
Fatty Acid Profile in the Liver of Mice with Early- and Late-Onset Forms of Huntington’s Disease

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
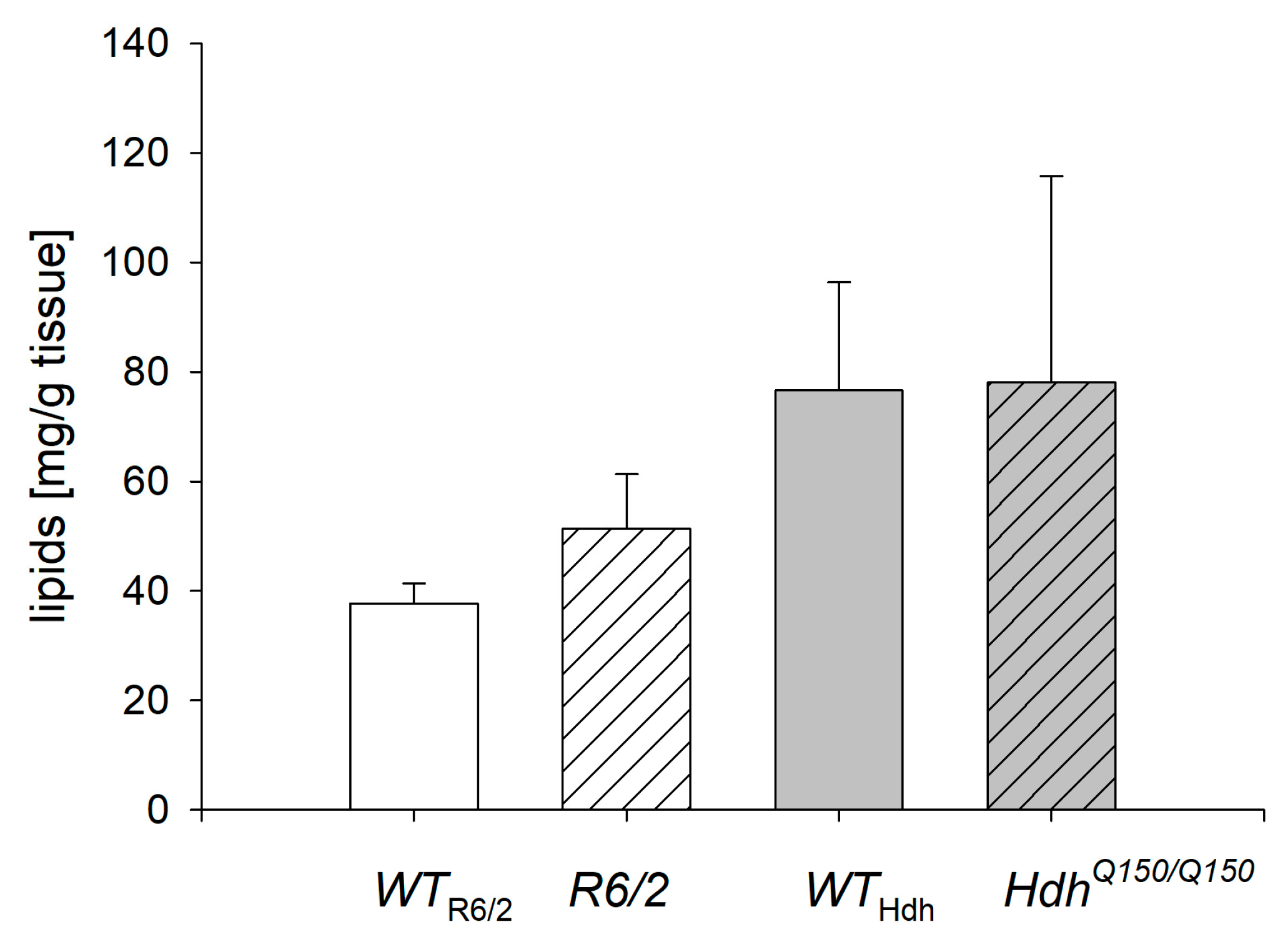
2.1. Lipid Concentration in the Liver
2.2. Fatty Acid Composition in the Liver
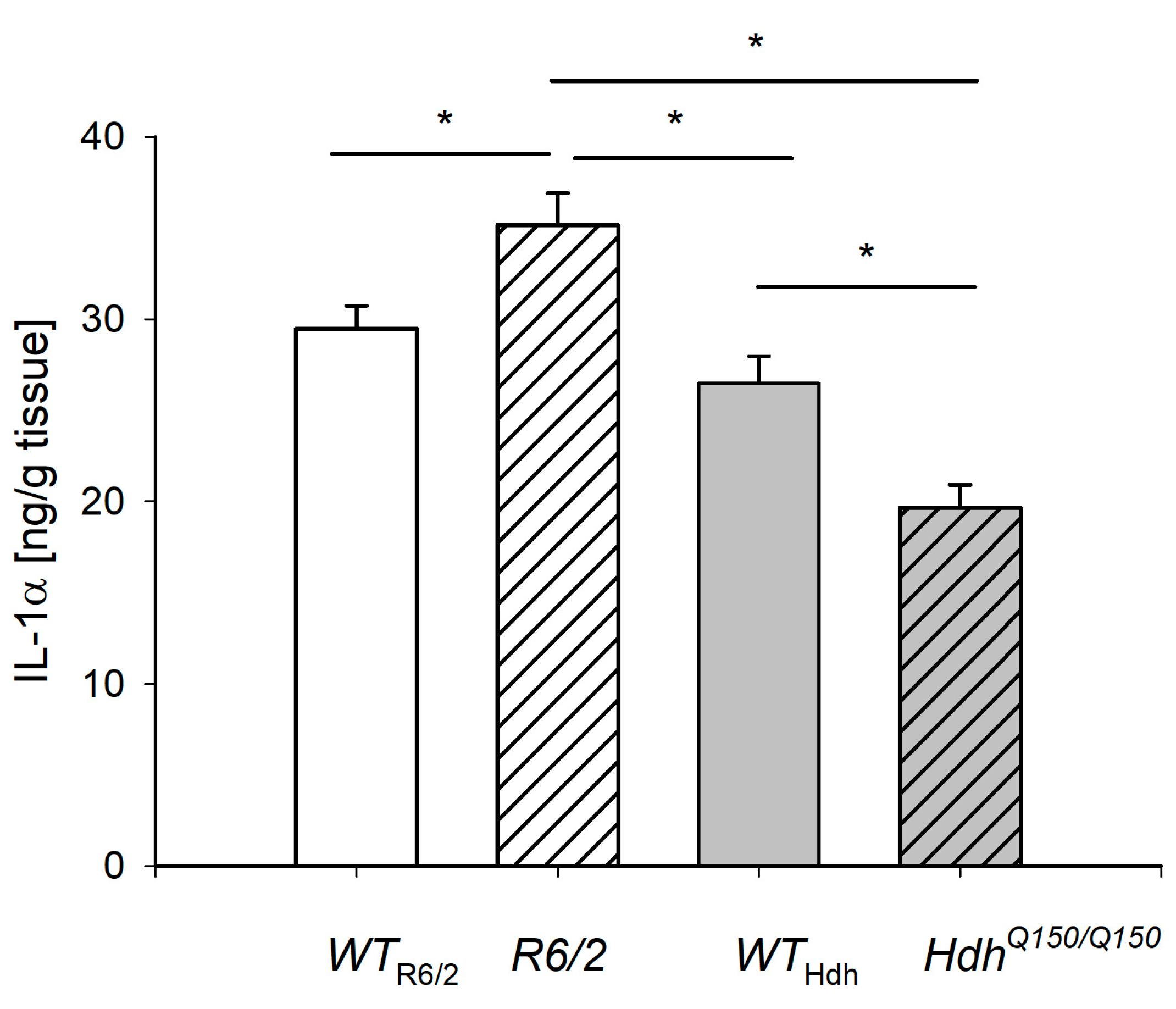
2.3. IL-1α Level in the Liver
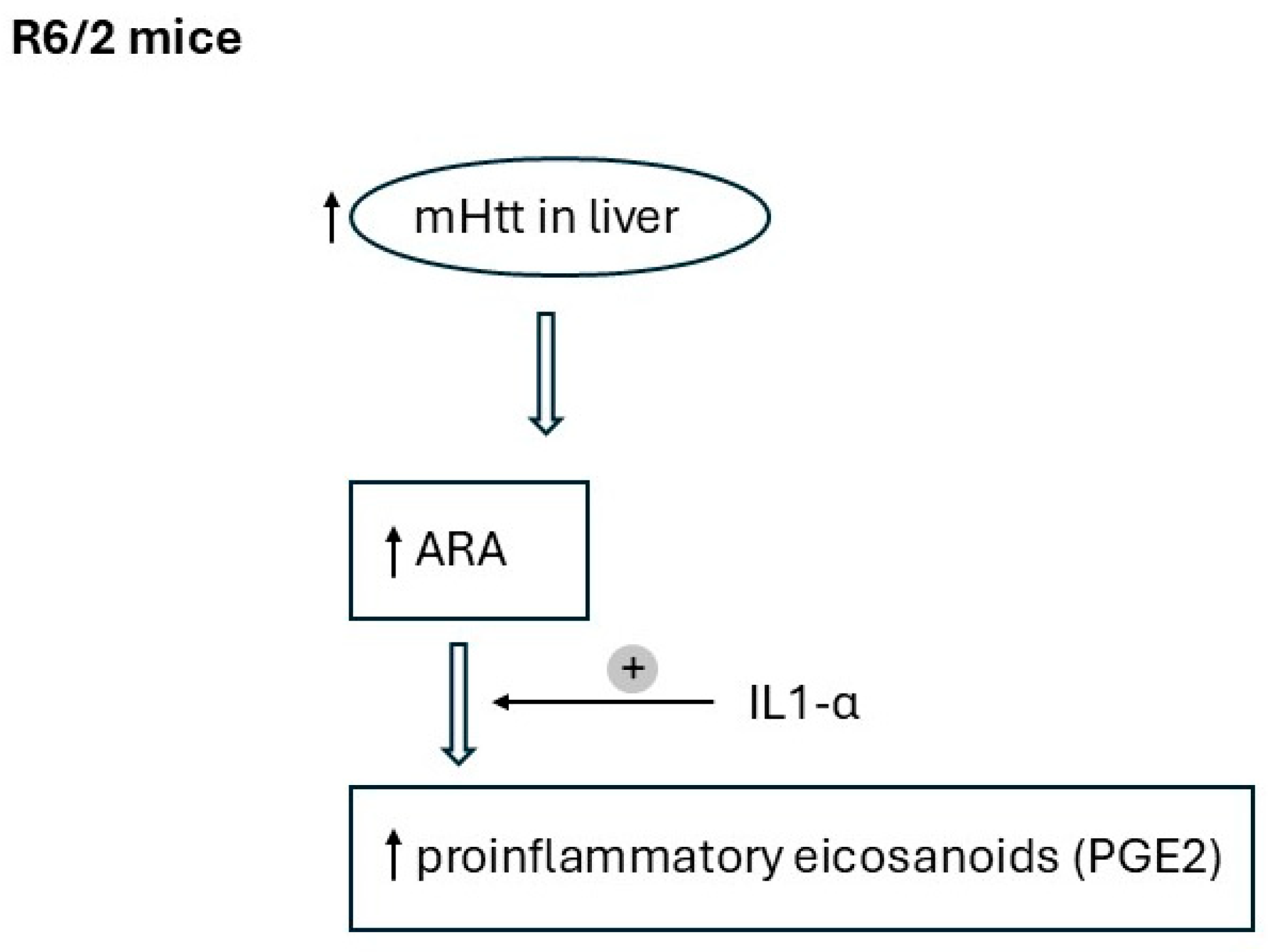
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Analysis of the Fatty Acids
4.3. Determination of IL-1α Level
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Chang, K.-H.; Cheng, M.-L.; Lo, C.-J.; Fan, C.-M.; Wu, Y.-R.; Chen, C.-M. Alterations of Lipoprotein Profiles in the Plasma as Biomarkers of Huntington’s Disease. Cells 2023, 12, 385. [Google Scholar] [CrossRef]
- Kim, A.; Lalonde, K.; Truesdell, A.; Welter, P.C.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New Avenues for the Treatment of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 8363. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, R.R.; Mezei, M.M.; Theilmann, J.; Almqvist, E.; Hayden, M.R. The Likelihood of Being Affected with Huntington Disease by a Particular Age, for Specific CAG Size. Am. J. Hum. Genet. 1997, 60, 1202–1210. [Google Scholar] [PubMed]
- Fusilli, C.; Migliore, S.; Mazza, T.; Consoli, F.; De Luca, A.; Barbagallo, G.; Ciammola, A.; Gatto, E.M.; Cesarini, M.; Etcheverry, J.L.; et al. Biological and clinical manifestations of juvenile Huntington’s disease a retrospective analysis. Lancet Neurol. 2018, 17, 986–993. [Google Scholar] [CrossRef]
- Squitieri, F.; Monti, L.; Graziola, F.; Colafati, G.S.; Sabatini, U. Early liver steatosis in children with pediatric Huntington disease and highly expanded CAG mutations. Park. Relat. Disord. 2022, 103, 102–104. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Leggo, J.; Coles, R.; Almqvist, E.; Biancalana, V.; Cassiman, J.-J.; Chotai, K.; Connarty, M.; Craufurd, D.; Curtis, A.; et al. Phenotypic Characterization of Individuals with 30-40 CAG Repeats in the Huntington Disease (HD) Gene Reveals HD Cases with 36 Repeats and Apparently Normal Elderly Individuals with 36-39 Repeats. Am. J. Hum. Genet. 1996, 59, 16–22. [Google Scholar]
