
HGF Overexpression in Mesenchymal Stromal Cell-Based Cell Sheets Enhances Autophagy-Dependent Cytoprotection and Proliferation to Guard the Epicardial Mesothelium
 , , , , ,
, , , , ,  , and
, and 
Abstract
1. Introduction
2. Results
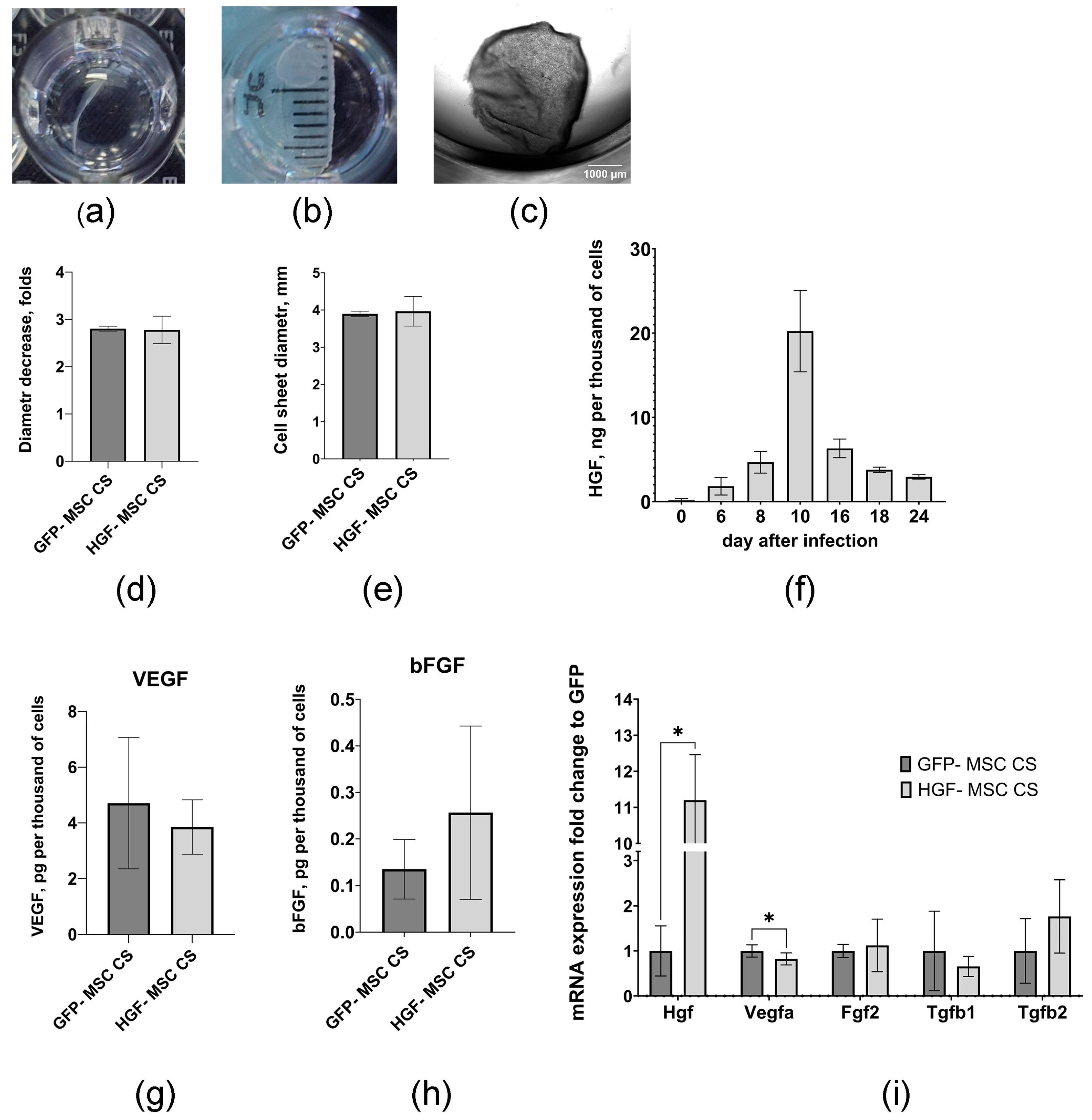
2.1. Genetically Modified MSCs Form Stable Cell Sheets and Are Characterized by a Prolonged Capacity to Produce HGF
2.2. HGF-MSC CSs Secretome Stimulates c-Met Receptor Phosphorylation and Downstream Signaling in Epicardium Cells
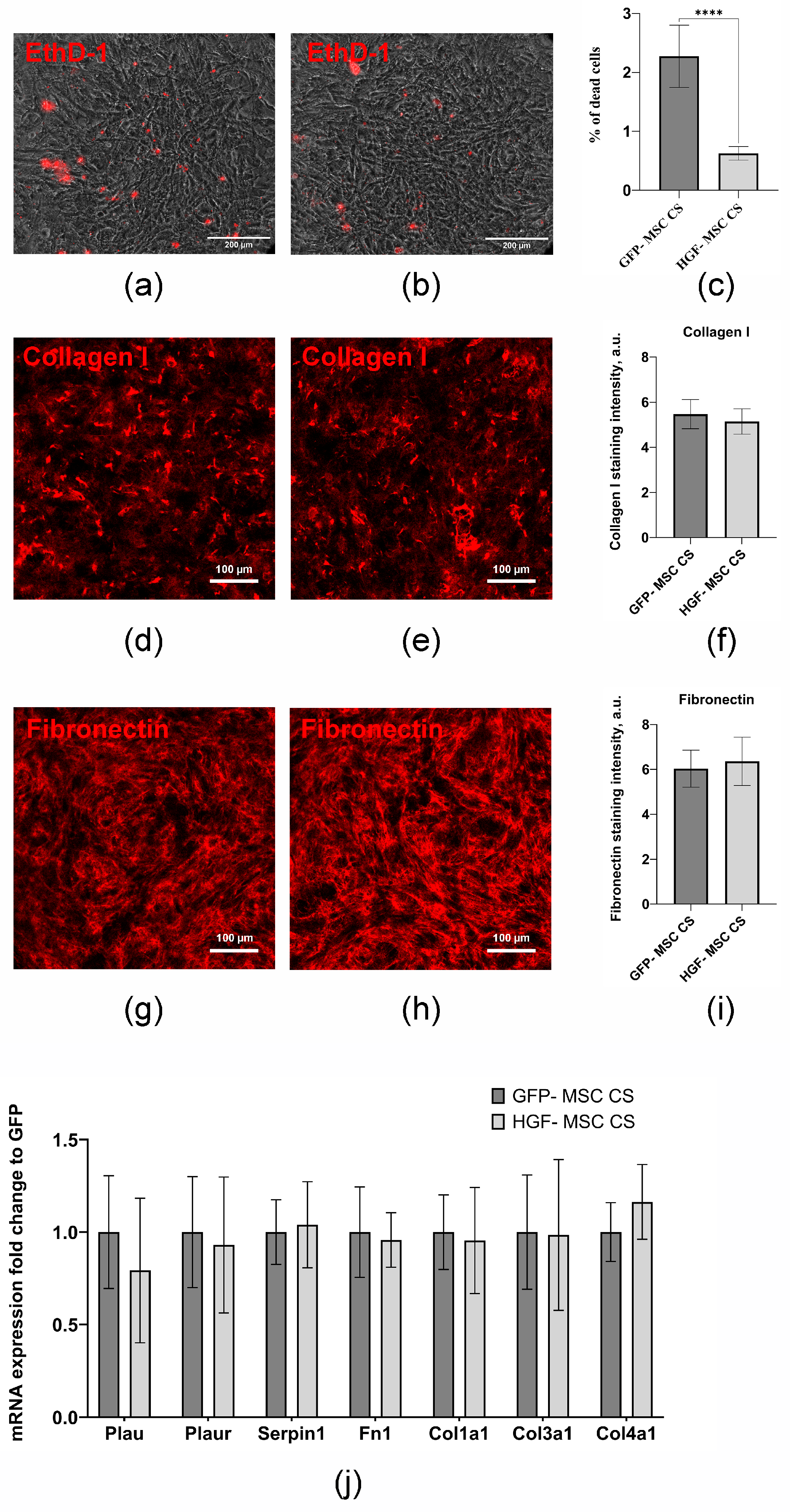
2.3. CSs Transplantation After Infarction Provides Epicardial Cell Recovery and Modulates Autophagy Levels
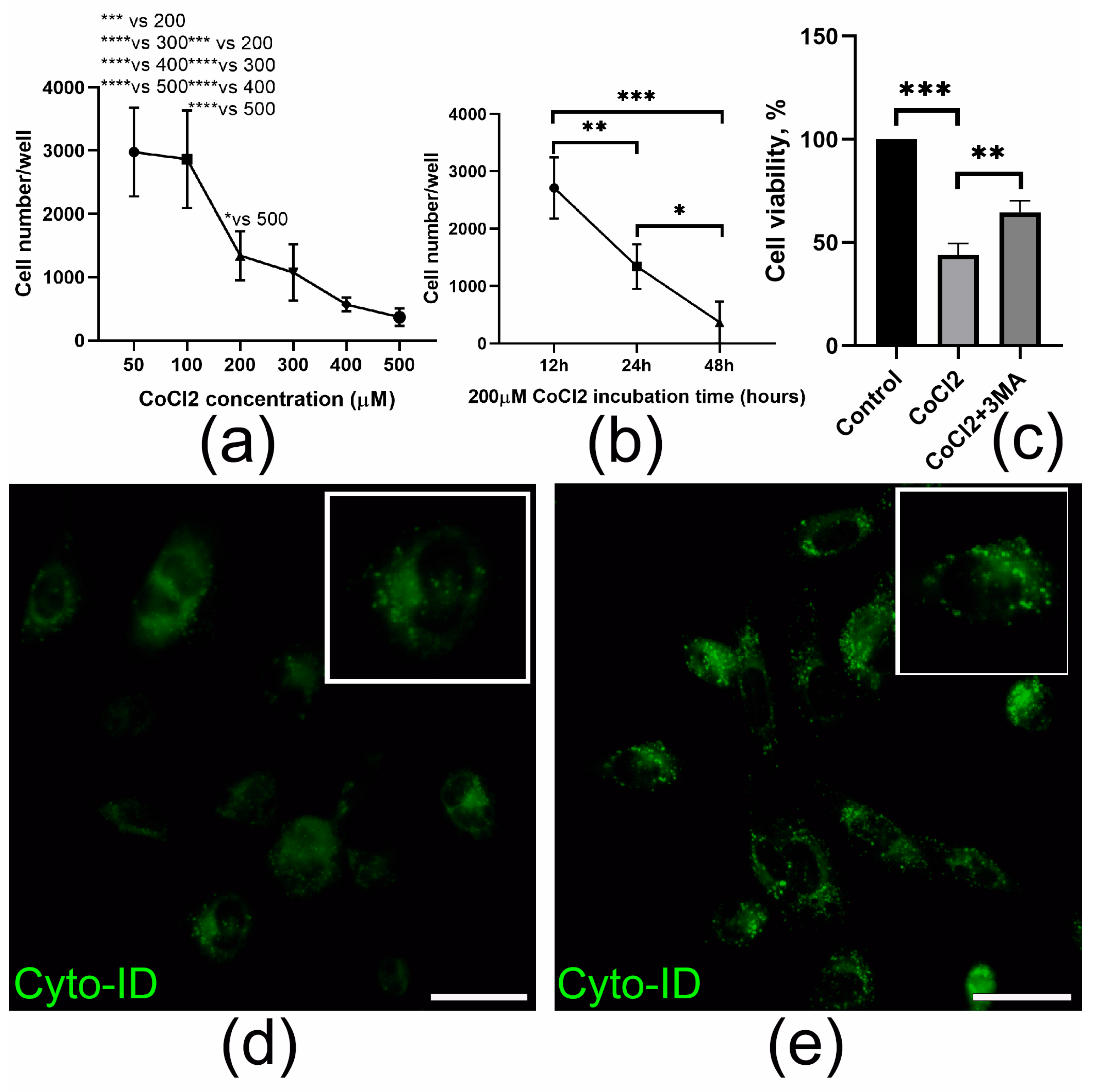
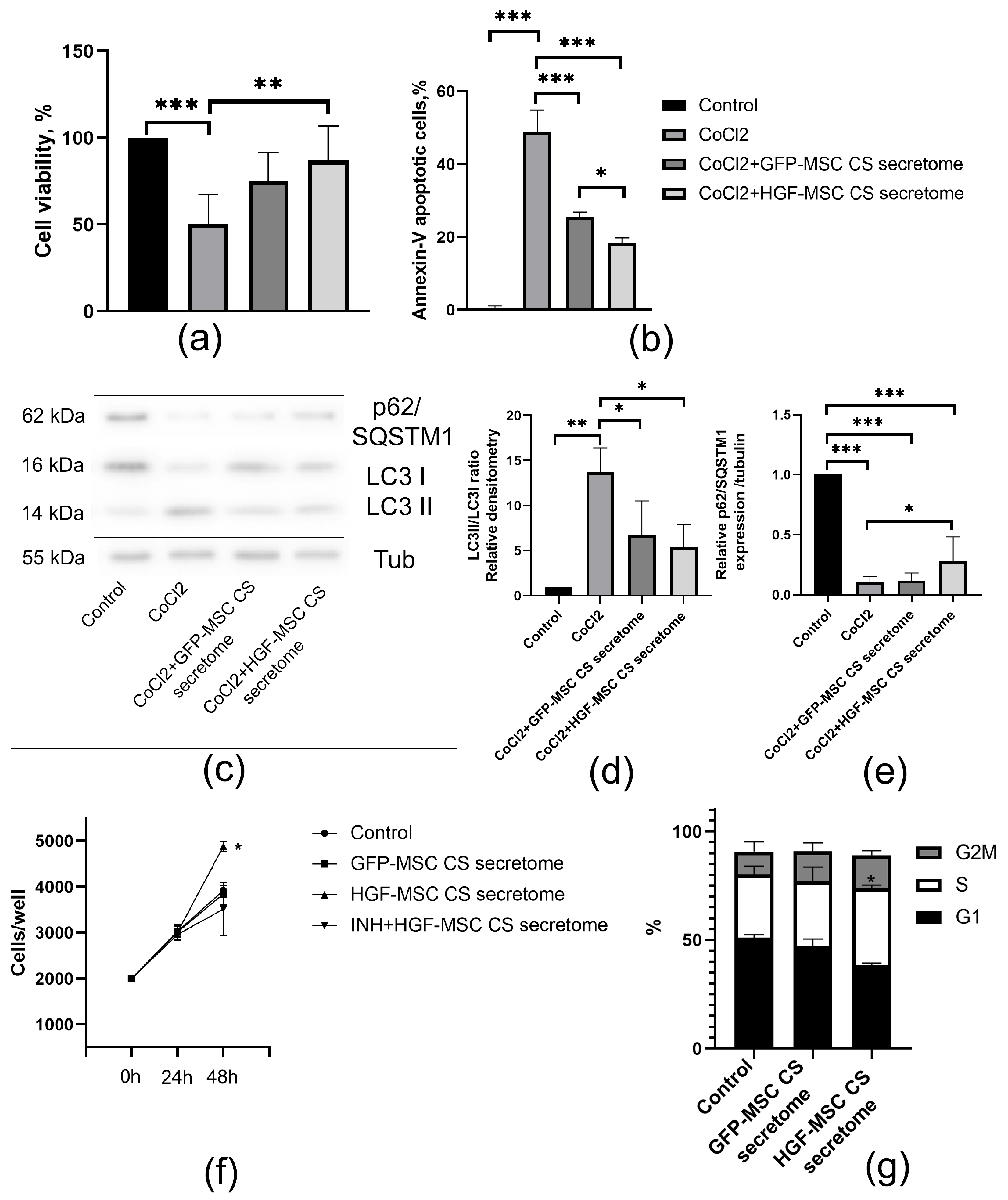
2.4. HGF-MSC CSs Secretome Protects Epicardial Cells from CoCl2-Induced Death and Stimulates Their Proliferation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. MSC Isolation and Cultivation
4.3. AAV Purification and MSC Transduction
4.4. CSs Assembly
4.5. ELISA
4.6. Analysis of Extracellular Matrix Proteins and Apoptosis in CSs
4.7. RNA Isolation, Reverse Transcription and Real-Time Quantitative PCR
4.8. Heart Cryoinjury Modeling and Cell Sheet Transplantation
4.9. Flow Cytometry Analysis
4.10. Prestoblue Assay
4.11. Cyto-ID Immunostaining
4.12. Western Blot Analysis
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar Singh, A.; Kumar Jat, R. Myocardial Infarction. Himal. J. Health Sci. 2022, 6, 16–32. [Google Scholar] [CrossRef]
- Al-Botaty, B.; El-Khoely, A.; Elsayed, E.M.; Eissa, A.A. Insight into the Pathophysiology of Myocardial Infarction. Adv. Pharm. Res. 2022, 6, 223–237. [Google Scholar] [CrossRef]
- Cao, J.; Poss, K.D. The Epicardium as a Hub for Heart Regeneration. Nat. Rev. Cardiol. 2018, 15, 631–647. [Google Scholar] [CrossRef]
- Dergilev, K.V.; Komova, A.V.; Tsokolaeva, Z.I.; Beloglazova, I.B.; Parfyonova, Y.V. Epicardium as a New Target for Regenerative Technologies in Cardiology. Genes Cells 2020, 15, 33–40. [Google Scholar] [CrossRef]
- Li, G.; Yan, Z.; Han, L.; Wu, S.; Wang, M.; Qi, A.; Zhou, Z.; Wang, N.; Sun, R.; Zhou, X. Research Progress on Epicardial Repair After Myocardial Injury. Cardiol. Rev. 2024. [Google Scholar] [CrossRef]
- Wong, D.; Martinez, J.; Quijada, P. Exploring the Function of Epicardial Cells Beyond the Surface. Circ. Res. 2024, 135, 353–371. [Google Scholar] [CrossRef]
- Stoker, M. Effect of Scatter Factor on Motility of Epithelial Cells and Fibroblasts. J. Cell. Physiol. 1989, 139, 565–569. [Google Scholar] [CrossRef]
