Abstract
The matrix metalloproteinases (MMPs) exhibit a broad array of activities, some catalytic and some non-catalytic in nature. An overall lack of selectivity has rendered small molecule, active site targeted MMP inhibitors problematic in execution. Inhibitors that favor few or individual members of the MMP family often take advantage of interactions outside the enzyme active site. We presently focus on peptide-based MMP inhibitors and probes that do not incorporate conventional Zn2+ binding groups. In some cases, these inhibitors and probes function by binding only secondary binding sites (exosites), while others bind both exosites and the active site. A myriad of MMP mediated-activities beyond selective catalysis can be inhibited by peptides, particularly cell adhesion, proliferation, motility, and invasion. Selective MMP binding peptides comprise highly customizable, unique imaging agents. Areas of needed improvement for MMP targeting peptides include binding affinity and stability.
1. Introduction
Matrix metalloproteinases (MMPs) are part of a super family of zinc-dependent endopeptidases [1]. There are at least 23 human MMPs [2,3,4]. Most MMPs share a common domain organization, consisting of propeptide, catalytic (CAT), linker/hinge, and hemopexin-like (HPX) domains [2]. MMP-1 (Figure 1) is an example of this prototype. Two members of the MMP family possess three fibronectin type II [FN(II)] inserts in their CAT domain. These MMPs [MMP-2 (Figure 2) and MMP-9 (Figure 3)] are often referred to as “gelatinases” due to their initial identification as proteases capable of catalyzing the degradation of denatured collagen (gelatin). Several MMP family members have transmembrane domains that facilitate cell surface binding, and thus these MMPs are referred to as membrane-type MMPs (MT-MMPs). MMP-14/MT1-MMP (Figure 4) is representative of the MT-MMPs.
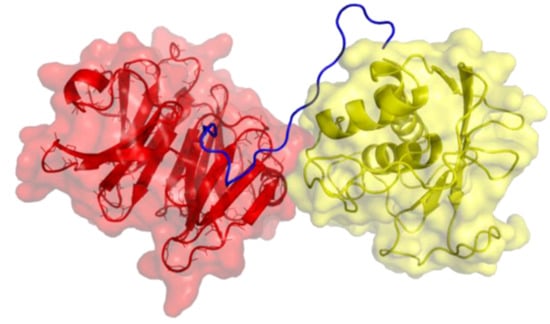
Figure 1.
Three-dimensional structure of MMP-1. The catalytic (CAT) domain is yellow, the linker/hinge is blue, and the hemopexin-like (HPX) domain is red. The structure is based on X-ray crystallographic analyses of full-length MMP-1 [5,6].
Figure 1.
Three-dimensional structure of MMP-1. The catalytic (CAT) domain is yellow, the linker/hinge is blue, and the hemopexin-like (HPX) domain is red. The structure is based on X-ray crystallographic analyses of full-length MMP-1 [5,6].
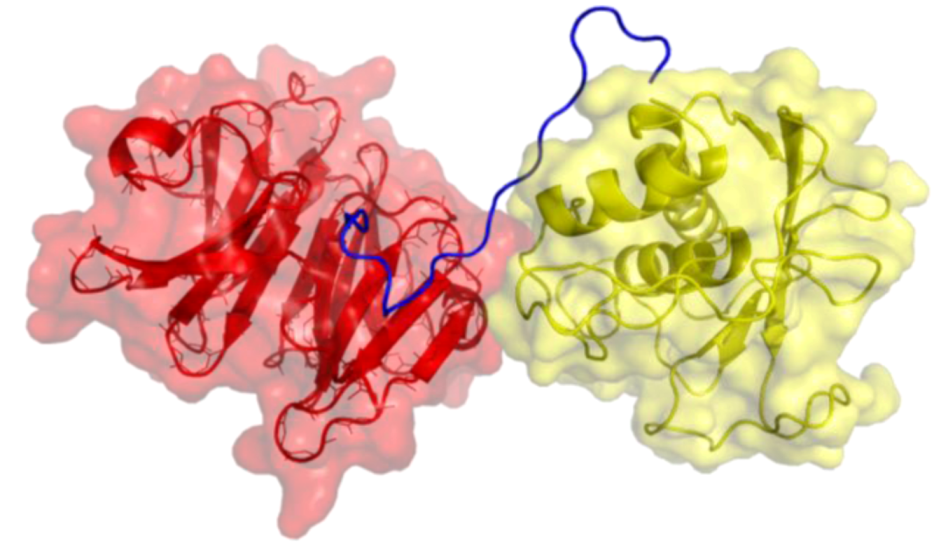
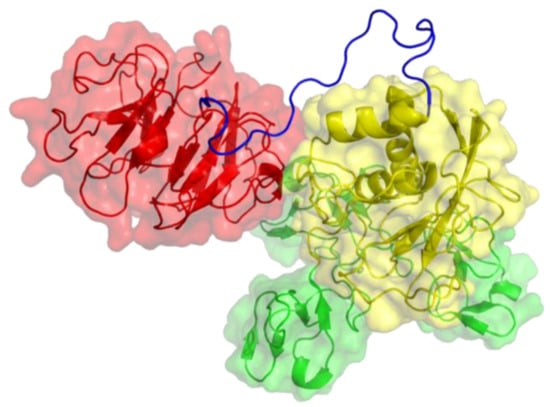
Figure 2.
Three-dimensional structure of MMP-2. The catalytic (CAT) domain is yellow, the linker/hinge is blue, the hemopexin-like (HPX) domain is red, and the fibronectin type II [FN(II)] inserts are green. The structure is based on X-ray crystallographic analysis of full-length MMP-2 [7].
Figure 2.
Three-dimensional structure of MMP-2. The catalytic (CAT) domain is yellow, the linker/hinge is blue, the hemopexin-like (HPX) domain is red, and the fibronectin type II [FN(II)] inserts are green. The structure is based on X-ray crystallographic analysis of full-length MMP-2 [7].
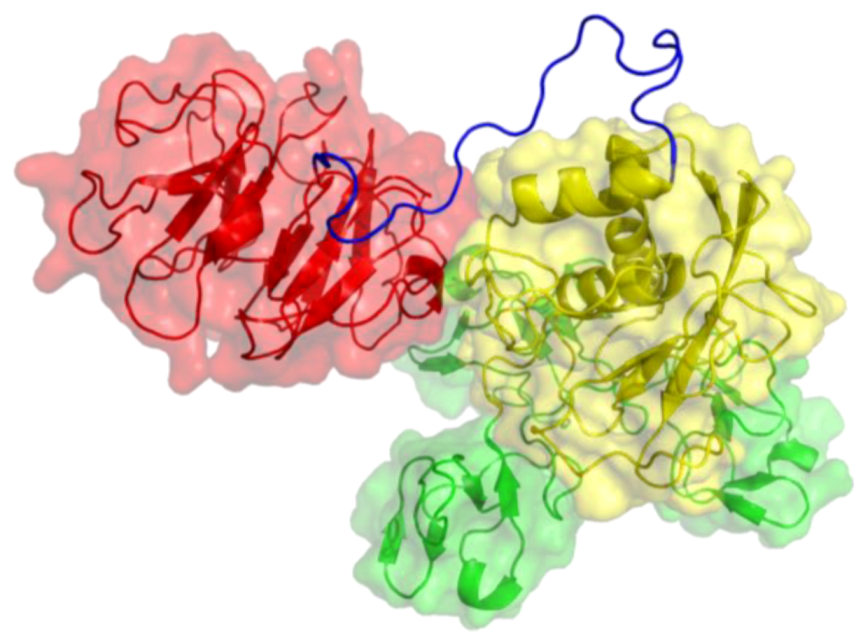
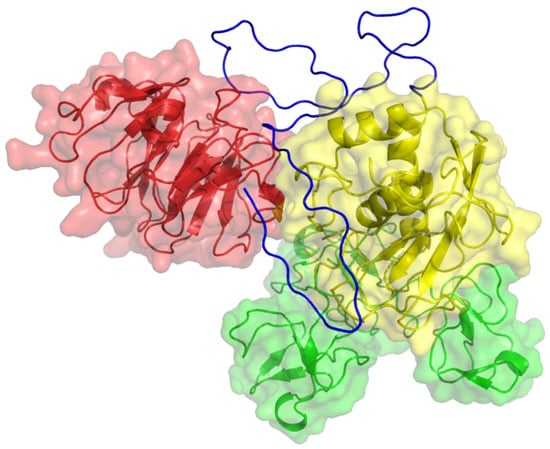
Figure 3.
Three-dimensional structure of MMP-9. The catalytic (CAT) domain is yellow, the linker/hinge is blue, the hemopexin-like (HPX) domain is red, and the fibronectin type II [FN(II)] inserts are green. The structure is based on X-ray crystallographic analyses of MMP-9 CAT and HPX domains [8,9].
Figure 3.
Three-dimensional structure of MMP-9. The catalytic (CAT) domain is yellow, the linker/hinge is blue, the hemopexin-like (HPX) domain is red, and the fibronectin type II [FN(II)] inserts are green. The structure is based on X-ray crystallographic analyses of MMP-9 CAT and HPX domains [8,9].
