
Genome Sequencing of a Fusarium Endophytic Isolate from Hazelnut: Phylogenetic and Metabolomic Implications

 ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Morphological Description
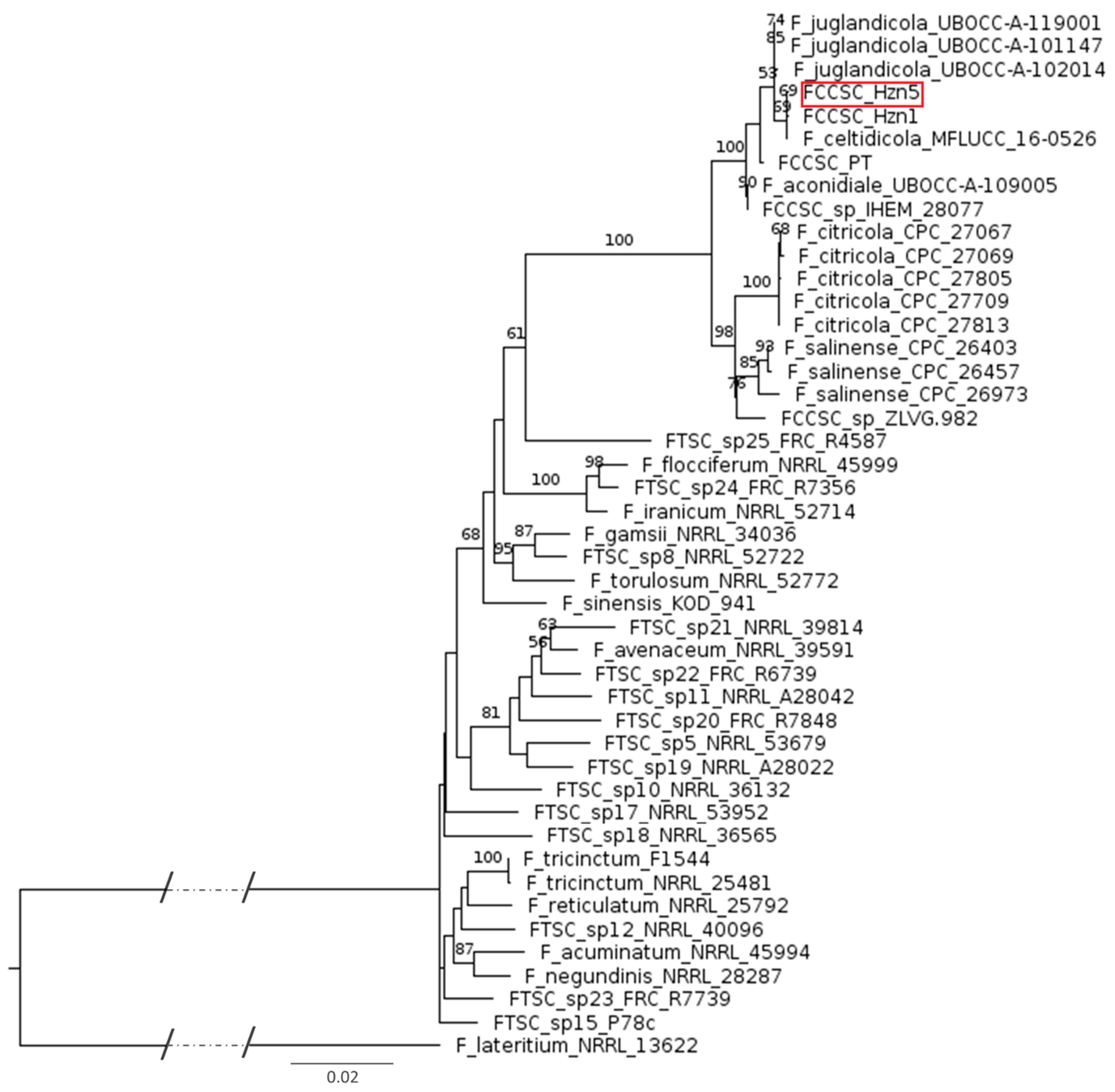
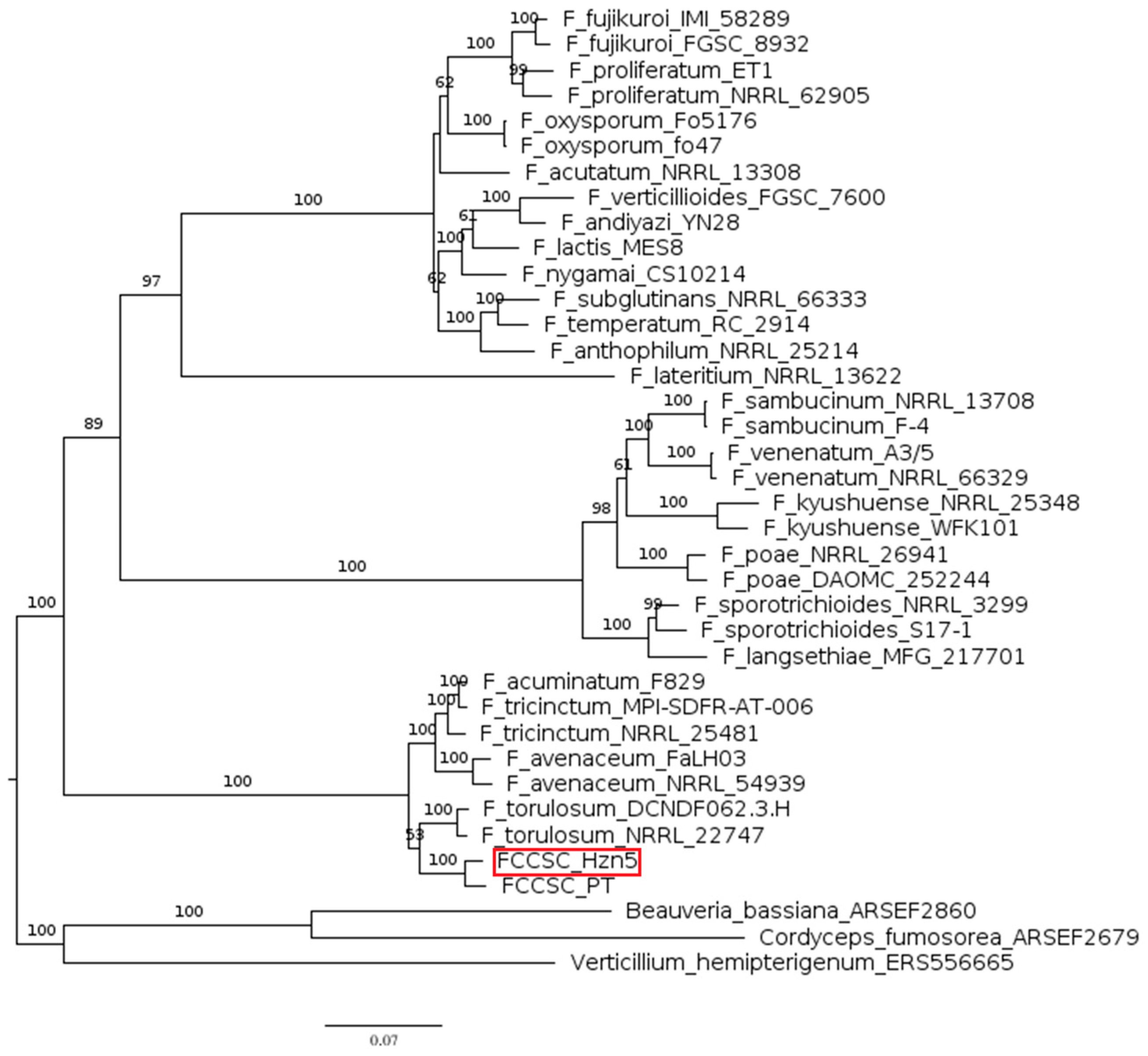
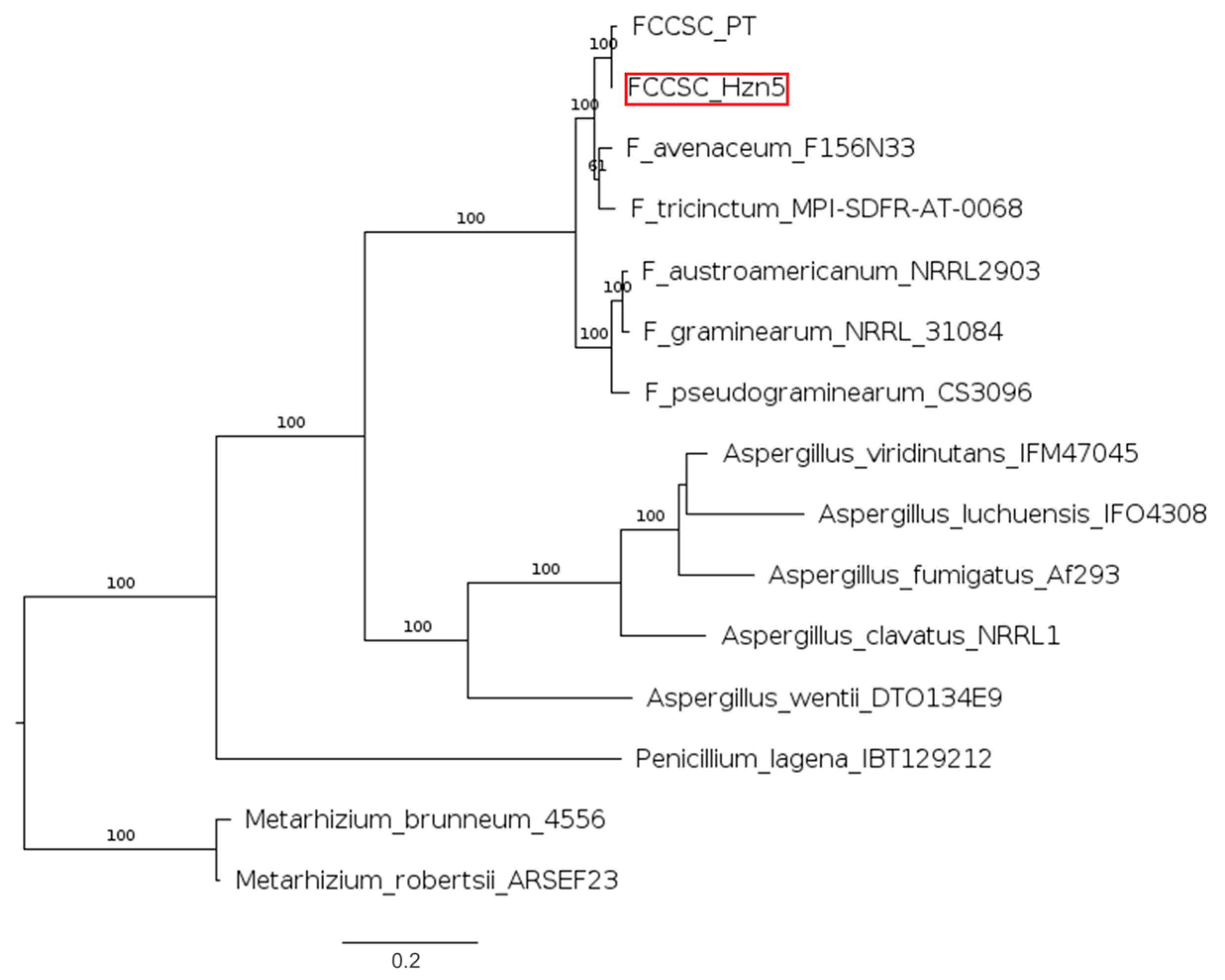
2.2. Phylogenesis
2.3. Genome De Novo Sequence Assembly
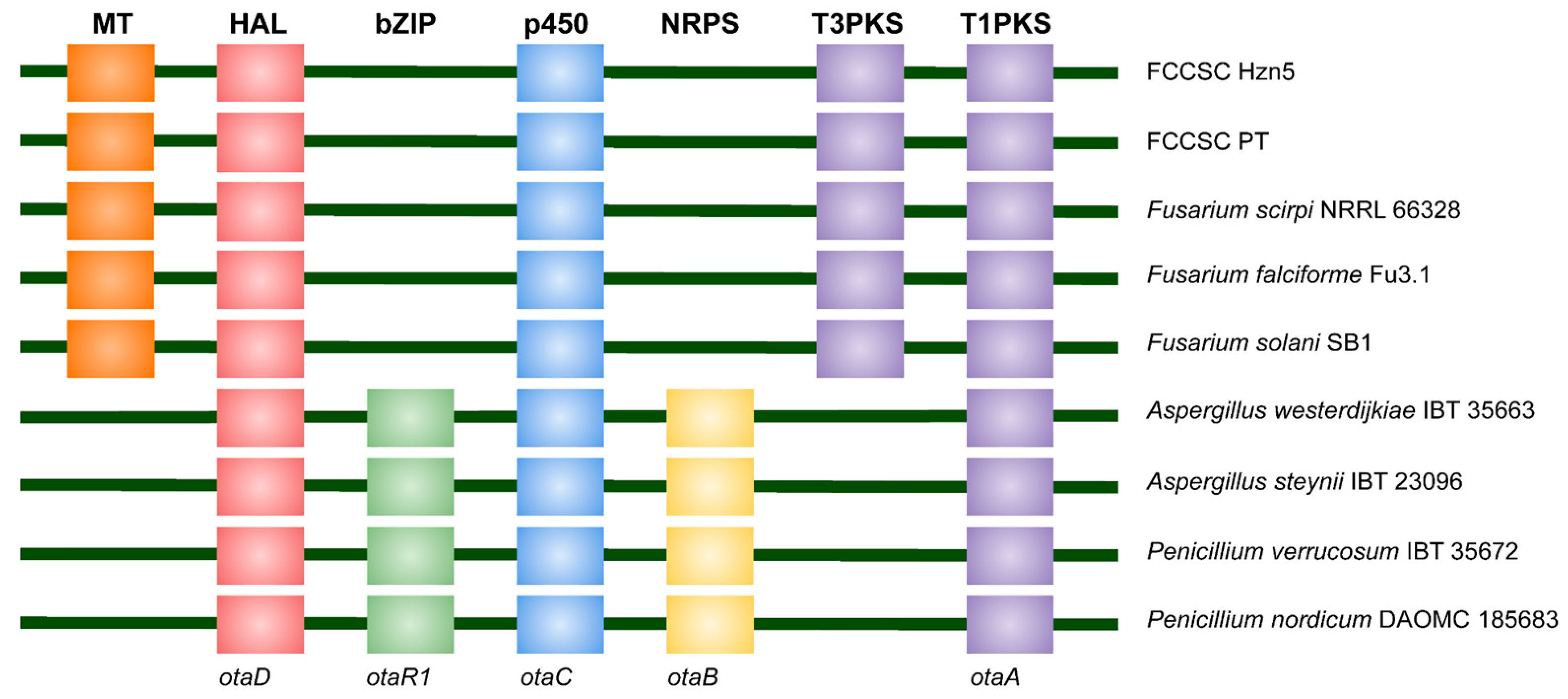
2.4. Annotation of Biosynthetic Gene Clusters Related to Secondary Metabolite Production
2.5. Phylogenetic Relationships Referred to Selected Secondary Metabolites
3. Discussion
4. Materials and Methods
4.1. Morphological Observations
4.2. Phylogenetic Analysis
4.3. Genome Sequencing and Assembly
4.3.1. Starting Material, DNA Extraction, and Sequencing
4.3.2. Genome Data Processing: De Novo Assembly and Annotation
4.3.3. Biosynthetic Gene Clusters Prediction and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crous, P.W.; Lombard, L.; Sandoval-Denis, M.; Seifert, K.A.; Schroers, H.J.; Chaverri, P.; Gené, J.; Guarro, J.; Hirooka, Y.; Bensch, K.; et al. Fusarium: More than a node or a foot-shaped basal cell. Stud. Mycol. 2021, 98, 100116. [Google Scholar] [CrossRef]
- O’Donnell, K.; Ward, T.J.; Robert, V.A.; Crous, P.W.; Geiser, D.M.; Kang, S. DNA sequence-based identification of Fusarium: Current status and future directions. Phytoparasitica 2015, 43, 583–595. [Google Scholar] [CrossRef]
- Zimowska, B.; Ludwiczuk, A.; Manganiello, G.; Wojtanowski, K.; Kot, I.; Staropoli, A.; Vinale, F.; Nicoletti, R. Fusarium and hazelnut: A story of twists and turns. Agriculture 2024, 14, 1080. [Google Scholar] [CrossRef]
- Turco, S.; Brugneti, F.; Giubilei, I.; Silvestri, C.; Petrović, M.; Drais, M.I.; Cristofori, V.; Speranza, S.; Mazzaglia, A.; Contarini, M.; et al. A bud’s life: Metabarcoding analysis to characterise hazelnut big buds microbiome biodiversity. Microbiol. Res. 2024, 287, 127851. [Google Scholar]
- Lombardi, S.J.; Pannella, G.; Tremonte, P.; Mercurio, I.; Vergalito, F.; Caturano, C.; Maiuro, L.; Iorizzo, M.; Succi, M.; Sorrentino, E.; et al. Fungi occurrence in ready-to-eat hazelnuts (Corylus avellana) from different boreal hemisphere areas. Front. Microbiol. 2022, 13, 900876. [Google Scholar] [CrossRef]
- Santori, A.; Vitale, S.; Luongo, L.; Belisario, A. First report of Fusarium lateritium as the agent of nut gray necrosis on hazelnut in Italy. Plant Dis. 2010, 94, 484. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.M.; Andolfi, A.; Nicoletti, R. Mycotoxin contamination in hazelnut: Current status, analytical strategies, and future prospects. Toxins 2023, 15, 99. [Google Scholar] [CrossRef]