- Naldini, L.; Weidner, K.M.; Vigna, E.; Gaudino, G.; Bardelli, A.; Ponzetto, C.; Narsimhan, R.P.; Hartmann, G.; Zarnegar, R.; Michalopoulos, G.K. Scatter Factor and Hepatocyte Growth Factor Are Indistinguishable Ligands for the MET Receptor. EMBO J. 1991, 10, 2867–2878. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET Signalling: Principles and Functions in Development, Organ Regeneration and Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Boldyreva, M.A.; Bondar, I.V.; Stafeev, I.S.; Makarevich, P.I.; Beloglazova, I.B.; Zubkova, E.S.; Shevchenko, E.K.; Molokotina, Y.D.; Karagyaur, M.N.; Ratner, E.I.; et al. Plasmid-Based Gene Therapy with Hepatocyte Growth Factor Stimulates Peripheral Nerve Regeneration after Traumatic Injury. Biomed. Pharmacother. 2018, 101, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Palomino-Durand, C.; Pauthe, E.; Gand, A. Fibronectin-Enriched Biomaterials, Biofunctionalization, and Proactivity: A Review. Appl. Sci. 2021, 11, 12111. [Google Scholar] [CrossRef]
- Zuo, X.; Xiao, Y.; Yang, J.; He, Y.; He, Y.; Liu, K.; Chen, X.; Guo, J. Engineering Collagen-Based Biomaterials for Cardiovascular Medicine. Collagen Leather 2024, 6, 33. [Google Scholar] [CrossRef]
- Rickert-Sperling, S.; Kelly, R.G.; Haas, N. (Eds.) Epicardium and Coronary Vessels. In Congenital Heart Diseases: The Broken Heart. Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2024; Volume 1441. [Google Scholar] [CrossRef]
- Wu, H.; Lan, Q.; He, Y.-X.; Xue, J.-Y.; Liu, H.; Zou, Y.; Liu, P.; Luo, G.; Chen, M.-T.; Liu, M.-N. Programmed Cardiomyocyte Death in Myocardial Infarction. Apoptosis 2025, 30, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, S.; Kizana, E. Mechanisms and Therapeutic Potential of Multiple Forms of Cell Death in Myocardial Ischemia–Reperfusion Injury. Int. J. Mol. Sci. 2024, 25, 13492. [Google Scholar] [CrossRef] [PubMed]
- Beilmann, M.; Odenthal, M.; Jung, W.; Vande Woude, G.F.; Dienes, H.-P.; Schirmacher, P. Neoexpression of the C-Met/Hepatocyte Growth Factor-Scatter Factor Receptor Gene in Activated Monocytes. Blood 1997, 90, 4450–4458. [Google Scholar] [CrossRef]
- Beilmann, M.; Vande Woude, G.F.; Dienes, H.-P.; Schirmacher, P. Hepatocyte Growth Factor–Stimulated Invasiveness of Monocytes. Blood 2000, 95, 3964–3969. [Google Scholar] [CrossRef]
- Rutella, S. Hepatocyte Growth Factor Favors Monocyte Differentiation into Regulatory Interleukin (IL)-10++IL-12low/Neg Accessory Cells with Dendritic-Cell Features. Blood 2006, 108, 218–227. [Google Scholar] [CrossRef]
- Coudriet, G.M.; He, J.; Trucco, M.; Mars, W.M.; Piganelli, J.D. Hepatocyte Growth Factor Modulates Interleukin-6 Production in Bone Marrow Derived Macrophages: Implications for Inflammatory Mediated Diseases. PLoS ONE 2010, 5, e15384. [Google Scholar] [CrossRef]
- Ding, S.; Merkulova-Rainon, T.; Han, Z.C.; Tobelem, G. HGF Receptor Up-Regulation Contributes to the Angiogenic Phenotype of Human Endothelial Cells and Promotes Angiogenesis in Vitro. Blood 2003, 101, 4816–4822. [Google Scholar] [CrossRef]
