
Omega-3 (n-3) Fatty Acid–Statin Interaction: Evidence for a Novel Therapeutic Strategy for Atherosclerotic Cardiovascular Disease


Abstract
1. Introduction
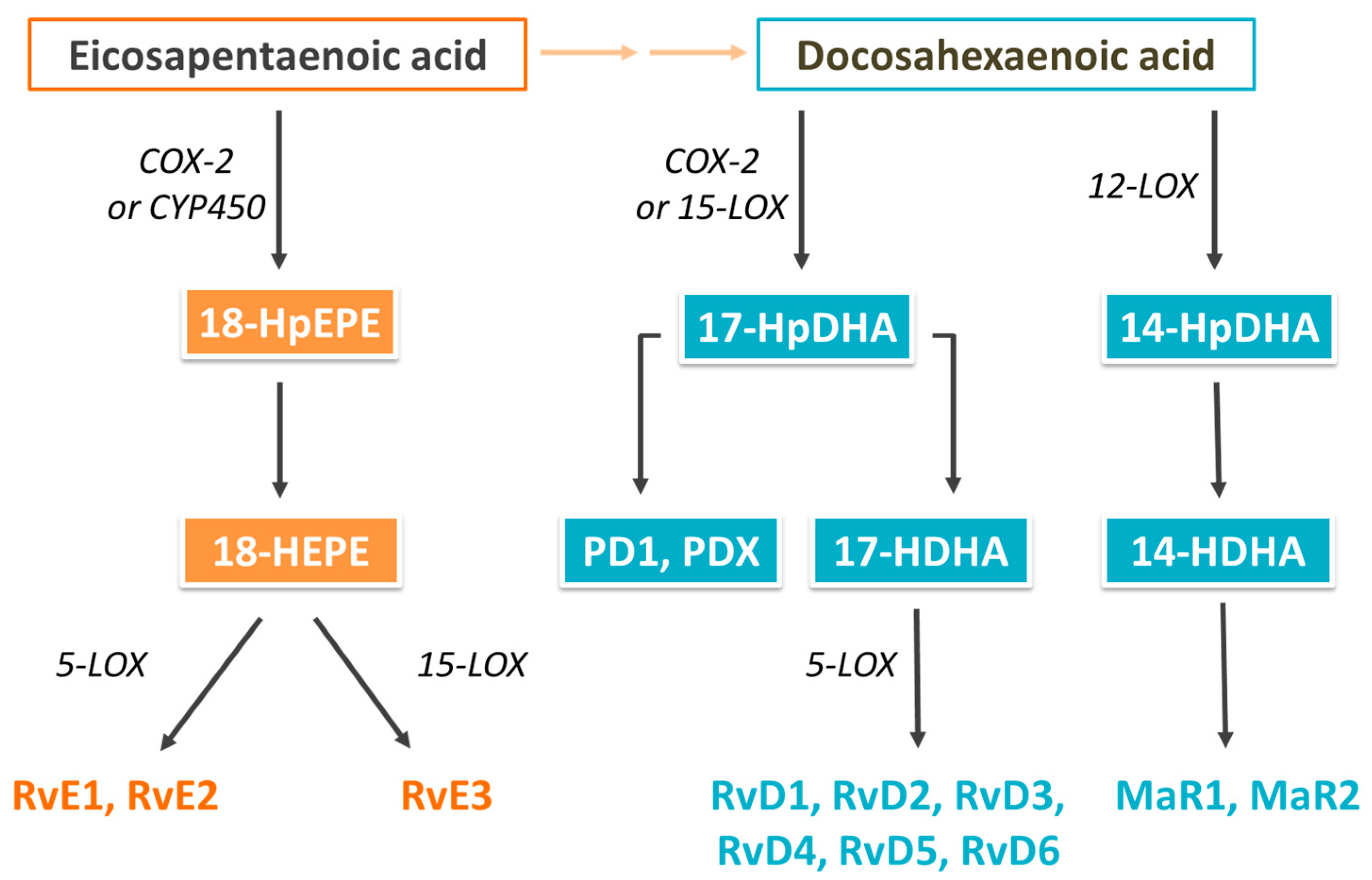
2. Biological Actions of Omega-3 (n-3) Fatty Acids and Their Oxylipin Metabolites
3. Oxylipins in Cardiovascular Diseases
4. The Pharmacology of Statins
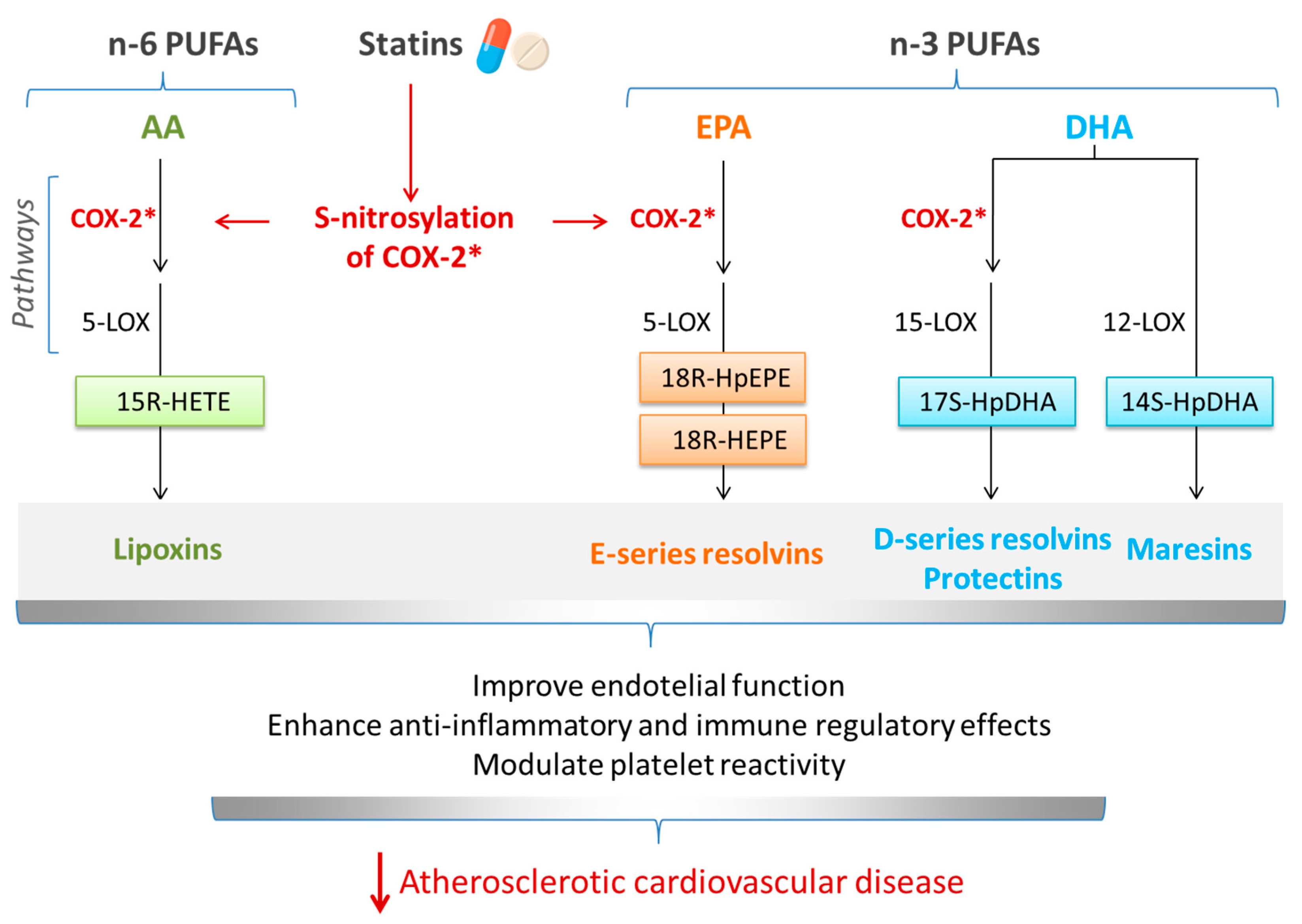
5. Interrelationship between Omega-3 (n-3) PUFAs and Statins in Cardiovascular Diseases: Involvement of SPMs
6. Omega-3 (n-3) Fatty Acids and Cardiovascular Diseases: Findings from Cohort Studies
7. Clinical Trials on Therapeutic Effects of Combined Statins and Omega-3 (n-3) Fatty Acids in Cardiovascular Diseases
8. Meta-Analyses of Clinical Trials of Statins and n-3 PUFAs and Blood Lipids and Cardiovascular Diseases
9. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Cardiovascular Diseases. Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1 (accessed on 19 January 2024).
- Pirillo, A.; Casula, M.; Olmastroni, E.; Norata, G.D.; Catapano, A.L. Global epidemiology of dyslipidaemias. Nat. Rev. Cardiol. 2021, 18, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Akivis, Y.; Alkaissi, H.; McFarlane, S.I.; Bukharovich, I. The role of triglycerides in atherosclerosis: Recent pathophysiologic insights and therapeutic implications. Curr. Cardiol. Rev. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Surma, S.; Toth, P.P. 2023: The year in cardiovascular disease—The year of new and prospective lipid lowering therapies. Can we render dyslipidemia a rare disease by 2024? Arch. Med. Sci. 2023, 19, 1602–1615. [Google Scholar] [CrossRef]
- Yanai, H.; Yoshida, H. Secondary dyslipidemia: Its treatments and association with atherosclerosis. Glob. Health Med. 2021, 3, 15–23. [Google Scholar] [CrossRef]
- Aygun, S.; Tokgozoglu, L. Comparison of current international guidelines for the management of dyslipidemia. J. Clin. Med. 2022, 11, 7249. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.-T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2019, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: Executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef]
- Chou, R.; Dana, T.; Blazina, I.; Daeges, M.; Jeanne, T.L. Statins for prevention of cardiovascular disease in adults: Evidence report and systematic review for the US Preventive Services Task Force. JAMA 2016, 316, 2008–2024. [Google Scholar] [CrossRef] [PubMed]
- Skulas-Ray, A.C.; Wilson, P.W.; Harris, W.S.; Brinton, E.A.; Kris-Etherton, P.M.; Richter, C.K.; Jacobson, T.A.; Engler, M.B.; Miller, M.; Robinson, J.G. Omega-3 fatty acids for the management of hypertriglyceridemia: A science advisory from the American Heart Association. Circulation 2019, 140, e673–e691. [Google Scholar] [CrossRef] [PubMed]
- Roche, H.M. Unsaturated fatty acids. Proc. Nutr. Soc. 1999, 58, 397–401. [Google Scholar] [CrossRef]
- Hodson, L.; Rosqvist, F.; Parry, S.A. The influence of dietary fatty acids on liver fat content and metabolism. Proc. Nutr. Soc. 2020, 79, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Omega-3 fatty acids and metabolic partitioning of fatty acids within the liver in the context of nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2022, 25, 248–255. [Google Scholar] [CrossRef]
- Shearer, G.C.; Savinova, O.V.; Harris, W.S. Fish oil—How does it reduce plasma triglycerides? Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2012, 1821, 843–851. [Google Scholar] [CrossRef]
- Rundblad, A.; Sandoval, V.; Holven, K.B.; Ordovás, J.M.; Ulven, S.M. Omega-3 fatty acids and individual variability in plasma triglyceride response: A mini-review. Redox Biol. 2023, 63, 102730. [Google Scholar] [CrossRef]
