hu, S.H.; Martin, J.L.; Sjogren, T.; Hajdu, J.; Bohlin, L.; Claeson, P.; Goransson, U.; Rosengren, K.J.; Tang, J.; et al. Combined X-ray and NMR analysis of the stability of the cyclotide cystine knot fold that underpins its insecticidal activity and potential use as a drug scaffold. J. Biol. Chem. 2009, 284, 10672–10683. [Google Scholar]
- Craik, D.J.; Daly, N.L. NMR as a tool for elucidating the structures of circular and knotted proteins. Mol. Biosyst. 2007, 3, 257–265. [Google Scholar]
- Colgrave, M.L.; Craik, D.J. Thermal, Chemical, And enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef]
- Jennings, C.; West, J.; Waine, C.; Craik, D.; Anderson, M. Biosynthesis and insecticidal properties of plant cyclotides: The cyclic knotted proteins from Oldenlandia affinis. Proc. Natl. Acad. Sci. USA 2001, 98, 10614–10619. [Google Scholar]
- Tam, J.P.; Lu, Y.A.; Yang, J.L.; Chiu, K.W. An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc. Natl. Acad. Sci. USA 1999, 96, 8913–8918. [Google Scholar] [CrossRef]
- Gustafson, K.R.; Sowder, R.C.; Henderson, L.E.; Parsons, I.C.; Kashman, Y.; Cardellina, J.H.; McMahon, J.B.; Buckheit, R.W.; Pannell, L.K.; Boyd, M.R. Circulins A and B, Novel human immunodeficiency virus (HIV)-inhibitory macrocyclic peptides from the tropical tree Chassalia parvifolia. J. Am. Chem. Soc. 1994, 116, 9337–9338. [Google Scholar]
- Svangard, E.; Goransson, U.; Hocaoglu, Z.; Gullbo, J.; Larsson, R.; Claeson, P.; Bohlin, L. Cytotoxic cyclotides from Viola tricolor. J. Nat. Prod. 2004, 67, 144–147. [Google Scholar] [CrossRef]
- Lindholm, P.; Goransson, U.; Johansson, S.; Claeson, P.; Gullbo, J.; Larsson, R.; Bohlin, L.; Backlund, A. Cyclotides: A novel type of cytotoxic agents. Mol. Cancer Ther. 2002, 1, 365–369. [Google Scholar] [CrossRef]
- Gustafson, K.R.; McKee, T.C.; Bokesch, H.R. Anti-HIV cyclotides. Curr. Protein Pept. Sci. 2004, 5, 331–340. [Google Scholar] [CrossRef]
- Hernandez, J.F.; Gagnon, J.; Chiche, L.; Nguyen, T.M.; Andrieu, J.P.; Heitz, A.; Trinh Hong, T.; Pham, T.T.; Le Nguyen, D. Squash trypsin inhibitors from Momordica cochinchinensis exhibit an atypical macrocyclic structure. Biochemistry 2000, 39, 5722–5730. [Google Scholar]
- Heitz, A.; Hernandez, J.F.; Gagnon, J.; Hong, T.T.; Pham, T.T.; Nguyen, T.M.; Le-Nguyen, D.; Chiche, L. Solution structure of the squash trypsin inhibitor MCoTI-II: A new family for cyclic knottins. Biochemistry 2001, 40, 7973–7983. [Google Scholar] [CrossRef]
- Avrutina, O.; Schmoldt, H.U.; Gabrijelcic-Geiger, D.; Le Nguyen, D.; Sommerhoff, C.P.; Diederichsen, U.; Kolmar, H. Trypsin inhibition by macrocyclic and open-chain variants of the squash inhibitor MCoTI-II. Biol. Chem. 2005, 386, 1301–1306. [Google Scholar]
- Kowalska, J.; Pszczola, K.; Wilimowska-Pelc, A.; Lorenc-Kubis, I.; Zuziak, E.; Lugowski, M.; Legowska, A.; Kwiatkowska, A.; Sleszynska, M.; Lesner, A.; et al. Trypsin inhibitors from the garden four o'clock (Mirabilis jalapa) and spinach (Spinacia oleracea) seeds: Isolation, Characterization and chemical synthesis. Phytochemistry 2007, 68, 1487–1496. [Google Scholar] [CrossRef]
