Function Prediction of Peptide Toxins with Sequence-Based Multi-Tasking PU Learning Method


Abstract
:1. Introduction
2. Results and Discussions
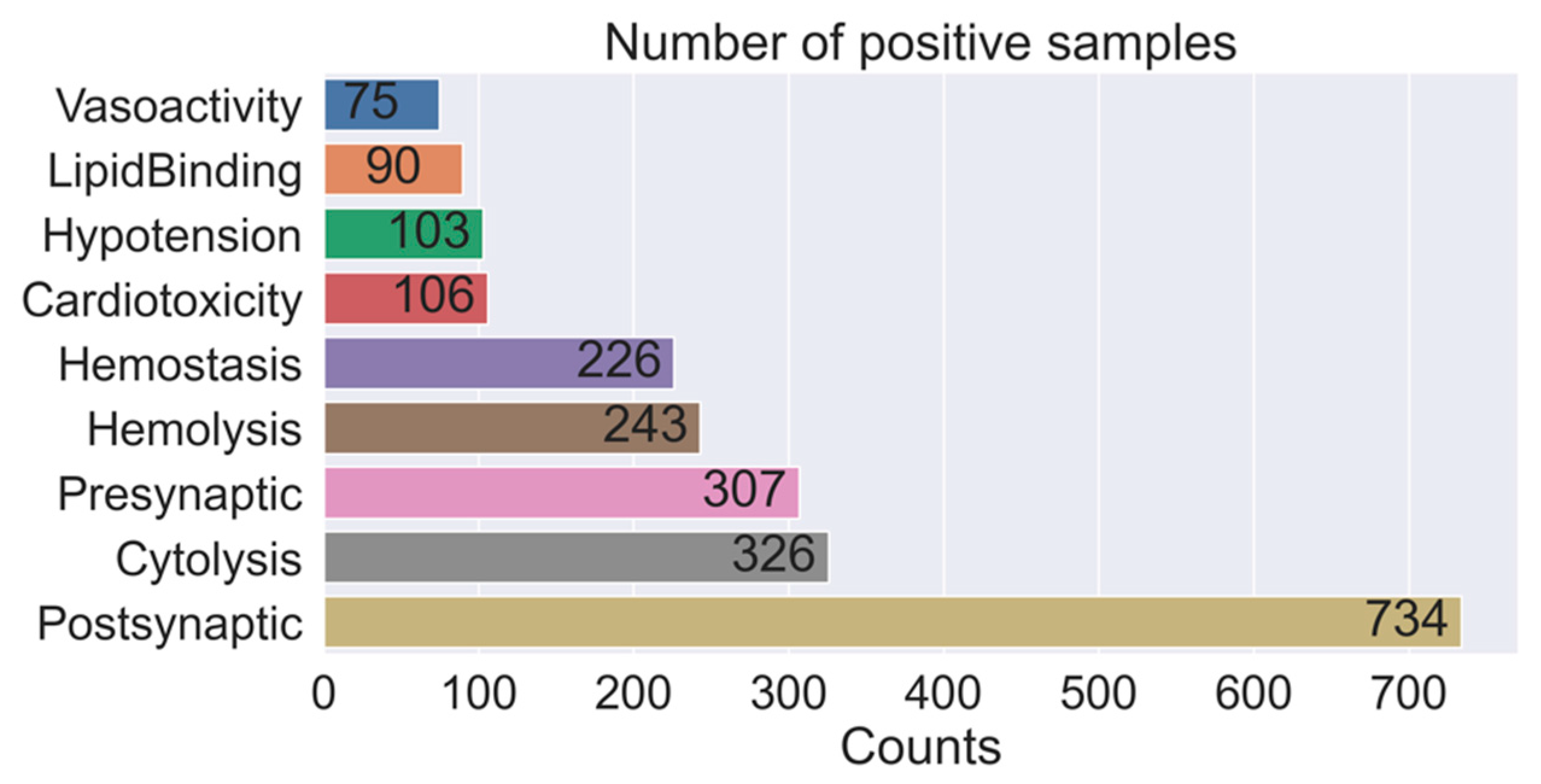
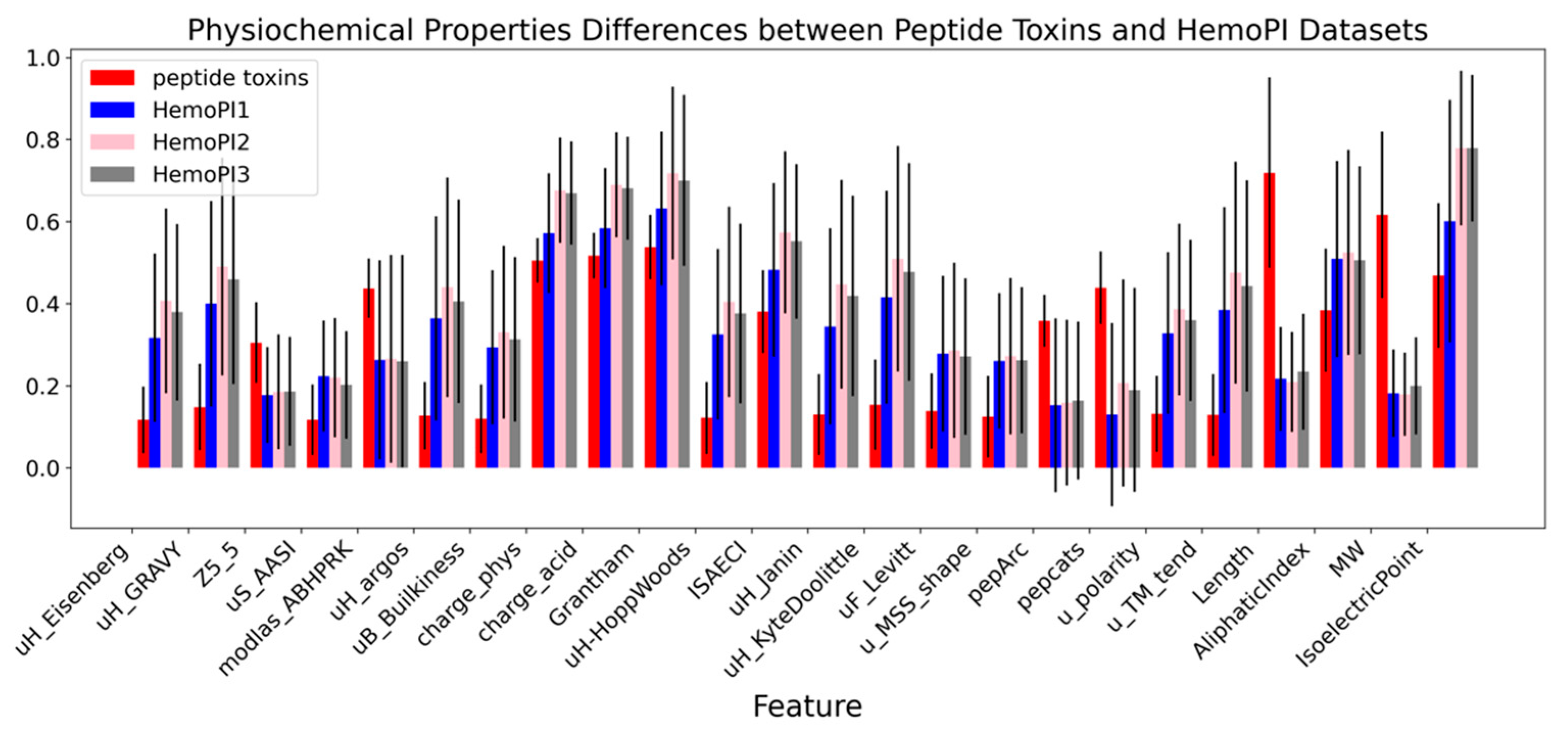
2.1. Data Distribution
2.2. Algorithm Selection
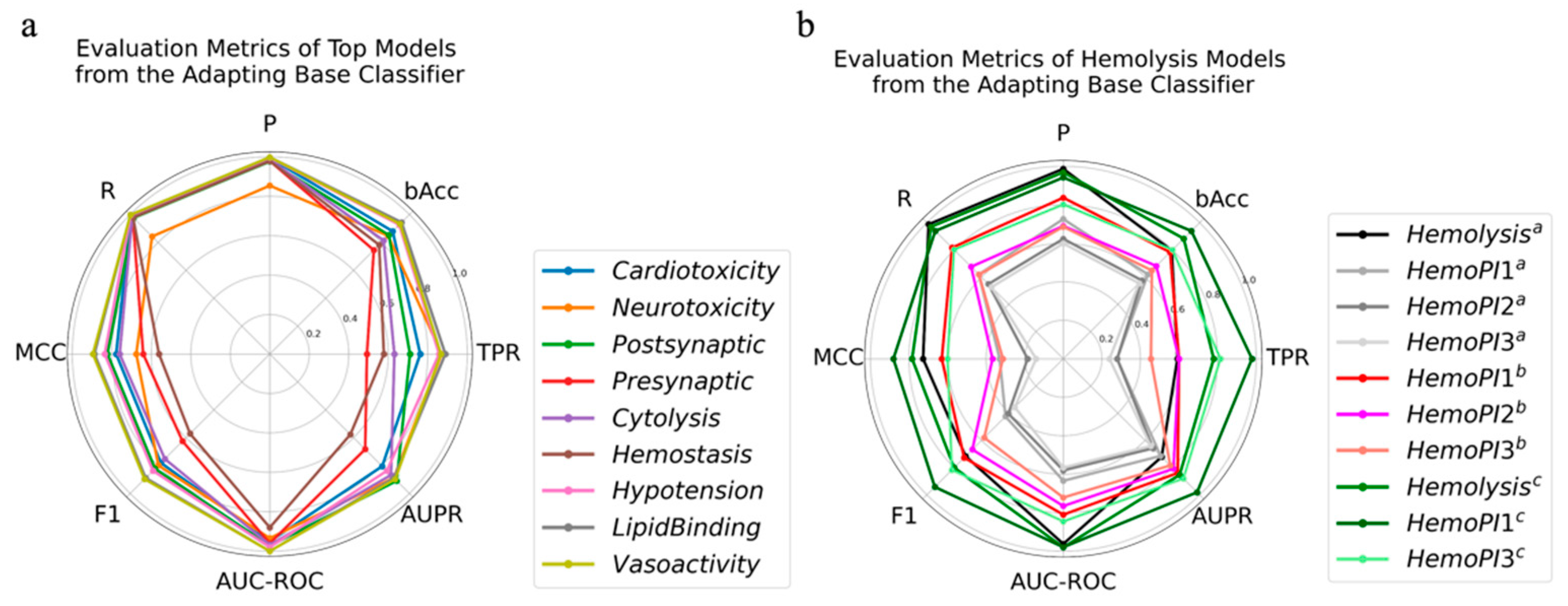
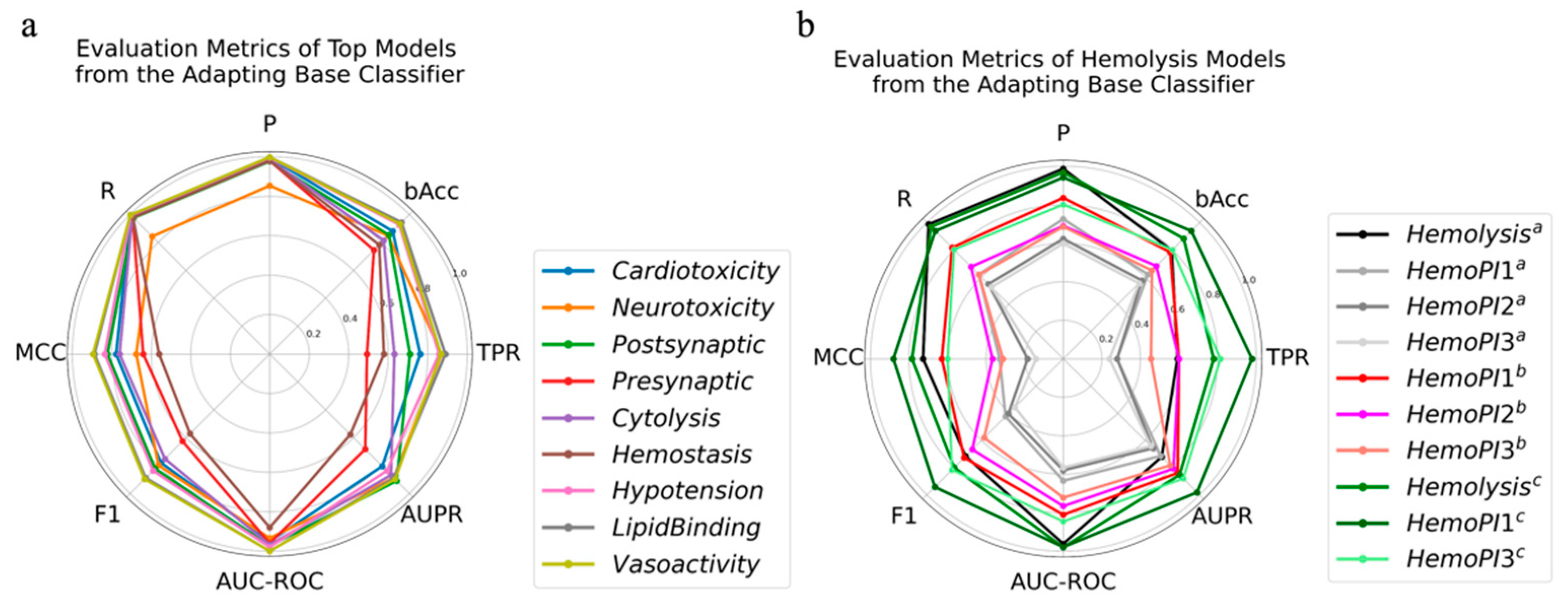
2.3. Adapting Base Classifier
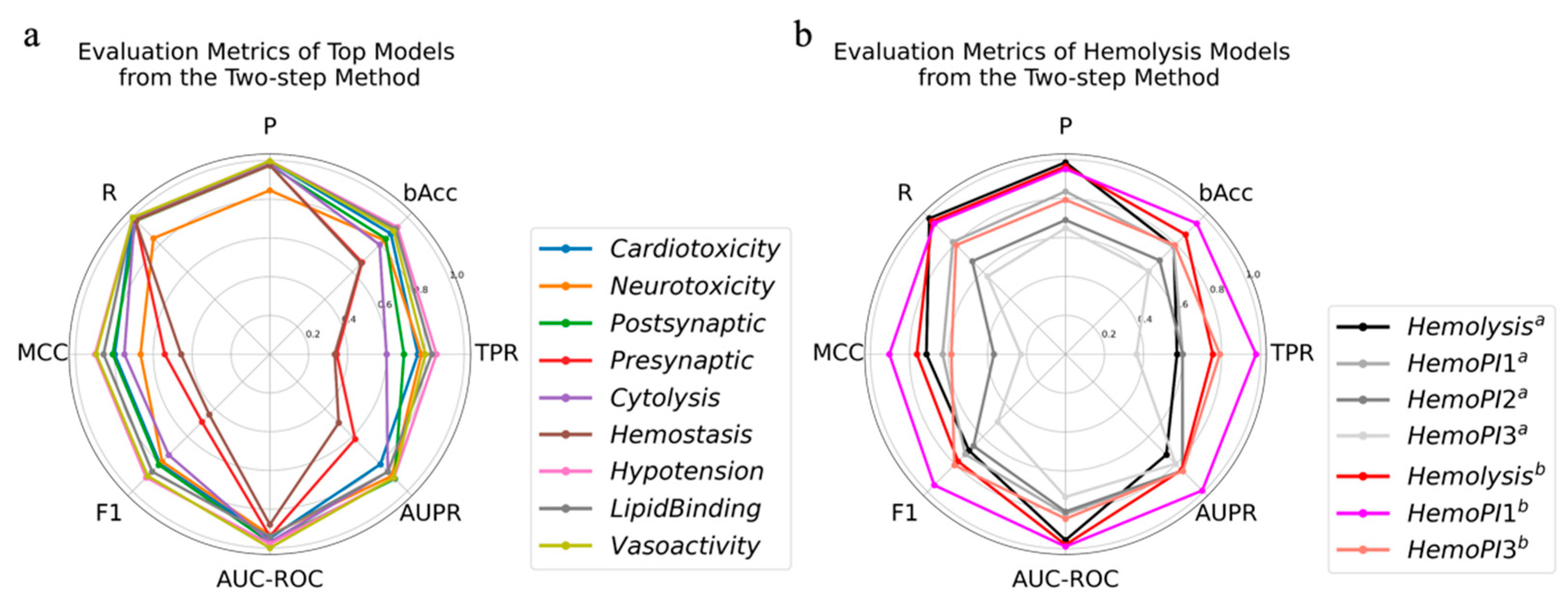
2.4. Two-Step Method
2.5. PU Bagging
2.6. Feature Selection
2.7. Hyperparameter Tuning
2.8. Model Validation
3. Conclusions
4. Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matsumura, K.; Yokogawa, M.; Osawa, M. Peptide Toxins Targeting KV Channels. In Pharmacology of Potassium Channels; Gamper, N., Wang, K., Eds.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2021; Volume 267, pp. 481–505. ISBN 978-3-030-84051-8. [Google Scholar]
- Schendel, V.; Rash, L.D.; Jenner, R.A.; Undheim, E.A.B. The Diversity of Venom: The Importance of Behavior and Venom System Morphology in Understanding Its Ecology and Evolution. Toxins 2019, 11, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madio, B.; Undheim, E.A.B.; King, G.F. Revisiting Venom of the Sea Anemone Stichodactyla Haddoni: Omics Techniques Reveal the Complete Toxin Arsenal of a Well-Studied Sea Anemone Genus. J. Proteom. 2017, 166, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Undheim, E. True Lies: Using Proteomics to Assess the Accuracy of Transcriptome-Based Venomics in Centipedes Uncovers False Positives and Reveals Startling Intraspecific Variation in Scolopendra Subspinipes. Toxins 2018, 10, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belting, M.; Almgren, P.; Manjer, J.; Hedblad, B.; Struck, J.; Wang, T.J.; Bergmann, A.; Melander, O. Vasoactive Peptides with Angiogenesis-Regulating Activity Predict Cancer Risk in Males. Cancer Epidemiol. Biomark. Prev. 2012, 21, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Rossetto, O.; Montecucco, C. Presynaptic Neurotoxins with Enzymatic Activities. In Pharmacology of Neurotransmitter Release; Südhof, T.C., Starke, K., Eds.; Handbook of Experimental Pharmacology; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2008; Volume 184, pp. 129–170. ISBN 978-3-540-74804-5. [Google Scholar]