- Pinto, R.M.; Arning, L.; Giordano, J.V.; Razghandi, P.; Andrew, M.A.; Gillis, T.; Correia, K.; Mysore, J.S.; Grote Urtubey, D.-M.; Parwez, C.R.; et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum. Mol. Genet. 2020, 29, 2551–2567. [Google Scholar] [CrossRef] [PubMed]
- Suopanki, J.; Götz, C.; Lutsch, G.; Schiller, J.; Harjes, P.; Herrmann, A.; Wanker, E.E. Interaction of huntingtin fragments with brain membranes—Clues to early dysfunction in Huntington’s disease. J. Neurochem. 2006, 96, 870–884. [Google Scholar] [CrossRef]
- Monaco, G.; Van Dam, S.; Ribeiro, J.L.C.N.; Larbi, A.; De Magalhães, J.P. A comparison of human and mouse gene co-expression networks reveals conservation and divergence at the tissue, pathway and disease levels. BMC Evol. Biol. 2015, 15, 259. [Google Scholar] [CrossRef]
- Watase, K.; Zoghbi, H.Y. Modelling brain diseases in mice: The challenges of design and analysis. Nat. Rev. Genet. 2003, 4, 296–307. [Google Scholar] [CrossRef]
- Nielsen, S.M.B.; Vinther-Jensen, T.; Jørgen, E.; Nielsen, J.E.; Nørremølle, A.; Hasholt, L.; Hjermind, L.E.; Josefsen, K. Liver function in Huntington’s disease assessed by blood biochemical analyses in a clinical setting. J. Neurol. Sci. 2016, 362, 326–332. [Google Scholar] [CrossRef]
- Stüwe, S.H.; Goetze, O.; Lukas, C.; Klotz, P.; Hoffmann, R.; Banasch, M.; Orth, M.; Schmidt, W.E.; Gold, R.; Saft, C. Hepatic mitochondrial dysfunction in manifest and premanifest Huntington disease. Neurology 2013, 80, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Farshbaf, M.J.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.C.C.R.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [PubMed]