- Heitz, A.; Chiche, L.; Le-Nguyen, D.; Castro, B. 1H 2D NMR and distance geometry study of the folding of Ecballium elaterium trypsin inhibitor, A member of the squash inhibitors family. Biochemistry 1989, 28, 2392–2398. [Google Scholar] [CrossRef]
- Favel, A.; Mattras, H.; Coletti-Previero, M.A.; Zwilling, R.; Robinson, E.A.; Castro, B. Protease inhibitors from Ecballium elaterium seeds. Int. J. Pept. Protein Res. 1989, 33, 202–208. [Google Scholar]
- Heitz, A.; Avrutina, O.; Le-Nguyen, D.; Diederichsen, U.; Hernandez, J.F.; Gracy, J.; Kolmar, H.; Chiche, L. Knottin cyclization: Impact on structure and dynamics. BMC Struct. Biol. 2008, 8, 54. [Google Scholar] [CrossRef]
- Clark, R.J.; Craik, D.J. Native chemical ligation applied to the synthesis and bioengineering of circular peptides and proteins. Biopolymers 2010, 94, 414–422. [Google Scholar] [CrossRef]
- Conlan, B.F.; Anderson, M.A. Circular micro-proteins and mechanisms of cyclization. Curr. Pharm. Des. 2011, 17, 4318–4328. [Google Scholar] [CrossRef]
- Craik, D.J. The folding of disulfide-rich proteins. Antioxid. Redox. Signal. 2011, 14, 61–64. [Google Scholar]
- Aboye, T.L.; Clark, R.J.; Burman, R.; Roig, M.B.; Craik, D.J.; Goransson, U. Interlocking disulfides in circular proteins: Toward efficient oxidative folding of cyclotides. Antioxid. Redox. Signal. 2011, 14, 77–86. [Google Scholar] [CrossRef]
- Schieck, A.; Muller, T.; Schulze, A.; Haberkorn, U.; Urban, S.; Mier, W. Solid-phase synthesis of the lipopeptide Myr-HBVpreS/2–78, A hepatitis B virus entry inhibitor. Molecules 2010, 15, 4773–4783. [Google Scholar] [CrossRef]
- Boulègue, C.; Musiol, H.J.; Prasas, V.; Moroder, L. Synthesis of cystine-rich peptides. Chem. Today 2006, 24, 24–36. [Google Scholar]
- Zhu, S.; Darbon, H.; Dyason, K.; Verdonck, F.; Tytgat, J. Evolutionary origin of inhibitor cystine knot peptides. FASEB J. 2003, 17, 1765–1767. [Google Scholar]
- Goransson, U.; Craik, D.J. Disulfide mapping of the cyclotide kalata B1: Chemical proof of the cystic cystine knot motif. J. Biol. Chem. 2003, 278, 48188–48196. [Google Scholar] [CrossRef]
- Daly, N.L.; Clark, R.J.; Craik, D.J. Disulfide folding pathways of cystine knot proteins: Tying the knot within the circular backbone of the cyclotides. J. Biol. Chem. 2003, 278, 6314–6322. [Google Scholar]
- Ireland, D.C.; Wang, C.K.; Wilson, J.A.; Gustafson, K.R.; Craik, D.J. Cyclotides as natural anti-HIV agents. Biopolymers 2008, 90, 51–60. [Google Scholar] [CrossRef]
- Craik, D.J.; Conibear, A.C. The chemistry of cyclotides. J. Org. Chem. 2011, 76, 4805–4817. [Google Scholar] [CrossRef]
- Austin, J.; Wang, W.; Puttamadappa, S.; Shekhtman, A.; Camarero, J.A. Biosynthesis and biological screening of a genetically encoded library based on the cyclotide MCoTI-I. ChemBioChem 2009, 10, 2663–2670. [Google Scholar] [CrossRef]