- Nakagami, H.; Morishita, R.; Yamamoto, K.; Taniyama, Y.; Aoki, M.; Kim, S.; Matsumoto, K.; Nakamura, T.; Higaki, J.; Ogihara, T. Anti-Apoptotic Action of Hepatocyte Growth Factor through Mitogen-Activated Protein Kinase on Human Aortic Endothelial Cells. J. Hypertens. 2000, 18, 1411–1420. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kohara, K.; Tabara, Y.; Miki, T. Association between Carotid Arterial Remodeling and Plasma Concentration of Circulating Hepatocyte Growth Factor. J. Hypertens. 2001, 19, 1975–1979. [Google Scholar] [CrossRef]
- Van Belle, E.; Witzenbichler, B.; Chen, D.; Silver, M.; Chang, L.; Schwall, R.; Isner, J.M. Potentiated Angiogenic Effect of Scatter Factor/Hepatocyte Growth Factor via Induction of Vascular Endothelial Growth Factor: The Case for Paracrine Amplification of Angiogenesis. Circulation 1998, 97, 381–390. [Google Scholar] [CrossRef]
- Nakamura, T.; Matsumoto, K.; Mizuno, S.; Sawa, Y.; Matsuda, H.; Nakamura, T. Hepatocyte Growth Factor Prevents Tissue Fibrosis, Remodeling, and Dysfunction in Cardiomyopathic Hamster Hearts. Am. J. Physiol.-Heart Circ. Physiol. 2005, 288, H2131–H2139. [Google Scholar] [CrossRef]
- Taniyama, Y.; Morishita, R.; Nakagami, H.; Moriguchi, A.; Sakonjo, H.; Shokei-Kim; Matsumoto, K.; Nakamura, T.; Higaki, J.; Ogihara, T. Potential Contribution of a Novel Antifibrotic Factor, Hepatocyte Growth Factor, to Prevention of Myocardial Fibrosis by Angiotensin II Blockade in Cardiomyopathic Hamsters. Circulation 2000, 102, 246–252. [Google Scholar] [CrossRef]
- Ueki, T.; Kaneda, Y.; Tsutsui, H.; Nakanishi, K.; Sawa, Y.; Morishita, R.; Matsumoto, K.; Nakamura, T.; Takahashi, H.; Okamoto, E.; et al. Hepatocyte Growth Factor Gene Therapy of Liver Cirrhosis in Rats. Nat. Med. 1999, 5, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Sasamura, H.; Mifune, M.; Shimizu-Hirota, R.; Kuroda, M.; Hayashi, M.; Saruta, T. Hepatocyte Growth Factor Regulates Proteoglycan Synthesis in Interstitial Fibroblasts. Kidney Int. 2003, 64, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Matsumoto, K.; Li, M.-Y.; Nakamura, T. HGF Reduces Advancing Lung Fibrosis in Mice: A Potential Role for MMP-dependent Myofibroblast Apoptosis. FASEB J. 2005, 19, 1–18. [Google Scholar] [CrossRef]
- Azuma, J.; Taniyama, Y.; Takeya, Y.; Iekushi, K.; Aoki, M.; Dosaka, N.; Matsumoto, K.; Nakamura, T.; Ogihara, T.; Morishita, R. Angiogenic and Antifibrotic Actions of Hepatocyte Growth Factor Improve Cardiac Dysfunction in Porcine Ischemic Cardiomyopathy. Gene Ther. 2006, 13, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Kucia, M.; Dawn, B.; Hunt, G.; Guo, Y.; Wysoczynski, M.; Majka, M.; Ratajczak, J.; Rezzoug, F.; Ildstad, S.T.; Bolli, R.; et al. Cells Expressing Early Cardiac Markers Reside in the Bone Marrow and Are Mobilized into the Peripheral Blood After Myocardial Infarction. Circ. Res. 2004, 95, 1191–1199. [Google Scholar] [CrossRef]