- Herzig, V.; Cristofori-Armstrong, B.; Israel, M.R.; Nixon, S.A.; Vetter, I.; King, G.F. Animal Toxins—Nature’s Evolutionary-Refined Toolkit for Basic Research and Drug Discovery. Biochem. Pharmacol. 2020, 181, 114096. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Baron, A.; Salinas, M.; Douguet, D.; Scarzello, S.; Dabert-Gay, A.-S.; Debayle, D.; Friend, V.; Alloui, A.; Lazdunski, M.; et al. Black Mamba Venom Peptides Target Acid-Sensing Ion Channels to Abolish Pain. Nature 2012, 490, 552–555. [Google Scholar] [CrossRef]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective Spider Toxins Reveal a Role for the Nav1.1 Channel in Mechanical Pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [Green Version]
- Moczydlowski, E.G. On the Natural and Unnatural History of the Voltage-Gated Na+ Channel. Curr. Top. Membr. 2016, 78, 3–36. [Google Scholar] [CrossRef]
- Nguyen, P.T.; Yarov-Yarovoy, V. Towards Structure-Guided Development of Pain Therapeutics Targeting Voltage-Gated Sodium Channels. Front. Pharmacol. 2022, 13, 842032. [Google Scholar] [CrossRef]
- Robinson, S.D.; Undheim, E.A.B.; Ueberheide, B.; King, G.F. Venom Peptides as Therapeutics: Advances, Challenges and the Future of Venom-Peptide Discovery. Expert Rev. Proteom. 2017, 14, 931–939. [Google Scholar] [CrossRef]
- Pennington, M.W.; Czerwinski, A.; Norton, R.S. Peptide Therapeutics from Venom: Current Status and Potential. Bioorg. Med. Chem. 2018, 26, 2738–2758. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.H.; Greene, L.J.; Alabaster, V.A.; Bakhle, Y.S.; Vane, J.R. Activity of Various Fractions of Bradykinin Potentiating Factor against Angiotensin I Converting Enzyme. Nature 1970, 225, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.P.; Keating, G.M. Eptifibatide: A Review of Its Use in Patients with Acute Coronary Syndromes and/or Undergoing Percutaneous Coronary Intervention. Drugs 2005, 65, 2009–2035. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.M.; Naughton, M.A.; Teng, W.; Rose, J.W.; Phillips, D.R.; Nannizzi, L.; Arfsten, A.; Campbell, A.M.; Charo, I.F. Design of Potent and Specific Integrin Antagonists. Peptide Antagonists with High Specificity for Glycoprotein IIb-IIIa. J. Biol. Chem. 1993, 268, 1066–1073. [Google Scholar] [CrossRef]
- Wermeling, D.P. Ziconotide, an Intrathecally Administered N-Type Calcium Channel Antagonist for the Treatment of Chronic Pain. Pharmacotherapy 2005, 25, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Miljanich, G.P. Ziconotide: Neuronal Calcium Channel Blocker for Treating Severe Chronic Pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kini, R.M. From Snake Venom Toxins to Therapeutics—Cardiovascular Examples. Toxicon 2012, 59, 497–506. [Google Scholar] [CrossRef]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-Venom Peptides as Therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef] [Green Version]
- Han, T.; Teichert, R.; Olivera, B.; Bulaj, G. Conus Venoms—A Rich Source of Peptide-Based Therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef]
- Bajaj, S.; Ong, S.T.; Chandy, K.G. Contributions of Natural Products to Ion Channel Pharmacology. Nat. Prod. Rep. 2020, 37, 703–716. [Google Scholar] [CrossRef]
- Rajendra, W.; Armugam, A.; Jeyaseelan, K. Neuroprotection and Peptide Toxins. Brain Res. Rev. 2004, 45, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting Antimicrobial Peptides with Improved Accuracy by Incorporating the Compositional, Physico-Chemical and Structural Features into Chou’s General PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plisson, F.; Ramírez-Sánchez, O.; Martínez-Hernández, C. Machine Learning-Guided Discovery and Design of Non-Hemolytic Peptides. Sci. Rep. 2020, 10, 16581. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Ptv, L. ACPP: A Web Server for Prediction and Design of Anti-Cancer Peptides. Int. J. Pept. Res. Ther. 2015, 21, 99–106. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.-C. IACP: A Sequence-Based Tool for Identifying Anticancer Peptides. Oncotarget 2016, 7, 16895–16909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. AIPpred: Sequence-Based Prediction of Anti-Inflammatory Peptides Using Random Forest. Front. Pharmacol. 2018, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Huo, H.; Li, T.; Wang, S.; Lv, Y.; Zuo, Y.; Yang, L. Prediction of Presynaptic and Postsynaptic Neurotoxins by Combining Various Chou’s Pseudo Components. Sci. Rep. 2017, 7, 5827. [Google Scholar] [CrossRef] [Green Version]
- Naamati, G.; Askenazi, M.; Linial, M. ClanTox: A Classifier of Short Animal Toxins. Nucleic Acids Res. 2009, 37, W363–W368. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Li, F.; Dong, S.; Leier, A.; Han, M.; Guo, X.; Xu, J.; Wang, X.; Pan, S.; Jia, C.; Zhang, Y.; et al. Positive-Unlabeled Learning in Bioinformatics and Computational Biology: A Brief Review. Brief. Bioinform. 2022, 23, bbab461. [Google Scholar] [CrossRef]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine Learning Methods in Drug Discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tan, J.; Han, D.; Zhu, H. From Machine Learning to Deep Learning: Progress in Machine Intelligence for Rational Drug Discovery. Drug Discov. Today 2017, 22, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Segal, M.R. Biological Sequence Classification Utilizing Positive and Unlabeled Data. Bioinformatics 2008, 24, 1198–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkan, C.; Noto, K. Learning Classifiers from Only Positive and Unlabeled Data. In Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining—KDD 08, Las Vegas, NV, USA, 24–27 August 2008; ACM Press: New York, NY, USA, 2008; p. 213. [Google Scholar]
- Kaboutari, A.; Bagherzadeh, J.; Kheradmand, F. An Evaluation of Two-Step Techniques for Positive-Unlabeled Learning in Text Classification. IJCATR 2014, 3, 592–594. [Google Scholar] [CrossRef]