- Camarero, J.A.; Kimura, R.H.; Woo, Y.H.; Shekhtman, A.; Cantor, J. Biosynthesis of a fully functional cyclotide inside living bacterial cells. ChemBioChem 2007, 8, 1363–1366. [Google Scholar] [CrossRef]
- Kimura, R.H.; Tran, A.T.; Camarero, J.A. Biosynthesis of the cyclotide Kalata B1 by using protein splicing. Angew. Chem. Int. Ed. Engl. 2006, 45, 973–976. [Google Scholar] [CrossRef]
- Puttamadappa, S.S.; Jagadish, K.; Shekhtman, A.; Camarero, J.A. Backbone dynamics of cyclotide MCoTI-I free and complexed with trypsin. Angew. Chem. Int. Ed. Engl. 2010, 49, 7030–7034. [Google Scholar] [CrossRef]
- Avrutina, O.; Schmoldt, H.U.; Gabrijelcic-Geiger, D.; Wentzel, A.; Frauendorf, H.; Sommerhoff, C.P.; Diederichsen, U.; Kolmar, H. Head-to-tail cyclized cystine-knot peptides by a combined recombinant and chemical route of synthesis. ChemBioChem 2008, 9, 33–37. [Google Scholar] [CrossRef]
- Thongyoo, P.; Jaulent, A.M.; Tate, E.W.; Leatherbarrow, R.J. Immobilized protease-assisted synthesis of engineered cysteine-knot microproteins. ChemBioChem 2007, 8, 1107–1109. [Google Scholar] [CrossRef]
- Thongyoo, P.; Roque-Rosell, N.; Leatherbarrow, R.J.; Tate, E.W. Chemical and biomimetic total syntheses of natural and engineered MCoTI cyclotides. Org. Biomol. Chem. 2008, 6, 1462–1470. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar]
- Lu, W.; Starovasnik, M.A.; Kent, S.B. Total chemical synthesis of bovine pancreatic trypsin inhibitor by native chemical ligation. FEBS Lett. 1998, 429, 31–35. [Google Scholar] [CrossRef]
- Avrutina, O.; Schmoldt, H.-U.; Kolmar, H.; Diederichsen, U. Fmoc-Assisted Synthesis of a 29-Residue Cystine-Knot Trypsin Inhibitor Containing a Guaninyl Amino Acid at the P1-Position. Eur. J. Org. Chem. 2004, 2004, 4931–4935. [Google Scholar] [CrossRef]
- Cemazar, M.; Daly, N.L.; Haggblad, S.; Lo, K.P.; Yulyaningsih, E.; Craik, D.J. Knots in rings, The circular knotted protein Momordica cochinchinensis trypsin inhibitor-II folds via a stable two-disulfide intermediate. J. Biol. Chem. 2006, 281, 8224–8232. [Google Scholar]
- Park, J.H.; Carlin, K.P.; Wu, G.; Ilyin, V.I.; Kyle, D.J. Cysteine racemization during the Fmoc solid phase peptide synthesis of the Nav1.7-selective peptide—protoxin II. J. Pept. Sci. 2012, 18, 442–448. [Google Scholar]
- Wentzel, A.; Christmann, A.; Kratzner, R.; Kolmar, H. Sequence requirements of the GPNG beta-turn of the Ecballium elaterium trypsin inhibitor II explored by combinatorial library screening. J. Biol. Chem. 1999, 274, 21037–21043. [Google Scholar] [CrossRef]
- Eliasen, R.; Andresen, T.L.; Conde-Frieboes, K.W. Handling a tricycle: Orthogonal versus random oxidation of the tricyclic inhibitor cystine knotted peptide gurmarin. Peptides 2012, 37, 144–149. [Google Scholar] [CrossRef]
- Gowd, K.H.; Yarotskyy, V.; Elmslie, K.S.; Skalicky, J.J.; Olivera, B.M.; Bulaj, G. Site-specific effects of diselenide bridges on the oxidative folding of a cystine knot peptide, omega-selenoconotoxin GVIA. Biochemistry 2010, 49, 2741–2752. [Google Scholar] [CrossRef]
