Occurrence of Ordered and Disordered Structural Elements in Postsynaptic Proteins Supports Optimization for Interaction Diversity

 ,
,
Abstract
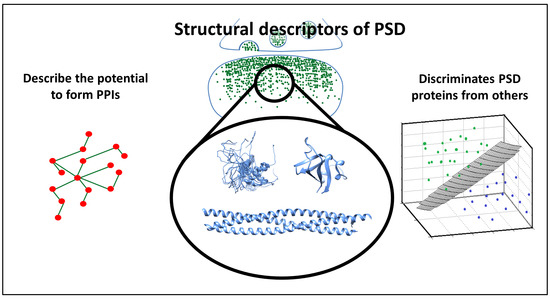
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Annotation and Prediction of Protein Properties
2.3. Statistical Analysis
2.4. Machine Learning
3. Results
3.1. Datasets
3.2. Sequences Feature a Mix of Disorder and Order Promoting Properties
3.3. PSD Proteins Exhibit a Diverse Range of Structured Elements
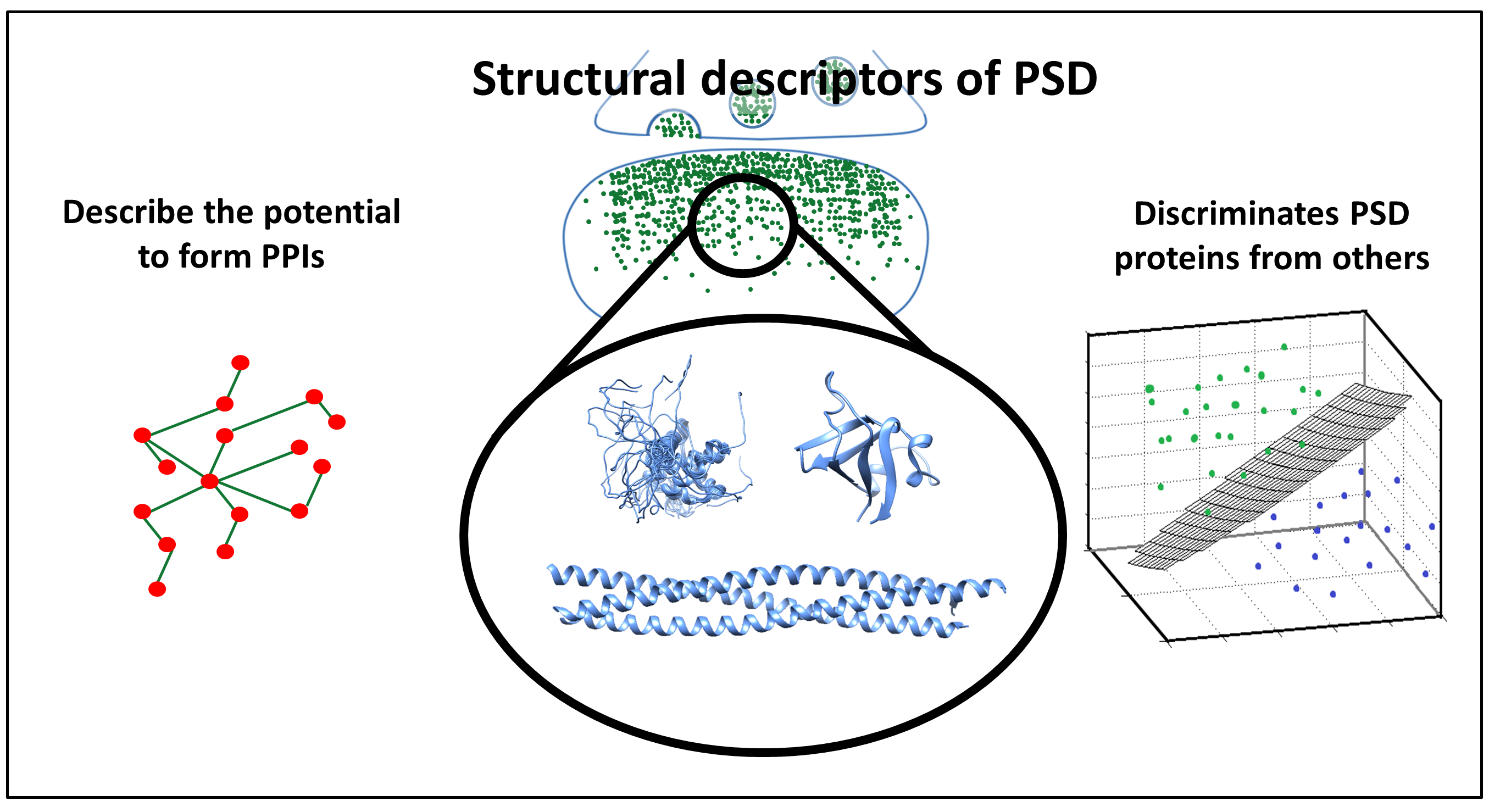
3.4. The Overlap between Protein-Protein Interactions and Post-Translational Modifications Hint a Tightly Regulated Protein Network in PSD
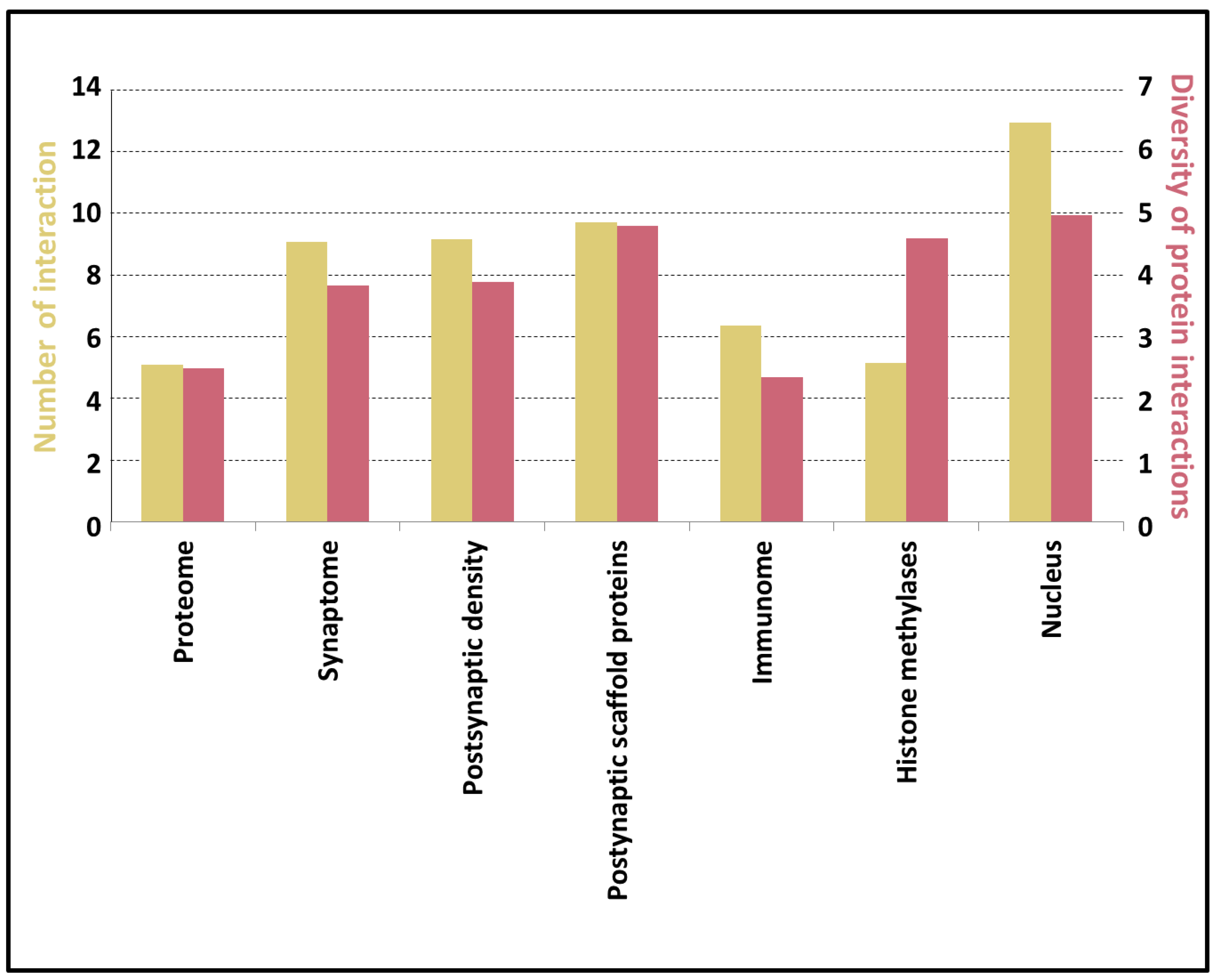
3.5. Potential of the Proteins to Be Engaged in Multivalent Interactions
3.6. Sequential and Structural Features Discriminate PSD Proteins from Other Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rudy, J. The Neurobiology of Learning and Memory; Sinauer Associates: Sunderland, MA, USA, 2008. [Google Scholar]
- Ho, V.M.; Lee, J.-A.; Martin, K.C. The Cell Biology of Synaptic Plasticity. Science 2011, 334, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, M. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. Neurosci. 2009, 10, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Dosemeci, A.; Tao-Cheng, J.H.; Vinade, L.; Winters, C.A.; Pozzo-Miller, L.; Reese, T.S. Glutamate-induced transient modification of the postsynaptic density. Proc. Natl. Acad. Sci. USA 2001, 98, 10428–10432. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, M.D. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 2003, 6, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Farley, M.M.; Swulius, M.T.; Waxham, M.N. Electron tomographic structure and protein composition of isolated rat cerebellar, hippocampal and cortical postsynaptic densities. Neuroscience 2015, 304, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Diering, G.H.; Nirujogi, R.S.; Roth, R.H.; Worley, P.F.; Pandey, A.; Huganir, R.L. Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 2017, 355, 511–515. [Google Scholar] [CrossRef] [PubMed]
- de Vivo, L.; Bellesi, M.; Marshall, W.; Bushong, E.A.; Ellisman, M.H.; Tononi, G.; Cirelli, C. Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 2017, 355, 507–510. [Google Scholar] [CrossRef]