- Mordelet, F.; Vert, J.-P. A Bagging SVM to Learn from Positive and Unlabeled Examples. Pattern Recogn. Lett. 2014, 37, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef] [Green Version]
- Hosmer, D.W.; Lemeshow, S.; Sturdivant, R.X. Applied Logistic Regression, Wiley Series in Probability and Statistics; 1st ed.; Wiley: Hoboken, NJ, USA, 2013; ISBN 978-0-470-58247-3. [Google Scholar]
- Tharwat, A. Linear vs. Quadratic Discriminant Analysis Classifier: A Tutorial. IJAPR 2016, 3, 145. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Thomas, M.C.; Peter, E.H. Nearest Neighbor Pattern Classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.F.; Golub, G.H.; LeVeque, R.J. Updating Formulae and a Pairwise Algorithm for Computing Sample Variances. In COMPSTAT 1982 5th Symposium Held at Toulouse 1982; Caussinus, H., Ettinger, P., Tomassone, R., Eds.; Physica-Verlag HD: Heidelberg, Germany, 1982; pp. 30–41. ISBN 978-3-7051-0002-2. [Google Scholar]
- Freund, Y.; Schapire, R.E. A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Menzies, T.; Kocagüneli, E.; Minku, L.; Peters, F.; Turhan, B. Using Goals in Model-Based Reasoning. In Sharing Data and Models in Software Engineering; Elsevier: Amsterdam, The Netherlands, 2015; pp. 321–353. ISBN 978-0-12-417295-1. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Liu, H. Data Mining and Processing for Train Unmanned Driving Systems. In Unmanned Driving Systems for Smart Trains; Elsevier: Amsterdam, The Netherlands, 2021; pp. 211–252. ISBN 978-0-12-822830-2. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; ACM: New York, NY, USA, 2016; pp. 785–794. [Google Scholar]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. LightGBM: A Highly Efficient Gradient Boosting Decision Tree. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; pp. 3149–3157. [Google Scholar]