- Leta Aboye, T.; Clark, R.J.; Craik, D.J.; Goransson, U. Ultra-stable peptide scaffolds for protein engineering-synthesis and folding of the circular cystine knotted cyclotide cycloviolacin O2. ChemBioChem 2008, 9, 103–113. [Google Scholar] [CrossRef]
- Wong, C.T.; Taichi, M.; Nishio, H.; Nishiuchi, Y.; Tam, J.P. Optimal oxidative folding of the novel antimicrobial cyclotide from Hedyotis biflora requires high alcohol concentrations. Biochemistry 2011, 50, 7275–7283. [Google Scholar] [CrossRef]
- McNulty, J.C.; Jackson, P.J.; Thompson, D.A.; Chai, B.; Gantz, I.; Barsh, G.S.; Dawson, P.E.; Millhauser, G.L. Structures of the agouti signaling protein. J. Mol. Biol. 2005, 346, 1059–1070. [Google Scholar] [CrossRef]
- Sommerhoff, C.P.; Avrutina, O.; Schmoldt, H.U.; Gabrijelcic-Geiger, D.; Diederichsen, U.; Kolmar, H. Engineered cystine knot miniproteins as potent inhibitors of human mast cell tryptase beta. J. Mol. Biol. 2010, 395, 167–175. [Google Scholar] [CrossRef]
- Kimura, R.H.; Jones, D.S.; Jiang, L.; Miao, Z.; Cheng, Z.; Cochran, J.R. Functional mutation of multiple solvent-exposed loops in the Ecballium elaterium trypsin inhibitor-II cystine knot miniprotein. PLoS One 2011, 6, e16112. [Google Scholar]
- Silverman, A.P.; Kariolis, M.S.; Cochran, J.R. Cystine-knot peptides engineered with specificities for alpha(IIb)beta(3) or alpha(IIb)beta(3) and alpha(v)beta(3) integrins are potent inhibitors of platelet aggregation. J. Mol. Recognit. 2011, 24, 127–135. [Google Scholar] [CrossRef]
- Blind, M.; Kolmar, H. Polypeptides comprising a knottin protein moiety. Patent EP 1,958,957,A1, 18 February 2008. [Google Scholar]
- Fabritz, S.; Horner, S.; Konning, D.; Empting, M.; Reinwarth, M.; Dietz, C.; Glotzbach, B.; Frauendorf, H.; Kolmar, H.; Avrutina, O. From pico to nano: Biofunctionalization of cube-octameric silsesquioxanes by peptides and miniproteins. Org. Biomol. Chem. 2012, 10, 6287–6293. [Google Scholar]
- Merrifield, R.B. Solid-Phase Peptide Synthesis. 3. An Improved Synthesis of Bradykinin. Biochemistry 1964, 3, 1385–1390. [Google Scholar] [CrossRef]
- Kates, S.A.; Albericio, F. Solid-Phase Synthesis: A Practical Guide, 1st ed.; CRC Press: Boca Raton, FL, USA, 2000; Volume 1, p. 848. [Google Scholar]
- Chang, C.D.; Meienhofer, J. Solid-phase peptide synthesis using mild base cleavage of N alpha-fluorenylmethyloxycarbonylamino acids, exemplified by a synthesis of dihydrosomatostatin. Int. J. Pept. Protein Res. 1978, 11, 246–249. [Google Scholar]
- Robertson, D. US FDA approves new class of HIV therapeutics. Nat. Biotechnol. 2003, 21, 470–471. [Google Scholar] [CrossRef]
- Kappe, C.O.; Stadler, A. Microwaves in Organic and Medicinal Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; p. 409. [Google Scholar]
- Hibino, H.; Nishiuchi, Y. 4-Methoxybenzyloxymethyl group, a racemization-resistant protecting group for cysteine in Fmoc solid phase peptide synthesis. Org. Lett. 2012, 14, 1926–1929. [Google Scholar]