- AbuMweis, S.; Jew, S.; Tayyem, R.; Agraib, L. Eicosapentaenoic acid and docosahexaenoic acid containing supplements modulate risk factors for cardiovascular disease: A meta-analysis of randomised placebo-control human clinical trials. J. Hum. Nutr. Diet. 2018, 31, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.K.; Calder, P.C.; Eggersdorfer, M. The role of n-3 long chain polyunsaturated fatty acids in cardiovascular disease prevention, and interactions with statins. Nutrients 2018, 10, 775. [Google Scholar] [CrossRef]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2015, 1851, 469–484. [Google Scholar] [CrossRef]
- Calder, P.C. n-3 PUFA and inflammation: From membrane to nucleus and from bench to bedside. Proc. Nutr. Soc. 2020, 79, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Eicosapentaenoic and docosahexaenoic acid derived specialised pro-resolving mediators: Concentrations in humans and the effects of age, sex, disease and increased omega-3 fatty acid intake. Biochimie 2020, 178, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Very long-chain n-3 fatty acids and human health: Fact, fiction and the future. Proc. Nutr. Soc. 2018, 77, 52–72. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.J.; Mann, N.J.; Lewis, J.L.; Milligan, G.C.; Sinclair, A.J.; Howe, P.R. Dietary intakes and food sources of omega-6 and omega-3 polyunsaturated fatty acids. Lipids 2003, 38, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Howe, P.; Meyer, B.; Record, S.; Baghurst, K. Dietary intake of long-chain omega-3 polyunsaturated fatty acids: Contribution of meat sources. Nutrition 2006, 22, 47–53. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority. Scientific opinion on dietary reference values for fats, including saturated fatty acids, polyunsaturated fatty acids, monounsaturated fatty acids, trans fatty acids and cholesterol. EFSA J. 2010, 8, 1461. [Google Scholar]
- Food and Agricultural Organisation of the United Nations. Fat and Fatty Acids in Human Nutrition: Report of an Expert Consultation; Food and Agricultural Organisation of the United Nations: Rome, Italy, 2010. [Google Scholar]
- Scientific Advisory Committee on Nutrition/Committee on Toxicity. Advice on Fish Consumption: Benefits and Risks; TSA: London, UK, 2004. [Google Scholar]
- Simopolous, A.P.; Leaf, A.; Salem, N. Essentiality and recommended dietary intakes for omega-6 and omega-3 fatty acids. Ann. Nutr. Metab. 1999, 43, 127–130. [Google Scholar] [CrossRef] [PubMed]
- National Health and Medical Research Council; Australian Government Department of Health and Ageing; New Zealand Ministry of Health. Nutrient Reference Values for Australia and New Zealand Including Recommended Dietary Intakes; National Health and Medical Research Council: Canberra, Australia, 2006. [Google Scholar]
- Sands, S.A.; Reid, K.J.; Windsor, S.L.; Harris, W.S. The impact of age, body mass index, and fish intake on the EPA and DHA content of human erythrocytes. Lipids 2005, 40, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Browning, L.M.; Walker, C.G.; Mander, A.P.; West, A.L.; Madden, J.; Gambell, J.M.; Young, S.; Wang, L.; Jebb, S.A.; Calder, P.C. Incorporation of eicosapentaenoic and docosahexaenoic acids into lipid pools when given as supplements providing doses equivalent to typical intakes of oily fish. Am. J. Clin. Nutr. 2012, 96, 748–758. [Google Scholar] [CrossRef]
- Djuricic, I.; Calder, P.C. Pros and cons of long-chain omega-3 polyunsaturated fatty acids in cardiovascular health. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 383–406. [Google Scholar] [CrossRef]
- Innes, J.K.; Calder, P.C. Marine omega-3 (n-3) fatty acids for cardiovascular health: An update for 2020. Int. J. Mol. Sci. 2020, 21, 1362. [Google Scholar] [CrossRef] [PubMed]
- Oscarsson, J.; Hurt-Camejo, E. Omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid and their mechanisms of action on apolipoprotein B-containing lipoproteins in humans: A review. Lipids Health Dis. 2017, 16, 149. [Google Scholar] [CrossRef] [PubMed]
- Grevengoed, T.J.; Trammell, S.A.; Svenningsen, J.S.; Makarov, M.V.; Nielsen, T.S.; Jacobsen, J.C.B.; Treebak, J.T.; Calder, P.C.; Migaud, M.E.; Cravatt, B.F. An abundant biliary metabolite derived from dietary omega-3 polyunsaturated fatty acids regulates triglycerides. J. Clin. Investig. 2021, 131, e143861. [Google Scholar] [CrossRef] [PubMed]