- Ravaut, G.; Legiot, A.; Bergeron, K.-F.; Mounier, C. Monounsaturated Fatty Acids in Obesity-Related Inflammation. Int. J. Mol. Sci. 2021, 22, 330. [Google Scholar] [CrossRef]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef]
- Herpen, N.A.; Schrauwen-Hinderling, V.B. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 2008, 94, 231–241. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Nilsen, D.W.T.; Myhre, P.L.; Kalstad, A.; Schmidt, E.B.; Arnesen, H.; Seljeflot, I. Serum Levels of Dihomo-Gamma (γ)-Linolenic Acid (DGLA) Are Inversely Associated with Linoleic Acid and Total Death in Elderly Patients with a Recent Myocardial Infarction. Nutrients 2021, 13, 3475. [Google Scholar] [CrossRef]
- Jacqueline, K.I.; Calder, P.C. Omega-6 fatty acids and inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2018, 132, 41–48. [Google Scholar] [CrossRef]
- Valadão, P.A.C.; Santos, K.B.S.; Ferreira, E.; Vieira, T.H.; Macedo, E.; Cordeiro, T.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s disease: A few new twists on an old tale. J. Neuroimmunol. 2020, 15, 577348–577380. [Google Scholar] [CrossRef]
- Mooli, R.G.R.; Ramakrishnan, S.K. Liver Steatosis is a Driving Factor of Inflammation. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1267–1270. [Google Scholar] [CrossRef]
- Mielcarek, M.; Toczek, M.; Smeets, C.J.L.M.; Franklin, S.A.; Bondulich, M.K.; Jolinon, N.; Muller, T.; Ahmed, M.; Dick, J.R.T.; Piotrowska, I.; et al. HDAC4-myogenin axis as an important marker of HD-related skeletal muscle atrophy. PLoS Genet. 2015, 11, e1005021. [Google Scholar] [CrossRef]
- Toczek, M.; Zielonka, D.; Zukowska, P.; Marcinkowski, J.T.; Slominska, E.; Isalan, M.; Smolenski, R.T.; Mielcarek, M. An impaired metabolism of nucleotides underpins a novel mechanism of cardiac remodeling leading to Huntington’s disease related cardiomyopathy. Biochim. Biophys. Acta 2016, 1862, 2147–2157. [Google Scholar] [CrossRef]