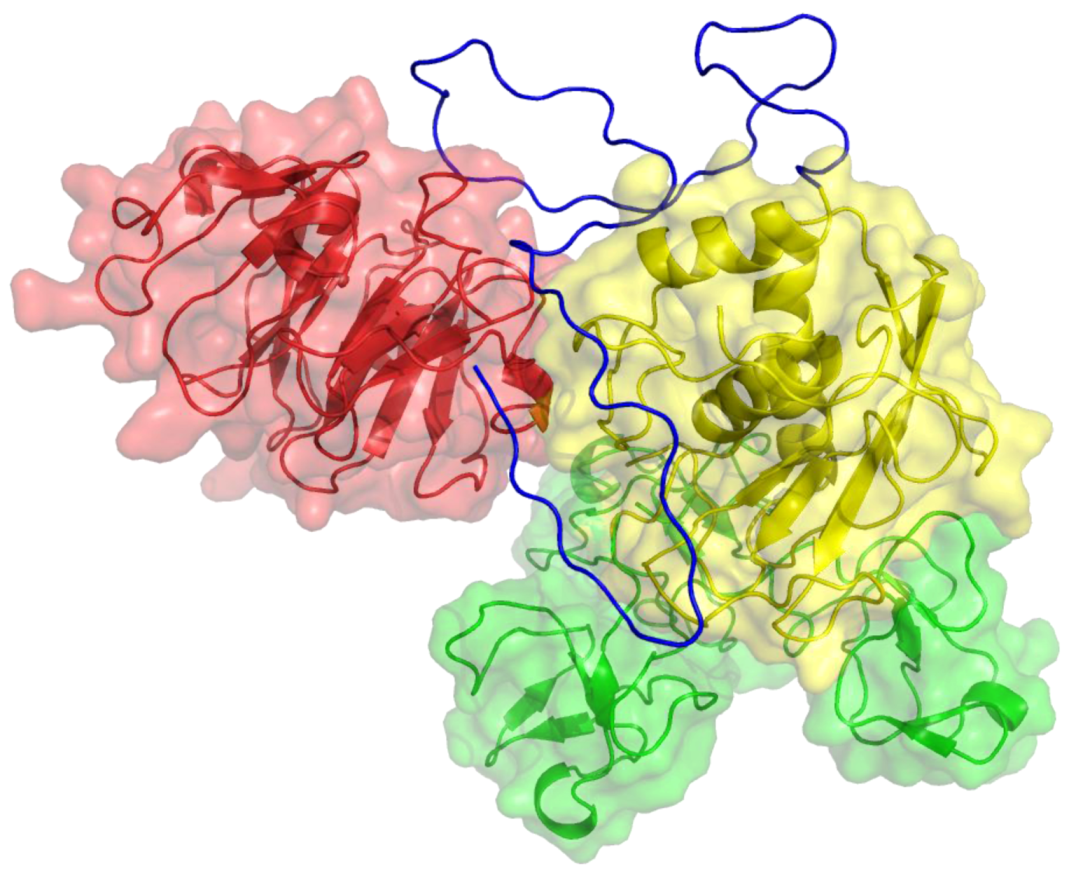
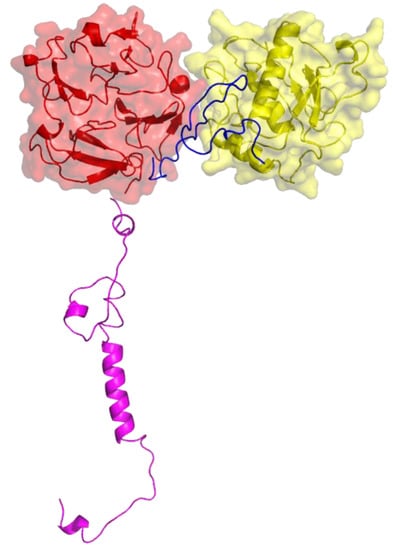
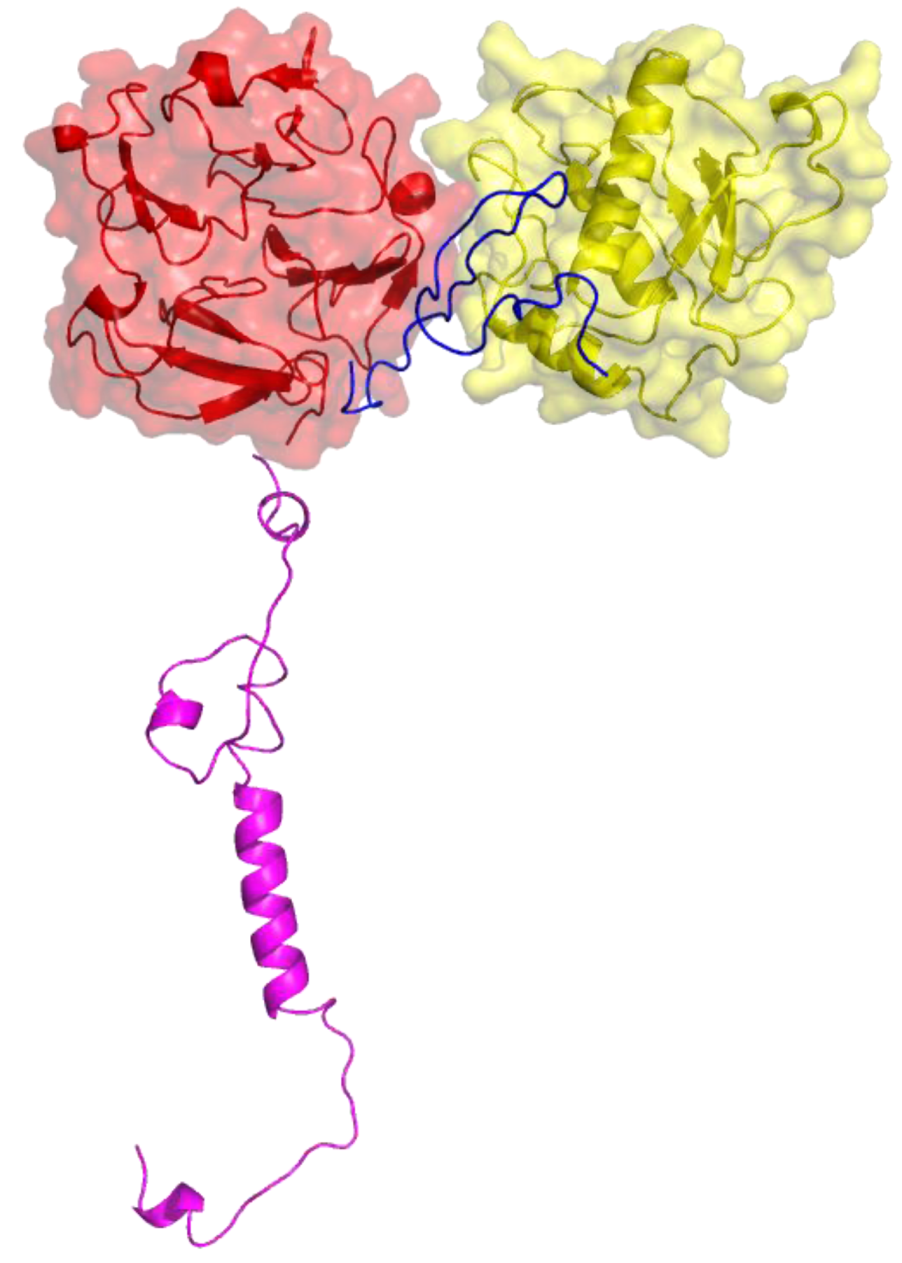
Figure 4.
Three-dimensional structure of MT1-MMP. The catalytic (CAT) domain is yellow, the linker/hinge is blue, the hemopexin-like (HPX) domain is red, and the transmembrane and cytoplasmic domains are purple. The structure is based on X-ray crystallographic analyses of MT1-MMP CAT and HPX domains [10,11] and modeling of the transmembrane and cytoplasmic domains by I-TASSER [12,13].
Figure 4.
Three-dimensional structure of MT1-MMP. The catalytic (CAT) domain is yellow, the linker/hinge is blue, the hemopexin-like (HPX) domain is red, and the transmembrane and cytoplasmic domains are purple. The structure is based on X-ray crystallographic analyses of MT1-MMP CAT and HPX domains [10,11] and modeling of the transmembrane and cytoplasmic domains by I-TASSER [12,13].
MMPs have long been recognized as potential targets for a variety of pathologies, including tumor angiogenesis and metastasis, osteoarthritis, inflammation, periodontitis, vascular diseases, post-myocardial infarction remodeling, neurodegenerative diseases, and neuropsychiatric disorders [14,15,16,17,18,19]. The development of MMP inhibitors has typically proceeded along the path of active site Zn2+ inhibition. The most common zinc-binding group used for this purpose is hydroxamic acid [20,21]. However, a primary reason hydroxamic acid-based inhibitors have not been successful in clinic trials is their lack of selectivity [21,22]. More recent work involves developing inhibitors that bind secondary binding sites (exosites) to pursue greater selectivity [23,24,25]. The present overview considers the use of peptides as MMP inhibitors and probes. Our discussion is restricted to peptides that interact with secondary binding sites of MMPs and/or exclude traditional zinc-interaction motifs.
2. Peptides That Target Gelatinase Members of the MMP Family (MMP-2 and MMP-9)
MMP-2 has been validated as an anticancer drug target in some aggressive tumors, while MMP-9 inhibition may have value in treating patients with early stage cancers [26]. MMP-9 has also been strongly implicated as a target for aberrant left ventricle remodeling post-myocardial infarction and multiple sclerosis progression [27,28]. Koivunen et al. used phage display peptide libraries to identify selective MMP-2 and MMP-9 inhibitors [29]. Peptide libraries such as CX9, CX5-8C, CX3CX4CX2C, etc. were displayed on filamentous phage and tested for their MMP inhibitory activity. From the screened and constructed peptides two cyclic sequences, CTTHWGFTLC (designated CTT; Table 1) and CRRHWGFEFC that contained a common HWGF motif, were found to be potent inhibitors of MMP-2 and MMP-9, respectively. The NMR-derived solution structure of CTT indicated a well-defined, saddle-shaped circular form [30]. In a gelatin degradation assay CTT inhibited MMP-2 with an IC50 value of 10 μM while CRRHWGFEFC preferentially inhibited MMP-9 relative to MMP-2 [29]. The latter peptide was not fully characterized due to solubility problems. Inhibitory effects were also studied for casein degradation, where CTT had an IC50 value of 5 μM for MMP-2 [29]. CTT did not inhibit MT1-MMP, MMP-8, or MMP-13 at concentrations up to 500 μM.