- Allaire, J.; Couture, P.; Leclerc, M.; Charest, A.; Marin, J.; Lépine, M.-C.; Talbot, D.; Tchernof, A.; Lamarche, B. A randomized, crossover, head-to-head comparison of eicosapentaenoic acid and docosahexaenoic acid supplementation to reduce inflammation markers in men and women: The Comparing EPA to DHA (ComparED) Study. Am. J. Clin. Nutr. 2016, 104, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Innes, J.K.; Calder, P.C. The differential effects of eicosapentaenoic acid and docosahexaenoic acid on cardiometabolic risk factors: A systematic review. Int. J. Mol. Sci. 2018, 19, 532. [Google Scholar] [CrossRef] [PubMed]
- Woodman, R.J.; Mori, T.A.; Burke, V.; Puddey, I.B.; Watts, G.F.; Beilin, L.J. Effects of purified eicosapentaenoic and docosahexaenoic acids on glycemic control, blood pressure, and serum lipids in type 2 diabetic patients with treated hypertension. Am. J. Clin. Nutr. 2002, 76, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.A.; Burke, V.; Puddey, I.B.; Watts, G.F.; O’Neal, D.N.; Best, J.D.; Beilin, L.J. Purified eicosapentaenoic and docosahexaenoic acids have differential effects on serum lipids and lipoproteins, LDL particle size, glucose, and insulin in mildly hyperlipidemic men. Am. J. Clin. Nutr. 2000, 71, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D.; Navar, A.M.; Thanassoulis, G. Apolipoprotein B vs low-density lipoprotein cholesterol and non–high-density lipoprotein cholesterol as the primary measure of apolipoprotein B lipoprotein-related risk: The debate is over. JAMA Cardiol. 2022, 7, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Sherratt, S.C.; Juliano, R.A.; Mason, R.P. Eicosapentaenoic acid (EPA) has optimal chain length and degree of unsaturation to inhibit oxidation of small dense LDL and membrane cholesterol domains as compared to related fatty acids in vitro. Biochim. Biophys. Acta BBA-Biomembr. 2020, 1862, 183254. [Google Scholar] [CrossRef]
- Sherratt, S.C.; Juliano, R.A.; Copland, C.; Bhatt, D.L.; Libby, P.; Mason, R.P. EPA and DHA containing phospholipids have contrasting effects on membrane structure. J. Lipid Res. 2021, 62, 100106. [Google Scholar] [CrossRef]
- Jacobs, M.L.; Faizi, H.A.; Peruzzi, J.A.; Vlahovska, P.M.; Kamat, N.P. EPA and DHA differentially modulate membrane elasticity in the presence of cholesterol. Biophys. J. 2021, 120, 2317–2329. [Google Scholar] [CrossRef]
- Mason, R.P.; Jacob, R.F.; Shrivastava, S.; Sherratt, S.C.; Chattopadhyay, A. Eicosapentaenoic acid reduces membrane fluidity, inhibits cholesterol domain formation, and normalizes bilayer width in atherosclerotic-like model membranes. Biochim. Biophys. Acta BBA-Biomembr. 2016, 1858, 3131–3140. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Blake, G.J.; Ridker, P.M. Novel clinical markers of vascular wall inflammation. Circ. Res. 2001, 89, 763–771. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Hallenbeck, J.M.; Hansson, G.K.; Becker, K.J. Immunology of ischemic vascular disease: Plaque to attack. Trends Immunol. 2005, 26, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Christie, W.W.; Harwood, J.L. Oxidation of polyunsaturated fatty acids to produce lipid mediators. Essays Biochem. 2020, 64, 401–421. [Google Scholar]
- Chiang, N.; Serhan, C.N. Specialized pro-resolving mediator network: An update on production and actions. Essays Biochem. 2020, 64, 443–462. [Google Scholar]
- Calder, P.C. Eicosanoids. Essays Biochem. 2020, 64, 423–441. [Google Scholar]
- Djuricic, I.; Calder, P.C. Beneficial outcomes of omega-6 and omega-3 polyunsaturated fatty acids on human health: An update for 2021. Nutrients 2021, 13, 2421. [Google Scholar] [CrossRef]