- Fry, B.G.; Winter, K.; Norman, J.A.; Roelants, K.; Nabuurs, R.J.A.; van Osch, M.J.P.; Teeuwisse, W.M.; van der Weerd, L.; Mcnaughtan, J.E.; Kwok, H.F.; et al. Functional and Structural Diversification of the Anguimorpha Lizard Venom System. Mol. Cell Proteom. 2010, 9, 2369–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, J.S.; Zdenek, C.N.; Hay, C.; Violette, A.; Fourmy, R.; Cochran, C.; Fry, B.G. Varanid Lizard Venoms Disrupt the Clotting Ability of Human Fibrinogen through Destructive Cleavage. Toxins 2019, 11, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naimuddin, M.; Kobayashi, S.; Tsutsui, C.; Machida, M.; Nemoto, N.; Sakai, T.; Kubo, T. Directed Evolution of a Three-Finger Neurotoxin by Using CDNA Display Yields Antagonists as Well as Agonists of Interleukin-6 Receptor Signaling. Mol. Brain 2011, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Kini, R.M.; Doley, R. Structure, Function and Evolution of Three-Finger Toxins: Mini Proteins with Multiple Targets. Toxicon 2010, 56, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, I.; Hwang, D.; Lee, H.; Yoon, W.; Chae, J.; Han, C.; Yum, S.; Kang, C.; Kim, E. Proteomic Analysis of Novel Components of Nemopilema Nomurai Jellyfish Venom: Deciphering the Mode of Action. Toxins 2019, 11, 153. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.T.; Gabernet, G.; Hiss, J.A.; Schneider, G. ModlAMP: Python for Antimicrobial Peptides. Bioinformatics 2017, 33, 2753–2755. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Chen, D.; Tantai, X.; Chang, X.; Tian, M.; Jia, T. Weakly Supervised Anomaly Detection Based on Two-Step Cyclic Iterative PU Learning Strategy. Neural Process. Lett. 2022, 54, 4409–4426. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activity | Model | Parameters | TPR | AUPR | R | bAcc | F1 | P | MCC | AUC-ROC |
|---|---|---|---|---|---|---|---|---|---|---|
| Cardiotoxicity | KNN | {‘n_neighbors’: 5, ‘p’: 1, ‘weights’: ‘distance’} | 0.81 | 0.83 | 0.81 | 0.90 | 0.83 | 0.85 | 0.83 | 0.95 |
| Neurotoxicity | LGBM | {‘max_depth’: 8, ‘num_leaves’: 40} | 0.89 | 0.90 | 0.86 | 0.81 | 0.74 | 0.85 | 0.70 | 0.94 |