- Tomczyk, M.; Glaser, T.; Slominska, E.M.; Ulrich, H.; Smolenski, R.T. Purine Nucleotides Metabolism and Signaling in Huntington’s Disease: Search for a Target for Novel Therapies. Int. J. Mol. Sci. 2021, 22, 6545. [Google Scholar] [CrossRef]
- Tomczyk, M.; Glaser, T.; Ulrich, H.; Slominska, E.M.; Smolenski, R.T. Huntingtin protein maintains balanced energetics in mouse cardiomyocytes. Nucleosides Nucleotides Nucleic Acids 2022, 41, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Valadão, P.A.C.; da Silva Oliveira, B.; Joviano-Santos, J.V.; Vieira, E.L.M.; Rocha, N.P.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammatory changes in peripheral organs in the BACHD murine model of Huntington’s disease. Life Sci. 2019, 232, 116653. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Chen, Y.C.; Chen, H.M.; Tu, P.H.; Chern, Y. A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum. Mol. Genet. 2013, 22, 1826–1842. [Google Scholar] [CrossRef] [PubMed]
- Risé, P.; Eligini, S.; Ghezzi, S.; Colli, S.; Galli, C. Fatty acid composition of plasma, blood cells and whole blood: Relevance for the assessment of the fatty acid status in humans. Prostaglandins Leukot. Essent. Fat. Acids 2007, 76, 363–369. [Google Scholar] [CrossRef]
- Block, R.C.; Dorsey, R.; Beck, C.A.; Brenna, J.T.; Shoulson, I. Altered cholesterol and fatty acid metabolism in Huntington disease. J. Clin. Lipidol. 2010, 4, 17–23. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, M.; Li, X.; Liu, J.; Li, H. Origin of the Autophagosome Membrane in Mammals Biomed. Res. Int. 2018, 1012789. [Google Scholar] [CrossRef]
- Iuliano, M.; Seeley, C.; Sapp, E.; Jones, E.L.; Martin, C.; Li, X.; DiFiglia, M.; Kegel-Gleason, K.B. Disposition of Proteins and Lipids in Synaptic Membrane Compartments Is Altered in Q175/Q7 Huntington’s Disease Mouse Striatum. Front. Synaptic. Neurosci. 2021, 13, 618391. [Google Scholar] [CrossRef]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef]