- Nicoletti, R.; Zimowska, B. Endophytic fungi of hazelnut (Corylus avellana). Plant Prot. Sci. 2023, 59, 107–123. [Google Scholar] [CrossRef]
- Samal, S.; Rai, S.; Upadhaya, R.S. Endophytic Fusarium and their association with plant growth. In Microbial Endophytes and Plant Growth; Solanki, M.K., Yadav, M.K., Singh, B.P., Gupta, V.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 259–268. [Google Scholar]
- Turco, S.; Grottoli, A.; Drais, M.I.; De Spirito, C.; Faino, L.; Reverberi, M.; Cristofori, V.; Mazzaglia, A. Draft genome sequence of a new Fusarium isolate belonging to Fusarium tricinctum species complex collected from hazelnut in Central Italy. Front. Plant Sci. 2021, 12, 788584. [Google Scholar] [CrossRef]
- Sandoval-Denis, M.; Guarnaccia, V.; Polizzi, G.; Crous, P.W. Symptomatic Citrus trees reveal a new pathogenic lineage in Fusarium and two new Neocosmospora species. Persoonia 2018, 40, 1–25. [Google Scholar] [CrossRef]
- Costa, M.M.; Sandoval-Denis, M.; Moreira, G.M.; Kandemir, H.; Kermode, A.; Buddie, A.G.; Ryan, M.J.; Becker, Y.; Yurkov, A.; Maier, W.; et al. Known from trees and the tropics: New insights into the Fusarium lateritium species complex. Stud. Mycol. 2024, 109, 403–450. [Google Scholar] [CrossRef]
- Laraba, I.; Busman, M.; Geiser, D.M.; O’Donnell, K. Phylogenetic diversity and mycotoxin potential of emergent phytopathogens within the Fusarium tricinctum species complex. Phytopathology 2022, 112, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Medema, M.H.; Weber, T. The antiSMASH database version 4: Additional genomes and BGCs, new sequence-based searches and more. Nucleic Acids Res. 2024, 52, D586–D589. [Google Scholar] [CrossRef]
- Hansen, F.T.; Gardiner, D.M.; Lysøe, E.; Fuertes, P.R.; Tudzynski, B.; Wiemann, P.; Sondergaard, T.E.; Giese, H.; Brodersen, D.E.; Sørensen, J.L. An update to polyketide synthase and non-ribosomal synthetase genes and nomenclature in Fusarium. Fungal Gen. Biol. 2015, 75, 20–29. [Google Scholar] [CrossRef]
- Nielsen, M.R.; Sondergaard, T.E.; Giese, H.; Sørensen, J.L. Advances in linking polyketides and non-ribosomal peptides to their biosynthetic gene clusters in Fusarium. Curr. Genet. 2019, 65, 1263–1280. [Google Scholar] [CrossRef]
- Janevska, S.; Tudzynski, B. Secondary metabolism in Fusarium fujikuroi: Strategies to unravel the function of biosynthetic pathways. Appl. Microbiol. Biotechnol. 2018, 102, 615–630. [Google Scholar]