- Son, B.; Marquez-Curtis, L.A.; Kucia, M.; Wysoczynski, M.; Turner, A.R.; Ratajczak, J.; Ratajczak, M.Z.; Janowska-Wieczorek, A. Migration of Bone Marrow and Cord Blood Mesenchymal Stem Cells In Vitro Is Regulated by Stromal-Derived Factor-1-CXCR4 and Hepatocyte Growth Factor-c-met Axes and Involves Matrix Metalloproteinases. Stem Cells 2006, 24, 1254–1264. [Google Scholar] [CrossRef]
- Cohen, M.E.; Fainstein, N.; Lavon, I.; Ben-Hur, T. Signaling through Three Chemokine Receptors Triggers the Migration of Transplanted Neural Precursor Cells in a Model of Multiple Sclerosis. Stem Cell Res. 2014, 13, 227–239. [Google Scholar] [CrossRef]
- Konoplyannikov, M.; Haider, K.H.; Lai, V.K.; Ahmed, R.P.H.; Jiang, S.; Ashraf, M. Activation of Diverse Signaling Pathways by Ex-Vivo Delivery of Multiple Cytokines for Myocardial Repair. Stem Cells Dev. 2013, 22, 204–215. [Google Scholar] [CrossRef]
- Liu, Y. Hepatocyte Growth Factor Promotes Renal Epithelial Cell Survival by Dual Mechanisms. Am. J. Physiol.-Ren. Physiol. 1999, 277, F624–F633. [Google Scholar] [CrossRef]
- Kitta, K.; Day, R.M.; Kim, Y.; Torregroza, I.; Evans, T.; Suzuki, Y.J. Hepatocyte Growth Factor Induces GATA-4 Phosphorylation and Cell Survival in Cardiac Muscle Cells. J. Biol. Chem. 2003, 278, 4705–4712. [Google Scholar] [CrossRef] [PubMed]
- Sekine, H.; Shimizu, T.; Dobashi, I.; Matsuura, K.; Hagiwara, N.; Takahashi, M.; Kobayashi, E.; Yamato, M.; Okano, T. Cardiac Cell Sheet Transplantation Improves Damaged Heart Function via Superior Cell Survival in Comparison with Dissociated Cell Injection. Tissue Eng. Part A 2011, 17, 2973–2980. [Google Scholar] [CrossRef] [PubMed]
- Dergilev, K.V.; Tsokolayeva, Z.I.; Beloglazova, I.B.; Ratner, E.I.; Parfyonova, E.V. Epicardial Transplantation of Cardiac Progenitor Cells Based Cells Sheets Is More Promising Method for Stimulation of Myocardial Regeneration, Than Conventional Cell Injections. Kardiologiia 2019, 59, 53–60. [Google Scholar] [CrossRef]
- Jensen, J.A.; Kosek, J.C.; Hunt, T.K.; Goodson, W.H.; Miller, D.C. Cardiac Cryolesions as an Experimental Model of Myocardial Wound Healing. Ann. Surg. 1987, 206, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Ciulla, M.M.; Paliotti, R.; Ferrero, S.; Braidotti, P.; Esposito, A.; Gianelli, U.; Busca, G.; Cioffi, U.; Bulfamante, G.; Magrini, F. Left Ventricular Remodeling after Experimental Myocardial Cryoinjury in Rats. J. Surg. Res. 2004, 116, 91–97. [Google Scholar] [CrossRef]
- Hirano, A.; Fujita, J.; Kanazawa, H.; Kawaguchi, S.; Handa, N.; Yamada, Y.; Okuda, S.; Hishikawa, S.; Teratani, T.; Kunita, S.; et al. Cryoinjury-Induced Acute Myocardial Infarction Model and Ameroid Constrictor-Induced Ischemic Heart Disease Model in Adult Micro-Mini Pigs for Preclinical Studies. Transl. Med. Commun. 2017, 2, 1. [Google Scholar] [CrossRef]
- Wünsch, M. In Situ Localization of Transforming Growth Factor Β1 in Porcine Heart: Enhanced Expression after Chronic Coronary Artery Constriction. J. Mol. Cell. Cardiol. 1991, 23, 1051–1062. [Google Scholar] [CrossRef]