- Rocha, N.P.; Ribeiro, F.M.; Furr-Stimming, E.; Teixeira, A.L. Neuroimmunology of Huntington’s Disease: Revisiting Evidence from Human Studies. Mediat. Inflamm. 2016, 8653132. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Nakajima, H.; Fujita, S.; Kakae, M.; Nagayasu, K.; Oh-Hora, M.; Shirakawa, H.; Kaneko, S. Orai2 channel regulates prostaglandin E2 production in TNFα/IL1α-stimulated astrocytes. Glia 2022, 70, 1666–1680. [Google Scholar] [CrossRef]
- Lin, D.; Lei, L.; Liu, Y.; Zhang, Y.; Hu, B.; Bao, G.; Song, Y.; Jin, Z.; Liu, C.; Mei, Y.; et al. Membrane IL1α Inhibits the Development of Hepatocellular Carcinoma via Promoting T- and NK-cell Activation. Cancer Res. 2016, 76, 3179–3188. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, H.; Jónasdóttir, H.S.; Kuipers, M.E.; Kwekkeboom, J.C.; Auger, J.L.; Gonzalez-Torres, M.; López-Vicario, C.; Clària, J.; Freysdottir, J.; Hardardottir, I. Anti-Inflammatory and Proresolving Effects of the Omega-6 Polyunsaturated Fatty Acid Adrenic Acid. J. Immunol. 2020, 205, 2840–2849. [Google Scholar] [CrossRef]
- Dyall, S.C.; Balas, L.; Bazan, N.G.; Brenna, J.T.; Chiang, N.; da Costa Souza, F.; Dalli, J.; Durand, T.; Galano, J.-M.; Lein, P.J.; et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog. Lipid Res. 2022, 86, 101165. [Google Scholar] [CrossRef] [PubMed]
- Gozdzik, P.; Czumaj, A.; Sledzinski, T.; Mika, A. Branched-chain fatty acids affect the expression of fatty acid synthase and C-reactive protein genes in the hepatocyte cell line. Biosci. Rep. 2023, 43, BSR20230114. [Google Scholar] [CrossRef]