- Fan, S.; Wang, Q.; Dai, J.; Jiang, J.; Hu, X.; Subbarao, K.V. The whole genome sequence of Fusarium redolens strain YP04, a pathogen that causes root rot of American ginseng. Phytopathology 2021, 111, 2130–2134. [Google Scholar] [CrossRef]
- He, T.; Li, X.; Iacovelli, R.; Hackl, T.; Haslinger, K. Genomic and metabolomic analysis of the endophytic fungus Fusarium sp. VM-40 isolated from the medicinal plant Vinca minor. J. Fungi 2023, 9, 704. [Google Scholar]
- Purayil, G.P.; Almarzooqi, A.Y.; El-Tarabily, K.A.; You, F.M.; AbuQamar, S.F. Fully resolved assembly of Fusarium proliferatum DSM106835 genome. Sci. Data 2023, 10, 705. [Google Scholar] [CrossRef]
- Rana, S.; Singh, S.K. Insights into the genomic architecture of a newly discovered endophytic Fusarium species belonging to the Fusarium concolor complex from India. Front. Microbiol. 2023, 14, 1266620. [Google Scholar] [CrossRef]
- Pokhrel, A.; Coleman, J.J. Inventory of the secondary metabolite biosynthetic potential of members within the terminal clade of the Fusarium solani species complex. J. Fungi 2023, 9, 799. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Chunduri, J.R. Genome-wide analysis and in silico screening of secondary metabolite potential of endophytic fungi Fusarium multiceps BPAL1 obtained in Mumbai, India. Egypt. J. Basic Appl. Sci. 2023, 10, 812–823. [Google Scholar] [CrossRef]
- Thomas, V.E.; Isakeit, T.; Antony-Babu, S. Draft genomes of five Fusarium oxysporum f. sp. niveum strains isolated from infected watermelon from Texas with temporal and spatial differences. PhytoFrontiers 2023, 3, 466–471. [Google Scholar]
- Gil-Serna, J.; Vázquez, C.; Patiño, B. The genomic regions that contain ochratoxin A biosynthetic genes widely differ in Aspergillus section Circumdati species. Toxins 2020, 12, 754. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.; Gallo, A.; Perrone, G.; Magistà, D.; Baker, S.E. Comparative genomic analysis of ochratoxin A biosynthetic cluster in producing fungi: New evidence of a cyclase gene involvement. Front. Microbiol. 2020, 11, 581309. [Google Scholar] [CrossRef]
- McCormick, S.P.; Alexander, N.J.; Harris, L.J. CLM1 of Fusarium graminearum encodes a longiborneol synthase required for culmorin production. Appl. Environ. Microbiol. 2010, 76, 136–141. [Google Scholar] [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Shi, X.S.; Yang, S.Q.; Li, X.M.; Li, Y.H.; Wang, D.J.; Li, X.; Meng, L.H.; Zhou, X.W.; Wang, B.G. Antimicrobial polyketides from the endophytic fungus Fusarium asiaticum QA-6 derived from medicinal plant Artemisia argyi. Phytochemistry 2025, 233, 114379. [Google Scholar] [CrossRef]