- Cemazar, M.; Gruber, C.W.; Craik, D.J. Oxidative folding of cyclic cystine knot proteins. Antioxid. Redox. Signal. 2008, 10, 103–111. [Google Scholar] [CrossRef]
- Daly, N.L.; Love, S.; Alewood, P.F.; Craik, D.J. Chemical synthesis and folding pathways of large cyclic polypeptides: Studies of the cystine knot polypeptide kalata B1. Biochemistry 1999, 38, 10606–10614. [Google Scholar] [CrossRef]
- Gunasekera, S.; Daly, N.L.; Clark, R.J.; Craik, D.J. Dissecting the oxidative folding of circular cystine knot miniproteins. Antioxid. Redox. Signal. 2009, 11, 971–980. [Google Scholar] [CrossRef]
- Muller, C.; Richter, S.; Rinas, U. Kinetics control preferential heterodimer formation of platelet-derived growth factor from unfolded A- and B-chains. J. Biol. Chem. 2003, 278, 18330–18335. [Google Scholar] [CrossRef]
- Cemazar, M.; Joshi, A.; Daly, N.L.; Mark, A.E.; Craik, D.J. The structure of a two-disulfide intermediate assists in elucidating the oxidative folding pathway of a cyclic cystine knot protein. Structure 2008, 16, 842–851. [Google Scholar] [CrossRef]
- Green, B.R.; Bulaj, G. Oxidative folding of conotoxins in immobilized systems. Protein Pept. Lett. 2006, 13, 67–70. [Google Scholar]
- Tam, J.P.; Lu, Y.A. A biomimetic strategy in the synthesis and fragmentation of cyclic protein. Protein Sci. 1998, 7, 1583–1592. [Google Scholar] [CrossRef]
- Steiner, A.M.; Bulaj, G. Optimization of oxidative folding methods for cysteine-rich peptides: A study of conotoxins containing three disulfide bridges. J. Pept. Sci. 2011, 17, 1–7. [Google Scholar]
- Isidro-Llobet, A.; Álvarez, M.; Albericio, F. Amino Acid-Protecting Groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef]
- Veber, D.F.; Milkowski, J.D.; Varga, S.L.; Denkewalter, R.G.; Hirschmann, R. Acetamidomethyl, A novel thiol protecting group for cysteine. J. Am. Chem. Soc. 1972, 94, 5456–5461. [Google Scholar]
- Zervas, L.; Photaki, I. On Cysteine and Cystine Peptides. I. New S-Protecting Groups for Cysteine. J. Am. Chem. Soc. 1962, 84, 3887–3897. [Google Scholar]
- Kamolkijkarn, P.; Prasertdee, T.; Netirojjanakul, C.; Sarnpitak, P.; Ruchirawat, S.; Deechongkit, S. Synthesis, Biophysical, And biological studies of wild-type and mutant psalmopeotoxins—anti-malarial cysteine knot peptides from Psalmopoeus cambridgei. Peptides 2010, 31, 533–540. [Google Scholar] [CrossRef]
- Raffa, R.B. Diselenium, instead of disulfide, bonded analogs of conotoxins: Novel synthesis and pharmacotherapeutic potential. Life Sci. 2010, 87, 451–456. [Google Scholar] [CrossRef]
- Dekan, Z.; Vetter, I.; Daly, N.L.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Alpha-Conotoxin ImI incorporating stable cystathionine bridges maintains full potency and identical three-dimensional structure. J. Am. Chem. Soc. 2011, 133, 15866–15869. [Google Scholar]
- Le-Nguyen, D.; Heitz, A.; Chiche, L.; el Hajji, M.; Castro, B. Characterization and 2D NMR study of the stable [9–21, 15–27] 2 disulfide intermediate in the folding of the 3 disulfide trypsin inhibitor EETI II. Protein Sci. 1993, 2, 165–174. [Google Scholar]