- Pan, G.; Zhang, P.; Yang, J.; Wu, Y. The regulatory effect of specialized pro-resolving mediators on immune cells. Biomed. Pharmacother. 2022, 156, 113980. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Discovery of specialized pro-resolving mediators marks the dawn of resolution physiology and pharmacology. Mol. Asp. Med. 2017, 58, 1–11. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol. Asp. Med. 2017, 58, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Leuti, A.; Maccarrone, M.; Chiurchiù, V. Proresolving lipid mediators: Endogenous modulators of oxidative stress. Oxidative Med. Cell. Longev. 2019, 2019, 8107265. [Google Scholar] [CrossRef] [PubMed]
- Bisicchia, E.; Sasso, V.; Catanzaro, G.; Leuti, A.; Besharat, Z.M.; Chiacchiarini, M.; Molinari, M.; Ferretti, E.; Viscomi, M.T.; Chiurchiù, V. Resolvin D1 halts remote neuroinflammation and improves functional recovery after focal brain damage via ALX/FPR2 receptor-regulated microRNAs. Mol. Neurobiol. 2018, 55, 6894–6905. [Google Scholar] [CrossRef] [PubMed]
- Recchiuti, A.; Serhan, C.N. Pro-resolving lipid mediators (SPMs) and their actions in regulating miRNA in novel resolution circuits in inflammation. Front. Immunol. 2012, 3, 298. [Google Scholar] [CrossRef]
- Dalli, J.; Gomez, E.A.; Jouvene, C.C. Utility of the specialized pro-resolving mediators as diagnostic and prognostic biomarkers in disease. Biomolecules 2022, 12, 353. [Google Scholar] [CrossRef]
- Jaén, R.I.; Sánchez-García, S.; Fernández-Velasco, M.; Boscá, L.; Prieto, P. Resolution-based therapies: The potential of lipoxins to treat human diseases. Front. Immunol. 2021, 12, 658840. [Google Scholar] [CrossRef]
- Fosshaug, L.E.; Colas, R.A.; Anstensrud, A.K.; Gregersen, I.; Nymo, S.; Sagen, E.L.; Michelsen, A.; Vinge, L.E.; Øie, E.; Gullestad, L. Early increase of specialized pro-resolving lipid mediators in patients with ST-elevation myocardial infarction. EBioMedicine 2019, 46, 264–273. [Google Scholar] [CrossRef]
- Halade, G.V.; Kain, V.; Dillion, C.; Beasley, M.; Dudenbostel, T.; Oparil, S.; Limdi, N.A. Race-based and sex-based differences in bioactive lipid mediators after myocardial infarction. ESC Heart Fail. 2020, 7, 1700–1710. [Google Scholar] [CrossRef]
- Schaller, M.S.; Zahner, G.J.; Gasper, W.J.; Harris, W.S.; Conte, M.S.; Hills, N.K.; Grenon, S.M. Relationship between the omega-3 index and specialized pro-resolving lipid mediators in patients with peripheral arterial disease taking fish oil supplements. J. Clin. Lipidol. 2017, 11, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Önal, M.A.; Fentoğlu, Ö.; Aksoy, F.; Calapoğlu, M.; Varol, E.; Orhan, H. Salivary levels of last generation specific pro-resolving lipid mediators (SPMs)(protectin and maresin) in patients with cardiovascular and periodontal disease: A case-control study. J. Periodontal Res. 2021, 56, 606–615. [Google Scholar] [CrossRef]
- Welty, F.K.; Schulte, F.; Alfaddagh, A.; Elajami, T.K.; Bistrian, B.R.; Hardt, M. Regression of human coronary artery plaque is associated with a high ratio of (18-hydroxy-eicosapentaenoic acid+ resolvin E1) to leukotriene B4. FASEB J. 2021, 35, e21448. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.J.; Spite, M.; Owens, C.D.; Lancero, H.; Kroemer, A.H.; Pande, R.; Creager, M.A.; Serhan, C.N.; Conte, M.S. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am. J. Pathol. 2010, 177, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Colas, R.A.; Souza, P.R.; Walker, M.E.; Burton, M.; Zasłona, Z.; Curtis, A.M.; Marques, R.M.; Dalli, J. Impaired production and diurnal regulation of vascular RvDn-3 DPA increase systemic inflammation and cardiovascular disease. Circ. Res. 2018, 122, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Pirela, D.; Nava, M.; Castro, A.; Angarita, L.; Parra, H.; Durán-Agüero, S.; Rojas-Gómez, D.M.; Galbán, N.; Añez, R. Specialized proresolving lipid mediators: A potential therapeutic target for atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3133. [Google Scholar] [CrossRef] [PubMed]