Table 1.
Peptide-based Inhibitors of MMP-Mediated Activities. ND = not determined.
| Enzyme Affected | Peptide Sequence | IC50 (μM) | Reference |
|---|---|---|---|
| MMP-2 | CTTHWGFTLC (CTT) | 5–10 | [29] |
| “ | H-β3-Phe-β-Ala-β3-Trp-β3-His-OH | 225 | [31] |
| “ | HWWQWPSSLQLRGGGS (M204C4) | 78.0 | [32] |
| “ | HNWTRWLLHPDRGGGS (M205C4) | 38.8 | [32] |
| “ | ISYGNDALMP (APP-IP) | 0.030 | [33,34] |
| “ | MCMPCFTTDHQMARKCDDCCGGKGRGKCYGPQCLCR (Cltx) | 0.200 | [35] |
| “ | CGAOGAOGSQGA (P713) | 30 | [36] |
| MMP-9 | CRRHWGFEFC | ND | [29] |
| “ | GACLRSGRGCG (TCTP-1) | ND | [37] |
| “ | NQVDQVGY (IVS4) | 50 a | [38] |
| “ | SRPQGPFL (IS4) | 12 a | [38] |
| “ | FPGVPLDTHDVFQYREK (P3a) | 109–279 a | [39] |
| “ | CQVTGALRSGRGKMLLC-NH2 (cyclic LRSG) | ND | [40] |
| “ | CRVYGPYLLC | 200 b | [41] |
| PRCBCGE (Regasepin1) | ~1 | [42,43] | |
| “ | RC-[D-B]-[D-R] | 0.75 | [44] |
| “ | NENLLRFFVAPFPEVFG | 50 | [45] |
| “ | CSCSDMTDKECLYFCMSEMS (STX-S4-CT) | 1.0 d | [46] |
| MT1-MMP | ISYGNDALMP (APP-IP) | 2.0 | [33,34] |
| “ | GACFSIAHECGA (Peptide G) | 150 | [47] |
| “ | AHQLH | 165 c | [48] |
| “ | acetyl-VMDGYPMP-NH2 (IS4) | ND | [49] |
| “ | acetyl-GYPKSALR-NH2 (IVS4) | ND | [49] |
| “ | VFDEASLEP | 238 | Present study |
| MMP-1 | CSCSDMTDKECLYFCMSEMS (STX-S4-CT) | 4.5 d | [46] |
a IC50 for cell migration; b IC50 for cell invasion; c IC50 for cell proliferation; d Ki value.
Using a transwell assay, CTT was shown to block migration of HT1080 fibrosarcoma, C8161 melanoma, SKOV-3 ovarian carcinoma, and KS1767 Kaposi sarcoma cell lines at significant levels in a dose dependent manner [29]. The cell migration of two endothelial cells (human Eahy926 and HUVEC) was also inhibited by the sequence. Control peptides did not affect cell migration. Due to the lack of a typical gelatinase substrate sequence, it was speculated that within the HWGF motif the Trp residue bound to the hydrophobic pocket of the enzyme while the His residue acted as a ligand for the catalytic zinc ion [29]. Complementing the CTT inhibitory properties, this sequence could be used to target tumors selectively as MMPs are over expressed in the tumor vasculature [29].
CTT has been utilized for radiopharmaceutical and imaging applications. 125I-[D-Tyr]-CTT exhibited an IC50 = 5–10 µM for MMP-2 [50]. Biodistribution studies in mice showed a tumor/blood ratio of 0.83 and tumor/organ ratios of 2.0, 2.56, and 6.6 for heart, muscle, and brain, respectively, at 120 min [50]. Large initial accumulation was observed in the liver and kidneys [50]. The modest tumor uptake and high concentrations in the liver and kidney was attributed to the high lipophilicity of 125I-[D-Tyr]-CTT. Ligand affinity may also have been diminished due to its monomeric nature, as opposed to the multivalent ligand presented in phage display. 125I-[D-Tyr]-CTT was 75% intact in serum after 10 min, with some release of 125I-D-Tyr [50]. Complete degradation occurred by 60 min.
125I-Ala-Ala-Tyr-CTT and 99mTc-CTT labeled CTT analogs have been prepared, where the first had the same MMP-2 inhibitory potency as the parent compound while the second was slightly less potent [30]. In mice bearing KS1767 Kaposi’s sarcoma xenografts, 125I-Ala-Ala-Tyr-CTT localized to the tumor, while 99mTc-CTT labeled liposomes were used for tumor imaging [30]. CTT coated liposomes had enhanced tumor binding compared with unmodified liposomes, but the blood, kidney, and liver had high levels of label.
64Cu-DOTA-CTT was found to inhibit MMP-2 (EC50 = 8.7 µM) and MMP-9 (EC50 = 18.2 µM) with similar affinity as CTT (EC50 = 13.2 and 11.0 µM, respectively), but in vivo was a poor tumor imaging agent due to its low affinity and instability [51]. 111In-DTPA-CTT (IC50 = 1026 µM for MMP-2) was found to accumulate in tumors possessing gelatinase activity, but offered low tumor contrast [52]. The stability of 111In-DTPA-CTT was ~85% in mouse serum after 3 h at 37 °C, so the affinity of the peptide appeared more problematic than the stability. Fusion of CTT with green fluorescent protein (GFP) allowed for fluorescence imaging of tongue carcinoma in vivo [53].
Incorporation of D-amino acids and certain secondary structural elements can help stabilize peptides against proteolytic degradation in vivo. A retro-inversion CTT, cyclic D-Cys-D-Leu-D-Thr-D-Phe-Gly-D-Trp-D-His-D-Thr-D-Thr-D-Cys, inhibited MMP-2 more efficiently than CTT [30]. Unfortunately, this peptide had limited water solubility. β-peptides have been shown to form stable secondary structures thereby emerging as a promising class of peptidomimetics [54,55,56,57]. The linear β-tetra peptide H-β3-Phe-β-Ala-β3-Trp-β3-His-OH was designed to mimic CTT. Mukai et al. found that H-β3-Phe-β-Ala-β3-Trp-β3-His-OH inhibited MMP-2 with an IC50 value of 225 µM, in comparison to CTT IC50 of 212 µM (as determined herein) [31]. Linear α-tetrapeptide had no significant contribution towards MMP inhibition, with an IC50 value of >1 mM [31]. While H-β3-Phe-β-Ala-β3-Trp-β3-His-OH has a similar affinity for MMP-2 as CTT, the new class of β-tetrapeptide is smaller in size and generally more stable, thereby enhancing its accumulation in target tissues.
Another peptide obtained from the above described phage display studies [29] was refined into an MMP-9 imaging agent [37]. Three modalities of the peptide TCTP-1 (GACLRSGRGCG) were produced; the linear form, a version with a disulfide bond between Cys3 and Cys10, and a version with a lactam. All three peptides had PEG(3)-DOTA added to the C-terminus and were labeled with 68Ga. The disulfide-bonded and lactam forms of TCTP-1 showed reasonable plasma stability, whereas the linear TCTP-1 was rapidly degraded. In a human melanoma xenograft model in rats, the disulfide-bonded TCTP-1 exhibited better tumor uptake than lactam TCTP-1. PET imaging demonstrated TCTP-1 uptake in the tumor, heart, liver, kidney, and urinary bladder. Overall, TCTP-1 tumor uptake was low, with weak correlation to MMP-9 expression in melanoma tumors.
A random 12-mer peptide phage display library composed of ~2.7 × 109 sequences was screened for binding to full-length MMP-2 [32]. Twenty two clones were isolated as binders, and synthetic peptides corresponding to the binding sequences were synthesized and tested as MMP-2 inhibitors. Peptides M204C4 (HWWQWPSSLQLRGGGS) and M205C4 (HNWTRWLLHPDRGGGS) were found to inhibit MMP-2 hydrolysis of Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 with IC50 values of 78.0 and 38.8 nM, respectively. Both peptides inhibited pancreatic tumor cell invasion of Matrigel and reduced tumor growth in a human xenograft pancreatic tumor mouse model. However, neither peptide was stable in plasma after 10 min, requiring local use of the peptides for tumor treatment. Cyclic versions of each peptide were much less effective MMP-2 inhibitors.
Amyloid precursor protein (APP) is an integral membrane protein expressed in many tissues and concentrated in the neurons. After APP is proteolytically cleaved, a soluble extracellular domain containing an MMP-2 inhibitor is released and is believed to help protect the extracellular matrix from MMP-2 degradation [58,59,60,61]. The β-amyloid precursor protein-derived inhibitory peptide (APP-IP; sequence ISYGNDALMP), corresponding to residues 586–595 of APP770, is a highly selective MMP-2 inhibitor, where selectivity is based on unique structural features [33,34]. An IC50 value of 30 nM was observed for MMP-2, while MMP-9 and MT1-MMP IC50 values were in the μM range. Tyr588, Asp591, and Leu593 were shown to be important residues for optimal interaction between MMP-2 and the peptide. The APP-IP Asp6 carboxylate group coordinated the Zn2+ in the CAT domain of the enzyme; substitution with other residues leads to loss of inhibitory activity [34,62]. The inhibitor bound in the inverse direction as substrate.