- Chen, D.; Liu, L.; Lu, Y.; Chen, S. Identification of fusarielin M as a novel inhibitor of Mycobacterium tuberculosis protein tyrosine phosphatase B (MptpB). Bioorg. Chem. 2021, 106, 104495. [Google Scholar] [CrossRef]
- Sørensen, J.L.; Akk, E.; Thrane, U.; Giese, H.; Sondergaard, T.E. Production of fusarielins by Fusarium. Int. J. Food Microbiol. 2013, 160, 206–211. [Google Scholar] [CrossRef]
- Nicoletti, R.; Trincone, A. Bioactive compounds produced by strains of Penicillium and Talaromyces of marine origin. Mar. Drugs 2016, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Zocher, R.; Keller, U.; Kleinkauf, H. Enniatin synthetase, a novel type of multifunctional enzyme catalyzing depsipeptide synthesis in Fusarium oxysporum. Biochemistry 1982, 21, 43–48. [Google Scholar]
- Pieper, R.; Kleinkauf, H.; Zocher, R. Enniatin synthetases from different Fusaria exhibiting distinct amino acid specificities. J. Antibiot. 1992, 45, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Stępień, Ł.; Waśkiewicz, A. Sequence divergence of the enniatin synthase gene in relation to production of beauvericin and enniatins in Fusarium species. Toxins 2013, 5, 537–555. [Google Scholar] [CrossRef]
- Liuzzi, V.C.; Mirabelli, V.; Cimmarusti, M.T.; Haidukowski, M.; Leslie, J.F.; Logrieco, A.F.; Caliandro, R.; Fanelli, F.; Mulè, G. Enniatin and beauvericin biosynthesis in Fusarium species: Production profiles and structural determinant prediction. Toxins 2017, 9, 45. [Google Scholar] [CrossRef]
- Olleik, H.; Nicoletti, C.; Lafond, M.; Courvoisier-Dezord, E.; Xue, P.; Hijazi, A.; Baydoun, E.; Perrier, J.; Maresca, M. Comparative structure–activity analysis of the antimicrobial activity, cytotoxicity, and mechanism of action of the fungal cyclohexadepsipeptides enniatins and beauvericin. Toxins 2019, 11, 514. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, M.; Stępień, Ł.; Uhlig, S. Evidence for naturally produced beauvericins containing N-methyl-tyrosine in Hypocreales fungi. Toxins 2019, 11, 182. [Google Scholar] [CrossRef]
- Urbaniak, M.; Waśkiewicz, A.; Stępień, Ł. Fusarium cyclodepsipeptide mycotoxins: Chemistry, biosynthesis, and occurrence. Toxins 2020, 12, 765. [Google Scholar] [CrossRef]
- Urbaniak, M.; Waskiewicz, A.; Trzebny, A.; Koczyk, G.; Stepien, L. Cyclodepsipeptide biosynthesis in Hypocreales fungi and sequence divergence of the non-ribosomal peptide synthase genes. Pathogens 2020, 9, 552. [Google Scholar] [CrossRef]