- Krause, S.; Schmoldt, H.U.; Wentzel, A.; Ballmaier, M.; Friedrich, K.; Kolmar, H. Grafting of thrombopoietin-mimetic peptides into cystine knot miniproteins yields high-affinity thrombopoietin antagonists and agonists. FEBS J. 2007, 274, 86–95. [Google Scholar] [CrossRef]
- Zhang, L.; Tam, J.P. Synthesis and Application of Unprotected Cyclic Peptides as Building Blocks for Peptide Dendrimers. J. Am. Chem. Soc. 1997, 119, 2363–2370. [Google Scholar]
- Camarero, J.A.; Muir, T.W. Chemoselective backbone cyclization of unprotected peptides. Chem. Commun. 1997, 1369–1370. [Google Scholar] [CrossRef]
- Camarero, J.A.; Cotton, G.J.; Adeva, A.; Muir, T.W. Chemical ligation of unprotected peptides directly from a solid support. J. Pept. Res. 1998, 51, 303–316. [Google Scholar]
- Tam, J.P.; Lu, Y.-A.; Yu, Q. Thia Zip Reaction for Synthesis of Large Cyclic Peptides: Mechanisms and Applications. J. Am. Chem. Soc. 1999, 121, 4316–4324. [Google Scholar] [CrossRef]
- Park, S.; Gunasekera, S.; Aboye, T.; Göransson, U. An Efficient Approach for the Total Synthesis of Cyclotides by Microwave Assisted Fmoc-SPPS. Int. J. Pept. Res. Ther. 2010, 16, 167–176. [Google Scholar] [CrossRef]
- Camarero, J.A.; Mitchell, A.R. Synthesis of proteins by native chemical ligation using Fmoc-based chemistry. Protein Pept. Lett. 2005, 12, 723–728. [Google Scholar] [CrossRef]
- Clippingdale, A.B.; Barrow, C.J.; Wade, J.D. Peptide thioester preparation by Fmoc solid phase peptide synthesis for use in native chemical ligation. J. Pept. Sci. 2000, 6, 225–234. [Google Scholar] [CrossRef]
- Ali, A.M.; Taylor, S.D. Efficient solid-phase synthesis of sulfotyrosine peptides using a sulfate protecting-group strategy. Angew. Chem. Int. Ed. Engl. 2009, 48, 2024–2026. [Google Scholar] [CrossRef]
- Camarero, J.A.; Hackel, B.J.; de Yoreo, J.J.; Mitchell, A.R. Fmoc-based synthesis of peptide alpha-thioesters using an aryl hydrazine support. J. Org. Chem. 2004, 69, 4145–4151. [Google Scholar] [CrossRef]
- Woo, Y.-H.; Mitchell, A.; Camarero, J. The Use of Aryl Hydrazide Linkers for the Solid Phase Synthesis of Chemically Modified Peptides. Int. J. Pept. Res. Ther. 2007, 13, 181–190. [Google Scholar] [CrossRef]
- Haase, C.; Seitz, O. Internal Cysteine Accelerates Thioester-Based Peptide Ligation. Eur. J. Org. Chem. 2009, 2009, 2096–2101. [Google Scholar] [CrossRef]
- Camarero, J.A.; Pavel, J.; Muir, T.W. Chemical Synthesis of a Circular Protein Domain: Evidence for Folding-Assisted Cyclization. Angew. Chem. Int. Ed. Engl. 1998, 37, 347–349. [Google Scholar] [CrossRef]
- Schmoldt, H.U.; Wentzel, A.; Becker, S.; Kolmar, H. A fusion protein system for the recombinant production of short disulfide bond rich cystine knot peptides using barnase as a purification handle. Protein Expr. Purif. 2005, 39, 82–89. [Google Scholar] [CrossRef]