- Hasturk, H.; Abdallah, R.; Kantarci, A.; Nguyen, D.; Giordano, N.; Hamilton, J.; Van Dyke, T.E. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1123–1133. [Google Scholar] [CrossRef]
- Viola, J.R.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Silvestre-Roig, C.; Dittmar, G.; Döring, Y.; Drechsler, M. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ. Res. 2016, 119, 1030–1038. [Google Scholar] [CrossRef]
- Akagi, D.; Chen, M.; Toy, R.; Chatterjee, A.; Conte, M.S. Systemic delivery of proresolving lipid mediators resolvin D2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J. 2015, 29, 2504. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Miyahara, T.; Nitta, J.; Miyahara, K.; Seo, A.; Kimura, M.; Suhara, M.; Akai, A.; Akagi, D.; Yamamoto, K. Proresolving lipid mediators resolvin D1 and protectin D1 isomer attenuate neointimal hyperplasia in the rat carotid artery balloon injury model. J. Surg. Res. 2019, 233, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Salic, K.; Morrison, M.C.; Verschuren, L.; Wielinga, P.Y.; Wu, L.; Kleemann, R.; Gjorstrup, P.; Kooistra, T. Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis 2016, 250, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E. Statin inhibition of HMG-CoA reductase: A 3-dimensional view. Atheroscler. Suppl. 2003, 4, 3–8. [Google Scholar] [CrossRef] [PubMed]
- German, C.A.; Liao, J.K. Understanding the molecular mechanisms of statin pleiotropic effects. Arch. Toxicol. 2023, 97, 1529–1545. [Google Scholar] [CrossRef]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, S1–S45. [Google Scholar] [CrossRef] [PubMed]
- He, W.B.; Ko, H.T.; Curtis, A.J.; Zoungas, S.; Woods, R.L.; Tonkin, A.; Neumann, J.T.; Turner, S.L.; Hopper, I. The effects of statins on cardiovascular and inflammatory biomarkers in primary prevention: A systematic review and meta-analysis. Heart Lung Circ. 2023, 32, 938–948. [Google Scholar] [CrossRef]
- Sheridan, A.; Wheeler-Jones, C.P.D.; Gage, M.C. The immunomodulatory effects of statins on macrophages. Immuno 2022, 2, 317–343. [Google Scholar] [CrossRef]
- Shahbaz, S.K.; Sadeghi, M.; Koushki, K.; Penson, P.E.; Sahebkar, A. Regulatory T cells: Possible mediators for the anti-inflammatory action of statins. Pharmacol. Res. 2019, 149, 104469. [Google Scholar] [CrossRef]
- Diamantis, E.; Kyriakos, G.; Victoria Quiles-Sanchez, L.; Farmaki, P.; Troupis, T. The anti-inflammatory effects of statins on coronary artery disease: An updated review of the literature. Curr. Cardiol. Rev. 2017, 13, 209–216. [Google Scholar] [CrossRef]
- Radbakhsh, S.; Katsiki, N.; Santos, R.D.; Mikhailidis, D.P.; Mantzoros, C.S.; Sahebkar, A. Effects of statins on specialized pro-resolving mediators: An additional pathway leading to resolution of inflammation. Metabolism 2022, 132, 155211. [Google Scholar] [CrossRef]
- Zhou, G.; Ge, S.; Liu, D.; Xu, G.; Zhang, R.; Yin, Q.; Zhu, W.; Chen, J.; Liu, X. Atorvastatin reduces plaque vulnerability in an atherosclerotic rabbit model by altering the 5-lipoxygenase pathway. Cardiology 2010, 115, 221–228. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Ye, Y.; Lin, Y.; Freeberg, S.Y.; Nishi, S.P.; Martinez, J.D.; Huang, M.-H.; Uretsky, B.F.; Perez-Polo, J.R. Augmentation of myocardial production of 15-epi-lipoxin-a4 by pioglitazone and atorvastatin in the rat. Circulation 2006, 114, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Lin, Y.; Perez-Polo, J.R.; Uretsky, B.F.; Ye, Z.; Tieu, B.C.; Birnbaum, Y. Phosphorylation of 5-lipoxygenase at ser523 by protein kinase A determines whether pioglitazone and atorvastatin induce proinflammatory leukotriene B4 or anti-inflammatory 15-epi-lipoxin a4 production. J. Immunol. 2008, 181, 3515–3523. [Google Scholar] [CrossRef]
- Ye, Y.; Nylander, S.; Birnbaum, Y. Unraveling the interaction of aspirin, ticagrelor, and rosuvastatin on the progression of atherosclerosis and inflammation in diabetic mice. Cardiovasc. Drugs Ther. 2017, 31, 489–500. [Google Scholar] [CrossRef]