- Suchiita, A.; Gupta, N.; Nandi, K.; Goswami, B. Navigating the Crossroads of Cell Death Interplay and Intersections among Ferroptosis, Apoptosis and Autophagy. Drug Metab. Pers. Ther. 2025, 40, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS Domains Form Energy-Sensing Modules Whose Binding of Adenosine Ligands Is Disrupted by Disease Mutations. J. Clin. Investig. 2004, 113, 274–284. [Google Scholar] [CrossRef]
- Cheung, P.C.F.; Salt, I.P.; Davies, S.P.; Hardie, D.G.; Carling, D. Characterization of AMP-Activated Protein Kinase γ-Subunit Isoforms and Their Role in AMP Binding. Biochem. J. 2000, 346, 659–669. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase Is a Direct Target of TSC2 GAP Activity and Regulates mTOR Signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.-L. Differential Regulation of Distinct Vps34 Complexes by AMPK in Nutrient Stress and Autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Sounidaki, M.; Antoniadis, N.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Preconditioning of Primary Human Renal Proximal Tubular Epithelial Cells without Tryptophan Increases Survival under Hypoxia by Inducing Autophagy. Int. Urol. Nephrol. 2017, 49, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Jeong, P.-S.; Sim, B.-W.; Kang, H.-G.; Kim, M.J.; Lee, S.; Yoon, S.-B.; Kang, P.; Park, Y.-H.; Kim, J.-S.; et al. Induction of Autophagy Protects against Extreme Hypoxia-Induced Damage in Porcine Embryo. Reproduction 2021, 161, 353–363. [Google Scholar] [CrossRef]
- Wang, D.; Weng, Q.; Zhang, L.; He, Q.; Yang, B. VEGF and Bcl-2 Interact Via MAPKs Signaling Pathway in the Response to Hypoxia in Neuroblastoma. Cell. Mol. Neurobiol. 2009, 29, 391–401. [Google Scholar] [CrossRef]
- Chen, N.; Chen, X.; Huang, R.; Zeng, H.; Gong, J.; Meng, W.; Lu, Y.; Zhao, F.; Wang, L.; Zhou, Q. BCL-xL Is a Target Gene Regulated by Hypoxia-Inducible Factor-1α. J. Biol. Chem. 2009, 284, 10004–10012. [Google Scholar] [CrossRef] [PubMed]
- Decker, R.S.; Wildenthal, K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am. J. Pathol. 1980, 98, 425–444. [Google Scholar]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef]
- Ciafrè, S.A.; Niola, F.; Giorda, E.; Farace, M.G.; Caporossi, D. CoCl2-Simulated Hypoxia in Skeletal Muscle Cell Lines: Role of Free Radicals in Gene up-Regulation and Induction of Apoptosis. Free. Radic. Res. 2007, 41, 391–401. [Google Scholar] [CrossRef] [PubMed]
- An, W.G.; Kanekal, M.; Simon, M.C.; Maltepe, E.; Blagosklonny, M.V.; Neckers, L.M. Stabilization of Wild-Type P53 by Hypoxia-Inducible Factor 1α. Nature 1998, 392, 405–408. [Google Scholar] [CrossRef]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-Dependent Regulation of Hypoxic Induction of the Cell Death Factors BNIP3 and NIX in Human Tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Valentim, L.; Laurence, K.M.; Townsend, P.A.; Carroll, C.J.; Soond, S.; Scarabelli, T.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Urocortin Inhibits Beclin1-Mediated Autophagic Cell Death in Cardiac Myocytes Exposed to Ischaemia/Reperfusion Injury. J. Mol. Cell. Cardiol. 2006, 40, 846–852. [Google Scholar] [CrossRef]