- Cascales, L.; Henriques, S.T.; Kerr, M.C.; Huang, Y.H.; Sweet, M.J.; Daly, N.L.; Craik, D.J. Identification and characterization of a new family of cell-penetrating peptides: Cyclic cell-penetrating peptides. J. Biol. Chem. 2011, 286, 36932–36943. [Google Scholar]
- Lovelace, E.S.; Armishaw, C.J.; Colgrave, M.L.; Wahlstrom, M.E.; Alewood, P.F.; Daly, N.L.; Craik, D.J. Cyclic MrIA: A stable and potent cyclic conotoxin with a novel topological fold that targets the norepinephrine transporter. J. Med. Chem. 2006, 49, 6561–6568. [Google Scholar] [CrossRef]
- Edman, P. A method for the determination of amino acid sequence in peptides. Arch. Biochem. 1949, 22, 475. [Google Scholar]
- Kratzner, R.; Debreczeni, J.E.; Pape, T.; Schneider, T.R.; Wentzel, A.; Kolmar, H.; Sheldrick, G.M.; Uson, I. Structure of Ecballium elaterium trypsin inhibitor II (EETI-II): A rigid molecular scaffold. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 1255–1262. [Google Scholar] [CrossRef]
- Craik, D.J.; Swedberg, J.E.; Mylne, J.S.; Cemazar, M. Cyclotides as a basis for drug design. Expert Opin. Drug Discov. 2012, 7, 179–194. [Google Scholar] [CrossRef]
- Contreras, J.; Elnagar, A.Y.; Hamm-Alvarez, S.F.; Camarero, J.A. Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J. Control. Release 2011, 155, 134–143. [Google Scholar] [CrossRef]
- Reiss, S.; Sieber, M.; Oberle, V.; Wentzel, A.; Spangenberg, P.; Claus, R.; Kolmar, H.; Losche, W. Inhibition of platelet aggregation by grafting RGD and KGD sequences on the structural scaffold of small disulfide-rich proteins. Platelets 2006, 17, 153–157. [Google Scholar] [CrossRef]
- Getz, J.A.; Rice, J.J.; Daugherty, P.S. Protease-resistant peptide ligands from a knottin scaffold library. ACS Chem. Biol. 2011, 6, 837–844. [Google Scholar] [CrossRef]
- Tanimoto, H.; Yan, Y.; Clarke, J.; Korourian, S.; Shigemasa, K.; Parmley, T.H.; Parham, G.P.; O'Brien, T.J. Hepsin, A cell surface serine protease identified in hepatoma cells, Is overexpressed in ovarian cancer. Cancer Res. 1997, 57, 2884–2887. [Google Scholar]
- Leytus, S.P.; Loeb, K.R.; Hagen, F.S.; Kurachi, K.; Davie, E.W. A novel trypsin-like serine protease (hepsin) with a putative transmembrane domain expressed by human liver and hepatoma cells. Biochemistry 1988, 27, 1067–1074. [Google Scholar]
- Saleem, M.; Adhami, V.M.; Zhong, W.; Longley, B.J.; Lin, C.Y.; Dickson, R.B.; Reagan-Shaw, S.; Jarrard, D.F.; Mukhtar, H. A novel biomarker for staging human prostate adenocarcinoma: overexpression of matriptase with concomitant loss of its inhibitor, hepatocyte growth factor activator inhibitor-1. Cancer Epidemiol. Biomark. Prev. 2006, 15, 217–227. [Google Scholar] [CrossRef]
- Lin, C.Y.; Anders, J.; Johnson, M.; Dickson, R.B. Purification and characterization of a complex containing matriptase and a Kunitz-type serine protease inhibitor from human milk. J. Biol. Chem. 1999, 274, 18237–18242. [Google Scholar] [CrossRef]
- Jiang, L.; Miao, Z.; Kimura, R.H.; Silverman, A.P.; Ren, G.; Liu, H.; Lu, H.; Cochran, J.R.; Cheng, Z. 111In-labeled cystine-knot peptides based on the Agouti-related protein for targeting tumor angiogenesis. J. Biomed. Biotechnol. 2012, 2012, 368075. [Google Scholar]