- Blanpied, T.A.; Kerr, J.M.; Ehlers, M.D. Structural plasticity with preserved topology in the postsynaptic protein network. Proc. Natl. Acad. Sci. USA 2008, 105, 12587–12592. [Google Scholar] [CrossRef] [PubMed]
- MacGillavry, H.D.; Song, Y.; Raghavachari, S.; Blanpied, T.A. Nanoscale Scaffolding Domains within the Postsynaptic Density Concentrate Synaptic AMPA Receptors. Neuron 2013, 78, 615–622. [Google Scholar] [CrossRef]
- Meyer, D.; Bonhoeffer, T.; Scheuss, V. Balance and Stability of Synaptic Structures during Synaptic Plasticity. Neuron 2014, 82, 430–443. [Google Scholar] [CrossRef]
- Sugase, K.; Dyson, H.J.; Wright, P.E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 2007, 447, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Tantos, A.; Han, K.-H.; Tompa, P. Intrinsic disorder in cell signaling and gene transcription. Mol. Cell. Endocrinol. 2012, 348, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Vucetic, S.; Xie, H.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional Anthology of Intrinsic Disorder. 2. Cellular Components, Domains, Technical Terms, Developmental Processes, and Coding Sequence Diversities Correlated with Long Disordered Regions. J. Proteome Res. 2007, 6, 1899–1916. [Google Scholar] [CrossRef] [PubMed]
- Csizmok, V.; Follis, A.V.; Kriwacki, R.W.; Forman-Kay, J.D. Dynamic Protein Interaction Networks and New Structural Paradigms in Signaling. Chem. Rev. 2016, 116, 6424–6462. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in scaffold proteins: getting more from less. Prog. Biophys. Mol. Biol. 2008, 98, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Schuman, E.M.; Dynes, J.L.; Steward, O. Synaptic Regulation of Translation of Dendritic mRNAs. J. Neurosci. 2006, 26, 7143–7146. [Google Scholar] [CrossRef]
- Songyang, Z.; Fanning, A.S.; Fu, C.; Xu, J.; Marfatia, S.M.; Chishti, A.H.; Crompton, A.; Chan, A.C.; Anderson, J.M.; Cantley, L.C. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 1997, 275, 73–77. [Google Scholar] [CrossRef]
- Sierralta, J.; Mendoza, C. PDZ-containing proteins: alternative splicing as a source of functional diversity. Brain Res. Rev. 2004, 47, 105–115. [Google Scholar] [CrossRef]