- Ma, S.; Wang, Y.; Chen, Y.; Cao, F. The Role of the Autophagy in Myocardial Ischemia/Reperfusion Injury. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 271–276. [Google Scholar] [CrossRef]
- Degtyarev, M.; De Mazière, A.; Orr, C.; Lin, J.; Lee, B.B.; Tien, J.Y.; Prior, W.W.; Van Dijk, S.; Wu, H.; Gray, D.C.; et al. Akt Inhibition Promotes Autophagy and Sensitizes PTEN-Null Tumors to Lysosomotropic Agents. J. Cell Biol. 2008, 183, 101–116. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Nave, B.T.; Ouwens, D.M.; Withers, D.J.; Alessi, D.R.; Shepherd, P.R. Mammalian Target of Rapamycin Is a Direct Target for Protein Kinase B: Identification of a Convergence Point for Opposing Effects of Insulin and Amino-Acid Deficiency on Protein Translation. Biochem. J. 1999, 344, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the Tuberous Sclerosis Complex-2 Tumor Suppressor Gene Product Tuberin as a Target of the Phosphoinositide 3-Kinase/Akt Pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The Serine/Threonine Kinase ULK1 Is a Target of Multiple Phosphorylation Events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-Mediated Regulation of Autophagy and Tumorigenesis Through Beclin 1 Phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef]
- Bell, E.S.; Coelho, P.P.; Ratcliffe, C.D.H.; Rajadurai, C.V.; Peschard, P.; Vaillancourt, R.; Zuo, D.; Park, M. LC3C-Mediated Autophagy Selectively Regulates the Met RTK and HGF-Stimulated Migration and Invasion. Cell Rep. 2019, 29, 4053–4068.e6. [Google Scholar] [CrossRef] [PubMed]
- Maddika, S.; Ande, S.R.; Wiechec, E.; Hansen, L.L.; Wesselborg, S.; Los, M. Akt-Mediated Phosphorylation of CDK2 Regulates Its Dual Role in Cell Cycle Progression and Apoptosis. J. Cell Sci. 2008, 121, 979–988. [Google Scholar] [CrossRef]
- Medema, R.H.; Kops, G.J.P.L.; Bos, J.L.; Burgering, B.M.T. AFX-like Forkhead Transcription Factors Mediate Cell-Cycle Regulation by Ras and PKB through P27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Parekh, P.; Motiwale, L.; Naik, N.; Rao, K.V.K. Downregulation of Cyclin D1 Is Associated with Decreased Levels of P38 MAP Kinases, Akt/PKB and Pak1 during Chemopreventive Effects of Resveratrol in Liver Cancer Cells. Exp. Toxicol. Pathol. 2011, 63, 167–173. [Google Scholar] [CrossRef]
- Accornero, P.; Miretti, S.; Starvaggi Cucuzza, L.; Martignani, E.; Baratta, M. Epidermal Growth Factor and Hepatocyte Growth Factor Cooperate to Enhance Cell Proliferation, Scatter, and Invasion in Murine Mammary Epithelial Cells. J. Mol. Endocrinol. 2010, 44, 115–125. [Google Scholar] [CrossRef]