Deshane et al. have shown that chlorotoxin (Cltx), a 36-amino acid peptide that was originally isolated from Leiurus quinquestriatus venom, inhibits the enzymatic activity of MMP-2 and causes a reduction in the surface expression of MMP-2 in glioma cells [63]. Immunohistochemical studies on Cltx revealed specific and selective binding of Cltx peptide to glioma cells but not to normal brain cells [64,65]. The remarkable specificity lead to the isolation and identification of a Cltx membrane receptor, which was MMP-2, co-purified as a major component of a stable macromolecular complex comprised of MMP-2, αvβ3 integrin, MT1-MMP, and tissue inhibitor of metalloproteinase 2 (TIMP-2) [35]. A stoichiometric balance was required to facilitate tumor cell migration and invasion, where MT1-MMP activated MMP-2 and the αvβ3 integrin promoted its maturation and release [66]. Western blotting analysis of Cltx treated with purified recombinant MMP-2, MMP-9, MMP-1, and MMP-3 resulted in only MMP-2 binding [35]. A dose-response curve was observed for MMP-2 inhibition, resulting in an IC50 of ~200 nM with complete inhibition in the presence of 300 nM Cltx. The Cltx binding site on MMP-2 is unknown. Cltx is a highly attractive therapeutic due to multiple tumors that express MMP-2, and the FDA has approved the use of Cltx in a Phase I/II clinical trial [65]. Cltx also has potential as an imaging agent; it been conjugated with Cy5.5 to allow imaging of glioma, medullolbastoma, prostate cancer, intestinal cancer, and sarcoma from adjacent non-neoplastic tissue in mouse models [67].
Unlike other MMPs, MMP-2 and MMP-9 have a collagen-binding domain (CBD) formed by three FN(II) modules. The CBD is the primary site of interaction for multiple collagens, gelatin, and elastin with MMP-2 and MMP-9 [68,69,70,71,72]. CBD-deleted mutants of MMP-2 had a 90% reduction in the rate of gelatin hydrolysis [73]. Shipley et al. demonstrated the inability of MMP-2 and MMP-9 to cleave elastin upon deletion of CBDs [70]. Using a combinatorial one peptide one bead library, Xu et al. determined that the CBD binds a short segment of the α1(I) collagen chain [36]. More specifically, CGAOGAOGSQGA (P713, where O = 4-hydroxyproline) was identified as an inhibitor of MMP-2 activity. P713 inhibited ~90% of MMP-2 gelatin cleavage (IC50 of ~30 µM), but less than 20% of the MMP-2 activity on a peptide substrate [NFF-1; Mca-Pro-Lys-Pro-Gln-Gln~Phe-Phe-Gly~Leu-Lys(Dnp)-Gly] which does not require the CBD for binding. To examine the specificity toward MMP-2, comparative inhibition assays were performed with MMP-8, with no alteration in MMP-8 activities observed upon P713 treatment [36].
Based on the single-stranded peptide model of the α1(I)715-721 collagen sequence identified above as a ligand for the MMP-2 FN(II) insert, our group assembled a triple-helical version of this ligand [α1(I)715-721 THP; (GPO)4-GAOGAOGSQGAO-(GPO)3-GPY-NH2] [74]. α1(I)715-721 THP inhibited MMP-2 and MMP-9 hydrolysis of α1(V)436-447 fTHP (Table 2), but did not inhibit MMP-2 or MMP-9 hydrolysis of a short, single-stranded substrate [Knight SSP; Mca-Lys-Pro-Leu-Gly-Leu-Lys(Dnp)-Ala-Arg-NH2] or a THP model of types I-III collagen (fTHP-15). To our knowledge, this demonstrated the first use of an exosite binder to selectively inhibit one collagen-based MMP activity (type V) but not another (types I–III).
Table 2.
Inhibition of MMPs by α1(I)715-721 THP [74]. NI = no inhibition.
| Enzyme | Substrate | Ki(app) (μM) |
|---|---|---|
| MMP-2 | DQ gelatin | 52.26 ± 5.110 |
| “ | Knight SSP a | NI |
| “ | fTHP-15 b | NI |
| “ | α1(V)436-447 fTHP c | 143.5 ± 11.40 |
| MMP-9 | DQ gelatin | 54.42 ± 7.616 |
| “ | Knight SSP a | NI |
| “ | fTHP-15 b | NI |
| “ | α1(V)436-447 fTHP c | 122.7 ± 5.83 |
a Knight SSP = Mca-Lys-Pro-Leu-Gly-Leu-Lys(Dnp)-Ala-Arg-NH2; b fTHP-15 = (Gly-Pro-Hyp)5-Gly-Pro-Lys(Mca)-Gly-Pro-Gln-Gly~Leu-Arg-Gly-Gln-Lys(Dnp)-Gly-Val-Arg-(Gly-Pro-Hyp)5-NH2; c α1(V)436-447 fTHP = (Gly-Pro-Hyp)5-Gly-Pro-Lys(Mca)-Gly-Pro-Pro-Gly~Val-Val-Gly-Glu-Lys(Dnp)-Gly-Glu-Gln-(Gly-Pro-Hyp)5-NH2.
Using confocal laser scanning and the multichannel character of existing DNA sequencers, Hu et al. screened several degenerate combinatorial peptide libraries and selected a novel MMP-9 inhibitor, regasepin1 (PRCBCGE, where B = biphenylalanine/Bip) [42,43,44]. Competitive enzyme inhibition assays using the substrate MIM-3b (POGPQGATGEOG), a fluorescent-labeled substrate, and gelatin, found regasepin1 inhibited MMP-9 in micromolar concentrations and in a dose-dependent manner. Regasepin1 was subsequently found to inhibit MMP-8, MMP-9, and ADAM17 equally well [42]. While the IC50 value for MMP-8 was 3 µM, MMP-1 and MMP-13 were inhibited with IC50 values of 100 µM. 3.1–7.5 mM regasepin1 protected mice against endotoxin shock [42].
The regasepin 1 backbone was used in a subsequent library that incorporated D-amino acids to develop relatively selective inhibitors of MMP-9 and ADAM17 [44]. The best inhibitor for MMP-9 was Arg-Cys-D-Bip-D-Arg (IC50 = 0.75 µM), while the best ADAM17 inhibitor was D-Pyr-D-Cys-Bip-D-Cys (IC50 = 0.6 µM). Modest selectivity was observed when comparing inhibitory potency between MMP-9, ADAM17, MMP-2, and MMP-3. For MMP-9, inhibition was by a mixed non-competitive mode.
Bacteria fermented milk has been shown to produce peptide inhibitors for proteases such as angiotensin converting enzyme (ACE), an enzyme involved in cardiovascular disorders [75,76]. Fermented milk is also thought to inhibit tumor growth directly or inhibit the proteolytic activity of tumor-associated stromal cells [77,78]. Juillerat-Jeanneret et al. evaluated peptides that had been described as ACE inhibitors for MMP inhibition. Two groups of peptides were synthesized and analyzed. These peptides incorporated the sequence -PFP- or -PLP- located at positions 42–44 of αS1-casein and 151–153 of β-casein, respectively. The peptide derived from αS1-casein inhibited MMP-2 and MMP-9 more efficiently than MMP-7. The most effective sequence for inhibiting MMP-9 from the -PFP-series was NENLLRFFVAPFPEVFG, with an IC50 value of 50 µM [45]. Peptides derived from β-casein had comparable IC50 values towards MMP-2, MMP-9, and MMP-7, with an increase in activity upon increasing the length of the N-terminus. The best inhibitor was VENLHLPLPLL, with IC50 values of 150, 250, and 300 µM for MMP-2, MMP-7, and MMP-9, respectively [45].
In most MMPs, the CAT domain is very similar, rendering it very difficult to construct selective inhibitors for each MMP. Diversity in the HPX domains among MMPs makes this exodomain a good target in the search for selective MMP inhibitors. The HPX domain is composed of four blades that consist of one α-helix and four antiparallel β-strands, giving them a propeller like structure. The MMP-9 X-ray crystallographic structure demonstrated that a homodimer is formed through blade IV of the HPX domain [9]. MMP-9 is presently thought to be the only secreted MMP that can form a homodimer, and the role of the homodimer is not well understood. Homodimer formation may be a prerequisite for MMP-9 induced cell migration, and therefore targeting homodimer formation was seen as a viable approach for inhibiting cell migration [79,80,81]. Using mammalian COS-1 cells, Dufour et al. tested the interaction interface of MMP-9 homodimer occurring through the outermost β-strand of the fourth blade of the HPX domain [38]. The outermost β-strand octapeptide (N688QVDQVGY695) was substituted with the corresponding region of the MMP-2 HPX domain to obtain a chimera MMP-9/IVS4. Furthermore, three additional mutants were made for the outermost β-strands of blade I, II, and III (MMP-9/IS4, MMP-9/IIS4, and MMP-9/IIIS4, respectively) to serve as controls. Using a co-immunoprecipitation assay, it was observed that mutants of blade I, II, and III had no effect on the homodimer formation while mutants of blade IV failed to dimerize. Evaluation of the four mutants in a transwell cell migration assay revealed mutant MMP-9/IVS4 failed to enhance cell migration in COS-1 cells in comparison to wild type MMP-9. Mutation of blade II and III had no effect on cell migration, while the blade I mutant MMP-9 failed to enhance cell migration. Inhibition assays for MMP-9 induced cell migration were carried out using NQVDQVGY (IVS4 peptide) and SRPQGPFL (IS4 peptide), designed to mimic the outermost β-strand of blade IV and blade I of the MMP-9 HPX domain, respectively. These peptides displayed a dose-dependent inhibition of MMP-9 induced cell migration, with IC50 values for IS4 and IVS4 of 12 and 50 µM, respectively. The IS4 peptide was found to disrupt interaction of MMP-9 with CD44.
The MMP-9 HPX domain was also found to bind to B chronic lymphocytic leukemia (B-CLL) cells via the α4β1 integrin [39]. Truncated variants of the MMP-9 HPX domain containing blades I and II (B1B2) or blades III and IV (B3B4) indicated that B3B4 interacted with the α4β1 integrin. Using peptides that span B3B4 (residues 621–707), peptide P3 from B4 (residues 654–674, PFPGVPLDTHDVFQYREKAYFC) inhibited cell adhesion to MMP-9. Asp660 and Asp663 mediated peptide P3 binding to the α4β1 integrin. A truncated version of peptide P3 (P3a, FPGVPLDTHDVFQYREK) bound to MEC-1 cells with a KD = 282 µM. Peptide P3a also inhibited B-CLL and MEC-1 cell adhesion to proMMP-9 with IC50 = 279 and 109 µM, respectively. Peptide P3a inhibited B-CLL cell transendothelial migration and intracellular survival signals.