- Lin, C.; Feng, X.L.; Liu, Y.; Li, Z.C.; Li, X.Z.; Qi, J. Bioinformatic analysis of secondary metabolite biosynthetic potential in pathogenic Fusarium. J. Fungi 2023, 9, 850. [Google Scholar] [CrossRef]
- Gautier, C.; Pinson-Gadais, L.; Richard-Forget, F. Fusarium mycotoxins enniatins: An updated review of their occurrence, the producing Fusarium species, and the abiotic determinants of their accumulation in crop harvests. J. Agric. Food Chem. 2020, 68, 4788–4798. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific opinion on the risks to human and animal health related to the presence of beauvericin and enniatins in food and feed. EFSA J. 2014, 12, 3802. [Google Scholar]
- De Felice, B.; Spicer, L.J.; Caloni, F. Enniatin B1: Emerging mycotoxin and emerging issues. Toxins 2023, 15, 383. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, P.; Sieber, C.M.K.; von Bargen, K.W.; Studt, L.; Niehaus, E.-M.; Espino, J.J.; Huß, K.; Michielse, C.B.; Albermann, S.; Wagner, D.; et al. Deciphering the cryptic genome: Genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathogens 2013, 9, e1003475. [Google Scholar] [CrossRef]
- Garello, M.; Piombo, E.; Buonsenso, F.; Prencipe, S.; Valente, S.; Meloni, G.R.; Marcet-Houben, M.; Gabaldón, T.; Spadaro, D. Several secondary metabolite gene clusters in the genomes of ten Penicillium spp. raise the risk of multiple mycotoxin occurrence in chestnuts. Food Microbiol. 2024, 122, 104532. [Google Scholar] [CrossRef] [PubMed]
- Atanasoff-Kardjalieff, A.K.; Studt, L. Secondary metabolite gene regulation in mycotoxigenic Fusarium species: A focus on chromatin. Toxins 2022, 14, 96. [Google Scholar] [CrossRef]
- Zaehle, C.; Gressler, M.; Shelest, E.; Geib, E.; Hertweck, C.; Brock, M. Terrein biosynthesis in Aspergillus terreus and its impact on phytotoxicity. Chem. Biol. 2014, 21, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Ugai, T.; Minami, A.; Tanaka, S.; Ozaki, T.; Liu, C.; Shigemori, H.; Hashimoto, M.; Oikawa, H. Biosynthetic machinery of 6-hydroxymellein derivatives leading to cyclohelminthols and palmaenones. ChemBioChem 2020, 21, 360–367. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Wu, F.; Liu, F.; Wang, Q.; Zhang, X.; Selvaraj, J.N.; Zhao, Y.; Xing, F.; Yin, W.B.; et al. A consensus ochratoxin A biosynthetic pathway: Insights from the genome sequence of Aspergillus ochraceus and a comparative genomic analysis. Appl. Environ. Microbiol. 2018, 84, e01009-18. [Google Scholar] [CrossRef]