- Feng, Z.; Zeng, M.; Chen, X.; Zhang, M. Neuronal Synapses: Microscale Signal Processing Machineries Formed by Phase Separation? Biochemistry 2018, 57, 2530–2539. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Chen, X.; Zeng, M.; Zhang, M. Phase separation as a mechanism for assembling dynamic postsynaptic density signalling complexes. Curr. Opin. Neurobiol. 2019, 57, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Shang, Y.; Araki, Y.; Guo, T.; Huganir, R.L.; Zhang, M. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 2016, 166, 1163–1175.e12. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Chen, X.; Guan, D.; Xu, J.; Wu, H.; Tong, P.; Zhang, M. Reconstituted Postsynaptic Density as a Molecular Platform for Understanding Synapse Formation and Plasticity. Cell 2018, 174, 1172–1187.e16. [Google Scholar] [CrossRef] [PubMed]
- Van Driesche, S.J.; Martin, K.C. New frontiers in RNA transport and local translation in neurons. Dev. Neurobiol. 2018, 78, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Szabó, B.; Schád, É.; Tantos, Á.; Rawan, A.; Tamás, H.; Murvai, N.; Kovács, O.P.; Kovács, M.; et al. PhaSePro: the database of proteins driving liquid-liquid phase separation. Nucleic Acids Res. 2020. submitted. [Google Scholar]
- Martin, E.W.; Mittag, T. Relationship of Sequence and Phase Separation in Protein Low-Complexity Regions. Biochemistry 2018, 57, 2478–2487. [Google Scholar] [CrossRef]
- Pritišanac, I.; Vernon, R.M.; Moses, A.M.; Forman Kay, J.D.; Pritišanac, I.; Vernon, R.M.; Moses, A.M.; Forman Kay, J.D. Entropy and Information within Intrinsically Disordered Protein Regions. Entropy 2019, 21, 662. [Google Scholar] [CrossRef]
- Bayés, À.; Collins, M.O.; Croning, M.D.R.; van de Lagemaat, L.N.; Choudhary, J.S.; Grant, S.G.N. Comparative Study of Human and Mouse Postsynaptic Proteomes Finds High Compositional Conservation and Abundance Differences for Key Synaptic Proteins. PLoS ONE 2012, 7, e46683. [Google Scholar] [CrossRef]
- Pirooznia, M.; Wang, T.; Avramopoulos, D.; Valle, D.; Thomas, G.; Huganir, R.L.; Goes, F.S.; Potash, J.B.; Zandi, P.P. SynaptomeDB: an ontology-based knowledgebase for synaptic genes. Bioinformatics 2012, 28, 897–899. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium, T. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [PubMed]
- Ortutay, C.; Vihinen, M. Immunome knowledge base (IKB): an integrated service for immunome research. BMC Immunol. 2009, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Frege, T.; Uversky, V.N. Intrinsically disordered proteins in the nucleus of human cells. Biochem. Biophys. Reports 2015, 1, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Lazar, T.; Schad, E.; Szabo, B.; Horvath, T.; Meszaros, A.; Tompa, P.; Tantos, A. Intrinsic protein disorder in histone lysine methylation. Biol. Direct 2016, 11, 30. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Kawashima, S.; Kanehisa, M. AAindex: amino acid index database. Nucleic Acids Res. 2000, 28, 374. [Google Scholar] [CrossRef]