- Laguna-Fernandez, A.; Checa, A.; Carracedo, M.; Artiach, G.; Petri, M.H.; Baumgartner, R.; Forteza, M.J.; Jiang, X.; Andonova, T.; Walker, M.E. ERV1/ChemR23 signaling protects against atherosclerosis by modifying oxidized low-density lipoprotein uptake and phagocytosis in macrophages. Circulation 2018, 138, 1693–1705. [Google Scholar] [CrossRef]
- Kromann, N.; Green, A. Epidemiological studies in the Upernavik district, Greenland. J. Intern. Med. 1980, 208, 401–406. [Google Scholar] [CrossRef]
- Bjerregaard, P.; Dyerberg, J. Mortality from ischaemic heart disease and cerebrovascular disease in Greenland. Int. J. Epidemiol. 1988, 17, 514–519. [Google Scholar] [CrossRef]
- Bang, H.; Dyerberg, J.; Hjørne, N. The composition of food consumed by Greenland Eskimos. J. Intern. Med. 1976, 200, 69–73. [Google Scholar] [CrossRef]
- Newman, W.; Middaugh, J.; Propst, M.; Rogers, D. Atherosclerosis in Alaska Natives and non-Natives. Lancet 1993, 341, 1056–1057. [Google Scholar] [CrossRef]
- Yano, K.; MacLean, C.J.; Reed, D.M.; Shimizu, Y.; Sasaki, H.; Kodama, K.; Kato, H.; Kagan, A. A comparison of the 12-year mortality and predictive factors of coronary heart disease among Japanese men in Japan and Hawaii. Am. J. Epidemiol. 1988, 127, 476–487. [Google Scholar] [CrossRef]
- Calder, P.C. n-3 Fatty acids and cardiovascular disease: Evidence explained and mechanisms explored. Clin. Sci. 2004, 107, 1–11. [Google Scholar] [CrossRef]
- Hu, F.; Bronner, L.; Willett, W.; Stampfer, M.; Rexrode, K.M.; Albert, C.; Hunter, D.; Manson, J. Fish and omega-3 fatty acid intake and risk of coronary heart disease in women. JAMA 2002, 287, 181. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, P.; He, W.; Chen, J.N.; Wang, W.Q.; Freedman, N.D.; Abnet, C.C.; Wang, J.B.; Jiao, J.J. Association of fish and long-chain omega-3 fatty acids intakes with total and cause-specific mortality: Prospective analysis of 421 309 individuals. J. Intern. Med. 2018, 284, 399–417. [Google Scholar] [CrossRef]
- Chowdhury, R.; Warnakula, S.; Kunutsor, S.; Crowe, F.; Ward, H.; Johnson, L.; Franco, O.; Butterworth, A.; Forouhi, N.; Thompson, S.; et al. Association of dietary, circulating, and supplement fatty acids with coronary risk: A systematic review and meta-analysis. Ann. Intern. Med. 2014, 160, 398–406. [Google Scholar] [CrossRef]
- Alexander, D.D.; Miller, P.E.; Van Elswyk, M.E.; Kuratko, C.N.; Bylsma, L.C. A meta-analysis of randomized controlled trials and prospective cohort studies of eicosapentaenoic and docosahexaenoic long-chain omega-3 fatty acids and coronary heart disease risk. Mayo Clin. Proc. 2017, 92, 15–29. [Google Scholar] [CrossRef]
- Albert, C.; Campos, H.; Stampfer, M.; Ridker, P.; Manson, J.; Willett, W.; Ma, J. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. N. Engl. J. Med. 2002, 346, 1113–1118. [Google Scholar] [CrossRef]
- Del Gobbo, L.C.; Imamura, F.; Aslibekyan, S.; Marklund, M.; Virtanen, J.K.; Wennberg, M.; Yakoob, M.Y.; Chiuve, S.E.; Dela Cruz, L.; Frazier-Wood, A.C.; et al. ω-3 Polyunsaturated fatty acid biomarkers and coronary heart disease: Pooling project of 19 cohort studies. JAMA Intern. Med. 2016, 176, 1155–1166. [Google Scholar] [CrossRef]
- Harris, W.S.; Del Gobbo, L.; Tintle, N.L. The omega-3 index and relative risk for coronary heart disease mortality: Estimation from 10 cohort studies. Atherosclerosis 2017, 262, 51–54. [Google Scholar] [CrossRef]
- Harris, W.S.; Tintle, N.L.; Imamura, F.; Qian, F.; Korat, A.V.A.; Marklund, M.; Djousse, L.; Bassett, J.K.; Carmichael, P.H.; Chen, Y.Y.; et al. Blood n-3 fatty acid levels and total and cause-specific mortality from 17 prospective studies. Nat. Commun. 2021, 12, 2329. [Google Scholar] [CrossRef]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef]