- Wipfler, R.; McCormick, S.P.; Proctor, R.H.; Teresi, J.M.; Hao, G.; Ward, T.J.; Alexander, N.; Vaughan, M.M. Synergistic phytotoxic effects of culmorin and trichothecene mycotoxins. Toxins 2019, 11, 555. [Google Scholar] [CrossRef]
- Bahadoor, A.; Schneiderman, D.; Gemmill, L.; Bosnich, W.; Blackwell, B.; Melanson, J.E.; McRae, G.; Harris, L.J. Hydroxylation of longiborneol by a Clm2-encoded CYP450 monooxygenase to produce culmorin in Fusarium graminearum. J. Nat. Prod. 2016, 79, 81–88. [Google Scholar] [CrossRef]
- Senatore, M.T.; Ward, T.J.; Cappelletti, E.; Beccari, G.; McCormick, S.P.; Busman, M.; Laraba, I.; O’Donnell, K.; Prodi, A. Species diversity and mycotoxin production by members of the Fusarium tricinctum species complex associated with Fusarium head blight of wheat and barley in Italy. Int. J. Food Microbiol. 2021, 358, 109298. [Google Scholar] [CrossRef] [PubMed]
- Laraba, I.; McCormick, S.P.; Vaughan, M.M.; Geiser, D.M.; O’Donnell, K. Phylogenetic diversity, trichothecene potential, and pathogenicity within Fusarium sambucinum species complex. PLoS ONE 2021, 16, e0245037. [Google Scholar] [CrossRef]
- Proctor, R.H.; McCormick, S.P.; Kim, H.S.; Cardoza, R.E.; Stanley, A.M.; Lindo, L.; Kelly, A.; Brown, D.W.; Lee, T.; Vaughan, M.M.; et al. Evolution of structural diversity of trichothecenes, a family of toxins produced by plant pathogenic and entomopathogenic fungi. PLoS Pathogens 2018, 14, e1006946. [Google Scholar] [CrossRef]
- Rokas, A.; Mead, M.E.; Steenwyk, J.L.; Raja, H.A.; Oberlies, N.H. Biosynthetic gene clusters and the evolution of fungal chemodiversity. Nat. Prod. Rep. 2020, 37, 868–878. [Google Scholar] [CrossRef]
- Witte, T.E.; Harris, L.J.; Nguyen, H.D.T.; Hermans, A.; Johnston, A.; Sproule, A.; Dettman, J.R.; Boddy, C.N.; Overy, D.P. Apicidin biosynthesis is linked to accessory chromosomes in Fusarium poae isolates. BMC Genomics 2021, 22, 591. [Google Scholar] [CrossRef] [PubMed]
- Witte, T.E.; Villeneuve, N.; Boddy, C.N.; Overy, D.P. Accessory chromosome-acquired secondary metabolism in plant pathogenic fungi: The evolution of biotrophs into host-specific pathogens. Front. Microbiol. 2021, 12, 664276. [Google Scholar] [CrossRef]