- Chiang, M.C.; Chern, Y.; Juo, C.G. The dysfunction of hepatic transcriptional factors in mice with Huntington’s Disease. Biochim. Biophys. Acta 2011, 1812, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Woodman, B.; Butler, R.; Landles, C.; Lupton, M.K.; Tse, J.; Hockly, E.; Moffitt, H.; Sathasivam, K.; Bates, G.P. The Hdh Q150/Q150 knock-in mouse model of HD and the R6/a exon 1 model develop comparable and widespread molecular phenotypes. Brain Res. Bull. 2007, 72, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Tallaksen-Greene, S.; Chien, W.M.; Cearley, J.A.; Jackson, W.S.; Crouse, A.B.; Ren, S.; Li, X.-J.; Albin, R.L.; Detloff, P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Martinez-Montoro, J.I.; Nunez-Sanchez, M.A.; Martinez-Sanchez, M.A.; Balaguer-Roman, A.; Fernandez-Ruiz, V.E.; Ferrer-Gomez, M.; Sledzinski, T.; Frutos, M.D.; Fernandez-Garcia, J.C.; Mika, A.; et al. Hepatic and serum branched-chain fatty acid profile in patients with nonalcoholic fatty liver disease: A case-control study. Obesity 2023, 31, 1064–1074. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LIVER FAs | |||
|---|---|---|---|
| WTR6/2 | R6/2 | p | |
| iso 14:0 | 0.002 ± 0.003 | 0.003 ± 0.004 | NS |
| iso 15:0 | 0.006 ± 0.005 | 0.009 ± 0.002 | NS |
| iso 16:0 | 0.017 ± 0.008 | 0.034 ± 0.010 | <0.05 |
| iso 17:0 | 0.023 ± 0.010 | 0.042 ± 0.008 | <0.01 |
| Total iso BCFA | 0.048 ± 0.023 | 0.088 ± 0.012 | <0.01 |
| anteiso 15:0 | 0.011 ± 0.012 | 0.007 ± 0.004 | NS |
| anteiso 17:0 | 0.009 ± 0.006 | 0.013 ± 0.003 | NS |
| anteiso 19:0 | 0.022 ± 0.009 | 0.045 ± 0.012 | <0.05 |
| Total anteiso BCFA | 0.042 ± 0.027 | 0.065 ± 0.012 | NS |
| Total BCFA | 0.090 ± 0.048 | 0.154 ± 0.019 | <0.05 |
| 16:0 | 11.5 ± 4.17 | 17.0 ± 4.85 | NS |
| 18:0 | 4.65 ± 0.99 | 9.96 ± 3.53 | <0.05 |
| Other ECFA | 0.49 ± 0.27 | 0.58 ± 0.16 | NS |
| Total ECFA | 16.6 ± 5.19 | 27.5 ± 8.44 | NS |
| Total OCFA | 0.25 ± 0.11 | 0.42 ± 0.13 | NS |
| 14:1 | 0.008 ± 0.008 | 0.011 ± 0.005 | NS |
| 16:1 | 1.46 ± 0.96 | 2.01 ± 0.40 | NS |
| 18:1 | 10.5 ± 6.75 | 25.0 ± 6.67 | <0.05 |
| 19:1 | 0.012 ± 0.006 | 0.035 ± 0.014 | <0.05 |