Phage display analyses revealed the CRVYGPYLLC sequence as an MMP-9 HPX domain binder that inhibited homodimerization [41]. This peptide inhibited MMP-9 binding to the α5β1 and αvβ5 integrins in fibrosarcoma cells and inhibited fibrosarcoma cell invasion (50% inhibition at 200 μM peptide). The peptide did not inhibit MMP-9 catalyzed gelatin hydrolysis, and thus was a non-catalytic inhibitor [41]. Conversely, the cyclic LRSG peptide [CQVTGALRSGRGKMLLC-NH2, derived from MMP-9 HPX domain residues 615–622] inhibited MMP-9 interaction with the α5β1 integrin, tumor cell invasion, and cell surface gelatinolytic activity [40].
3. Inhibitors and Probes of the Membrane-Bound MMPs (MT1-MMP/MMP-14)
MT1-MMP is a critical protein in cancer invasion and metastasis [82]. MT1-MMP is up regulated in metastatic breast cancer patients in comparison to normal patients [83] and high expression of MT1-MMP has been correlated with low patient survival [84,85]. Using phage display libraries, the search for an MMP-2/MMP-9 inhibitor led to the preparation of a cyclic peptide with the sequence GACFSIAHECGA (Peptide G) as a control peptide due to its non-inhibitory properties [29]. Peptide G turned out to uniquely inhibit MT1-MMP. Peptide G inhibited MT1-MMP-mediated proteolysis of β-casein by 37% at 500 μM concentration. In the Quantizyme assay MT1-MMP inhibition by Peptide G was dose dependent with an IC50 value of 150 µM. The linear, scrambled peptide CGAAPEACGIHS had no effect on MT1-MMP activity. Peptide G had no inhibitory activity towards MMP-1, MMP-3, MMP-7, MMP-8, MMP-9, MMP-10, MMP-11, MMP-12, MMP-13, MMP-15, MMP-17, or MMP-20.
Peptide G also inhibited MT1-MMP-mediated proteolysis of the laminin Ln-332 γ2-chain by 64% at 100 µM. As the Ln-332 γ2-chain is hydrolyzed by a variety of carcinoma cells and linked to tumor progression [86,87], Peptide G inhibition of this process is an indication of its potential usefulness in the invasive phase of cancer. In transwell and matrigel assays, 100 µM Peptide G inhibited HT1080 fibrosarcoma and C8161 melanoma cell migration on fibronectin and invasion by 70 and 51%, respectively [47]. Peptide G inhibited soluble MT1-MMP activity in HSC-3 cell culture medium and increased the survival of mice bearing HSC-3 xenografts [47]. The potent nature of the peptide was attributed to its disulfide bond and restrained conformation. The full activity of peptide G was lost when the Cys residues were replaced by Ser or when Phe4 was altered. D-amino acid replacements led to a drop in activity, suggesting a strong conformational effect essential for efficient MT1-MMP binding.
Subtractive cell surface panning from phage random peptide libraries was used to identify peptides that targeted induced MT1-MMP and metal ions on MG-63 cells [88]. A prior study from this group using a phage dodecapeptide library and the CAT domain of MT1-MMP identified 13 binding peptides [48]. These peptides were combined with those obtained in the present study for a total of 35 peptides. Consensus sequences were AHQ/SLH/P, L/I/EPLL/I, T/Q/DARH/FQ, and MK/PSR. Representative peptides AHQLH, LPLL, DTARFQ, and MKPSR docked with MT1-MMP at residues 120–125, where Zn2+ binding occurs. MG-63 and HepG2 tumor cell proliferation was examined in the presence of the representative peptides. All four peptides inhibited cell proliferation, with the maximum inhibition of 54% obtained with 100 µg/mL (~165 µM) AHQLH against MG63 cells.
MT1-MMP associates with CD44 and homodimerizes through its HPX domain, similarly to MMP-9. These associations lead to cell migration and invasion [89,90]. Detailed analysis of regions of the MT1-MMP HPX domain thought to promote cell migration and invasion led to the identification of sequences comprising the outer-strand of the individual HPX domain blades. Zarabbi et al. generated MT1-MMP chimeras by replacing the four outermost strands (S4) of the HPX domain with the corresponding regions of MMP-1 and examined the chimeras for their ability to induce cell migration [49]. Based on gelatin zymography, chimeras IS4 and IVS4 exhibited complete disruption of MT1-MMP activation of proMMP-2. From immunoprecipitation and Western blotting [38], the IVS4 chimera failed to bind wild-type MT1-MMP, suggesting homodimer formation required the outermost strand of blade IV. The same approach revealed that the interaction of CD44 with MT1-MMP required the outermost strand of blade I. A transwell chamber migration assay was performed to determine whether peptide sequences derived from MT1-MMP could interfere with MT1-MMP homodimer and CD44/MT1-MMP heterodimer formation. MT1-MMP mediated cell migration was significantly inhibited by the IS4 peptide acetyl-VMDGYPMP-NH2 and IVS4 peptide acetyl-GYPKSALR-NH2 while IIS4, IIIS4, and scrambled peptide had no effect on cell migration. IS4 and IVS4 were also found to inhibit metastasis in vivo. IS4 and IVS4 represented non-catalytic inhibitors, as catalytic inactivation of MT1-MMP had no effect on cell migration [91]. Interestingly, X-ray crystallographic analysis indicated that the MT1-MMP dimer interface involved interactions between blades II and III of one HPX domain with blades III and II of the other HPX domain [11]. Important residues for homodimerization were Asp385, Lys386, Thr412, Lys434, and Tyr436. The contrasting conclusions of the MT1-MMP dimerization studies, where interaction occurred between either HPX domain blades IV [49] or blades II and III [11], could have arisen from the different conditions that each study was performed under.
The outer blade regions of the MT1-MMP HPX domain have non-homologous loop sequences compared to other members of the MMP family. Our laboratory synthesized peptide models of the 5 MT1-MMP HPX domain loops (blade I strand 4, blade II strand 2, blade II strands 3–4, blade III strand 1, and blade IV strand 4), and examined their inhibitory activity for MT1-MMP processing of a triple-helical substrate (fTHP-17) (Table 3). Two peptides were micromolar inhibitors of MT1-MMP but did not inhibit MMP-1. VFDEASLEP-NH2, from blade II strand 3–4, had the best IC50 value, 238 µM. This peptide may directly or indirectly inhibit MT1-MMP dimerization (see above) or interaction of MT1-MMP with the substrate triple-helix. VRNNQV-[Nle]-DGYP-[Nle]-P-NH2, which is an N-terminal extension and modified version of the IVS4 peptide described above, was more effective than IVS4 for MT1-MMP inhibition of triple-helical peptidase activity (Table 3). When the complete cleavage reaction solutions were run over HPLC, peptides LFW-[Nle]-PNG-NH2 and RKDGKFV-NH2 both showed additional peaks indicating peptide processing by MT1-MMP.
Table 3.
Inhibition of MMP Triple-helical Peptidase Activity by MT1-MMP HPX Domain Loop Peptides a.
| Enzyme | Loop Peptide Sequence | IC50 (μM) b |
|---|---|---|
| MT1-MMP | LFW-[Nle]-PNG-NH2 (bIIIs1) c | 15,400 |
| “ | VFDEASLEP-NH2 (bIIs3-4) | 240 |
| “ | RKDGKFV-NH2 (bIIs2) | 3,000 |
| “ | VRNNQV-[Nle]-DGYP-[Nle]-P-NH2 (bIs4) c | 670 |
| “ | NNQKLKVEPGYPKSALRD-NH2 (bIVs4) | 5,400 |
| “ | acetyl-VMDGYPMP-NH2 (IS4) | 3,400 |
| MMP-1 | VFDEASLEP-NH2 (bIIs3-4) | NI |
| “ | VRNNQV-[Nle]-DGYP-[Nle]-P-NH2 (bIs4) c | NI |
a Concentrated peptide stocks were prepared, 80 µL of which was placed in a well of an opaque white 384 well plate, and serially diluted 1:2 nine times in triplicate. A buffer sample was included for 100% activity. Activated MT1-MMP or MMP-1 was added to each well at a concentration of 10 nM, shaken for 60 sec in a BioTEX plate reader, and incubated for 10 min at room temperature. 40 µL fTHP-17 [(Gly-Pro-Hyp)4-Gly-Pro-Lys(Mca)-Gly-Pro-Gln-Gly~Leu-Arg-Gly-Gln-Lys(Dnp)-Gly-Val-Arg-Gly-Leu-Hyp-Gly-Gln-Arg-Gly-Glu-Arg-(Gly-Pro-Hyp)4-NH2] was added to each reaction, the plate was shaken for 30 s in a BioTEK plate reader, and fluorescent readings were taken every 10 s for 10 min. Plates were sealed and placed in the dark at room temperature for a minimum of 24 h, and then read again for a fluorescent endpoint reading to facilitate enzyme activity calculation. Enzyme activity was calculated from the slope of the initial rate, and normalized as a percentage of the uninhibited MMP/fTHP-17 reaction. These normalized activities were plotted versus the log of micromolar inhibitor concentration in GraphPad Prism to determine approximate IC50 values for each peptide. 10 µL of each 250 µM reaction mixture was analyzed by C18 reversed-phase HPLC column to determine if the inhibitory peptide had been cleaved by MT1-MMP; b NI = no inhibition; c Met residues were replaced by norleucine (Nle).