- Hoogendoorn, K.; Barra, L.; Waalwijk, C.; Dickschat, J.S.; Van der Lee, T.A.; Medema, M.H. Evolution and diversity of biosynthetic gene clusters in Fusarium. Front. Microbiol. 2018, 9, 1158. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.; Proctor, R.H.; Kim, H.S.; Brown, D.W.; Logrieco, A.F.; Amatulli, M.T.; Moretti, A.; Susca, A. Variation in secondary metabolite production potential in the Fusarium incarnatum-equiseti species complex revealed by comparative analysis of 13 genomes. BMC Genomics 2019, 20, 314. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Zerbino, D.R. Using the Velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinform. 2010, 31, 11.5.1–11.5.12. [Google Scholar]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L.; et al. ABySS 2.0: Resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 2017, 27, 768–777. [Google Scholar] [CrossRef]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contig | Region | Type | From | To | Most Similar Known BGCs | Similarity (%) | |
|---|---|---|---|---|---|---|---|
| Hzn5 | PT | ||||||
| 16265 | 18.1 | NRPS | 64,724 | 127,601 | |||
| 16275 | 22.1 | T1PKS | 754,845 | 804,388 | orcinol/orsellinic acid | 55 | 55 |
| 16279 | 23.1 | NRPS-like, T1PKS | 137,159 | 195,811 | fusarielin H | 62 | 50 |
| 16279 | 23.2 | terpene | 325,453 | 348,561 | gibberellin | 28 | 42 |
| 16265 | 33.1 | betalactone | 270,136 | 303,018 | |||
| 16265 | 33.2 | NRPS-like | 667,535 | 711,386 | choline | 100 | 100 |
| 16265 | 33.3 | NRPS-like | 787,157 | 832,949 | |||
| 16265 | 33.4 | T1PKS, NRPS | 979,234 | 1,031,349 | fusaridione A | 18 | 18 |
| 16257 | 37.1 | T1PKS, NRPS | 25,277 | 136,354 | |||
| 16257 | 37.2 | T3PKS | 382,522 | 423,836 | 6-hydroxymellein | 33 | nd |
| 16257 | 37.3 | NRPS-like | 470,544 | 513,917 | |||
| 16257 | 37.4 | T1PKS, NRPS | 1,879,135 | 1,931,246 | ilicicolin H,J/8-epi-ilicicolin H | 60 | 60 |
| 16257 | 37.5 | fungal-RiPP-like | 2,346,099 | 2,409,176 | |||
| 16277 | 39.1 | T1PKS | 1 | 40,460 | fujikurins A−D | 50 | 83 |
| 16277 | 40.1 | NRPS | 119,297 | 175,596 | |||
| 16277 | 40.2 | terpene | 247,516 | 268,206 | |||
| 16277 | 40.3 | NRPS-like | 444,480 | 488,237 | |||
| 16277 | 40.4 | NRPS | 1,135,033 | 1,228,604 | |||
| 16277 | 40.5 | fungal-RiPP-like | 1,534,027 | 1,594,923 | |||
| 16293 | 40.6 | T1PKS | 1,617,087 | 1,665,688 | gibepyrone A | 40 | 40 |
| 16293 | 40.7 | NRPS-like | 3,606,132 | 3,649,956 | bassianolide | 13 | 13 |
| 16293 | 40.8 | terpene | 4,308,961 | 4,329,877 | CLM1, CLM2 1 | ||
| 16283 | 40.9 | NRPS | 4,372,229 | 4,420,244 | chrysogine | 83 | 83 |
| 16283 | 40.10 | NRPS | 4,435,111 | 4,504,467 | |||
| 16248 | 42.1 | NRPS, T1PKS | 40,368 | 103,235 | fusaristatin A | 100 | 80 |
| 16247 | 47.1 | phosphonate | 275,063 | 296,779 | fosfonochlorin | 53 | 61 |
| 16247 | 47.2 | T1PKS | 376,594 | 423,598 | bikaverin | 42 | 42 |
| 16247 | 47.3 | T3PKS | 978,133 | 1,019,603 | |||