- Imura, Y.; Nakai, T.; Yamada, S.; Outani, H.; Takenaka, S.; Hamada, K.; Araki, N.; Itoh, K.; Yoshikawa, H.; Naka, N. Functional and Therapeutic Relevance of Hepatocyte Growth Factor/c-MET Signaling in Synovial Sarcoma. Cancer Sci. 2016, 107, 1867–1876. [Google Scholar] [CrossRef]
- Pilotto, S.; Carbognin, L.; Karachaliou, N.; Ma, P.C.; Rosell, R.; Tortora, G.; Bria, E. Tracking MET De-Addiction in Lung Cancer: A Road towards the Oncogenic Target. Cancer Treat. Rev. 2017, 60, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-C.; Wang, C.-T.; Hung, H.-C.; Wu, W.-J.; Wu, D.-C.; Chang, M.-C.; Sung, P.-J.; Chou, Y.-W.; Wen, Z.-H.; Tai, M.-H. Heteronemin Is a Novel C-Met/STAT3 Inhibitor Against Advanced Prostate Cancer Cells: Heteronemin, a Novel c-Met/STAT3 Inhibitor, for Prostate Cancer. Prostate 2016, 76, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Safaie Qamsari, E.; Safaei Ghaderi, S.; Zarei, B.; Dorostkar, R.; Bagheri, S.; Jadidi-Niaragh, F.; Somi, M.H.; Yousefi, M. The C-Met Receptor: Implication for Targeted Therapies in Colorectal Cancer. Tumour Biol. 2017, 39, 101042831769911. [Google Scholar] [CrossRef] [PubMed]
- Makarevich, P.I.; Boldyreva, M.A.; Gluhanyuk, E.V.; Efimenko, A.Y.; Dergilev, K.V.; Shevchenko, E.K.; Sharonov, G.V.; Gallinger, J.O.; Rodina, P.A.; Sarkisyan, S.S.; et al. Enhanced Angiogenesis in Ischemic Skeletal Muscle after Transplantation of Cell Sheets from Baculovirus-Transduced Adipose-Derived Stromal Cells Expressing VEGF165. Stem Cell Res. Ther. 2015, 6, 204. [Google Scholar] [CrossRef]
- Dergilev, K.V.; Makarevich, P.I.; Tsokolaeva, Z.I.; Boldyreva, M.A.; Beloglazova, I.B.; Zubkova, E.S.; Menshikov, M.Y.; Parfyonova, Y.V. Comparison of Cardiac Stem Cell Sheets Detached by Versene Solution and from Thermoresponsive Dishes Reveals Similar Properties of Constructs. Tissue Cell 2017, 49, 64–71. [Google Scholar] [CrossRef]
- Wang, D.; Tediashvili, G.; Hu, X.; Gravina, A.; Marcus, S.G.; Zhang, H.; Olgin, J.E.; Deuse, T.; Schrepfer, S. A Cryoinjury Model to Study Myocardial Infarction in the Mouse. JoVE 2019, 151, 59958. [Google Scholar] [CrossRef]
- Dergilev, K.; Zubkova, E.; Guseva, A.; Tsokolaeva, Z.; Goltseva, Y.; Beloglazova, I.; Ratner, E.; Andreev, A.; Partigulov, S.; Lepilin, M.; et al. Tumor Necrosis Factor-Alpha Induces Proangiogenic Profiling of Cardiosphere-Derived Cell Secretome and Increases Its Ability to Stimulate Angiogenic Properties of Endothelial Cells. Int. J. Mol. Sci. 2023, 24, 16575. [Google Scholar] [CrossRef]
- Dergilev, K.; Tsokolaeva, Z.; Goltseva, Y.; Beloglazova, I.; Ratner, E.; Parfyonova, Y. Urokinase-Type Plasminogen Activator Receptor Regulates Prosurvival and Angiogenic Properties of Cardiac Mesenchymal Stromal Cells. Int. J. Mol. Sci. 2023, 24, 15554. [Google Scholar] [CrossRef]