- Martin, L.; Stricher, F.; Misse, D.; Sironi, F.; Pugniere, M.; Barthe, P.; Prado-Gotor, R.; Freulon, I.; Magne, X.; Roumestand, C.; et al. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat. Biotechnol. 2003, 21, 71–76. [Google Scholar] [CrossRef]
- Stricher, F.; Huang, C.C.; Descours, A.; Duquesnoy, S.; Combes, O.; Decker, J.M.; Kwon, Y.D.; Lusso, P.; Shaw, G.M.; Vita, C.; et al. Combinatorial optimization of a CD4-mimetic miniprotein and cocrystal structures with HIV-1 gp120 envelope glycoprotein. J. Mol. Biol. 2008, 382, 510–524. [Google Scholar] [CrossRef]
- Vita, C.; Drakopoulou, E.; Vizzavona, J.; Rochette, S.; Martin, L.; Menez, A.; Roumestand, C.; Yang, Y.S.; Ylisastigui, L.; Benjouad, A.; et al. Rational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 1999, 96, 13091–13096. [Google Scholar]
- Drakopoulou, E.; Vizzavona, J.; Vita, C. Engineering a CD4 mimetic inhibiting the binding of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein gp120 to human lymphocyte CD4 by the transfer of a CD4 functional site to a small natural scaffold. Lett. Pept. Sci. 1998, 5, 241–245. [Google Scholar]
- Gunasekera, S.; Foley, F.M.; Clark, R.J.; Sando, L.; Fabri, L.J.; Craik, D.J.; Daly, N.L. Engineering stabilized vascular endothelial growth factor-A antagonists: Synthesis, Structural Characterization, And bioactivity of grafted analogues of cyclotides. J. Med. Chem. 2008, 51, 7697–7704. [Google Scholar] [CrossRef]
- Pranting, M.; Loov, C.; Burman, R.; Goransson, U.; Andersson, D.I. The cyclotide cycloviolacin O2 from Viola odorata has potent bactericidal activity against Gram-negative bacteria. J. Antimicrob. Chemother. 2010, 65, 1964–1971. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reinwarth, M.; Nasu, D.; Kolmar, H.; Avrutina, O. Chemical Synthesis, Backbone Cyclization and Oxidative Folding of Cystine-knot Peptides — Promising Scaffolds for Applications in Drug Design. Molecules 2012, 17, 12533-12552. https://doi.org/10.3390/molecules171112533
Reinwarth M, Nasu D, Kolmar H, Avrutina O. Chemical Synthesis, Backbone Cyclization and Oxidative Folding of Cystine-knot Peptides — Promising Scaffolds for Applications in Drug Design. Molecules. 2012; 17(11):12533-12552. https://doi.org/10.3390/molecules171112533
Chicago/Turabian StyleReinwarth, Michael, Daichi Nasu, Harald Kolmar, and Olga Avrutina. 2012. "Chemical Synthesis, Backbone Cyclization and Oxidative Folding of Cystine-knot Peptides — Promising Scaffolds for Applications in Drug Design" Molecules 17, no. 11: 12533-12552. https://doi.org/10.3390/molecules171112533
APA StyleReinwarth, M., Nasu, D., Kolmar, H., & Avrutina, O. (2012). Chemical Synthesis, Backbone Cyclization and Oxidative Folding of Cystine-knot Peptides — Promising Scaffolds for Applications in Drug Design. Molecules, 17(11), 12533-12552. https://doi.org/10.3390/molecules171112533