| 16247 | 47.4 | T1PKS | 2,304,527 | 2,352,015 | |||
| 16297 | 49.1 | fungal-RiPP-like | 137,098 | 198,224 | |||
| 16290 | 49.2 | NRPS, T1PKS | 404,613 | 456,534 | ACT-toxin II | 100 | 100 |
| 16290 | 49.3 | T1PKS | 570,192 | 616,346 | bikaverin | 57 | 57 |
| 16276 | 50.1 | T1PKS | 364,207 | 411,510 | oxyjavanicin | 62 | 50 |
| 16276 | 50.2 | terpene | 1,034,967 | 1,056,515 | squalestatin S1 | 40 | 40 |
| 16276 | 50.3 | NRP-metallophore, NRPS | 2,607,962 | 2,671,361 | |||
| 16303 | 51.1 | T1PKS | 1 | 28,837 | |||
| 16303 | 51.2 | NRPS-like | 234,494 | 277,357 | |||
| 16303 | 51.3 | terpene | 554,993 | 579,071 | |||
| 16303 | 51.4 | terpene | 670,601 | 691,870 | α-acorenol | 100 | 100 |
| 16303 | 51.5 | NRPS | 1,179,738 | 1,228,091 | |||
| 16320 | 52.1 | terpene | 323,302 | 345,735 | |||
| 16319 | 52.2 | T1PKS | 2,037,563 | 2,085,451 | |||
| 16320 | 53.1 | T1PKS | 97,452 | 147,762 | |||
| 16320 | 53.2 | fungal-RiPP-like, T1PKS | 148,668 | 215,872 | fusarubin/1233A–B/NG-391/lucilactaene | 28 | nd |
| 16320 | 53.3 | NRPS | 592,686 | 640,553 | |||
| 16254 | 54.1 | terpene | 719,300 | 741,312 | |||
| 16254 | 54.2 | terpene | 1,138,511 | 1,160,268 | koraiol | 100 | 100 |
| 16254 | 54.3 | fungal-RiPP-like, isocyanide | 1,269,111 | 1,347,781 | |||
| 16321 | 55.1 | NRPS | 268,291 | 317,683 | beauvericin 2 | 20 | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becchimanzi, A.; Zimowska, B.; Calandrelli, M.M.; De Masi, L.; Nicoletti, R. Genome Sequencing of a Fusarium Endophytic Isolate from Hazelnut: Phylogenetic and Metabolomic Implications. Int. J. Mol. Sci. 2025, 26, 4377. https://doi.org/10.3390/ijms26094377
Becchimanzi A, Zimowska B, Calandrelli MM, De Masi L, Nicoletti R. Genome Sequencing of a Fusarium Endophytic Isolate from Hazelnut: Phylogenetic and Metabolomic Implications. International Journal of Molecular Sciences. 2025; 26(9):4377. https://doi.org/10.3390/ijms26094377
Chicago/Turabian StyleBecchimanzi, Andrea, Beata Zimowska, Marina Maura Calandrelli, Luigi De Masi, and Rosario Nicoletti. 2025. "Genome Sequencing of a Fusarium Endophytic Isolate from Hazelnut: Phylogenetic and Metabolomic Implications" International Journal of Molecular Sciences 26, no. 9: 4377. https://doi.org/10.3390/ijms26094377
APA StyleBecchimanzi, A., Zimowska, B., Calandrelli, M. M., De Masi, L., & Nicoletti, R. (2025). Genome Sequencing of a Fusarium Endophytic Isolate from Hazelnut: Phylogenetic and Metabolomic Implications. International Journal of Molecular Sciences, 26(9), 4377. https://doi.org/10.3390/ijms26094377