- Jo, S.-H.; Han, S.H.; Kim, S.-H.; Eckel, R.H.; Koh, K.K. Cardiovascular effects of omega-3 fatty acids: Hope or hype? Atherosclerosis 2021, 322, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Budoff, M.J.; Bhatt, D.L.; Kinninger, A.; Lakshmanan, S.; Muhlestein, J.B.; Le, V.T.; May, H.T.; Shaikh, K.; Shekar, C.; Roy, S.K. Effect of icosapent ethyl on progression of coronary atherosclerosis in patients with elevated triglycerides on statin therapy: Final results of the EVAPORATE trial. Eur. Heart J. 2020, 41, 3925–3932. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Lincoff, A.M.; Garcia, M.; Bash, D.; Ballantyne, C.M.; Barter, P.J.; Davidson, M.H.; Kastelein, J.J.; Koenig, W.; McGuire, D.K. Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: The STRENGTH randomized clinical trial. JAMA 2020, 324, 2268–2280. [Google Scholar] [CrossRef]
- Nishizaki, Y.; Miyauchi, K.; Iwata, H.; Inoue, T.; Hirayama, A.; Kimura, K.; Ozaki, Y.; Murohara, T.; Ueshima, K.; Kuwabara, Y. Study protocol and baseline characteristics of Randomized trial for Evaluation in Secondary Prevention Efficacy of Combination Therapy–Statin and Eicosapentaenoic Acid: RESPECT-EPA, the combination of a randomized control trial and an observational biomarker study. Am. Heart J. 2023, 257, 1–8. [Google Scholar] [PubMed]
- Nelson, J.; Raskin, S. The eicosapentaenoic acid: Arachidonic acid ratio and its clinical utility in cardiovascular disease. Postgrad. Med. 2019, 131, 268–277. [Google Scholar] [CrossRef]
- Abe, S.; Sugimura, H.; Watanabe, S.; Murakami, Y.; Ebisawa, K.; Ioka, T.; Takahashi, T.; Ando, T.; Kono, K.; Inoue, T. Eicosapantaenoic acid treatment based on the EPA/AA ratio in patients with coronary artery disease: Follow-up data from the Tochigi Ryomo EPA/AA Trial in Coronary Artery Disease (TREAT-CAD) study. Hypertens. Res. 2018, 41, 939–946. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, X.; Wang, L.; Lu, X.; Huang, J.; Cao, J.; Li, H.; Gu, D. Effect of omega-3 fatty acids supplementation on endothelial function: A meta-analysis of randomized controlled trials. Atherosclerosis 2012, 221, 536–543. [Google Scholar] [CrossRef]
- Lin, N.; Shi, J.J.; Li, Y.M.; Zhang, X.Y.; Chen, Y.; Calder, P.C.; Tang, L.J. What is the impact of n-3 PUFAs on inflammation markers in Type 2 diabetic mellitus populations?: A systematic review and meta-analysis of randomized controlled trials. Lipids Health Dis. 2016, 15, 133. [Google Scholar] [CrossRef]
- O’Mahoney, L.L.; Matu, J.; Price, O.J.; Birch, K.M.; Ajjan, R.A.; Farrar, D.; Tapp, R.; West, D.J.; Deighton, K.; Campbell, M.D. Omega-3 polyunsaturated fatty acids favourably modulate cardiometabolic biomarkers in type 2 diabetes: A meta-analysis and meta-regression of randomized controlled trials. Cardiovasc. Diabetol. 2018, 17, 98. [Google Scholar] [CrossRef] [PubMed]
- Sedighiyan, M.; Djafarian, K.; Dabiri, S.; Abdolahi, M.; Shab-Bidar, S. The effects of omega-3 supplementation on the expanded disability status scale and inflammatory cytokines in multiple sclerosis patients: A systematic review and meta-analysis. CNS Neurol. Disord. Drug Targets 2019, 18, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.F.; Li, K.L.; Li, J.M.; Li, D. Effects of EPA and DHA on blood pressure and inflammatory factors: A meta-analysis of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 2019, 59, 3380–3393. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Hu, F.B.; Manson, J.E. Marine omega-3 supplementation and cardiovascular disease: An updated meta-analysis of 13 randomized controlled trials involving 127,477 participants. J. Am. Heart Assoc. 2019, 8, e013543. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, A.A.; Wiest, M.M.; Lavie, C.J.; Milani, R.V.; Laukkanen, J.A. Effect of omega-3 dosage on cardiovascular outcomes: An updated meta-analysis and meta-regression of interventional trials. Mayo Clin. Proc. 2021, 96, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.D.; Chae, S.M. Comparison of efficacy and safety of combination therapy with statins and omega-3 fatty acids versus statin monotherapy in patients with dyslipidemia: A systematic review and meta-analysis. Medicine 2018, 97, e13593. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Deng, W.; Wang, Y.; Li, T.; Chen, Y.; Long, C.; Wen, Q.; Wu, Y.; Chen, Q. The effect of omega-3 fatty acids and its combination with statins on lipid profile in patients with hypertriglyceridemia: A systematic review and meta-analysis of randomized controlled trials. Front. Nutr. 2022, 9, 1039056. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.; Kim, J. Comparative effect of statins and omega-3 supplementation on cardiovascular events: Meta-analysis and network meta-analysis of 63 randomized controlled trials including 264,516 participants. Nutrients 2020, 12, 2218. [Google Scholar] [CrossRef]
- Fan, H.; Zhou, J.; Yuan, Z. Meta-analysis comparing the effect of combined omega-3+ statin therapy versus statin therapy alone on coronary artery plaques. Am. J. Cardiol. 2021, 151, 15–24. [Google Scholar] [CrossRef]
- Reijnders, E.; van der Laarse, A.; Jukema, J.W.; Cobbaert, C.M. High residual cardiovascular risk after lipid-lowering: Prime time for predictive, preventive, personalized, participatory, and psycho-cognitive medicine. Front. Cardiovasc. Med. 2023, 10, 1264319. [Google Scholar] [CrossRef]
- Arnold, N.; Lechner, K.; Waldeyer, C.; Shapiro, M.D.; Koenig, W. Inflammation and cardiovascular disease: The future. Eur. Cardiol. Rev. 2021, 16, e20. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Sata, M.; Fukuda, D.; Tanaka, K.; Soma, M.; Hirata, Y.; Nagai, R. Orally administered eicosapentaenoic acid reduces and stabilizes atherosclerotic lesions in ApoE-deficient mice. Atherosclerosis 2008, 197, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Thies, F.; Garry, J.M.; Yaqoob, P.; Rerkasem, K.; Williams, J.; Shearman, C.P.; Gallagher, P.J.; Calder, P.C.; Grimble, R.F. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: A randomised controlled trial. Lancet 2003, 361, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Cawood, A.L.; Ding, R.; Napper, F.L.; Young, R.H.; Williams, J.A.; Ward, M.J.; Gudmundsen, O.; Vige, R.; Payne, S.P.; Ye, S. Eicosapentaenoic acid (EPA) from highly concentrated n-3 fatty acid ethyl esters is incorporated into advanced atherosclerotic plaques and higher plaque EPA is associated with decreased plaque inflammation and increased stability. Atherosclerosis 2010, 212, 252–259. [Google Scholar] [CrossRef]
- Fredman, G.; Serhan, C.N. Specialized pro-resolving mediators in vascular inflammation and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2024, in press. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Low-Intensity | Moderate-Intensity | High-Intensity |
|---|---|---|
| Simvastatin (10 mg) | Atorvastatin (10–20 mg) | Atorvastatin (40–80 mg) |
| Pravastatin (10–20 mg) | Rosuvastatin (5–10 mg) | Rosuvastatin (20–40 mg) |
| Lovastatin (20 mg) | Simvastatin (20–40 mg) | |
| Fluvastatin (20–40 mg) | Pravastatin (40–80 mg) | |
| Pitavastatin (1 mg) | Lovastatin (40 mg) | |
| Fluvastatin XL (80 mg) | ||
| Fluvastatin (40 mg twice daily) | ||
| Pitavastatin (2–4 mg) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djuricic, I.; Calder, P.C. Omega-3 (n-3) Fatty Acid–Statin Interaction: Evidence for a Novel Therapeutic Strategy for Atherosclerotic Cardiovascular Disease. Nutrients 2024, 16, 962. https://doi.org/10.3390/nu16070962
Djuricic I, Calder PC. Omega-3 (n-3) Fatty Acid–Statin Interaction: Evidence for a Novel Therapeutic Strategy for Atherosclerotic Cardiovascular Disease. Nutrients. 2024; 16(7):962. https://doi.org/10.3390/nu16070962
Chicago/Turabian StyleDjuricic, Ivana, and Philip C. Calder. 2024. "Omega-3 (n-3) Fatty Acid–Statin Interaction: Evidence for a Novel Therapeutic Strategy for Atherosclerotic Cardiovascular Disease" Nutrients 16, no. 7: 962. https://doi.org/10.3390/nu16070962
APA StyleDjuricic, I., & Calder, P. C. (2024). Omega-3 (n-3) Fatty Acid–Statin Interaction: Evidence for a Novel Therapeutic Strategy for Atherosclerotic Cardiovascular Disease. Nutrients, 16(7), 962. https://doi.org/10.3390/nu16070962