An MT1-MMP near infrared probe was designed based on a peptide sequence identified in a phage display substrate library. The MT loop region of MT1-MMP is located within the CAT domain and contains an eight amino acid insertion unique to MT-MMPs (MT1-, 2-, 3-, and 5-MMP). To enhance selectivity, the phage display library was screened against this MT1-MMP sequence (160Arg-Glu-Val-Pro-Tyr-Ala-Tyr-Ile-Arg-Glu-Gly-His-Glu-Lys-Gln174, designated MT1-160p) rather than the complete sequence of MT1-MMP or its CAT domain. The non-substrate peptide MT1-AF7p (HWKHLHNTKTFL) displayed the highest affinity towards MT1-160p (KD = 0.075 nM). MT1-AF7p bound via hydrogen bonding and hydrophobic interactions [92]. MT1-AF7p was labeled with Cy5.5 (Cy5.5-MT1-AF7p) and chosen for further validation in vivo [92]. The evaluation was performed in mice carrying MDA-MB-435 breast cancer xenografts (expressing high levels of MT1-MMP) or A549 xenografts (low MT1-MMP levels). MDA-MB-435 xenografts had significantly higher signal accumulation and better tumor contrast than the A549 xenografts. However, more precise quantitative data on tumor uptake and pharmacokinetics will be needed to determine the further utility of this probe.
4. Inhibitors of Matrix Metalloproteinase 1
MMP-1 is another MMP family member that has been validated as a cancer target [93]. TIMPs are well-characterized, high affinity, natural inhibitors of MMPs [94,95], and the TIMP scaffold has been utilized to pursue MMP-1 selective inhibitors. Modification of TIMPs can improve their selectivity within the MMP family [96,97,98]. Mini-TIMPs have been constructed using the sarafotoxin 6b fold [46], with the sarafotoxin 6b C-terminal residues VIW deleted to remove vasopressive activity. Substitutions were incorporated to make the sarafotoxin 6b sequence more “TIMP-like.” The best inhibitor was STX-S4-CT (CSCSDMTDKECLYFCMSEMS), which inhibited MMP-1 and MMP-9 with Ki = 4.5 and 1.0 μM, respectively. STX-S4-CT offered intriguing selectivity, as it did not inhibit MMP-3, MT1-MMP, or ADAM17. Binding simulations indicated complex interactions; both active site and exosite binding of STX-S4-CT with MMP-1 in ways reminiscent of TIMP binding and ways unique to STX-S4-CT.
In related work from the same laboratory, phage display was utilized with TIMP-2 to identify selective inhibitors of MMP-1 [99]. Three regions of TIMP-2 underwent randomization, residues 2–6 (excluding residue 3), 34–40, and 67–70. TM8, which possessed Ser2 to Asp and Ser4 to Ala substitutions, offered the best selectivity. TM8 inhibited MMP-1, MMP-8, MMP-9, and MMP-13 with Ki values of 10, 28, 1.5, and 12 nM, respectively, while poorly inhibiting MMP-2, MMP-3, MMP-7, and MT1-MMP. Wild-type TIMP-2 effectively inhibits all of the aforementioned enzymes. These results reveal great potential for the design of MMP-selective mini-TIMPs.
5. Conclusions
MMP peptide-based approaches have yielded intriguing probes, including inhibitors that function on non-catalytic activities while leaving catalytic activities unaffected. For example, peptides based on MMP-9 and MT1-MMP HPX domain blade I strand 4 inhibit enzyme binding to CD44 while peptides based on blade IV strand 4 inhibit enzyme homodimerization. In turn, a peptide based on HPX domain blade II strands 3–4 inhibits MT1-MMP triple-helical peptidase activity. The main drawbacks of these probes have been their affinity and/or stability. In general, cyclization improves peptide stability, while sometimes offering improved affinity. Recent advances in phage display utilized mRNA to identify cyclic peptide inhibitors of thrombin with low nM affinities [100]. mRNA-display technology is further enhanced by incorporation of 12 unnatural amino acids. Thus, recent display technologies, in combination with methods for producing modified peptides, holds great promise for the future development of selective MMP inhibitors.
Acknowledgments
References
- Barrett, A.J.; Rawlings, N.D.; Woessner, J.F. Handbook of Proteolytic Enzymes, 2nd ed; Elsevier/Academic Press: Amsterdam, The Netherlands, 2004; Volume 1. [Google Scholar]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Jackson, B.C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse matrix metalloproteinase families. Hum. Genomics 2010, 4, 194–201. [Google Scholar]
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Aspects Med. 2008, 29, 290–308. [Google Scholar] [CrossRef]
- Li, J.; Brick, P.; O’Hare, M.C.; Skarzynski, T.; Lloyd, L.F.; Curry, V.A.; Clark, I.M.; Bigg, H.F.; Hazleman, B.L.; Cawston, T.E.; et al. Structure of full-length porcine synovial collagenase reveals a C-terminal domain containing a calcium-linked, four bladed β-propeller. Structure 1995, 15, 541–549. [Google Scholar]
- Iyer, S.; Visse, R.; Nagase, H.; Acharya, K.R. Crystal structure of an active form of human MMP-1. J. Mol. Biol. 2006, 362, 78–88. [Google Scholar] [CrossRef]
- Morgunova, E.; Tuuttila, A.; Bergmann, U.; Isupov, M.; Lindqvist, Y.; Schneider, G.; Tryggvason, K. Structure of human pro-matrix metalloproteinase-2: Activation mechanism revealed. Science 1999, 284, 1667–1670. [Google Scholar]
- Elkins, P.A.; Ho, S.H.; Smith, W.W.; Janson, C.A.; D'Alessio, K.J.; McQueney, M.S.; Cummings, M.D.; Romanic, A.M. Structure of the C-teminally truncated human proMMP9, a gelatin-binding matrix metalloproteinase. Acta Cryst. 2002, D58, 1182–1192. [Google Scholar]
- Cha, H.; Kopetzki, E.; Huber, R.; Lanzendorfer, M.; Brandstetter, H. Structural basis of the adaptive molecular recognition by MMP-9. J. Mol. Biol. 2002, 320, 1065–1079. [Google Scholar]
- Grossman, M.; Tworowski, D.; Dym, O.; Lee, M.H.; Levy, Y.; Murphy, G.; Sagi, I. The intrinsic protein flexibility of endogenous protease inhibitor TIMP-1 controls its binding interface and affects its function. Biochemistry 2010, 49, 6184–6192. [Google Scholar]
- Tochowicz, A.; Goettig, P.; Evans, R.; Visse, R.; Shitomi, Y.; Palmisano, R.; Ito, N.; Richter, K.; Maskos, K.; Franke, D.; et al. The dimer interface of the membrane type 1 matrix metalloproteinase hemopexin domain: Crystal structure and biological functions. J. Biol. Chem. 2011, 286, 7587–7600. [Google Scholar]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protocols 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008, 9, 40. [Google Scholar]
- Rosenberg, G.A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet 2009, 8, 205–216. [Google Scholar]
- Hu, J.; Van den Steen, P.E.; Sang, Q.-X.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar]
- Lindsey, M.L.; Zamilpa, R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc. Ther. 2012, 30, 31–41. [Google Scholar] [CrossRef]
- Newby, A.C. Matrix metalloproteinase inhibition therapy for vascular diseases. Vascul. Pharmacol. 2012, 56, 232–244. [Google Scholar] [CrossRef]
- Rivera, S.; Khrestchatisky, M.; Kaczmarek, L.; Rosenberg, G.A.; Jaworski, D.M. Metzincin proteases and their inhibitors: Foes or friends in nervous system physiology? J. Neurosci. 2010, 30, 15337–15357. [Google Scholar]
- Whittaker, M.; Floyd, C.D.; Brown, P.; Gearing, A.J.H. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999, 99, 2735–2776. [Google Scholar] [CrossRef]
- Jacobsen, J.A.; Jourden, J.L.M.; Miller, M.T.; Cohen, S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta 2010, 1803, 72–94. [Google Scholar]
- Saghatelian, A.; Jessani, N.; Joseph, A.; Humphrey, M.; Cravatt, B.F. Activity-based probes for the proteomic profiling of metalloproteases. Proc. Natl. Acad. Sci. USA 2004, 101, 10000–10005. [Google Scholar]
- Morrison, C.J.; Butler, G.S.; Rodríguez, D.; Overall, C.M. Matrix metalloproteinase proteomics: Substrates, targets, and therapy. Curr. Opin. Cell Biol. 2009, 21, 645–653. [Google Scholar]
- Sela-Passwell, N.; Trahtenherts, A.; Krüger, A.; Sagi, I. New opportunities in drug design of metalloproteinase inhibitors: Combination between structure-function experimental approaches and systems biology. Expert Opin. Drug Discov. 2011, 6, 527–542. [Google Scholar] [CrossRef]
- Sela-Passwell, N.; Rosenblum, G.; Shoham, T.; Sagi, I. Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim. Biophys. Acta 2010, 1803, 29–38. [Google Scholar]
- Bergers, G.; Brekken, R.A.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biol. 2000, 2, 737–744. [Google Scholar]