| Postsynaptic | LGBM | {‘max_depth’: 8, ‘num_leaves’: 40} | 0.91 | 0.82 | 0.91 | 0.86 | 0.90 | 0.98 | 0.85 | 0.97 |
| Presynaptic | LGBM | {‘max_depth’: 8, ‘num_leaves’: 40} | 0.70 | 0.66 | 0.71 | 0.85 | 0.65 | 0.61 | 0.64 | 0.95 |
| Cytolysis | LGBM | {‘max_depth’: 7, ‘num_leaves’: 30} | 0.80 | 0.87 | 0.80 | 0.90 | 0.85 | 0.90 | 0.84 | 0.97 |
| Hemostasis | KNN | {‘n_neighbors’: 3, ‘p’: 4, ‘weights’: ‘distance’} | 0.60 | 0.58 | 0.60 | 0.80 | 0.61 | 0.63 | 0.60 | 0.88 |
| Vasoactivity | KNN | {‘n_neighbors’: 5, ‘p’: 2, ‘weights’: ‘distance’} | 0.93 | 0.99 | 0.93 | 0.97 | 0.93 | 0.93 | 0.93 | 1.00 |
| Hypotension | KNN | {‘n_neighbors’: 6, ‘p’: 1, ‘weights’: ‘distance’} | 0.86 | 0.89 | 0.86 | 0.93 | 0.88 | 0.90 | 0.88 | 0.97 |
| Lipid binding | LGBM | {‘max_depth’: 8, ‘num_leaves’: 20} | 0.94 | 0.87 | 0.94 | 0.97 | 0.85 | 0.77 | 0.85 | 0.99 |
| Hemolysis a | LGBM | {‘max_depth’: 7, ‘num_leaves’: 20} | 0.82 | 0.84 | 0.82 | 0.90 | 0.78 | 0.74 | 0.76 | 0.98 |
| HemoPI1 b | 0.99 | 0.96 | 0.99 | 0.85 | 0.87 | 0.78 | 0.73 | 0.95 | ||
| HemoPI3 b | 0.87 | 0.88 | 0.87 | 0.78 | 0.82 | 0.77 | 0.57 | 0.84 |
| Activity | Model | Parameters | TPR | AUPR | R | bAcc | F1 | P | MCC | AUC-ROC |
|---|---|---|---|---|---|---|---|---|---|---|
| Cardiotoxicity | KNN | {‘n_neighbors’: 2, ‘weights’: ‘uniform’} | 0.81 | 0.87 | 1.00 | 0.90 | 0.85 | 1.00 | 0.85 | 0.95 |
| Neurotoxicity | LGBM | {‘max_depth’: 8, ‘num_leaves’: 40} | 0.85 | 0.90 | 0.86 | 0.86 | 0.81 | 0.86 | 0.70 | 0.94 |
| Postsynaptic | LGBM | {‘max_depth’: 8, ‘num_leaves’: 40} | 0.80 | 0.91 | 0.98 | 0.90 | 0.86 | 0.98 | 0.86 | 0.97 |
| Presynaptic | LGBM | {‘max_depth’: 6, ‘num_leaves’: 40} | 0.51 | 0.64 | 0.98 | 0.75 | 0.57 | 0.97 | 0.56 | 0.94 |
| Cytolysis | LGBM | {‘max_depth’: 6, ‘num_leaves’: 30} | 0.77 | 0.87 | 0.99 | 0.88 | 0.81 | 0.99 | 0.80 | 0.97 |
| Hemostasis | KNN | {‘n_neighbors’: 4, ‘p’: 2, ‘weights’: ‘distance’} | 0.36 | 0.55 | 0.98 | 0.68 | 0.48 | 0.98 | 0.50 | 0.87 |
| Vasoactivity | KNN | {‘n_neighbors’: 8, ‘p’: 2, ‘weights’: ‘distance’} | 0.87 | 1.00 | 1.00 | 0.93 | 0.93 | 1.00 | 0.93 | 1.00 |