- Ward, J.H. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Gáspári, Z. Is Five Percent Too Small? Analysis of the Overlaps between Disorder, Coiled Coil and Collagen Predictions in Complete Proteomes. Proteomes 2014, 2, 72–83. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins Struct. Funct. Bioinforma. 2005, 61, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A.; Van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Berger, B.; Wilson, D.B.; Wolf, E.; Tonchev, T.; Milla, M.; Kim, P.S. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. USA 1995, 92, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Kovács, Á.; Dudola, D.; Nyitray, L.; Tóth, G.; Nagy, Z.; Gáspári, Z. Detection of single alpha-helices in large protein sequence sets using hardware acceleration. J. Struct. Biol. 2018, 204, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Wootton, J.C. Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput. Chem. 1994, 18, 269–285. [Google Scholar] [CrossRef]
- Dobson, L.; Reményi, I.; Tusnády, G.E. The Human Transmembrane Proteome. Biol. Direct 2015. submitted. [Google Scholar] [CrossRef]
- Dobson, L.; Reményi, I.; Tusnády, G.E. CCTOP: a Consensus Constrained TOPology prediction web server. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Gouw, M.; Sámano-Sánchez, H.; Van Roey, K.; Diella, F.; Gibson, T.J.; Dinkel, H. Exploring Short Linear Motifs Using the ELM Database and Tools. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; Volume 58, pp. 8.22.1–8.22.35. [Google Scholar]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C.; VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 5 August 2019).
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Dobson, L.; Fichó, E.; Tusnády, G.E.; Dosztányi, Z.; Simon, I. How folding and binding intertwine during protein complex formation provides an additional layer of functional regulation. J. Mol. Biol. 2019, in press. [Google Scholar]
- Tusnády, G.E.; Dobson, L.; Tompa, P. Disordered regions in transmembrane proteins. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, T.; Zagotta, W.N.; Aldrich, R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 1990, 250, 533–538. [Google Scholar] [CrossRef]