| 20:1 | 0.20 ± 0.12 | 0.54 ± 0.11 | <0.05 |
| 22:1 | 0.024 ± 0.007 | 0.026 ± 0.009 | NS |
| 24:1 | 0.053 ± 0.017 | 0.097 ± 0.038 | NS |
| Total MUFA | 12.3 ± 7.85 | 27.7 ± 7.18 | <0.05 |
| 18:3n-3 | 0.062 ± 0.021 | 0.072 ± 0.022 | NS |
| 20:4n-3 | 0.020 ± 0.009 | 0.066 ± 0.008 | <0.01 |
| 20:5n-3 | 0.28 ± 0.10 | 0.61 ± 0.20 | <0.05 |
| 22:5n-3 | 0.32 ± 0.11 | 0.44 ± 0.18 | NS |
| 22:6n-3 | 3.36 ± 0.77 | 5.19 ± 1.90 | NS |
| Total PUFAn-3 | 4.05 ± 1.00 | 6.37 ± 2.25 | NS |
| 16:2n-6 | 0.014 ± 0.008 | 0.017 ± 0.05 | NS |
| 18:2n-6 | 8.89 ± 3.00 | 14.60 ± 3.61 | <0.05 |
| 20:2n-6 | 0.16 ± 0.064 | 0.30 ± 0.057 | <0.01 |
| 20:3n-6 | 0.57 ± 0.19 | 0.86 ± 0.18 | <0.05 |
| 20:4n-6 | 3.68 ± 0.79 | 6.23 ± 1.66 | <0.05 |
| 22:4n-6 | 0.093 ± 0.027 | 0.164 ± 0.050 | <0.05 |
| 22:5n-6 | 0.028 ± 0.016 | 0.047 ± 0.013 | NS |
| Total PUFAn-6 | 13.6 ± 4.07 | 22.5 ± 5.39 | <0.05 |
| LIVER FAs | |||
|---|---|---|---|
| WTHdh | HdhQ150/Q150 | p | |
| iso 14:0 | 0.009 ± 0.007 | 0.004 ± 0.003 | NS |
| iso 15:0 | 0.012 ± 0.14 | 0.008 ± 0.005 | NS |
| iso 16:0 | 0.04 ± 0.025 | 0.046 ± 0.030 | NS |
| iso 17:0 | 0.045 ± 0.021 | 0.038 ± 0.012 | NS |
| Total isoBCFA | 0.11 ± 0.06 | 0.096 ± 0.041 | NS |
| anteiso 15:0 | 0.024 ± 0.019 | 0.011 ± 0.004 | NS |
| anteiso 17:0 | 0.025 ± 0.012 | 0.014 ± 0.01 | NS |
| anteiso 19:0 | 0.064 ± 0.03 | 0.081 ± 0.100 | NS |
| Total anteiso BCFA | 0.11 ± 0.06 | 0.11 ± 0.11 | NS |
| Total BCFA | 0.22 ± 0.11 | 0.20 ± 0.16 | NS |
| 16:0 | 29.1 ± 12.9 | 21.5 ± 6.24 | NS |
| 18:0 | 12.5 ± 5.14 | 7.86 ± 0.74 | NS |
| Other ECFA | 0.87 ± 0.50 | 0.99 ± 0.64 | NS |
| Total ECFA | 42.5 ± 18.4 | 30.4 ± 6.95 | NS |
| Total OCFA | 0.67 ± 0.30 | 0.50 ± 0.14 | NS |
| 14:1 | 0.031 ± 0.037 | 0.015 ± 0.005 | NS |
| 16:1 | 2.62 ± 1.43 | 2.66 ± 2.08 | NS |
| 18:1 | 26.4 ± 14.4 | 24.7 ± 23.2 | NS |
| 19:1 | 0.021 ± 0.010 | 0.021 ± 0.024 | NS |
| 20:1 | 0.37 ± 0.18 | 0.71 ± 0.79 | NS |
| 22:1 | 0.032 ± 0.018 | 0.035 ± 0.009 | NS |