| Hypotension | KNN | {‘n_neighbors’: 1, ‘weights’: ‘uniform’} | 0.86 | 0.86 | 1.00 | 0.93 | 0.86 | 1.00 | 0.86 | 0.93 |
| Lipid binding | SVM | {‘C’: 100.0, ‘gamma’: 1.0, ‘kernel’: ‘rbf’} | 0.94 | 0.92 | 1.00 | 0.97 | 0.90 | 1.00 | 0.90 | 0.99 |
| Hemolysis a | LGBM | {‘max_depth’: 8, ‘num_leaves’: 40} | 0.82 | 0.85 | 0.96 | 0.90 | 0.78 | 0.97 | 0.76 | 0.98 |
| HemoPI1 b | 0.99 | 0.98 | 0.87 | 0.87 | 0.88 | 0.89 | 0.75 | 0.98 | ||
| HemoPI3 b | 0.85 | 0.85 | 0.79 | 0.78 | 0.81 | 0.79 | 0.57 | 0.85 |
| Adaptive Base Classifier | Two-Step Method | |||||||
|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | TP | FP | FN | TN | |
| Cytolysis | 7 | 3 | 0 | 51 | 6 | 1 | 1 | 53 |
| Cardiotoxicity | 7 | 0 | 0 | 54 | 7 | 0 | 0 | 54 |
| Neurotoxicity | 31 | 19 | 0 | 11 | 30 | 18 | 1 | 12 |
| Presynaptic | 0 | 0 | 0 | 61 | 0 | 0 | 0 | 61 |
| Postsynaptic | 22 | 7 | 8 | 24 | 22 | 7 | 8 | 24 |
| Lipid binding | 0 | 0 | 0 | 61 | 0 | 0 | 0 | 61 |
| Vasoactivity | 0 | 0 | 0 | 61 | 0 | 0 | 0 | 61 |
| Hypotension | 0 | 0 | 0 | 61 | 0 | 1 | 0 | 60 |
| Hemolysis | 0 | 0 | 2 | 59 | 0 | 0 | 2 | 59 |
| Hemostasis | 0 | 2 | 0 | 59 | 0 | 1 | 0 | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, Y.; Zhang, H.; Zhang, L. Function Prediction of Peptide Toxins with Sequence-Based Multi-Tasking PU Learning Method. Toxins 2022, 14, 811. https://doi.org/10.3390/toxins14110811
Chu Y, Zhang H, Zhang L. Function Prediction of Peptide Toxins with Sequence-Based Multi-Tasking PU Learning Method. Toxins. 2022; 14(11):811. https://doi.org/10.3390/toxins14110811
Chicago/Turabian StyleChu, Yanyan, Huanhuan Zhang, and Lei Zhang. 2022. "Function Prediction of Peptide Toxins with Sequence-Based Multi-Tasking PU Learning Method" Toxins 14, no. 11: 811. https://doi.org/10.3390/toxins14110811
APA StyleChu, Y., Zhang, H., & Zhang, L. (2022). Function Prediction of Peptide Toxins with Sequence-Based Multi-Tasking PU Learning Method. Toxins, 14(11), 811. https://doi.org/10.3390/toxins14110811