- Magidovich, E.; Orr, I.; Fass, D.; Abdu, U.; Yifrach, O. Intrinsic disorder in the C-terminal domain of the Shaker voltage-activated K+ channel modulates its interaction with scaffold proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 13022–13027. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Mészáros, B.; Simon, I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef]
- Gasparini, A.; Tosatto, S.C.E.; Murgia, A.; Leonardi, E. Dynamic scaffolds for neuronal signaling: in silico analysis of the TANC protein family. Sci. Rep. 2017, 7, 6829. [Google Scholar] [CrossRef]
- Myrum, C.; Baumann, A.; Bustad, H.J.; Flydal, M.I.; Mariaule, V.; Alvira, S.; Cuéllar, J.; Haavik, J.; Soulé, J.; Valpuesta, J.M.; et al. Arc is a flexible modular protein capable of reversible self-oligomerization. Biochem. J. 2015, 468, 145–158. [Google Scholar] [CrossRef]
- Pastuzyn, E.D.; Day, C.E.; Kearns, R.B.; Kyrke-Smith, M.; Taibi, A.V.; McCormick, J.; Yoder, N.; Belnap, D.M.; Erlendsson, S.; Morado, D.R.; et al. The Neuronal Gene Arc Encodes a Repurposed Retrotransposon Gag Protein that Mediates Intercellular RNA Transfer. Cell 2018, 172, 275–288.e18. [Google Scholar] [CrossRef] [PubMed]
- Gáspári, Z.; Nyitray, L. Coiled coils as possible models of protein structure evolution. Biomol. Concepts 2011, 2, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.K.; Ames, H.M.; Hayashi, Y. Tetrameric Hub Structure of Postsynaptic Scaffolding Protein Homer. J. Neurosci. 2006, 26, 8492–8501. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.K.; Tang, C.; Verpelli, C.; Narayanan, R.; Stearns, M.H.; Xu, R.-M.; Li, H.; Sala, C.; Hayashi, Y. The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell 2009, 137, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sun, M.; Bernard, L.P.; Zhang, H. Postsynaptic density 95 (PSD-95) serine 561 phosphorylation regulates a conformational switch and bidirectional dendritic spine structural plasticity. J. Biol. Chem. 2017, 292, 16150–16160. [Google Scholar] [CrossRef] [PubMed]
- Serber, Z.; Ferrell, J.E. Tuning Bulk Electrostatics to Regulate Protein Function. Cell 2007, 128, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Jackson, R.A.; Ng, C.; Guy, G.R.; Sivaraman, J. Additional Serine/Threonine Phosphorylation Reduces Binding Affinity but Preserves Interface Topography of Substrate Proteins to the c-Cbl TKB Domain. PLoS ONE 2010, 5, e12819. [Google Scholar] [CrossRef] [PubMed]