- Guo, S.; Liang, Y.; Murphy, S.F.; Huang, A.; Shen, H.; Kelly, D.F.; Sobrado, P.; Sheng, Z. A Rapid and High Content Assay That Measures Cyto-ID-Stained Autophagic Compartments and Estimates Autophagy Flux with Potential Clinical Applications. Autophagy 2015, 11, 560–572. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward | Reversed |
|---|---|---|
| Hgf | AACAGGGGCTTTACGTTCACT | CGTCCCTTTATAGCTGCCTCC |
| Vegfa | GGAGACTCTTCGAGGAGCACTT | GGCGATTTAGCAGCAGATATAAGAA |
| Fgf2 | GCGACCCACACGTCAAACTA | CCGTCCATCTTCCTTCATAGC |
| Tgfb1 | TGGAGCAACATGTGGAACTC | GTCAGCAGCCGGTTACCA |
| Tgfb2 | TTGTTGCCCTCCTACAGACTGG | GTAAAGAGGGCGAAGGCAGCAA |
| Plau | ATGGAAATGGTGACTCTTACCGA | TGG GCA TTG TAG GGT TTC TGA |
| Plaur | CGCCACAAACCTCTGCAAC | CTCTGTAGGATAGCGGCATTG |
| Serpin1 | TGCAAAAGGTCAGGATCGAGG | ATGAAGAGGATTGTCTCTGTCGG |
| Fn1 | GGAATGGACCTGCAAACCTA | GTAGGGCTTTTCCCAGGTCT |
| Col1a1 | CCGCTGGTCAAGATGGTC | CTCCAGCCTTTCCAGGTTCT |
| Col3a1 | CTGTAACATGGAAACTGGGGAAA | CCATAGCTGAACTGAAAACCACC |
| Col4a1 | CTGGCACAAAAGGGACGAG | ACGTGGCCGAGAATTTCACC |
| Actb | GGCTGTATTCCCCTCCATCG | CCAGTTGGTAACAATGCCATGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dergilev, K.; Beloglazova, I.; Tsokolaeva, Z.; Azimova, E.; Dolgodvorova, A.; Goltseva, Y.; Boldyreva, M.; Menshikov, M.; Penkov, D.; Parfyonova, Y. HGF Overexpression in Mesenchymal Stromal Cell-Based Cell Sheets Enhances Autophagy-Dependent Cytoprotection and Proliferation to Guard the Epicardial Mesothelium. Int. J. Mol. Sci. 2025, 26, 7298. https://doi.org/10.3390/ijms26157298
Dergilev K, Beloglazova I, Tsokolaeva Z, Azimova E, Dolgodvorova A, Goltseva Y, Boldyreva M, Menshikov M, Penkov D, Parfyonova Y. HGF Overexpression in Mesenchymal Stromal Cell-Based Cell Sheets Enhances Autophagy-Dependent Cytoprotection and Proliferation to Guard the Epicardial Mesothelium. International Journal of Molecular Sciences. 2025; 26(15):7298. https://doi.org/10.3390/ijms26157298
Chicago/Turabian StyleDergilev, Konstantin, Irina Beloglazova, Zoya Tsokolaeva, Ekaterina Azimova, Aleria Dolgodvorova, Yulia Goltseva, Maria Boldyreva, Mikhail Menshikov, Dmitry Penkov, and Yelena Parfyonova. 2025. "HGF Overexpression in Mesenchymal Stromal Cell-Based Cell Sheets Enhances Autophagy-Dependent Cytoprotection and Proliferation to Guard the Epicardial Mesothelium" International Journal of Molecular Sciences 26, no. 15: 7298. https://doi.org/10.3390/ijms26157298
APA StyleDergilev, K., Beloglazova, I., Tsokolaeva, Z., Azimova, E., Dolgodvorova, A., Goltseva, Y., Boldyreva, M., Menshikov, M., Penkov, D., & Parfyonova, Y. (2025). HGF Overexpression in Mesenchymal Stromal Cell-Based Cell Sheets Enhances Autophagy-Dependent Cytoprotection and Proliferation to Guard the Epicardial Mesothelium. International Journal of Molecular Sciences, 26(15), 7298. https://doi.org/10.3390/ijms26157298