- Zamilpa, R.; Lopez, E.F.; Chiao, Y.A.; Dai, Q.; Escobar, G.P.; Hakala, K.; Weintraub, S.T.; Lindsey, M.L. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics 2010, 10, 2214–2223. [Google Scholar]
- Fernandes, K.S.; Brum, D.G.; Palei, A.C.; Sandrim, V.C.; Guerreiro, C.T.; Tanus-Santos, J.E.; Barreira, A.A. Functional MMP-9 polymorphisms modulate plasma MMP-9 levels in multiple sclerosis patients. J. Neuroimmunol. 2012, 249, 56–59. [Google Scholar] [CrossRef]
- Koivunen, E.; Arap, W.; Valtanen, H.; Rainisalo, A.; Medina, O.P.; Heikkila, P.; Kantor, C.; Gahmberg, C.G.; Salo, T.; Konttinen, Y.T.; et al. Tumor targeting with a selective gelatinase inhibitor. Nat. Biotech. 1999, 17, 768–774. [Google Scholar] [CrossRef]
- Medina, O.P.; Kairemo, K.; Valtanen, H.; Kangasniemi, A.; Kaukinen, S.; Ahonen, I.; Permi, P.; Annila, A.; Sneck, M.; Holopainen, J.M.; et al. Radionuclide imaging of tumor xenografts in mice using a gelatinase-targeting peptide. Anticancer Res. 2005, 25, 33–42. [Google Scholar]
- Mukai, T.; Suganuma, N.; Soejima, K.; Sasaki, J.; Yamamoto, F.; Maeda, M. Synthesis of a beta-tetrapeptide analog as a mother compound for the development of matrix metalloproteinase-2-imaging agents. Chem. Pharm. Bull. 2008, 56, 260–265. [Google Scholar]
- Lu, G.; Zheng, M.; Zhu, Y.; Sha, M.; Wu, Y.; Han, X. Selection of peptide inhibitor to matrix metalloproteinase-2 using phage display and its effects on pancreatic cancer cell lines PANC-1 and CFPAC-1. Int. J. Biol. Sci. 2012, 8, 650–662. [Google Scholar]
- Hashimoto, H.; Takeuchi, T.; Komatsu, K.; Miyazaki, K.; Sato, M.; Higashi, S. Structural basis for matrix metalloproteinase-2 (MMP-2)-selective inhibitory action of beta-amyloid precursor protein-derived inhibitor. J. Biol. Chem. 2011, 286, 33236–33243. [Google Scholar]
- Higashi, S.; Miyazaki, K. Identification of a region of β-amyloid precursor protein essential for its gelatinase A inhibitory activity. J. Biol. Chem. 2003, 278, 14020–14028. [Google Scholar] [CrossRef]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar]
- Xu, X.; Chen, Z.; Wang, Y.; Bonewald, L.; Steffensen, B. Inhibition of MMP-2 gelatinolysis by targeting exodomain-substrate interactions. Biochem. J. 2007, 406, 147–155. [Google Scholar] [CrossRef]
- Ujula, T.; Huttunen, M.; Luoto, P.; Peräkylä, H.; Simpura, I.; Wilson, I.; Bergman, M.; Roivainen, A. Matrix metalloproteinase 9 targeting peptides: Syntheses, 68Ga-labeling, and preliminary evaluation in a rat melanoma xenograft model. Bioconjugate Chem. 2010, 21, 612–1621. [Google Scholar]
- Dufour, A.; Zucker, S.; Sampson, N.S.; Kuscu, C.; Cao, J. Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J. Biol. Chem. 2010, 285, 35944–35956. [Google Scholar]
- Ugarte-Berzal, E.; Bailón, E.; Amigo-Jiménez, I.; Vituri, C.L.; del Cerro, H.; Terol, M.J.; Albar, J.P.; Rivas, G.; García-Marco, J.A.; Garcío-Pardo, A. A 17-residue sequence from the matrix metalloproteinase-9 (MMP-9) hemopexin domain binds α4β1 integrin and inhibits MMP-9-induced functions in chronic lymphocytic leukemia B cells. J. Biol. Chem. 2012, 287, 27601–27613. [Google Scholar]
- Radjabi, A.R.; Sawada, K.; Jagadeeswaran, S.; Eichbichler, A.; Kenny, H.A.; Montag, A.; Bruno, K.; Lengyel, E. Thrombin induces tumor invasion through the induction and association of matrix metalloproteinase-9 and beta1-integrin on the cell surface. J. Biol. Chem. 2008, 283, 2822–2834. [Google Scholar]
- Björklund, M.; Heikkilä, P.; Koivunen, E. Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J. Biol. Chem. 2004, 279, 29589–29597. [Google Scholar]
- Hu, J.; Van den Steen, P.E.; Dillen, C.; Opdenakker, G. Targeting neutrophil collagenase/matrix metalloproteinase-8 and gelatinase B/matrix metalloproteinase-9 with a peptidomimetic inhibitor protects against endotoxin shock. Biochem. Pharmacol. 2005, 70, 535–544. [Google Scholar]
- Hu, J.; Fiten, P.; Van den Steen, P.E.; Chaltin, P.; Opdenakker, G. Simulation of evolution-selected propeptide by high-throughput selection of a peptidomimetic inhibitor on a capillary DNA sequencer platform. Anal. Chem. 2005, 77, 2116–2124. [Google Scholar] [CrossRef]
- Qiu, Z.; Yan, M.; Li, Q.; Liu, D.; Van den Steen, P.E.; Wang, M.; Opdenakker, G.; Hu, J. Definition of peptide inhibitors from a synthetic peptide library by targeting gelatinase B/matrix metalloproteinase-9 (MMP-9) and TNF-alpha converting enzyme (TACE/ADAM-17). J. Enzym. Inhib. Med. Chem. 2012, 27, 533–540. [Google Scholar]
- Juillerat-Jeanneret, L.; Robert, M.C.; Juillerat, M.A. Peptides from Lactobacillus hydrolysates of bovine milk caseins inhibit prolyl-peptidases of human colon cells. J. Agric. Food Chem. 2011, 59, 370–377. [Google Scholar] [CrossRef]
- Lauer-Fields, J.L.; Marí, F.; Wei, S.; Fields, G.B.; Brew, K. Engineered Sarafotoxins as TIMP-like MMP Inhibitors. J. Biol. Chem. 2007, 282, 26948–26955. [Google Scholar]
- Suojanen, J.; Salo, T.; Koivunen, E.; Sorsa, T.; Pirila, E. A novel and selective membrane type-1 matrix metalloproteinase (MT1-MMP) inhibitor reduces cancer cell motility and tumor growth. Cancer Biol. Ther. 2009, 8, 2362–2370. [Google Scholar]
- Liang, Z.J.; Huang, J.S.; Cui, J.; Xiong, L.L.; Mao, C.Q. Selection and molecular simulation of binding peptides dual-targeting MMP-14 and metal ions. Chin. J. Biochem. Mol. Biol. 2011, 27, 341–349. [Google Scholar]
- Zarrabi, K.; Dufour, A.; Li, J.; Kuscu, C.; Pulkoski-Gross, A.; Zhi, J.; Hu, Y.; Sampson, N.S.; Zucker, S.; Cao, J. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J. Biol. Chem. 2011, 286, 33167–33177. [Google Scholar]
- Kuhnast, B.; Bodenstein, C.; Haubner, R.; Wester, H.J.; Senekowitsch-Schmidtke, R.; Schwaiger, M.; Weber, W.A. Targeting of gelatinase activity with a radiolabeled cyclic HWGF peptide. Nucl. Med. Biol. 2004, 31, 337–344. [Google Scholar] [CrossRef]
- Sprague, J.E.; Li, W.P.; Liang, K.; Achilefu, S.; Anderson, C.J. In vitro and in vivo investigation of matrix metalloproteinase expression in metastatic tumor models. Nucl. Med. Biol. 2006, 33, 227–237. [Google Scholar]
- Hanaoka, H.; Mukai, T.; Habashita, S.; Asano, D.; Ogawa, K.; Kuroda, Y.; Akizawa, H.; Iida, Y.; Endo, K.; Saga, T.; et al. Chemical design of a radiolabeled gelatinase inhibitor peptide for the imaging of gelatinase activity in tumors. Nucl. Med. Biol. 2007, 34, 503–510. [Google Scholar]
- Suojanen, J.; Vilen, S.-T.; Nyberg, P.; Heikkilä, P.; Penate-Medina, O.; Saris, P.E.J.; Hagström, J.; Ranta, T.-M.; Salo, T.; Sorsa, T.; et al. Selective gelatinase inhibitor peptide is effective in targeting tongue carcinoma cell tumors in vivo. Anticancer Res. 2011, 31, 3659–3664. [Google Scholar]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-Peptides: From structure to function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar]
- Gademann, K.; Kimmerlin, T.; Hoyer, D.; Seebach, D. Peptide folding induces high and selective affinity of a linear and small beta-peptide to the human somatostatin receptor 4. J. Med. Chem. 2001, 44, 2460–2468. [Google Scholar] [CrossRef]
- Seebach, D.; Beck, A.K.; Bierbaum, D.J. The world of β- and γ-peptides comprised of homologated proteinogenic amino acids and other components. Chem. Biodiver. 2004, 1, 1111–1239. [Google Scholar] [CrossRef]
- Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguilar, M.I. β-amino acids: Versatile peptidomimetics. Curr. Med. Chem. 2002, 9, 811–822. [Google Scholar] [CrossRef]
- Sisodia, S.S.; Koo, E.H.; Beyreuther, K.; Unterbeck, A.; Price, D.L. Evidence that β-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science 1990, 248, 492–495. [Google Scholar]
- Esch, F.S.; Keim, P.S.; Beattie, E.C.; Blacher, R.W.; Culwell, A.R.; Oltersdorf, T.; McClure, D.; Ward, P.J. Cleavage of amyloid β peptide during constitutive processing of its precursor. Science 1990, 248, 1122–1124. [Google Scholar]
- Miyazaki, K.; Hasegawa, M.; Funahashi, K.; Umeda, M. A metalloproteinase inhibitor domain in Alzheimer amyloid protein precursor. Nature 1993, 362, 839–841. [Google Scholar]
- Higashi, S.; Miyazaki, K. Novel processing of β-amyloid precursor protein catalyzed by membrane type 1 matrix metalloproteinase releases a fragment lacking the inhibitor domain against gelatinase A. Biochemistry 2003, 42, 6514–6526. [Google Scholar] [CrossRef]
- Higashi, S.; Miyazaki, K. Identification of amino acid residues of the matrix metalloproteinase-2 essential for its selective inhibition by β-amyloid precursor protein-derived inhibitor. J. Biol. Chem. 2008, 283, 10068–10078. [Google Scholar] [CrossRef]