- Coba, M.P. Regulatory mechanisms in postsynaptic phosphorylation networks. Curr. Opin. Struct. Biol. 2019, 54, 86–94. [Google Scholar] [CrossRef]
- Davey, N.E.; Cyert, M.S.; Moses, A.M. Short linear motifs—Ex nihilo evolution of protein regulation. Cell Commun. Signal. 2015, 13, 43. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Proteins | Proportion | |||
|---|---|---|---|---|
| Proteome | PSD | Proteome | PSD | |
| IDR * | 2851 | 128 | 0.14 | 0.07 |
| CC * | 1187 | 231 | 0.06 | 0.13 |
| TM * | 3460 | 154 | 0.17 | 0.08 |
| DOMAIN * | 2614 | 187 | 0.13 | 0.11 |
| IDR + CC * | 1194 | 132 | 0.06 | 0.07 |
| IDR + TM | 1224 | 92 | 0.06 | 0.05 |
| IDR + DOMAIN * | 2288 | 221 | 0.11 | 0.13 |
| IDR + CC + DOMAIN * | 1088 | 229 | 0.05 | 0.13 |
| ALL * | 125 | 21 | 0.01 | 0.01 |
| All other combination of ordered domains * (CC, TM and Domain) | 3884 | 366 | 0.19 | 0.21 |
| Cross-Validation | Independent Dataset | |||||
|---|---|---|---|---|---|---|
| MCC | BAC | AUC | MCC | BAC | AUC | |
| All features | 0.54 ± 0.09 | 0.77 ± 0.04 | 0.85 ± 0.03 | 0.52 | 0.76 | 0.84 |
| Intrinsic features | 0.32 ± 0.08 | 0.66 ± 0.04 | 0.75 ± 0.03 | 0.38 | 0.68 | 0.76 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiss-Tóth, A.; Dobson, L.; Péterfia, B.; Ángyán, A.F.; Ligeti, B.; Lukács, G.; Gáspári, Z. Occurrence of Ordered and Disordered Structural Elements in Postsynaptic Proteins Supports Optimization for Interaction Diversity. Entropy 2019, 21, 761. https://doi.org/10.3390/e21080761
Kiss-Tóth A, Dobson L, Péterfia B, Ángyán AF, Ligeti B, Lukács G, Gáspári Z. Occurrence of Ordered and Disordered Structural Elements in Postsynaptic Proteins Supports Optimization for Interaction Diversity. Entropy. 2019; 21(8):761. https://doi.org/10.3390/e21080761
Chicago/Turabian StyleKiss-Tóth, Annamária, Laszlo Dobson, Bálint Péterfia, Annamária F. Ángyán, Balázs Ligeti, Gergely Lukács, and Zoltán Gáspári. 2019. "Occurrence of Ordered and Disordered Structural Elements in Postsynaptic Proteins Supports Optimization for Interaction Diversity" Entropy 21, no. 8: 761. https://doi.org/10.3390/e21080761
APA StyleKiss-Tóth, A., Dobson, L., Péterfia, B., Ángyán, A. F., Ligeti, B., Lukács, G., & Gáspári, Z. (2019). Occurrence of Ordered and Disordered Structural Elements in Postsynaptic Proteins Supports Optimization for Interaction Diversity. Entropy, 21(8), 761. https://doi.org/10.3390/e21080761