| 24:1 | 0.11 ± 0.036 | 0.07 ± 0.01 | NS |
| Total MUFA | 29.6 ± 15.9 | 28.2 ± 26.0 | NS |
| 18:3n-3 | 0.20 ± 0.09 | 0.10 ± 0.07 | NS |
| 20:4n-3 | 0.06 ± 0.05 | 0.03 ± 0.008 | NS |
| 20:5n-3 | 0.79 ± 0.32 | 0.38 ± 0.14 | <0.05 |
| 22:5n-3 | 0.96 ± 0.40 | 0.44 ± 0.15 | <0.05 |
| 22:6n-3 | 8.47 ± 3.45 | 5.08 ± 1.82 | NS |
| Total PUFAn-3 | 10.5 ± 4.20 | 6.03 ± 2.14 | NS |
| 16:2n-6 | 0.04 ± 0.032 | 0.04 ± 0.025 | NS |
| 18:2n-6 | 24.2 ± 10.8 | 20.9 ± 13.1 | NS |
| 20:2n-6 | 0.39 ± 0.15 | 0.30 ± 0.10 | NS |
| 20:3n-6 | 1.01 ± 0.42 | 0.79 ± 0.21 | NS |
| 20:4n-6 | 8.31 ± 3.52 | 5.63 ± 1.79 | NS |
| 22:4n-6 | 0.30 ± 0.11 | 0.16 ± 0.02 | <0.05 |
| 22:5n-6 | 0.060 ± 0.029 | 0.040 ± 0.011 | NS |
| Total PUFAn-6 | 34.7 ± 14.4 | 28.1 ± 11.5 | NS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gregorczyk, M.; Mika, A.; Śledziński, T.; Tomczyk, M.; Rybakowska, I. Fatty Acid Profile in the Liver of Mice with Early- and Late-Onset Forms of Huntington’s Disease. Int. J. Mol. Sci. 2025, 26, 7304. https://doi.org/10.3390/ijms26157304
Gregorczyk M, Mika A, Śledziński T, Tomczyk M, Rybakowska I. Fatty Acid Profile in the Liver of Mice with Early- and Late-Onset Forms of Huntington’s Disease. International Journal of Molecular Sciences. 2025; 26(15):7304. https://doi.org/10.3390/ijms26157304
Chicago/Turabian StyleGregorczyk, Magdalena, Adriana Mika, Tomasz Śledziński, Marta Tomczyk, and Iwona Rybakowska. 2025. "Fatty Acid Profile in the Liver of Mice with Early- and Late-Onset Forms of Huntington’s Disease" International Journal of Molecular Sciences 26, no. 15: 7304. https://doi.org/10.3390/ijms26157304
APA StyleGregorczyk, M., Mika, A., Śledziński, T., Tomczyk, M., & Rybakowska, I. (2025). Fatty Acid Profile in the Liver of Mice with Early- and Late-Onset Forms of Huntington’s Disease. International Journal of Molecular Sciences, 26(15), 7304. https://doi.org/10.3390/ijms26157304