- DeBin, J.A.; Strichartz, G.R. Chloride channel inhibition by the venom of the scorpion Leiurus quinquestriatus. Toxicon 1991, 29, 1403–1408. [Google Scholar] [CrossRef]
- Soroceanu, L.; Gillespie, Y.; Khazaeli, M.B.; Sontheimer, H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998, 58, 4871–4879. [Google Scholar]
- Lyons, S.A.; O’Neal, J.; Sontheimer, H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia 2002, 39, 162–173. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Ratnikov, B.; Monosov, E.; Postnova, T.I.; DiScipio, R.; Smith, J.W.; Strongin, A.Y. MT1-MMP initiates activation of pro-MMP-2 and integrin avb3 promotes maturation of MMP-2 in breast carcinoma cells. Exp. Cell Res. 2001, 263, 209–223. [Google Scholar] [CrossRef]
- Veiseh, M.; Gabikian, P.; Bahrami, S.-B.; Veiseh, O.; Zhang, M.; Hackman, R.C.; Ravanpay, A.C.; Stroud, M.R.; Kusuma, Y.; Hansen, S.J.; et al. Tumor paint: A chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007, 67, 6882–6888. [Google Scholar]
- Banyai, L.; Patthy, L. Evidence for the involvement of type II domains in collagen binding by 72 kDa type IV procollagenase. FEBS Lett. 1991, 282, 23–25. [Google Scholar] [CrossRef]
- Senior, R.M.; Griffin, G.L.; Fliszar, C.J.; Shapiro, S.D.; Goldberg, G.I.; Welgus, H.G. Human 92- and 72-kilodalton type IV collagenases are elastases. J. Biol. Chem. 1991, 266, 7870–7875. [Google Scholar]
- Shipley, J.M.; Doyle, G.A.; Fliszar, C.J.; Ye, Q.Z.; Johnson, L.L.; Shapiro, S.D.; Welgus, H.G.; Senior, R.M. The structural basis for the elastolytic activity of the 92-kDa and 72-kDa gelatinases. Role of the fibronectin type II-like repeats. J. Biol. Chem. 1996, 271, 4335–4341. [Google Scholar]
- Steffensen, B.; Bigg, H.F.; Overall, C.M. The involvement of the fibronectin type II-like modules of human gelatinase A in cell surface localization and activation. J. Biol. Chem. 1998, 273, 20622–20628. [Google Scholar] [CrossRef]
- Steffensen, B.; Wallon, U.M.; Overall, C.M. Extracellular matrix binding properties of recombinant fibronectin type II-like modules of human 72-kDa gelatinase/type IV collagenase. High affinity binding to native type I collagen but not native type IV collagen. J. Biol. Chem. 1995, 270, 11555–11566. [Google Scholar] [CrossRef]
- Murphy, G.; Nguyen, Q.; Cockett, M.I.; Atkinson, S.J.; Allan, J.A.; Knight, C.G.; Willenbrock, F.; Docherty, A.J. Assessment of the role of the fibronectin-like domain of gelatinase A by analysis of a deletion mutant. J. Biol. Chem. 1994, 269, 6632–6636. [Google Scholar]
- Lauer-Fields, J.L.; Whitehead, J.K.; Li, S.; Hammer, R.P.; Brew, K.; Fields, G.B. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J. Biol. Chem. 2008, 283, 20087–20095. [Google Scholar]
- Tauzin, J.; Miclo, L.; Gaillard, J.L. Angiotensin-I-converting enzyme inhibitory peptides from tryptic hydrolysate of bovine αS2-casein. FEBS Lett. 2002, 531, 369–374. [Google Scholar]
- Robert, M.C.; Razaname, A.; Mutter, M.; Juillerat, M.A. Identification of angiotensin-I-converting enzyme inhibitory peptides derived from sodium caseinate hydrolysates produced by Lactobacillus helveticus NCC 2765. J. Agric. Food Chem. 2004, 52, 6923–6931. [Google Scholar] [CrossRef]
- Mook, O.R.; Frederiks, W.M.; Van Noorden, C.J. The role of gelatinases in colorectal cancer progression and metastasis. Biochim. Biophys. Acta 2004, 1705, 69–89. [Google Scholar]
- Basset, P.; Okada, A.; Chenard, M.P.; Kannan, R.; Stoll, I.; Anglard, P.; Bellocq, J.P.; Rio, M.C. Matrix metalloproteinases as stromal effectors of human carcinoma progression: therapeutic implications. Matrix Biol. 1997, 15, 535–541. [Google Scholar] [CrossRef]
- Redondo-Munoz, J.; Ugarte-Berzal, E.; Garcia-Marco, J.A.; del Cerro, M.H.; Van den Steen, P.E.; Opdenakker, G.; Terol, M.J.; Garcia-Pardo, A. α4β1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood 2008, 112, 169–178. [Google Scholar] [CrossRef]
- Dufour, A.; Sampson, N.S.; Zucker, S.; Cao, J. Role of the hemopexin domain of matrix metalloproteinases in cell migration. J. Cell. Physiol. 2008, 217, 643–651. [Google Scholar]
- Stefanidakis, M.; Karjalainen, K.; Jaalouk, D.E.; Gahmberg, C.G.; O’Brien, S.; Pasqualini, R.; Arap, W.; Koivunen, E. Role of leukemia cell invadosome in extramedullary infiltration. Blood 2009, 114, 3008–3017. [Google Scholar] [CrossRef]
- Sabeh, F.; Li, X.Y.; Saunders, T.L.; Rowe, R.G.; Weiss, S.J. Secreted versus membrane-anchored collagenases: Relative roles in fibroblast-dependent collagenolysis and invasion. J. Biol. Chem. 2009, 284, 23001–23011. [Google Scholar] [CrossRef]
- Van de Vijver, M.J.; He, Y.D.; van’t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. New Eng. J. Med. 2002, 347, 1999–2009. [Google Scholar]
- Wang, Y.; Klijn, J.G.; Zhang, Y.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J.; et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar]
- Pawitan, Y.; Bjohle, J.; Amler, L.; Borg, A.L.; Egyhazi, S.; Hall, P.; Han, X.; Holmberg, L.; Huang, F.; Klaar, S.; et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005, 7, R953–R964. [Google Scholar]
- Seftor, R.E.; Seftor, E.A.; Kirschmann, D.A.; Hendrix, M.J. Targeting the tumor microenvironment with chemically modified tetracyclines: inhibition of laminin 5 γ2 chain promigratory fragments and vasculogenic mimicry. Mol. Cancer Ther. 2002, 1, 1173–1179. [Google Scholar]
- Oku, N.; Sasabe, E.; Ueta, E.; Yamamoto, T.; Osaki, T. Tight junction protein claudin-1 enhances the invasive activity of oral squamous cell carcinoma cells by promoting cleavage of laminin-5 γ2 chain via matrix metalloproteinase (MMP)-2 and membrane-type MMP-1. Cancer Res. 2006, 66, 5251–5257. [Google Scholar] [CrossRef]
- Liang, Z.; Huang, J.; Huang, T.; Cui, J.; Zeng, L.; Xiong, L.; Wu, F.; Mao, C. Selection and finding of lead peptides dual-targeting MMP-14 and metal ions by subtractive cell surface panning and molecular docking. Int. J. Pept. Res. Ther. 2012, 18, 31–40. [Google Scholar] [CrossRef]
- Mori, H.; Tomari, T.; Koshikawa, N.; Kajita, M.; Itoh, Y.; Sato, H.; Tojo, H.; Yana, I.; Seiki, M. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002, 21, 3949–3959. [Google Scholar] [CrossRef]
- Lichte, A.; Kolkenbrock, H.; Tschesche, H. The recombinant catalytic domain of membrane-type matrix metalloproteinase-1 (MT1-MMP) induces activation of progelatinase A and progelatinase A complexed with TIMP-2. FEBS Lett. 1996, 397, 277–282. [Google Scholar]
- Cao, J.; Kozarekar, P.; Pavlaki, M.; Chiarelli, C.; Bahou, W.F.; Zucker, S. Distinct roles for the catalytic and hemopexin domains of membrane type 1-matrix metalloproteinase in substrate degradation and cell migration. J. Biol. Chem. 2004, 279, 14129–14139. [Google Scholar]
- Zhu, L.; Wang, H.; Wang, L.; Wang, Y.; Jiang, K.; Li, C.; Ma, Q.; Gao, S.; Wang, L.; Li, W.; et al. High-affinity peptide against MT1-MMP for in vivo tumor imaging. J. Control. Release 2011, 150, 248–255. [Google Scholar] [CrossRef]
- Overall, C.M.; Kleifeld, O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases (TIMPs): evolution, structure and function. Biochem. Biophys. Acta 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 2010, 55–71. [Google Scholar] [CrossRef]
- Nagase, H.; Brew, K. Engineering of tissue inhibitor of metalloproteinases mutants as potential therapeutics. Arthritis Res. 2002, 4 (Suppl. 3), S51–S61. [Google Scholar]
- Wei, S.; Chen, Y.; Chung, L.; Nagase, H.; Brew, K. Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP-1) inhibitory domain. J. Biol. Chem. 2003, 278, 9831–9834. [Google Scholar]
- Hamze, A.B.; Wei, S.; Bahudhanapati, H.; Kota, S.; Acharya, K.R.; Brew, K. Constraining specificity in the N-domain of tissue inhibitor of metalloproteinases-1; gelatinase-selective inhibitors. Protein Sci. 2007, 16, 1905–1913. [Google Scholar]
- Bahudhanapati, H.; Zhang, Y.; Sidhu, S.S.; Brew, K. Phage display of tissue inhibitor of metalloproteinases-2 (TIMP-2). J. Biol. Chem. 2011, 286, 31761–31770. [Google Scholar]
- Schlippe, Y.V.G.; Hartman, M.C.T.; Josephson, K.; Szostak, J.W. In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. J. Am. Chem. Soc. 2012, 134, 10469–10477. [Google Scholar]
- Sample Availability: Contact the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).