

Target Validation Studies of PS48, a PDK-1 Allosteric Agonist, for the Treatment of Alzheimer’s Disease Phenotype in APP/PS1 Transgenic Mice
 , , and
, , and
Abstract

1. Introduction
2. Results
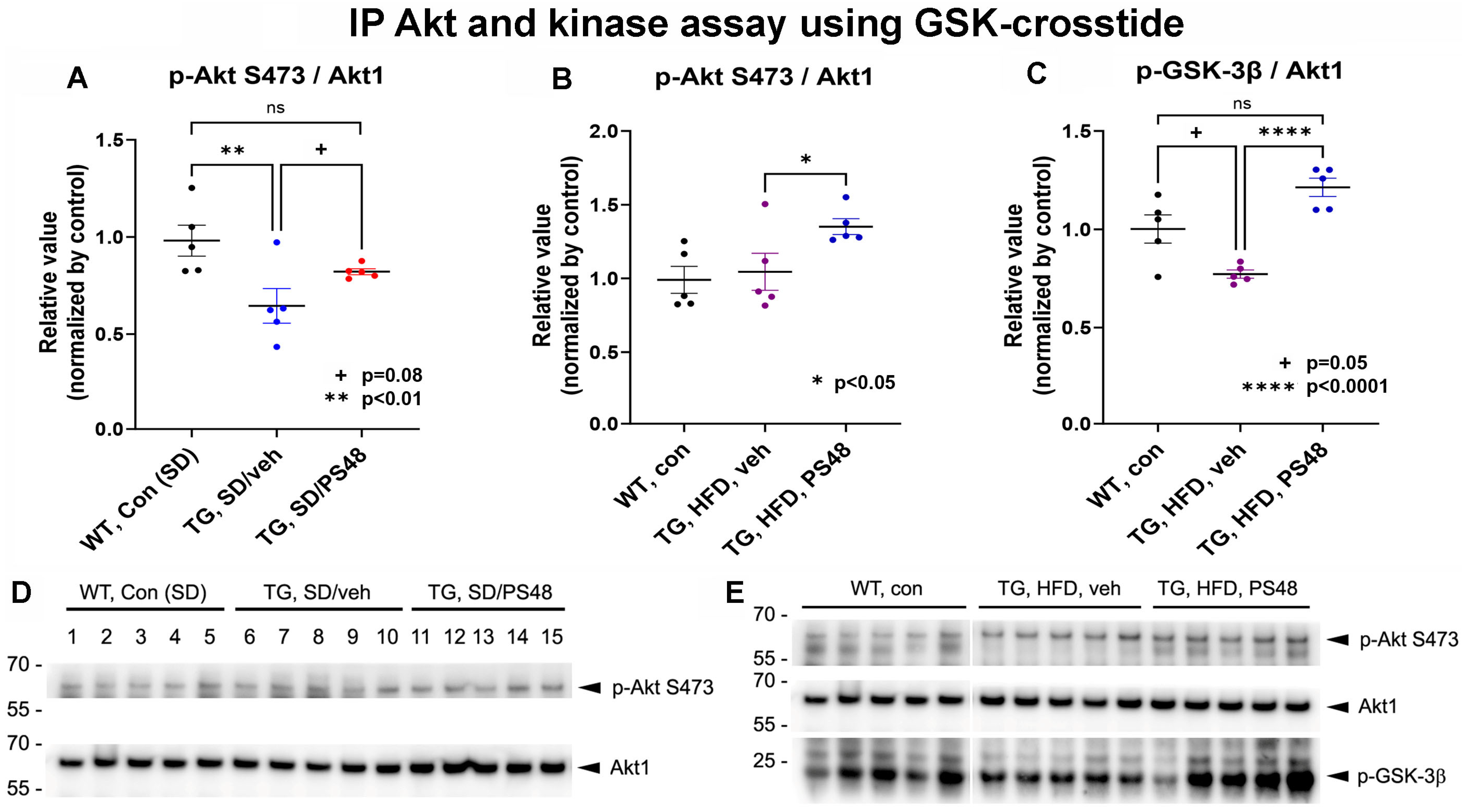
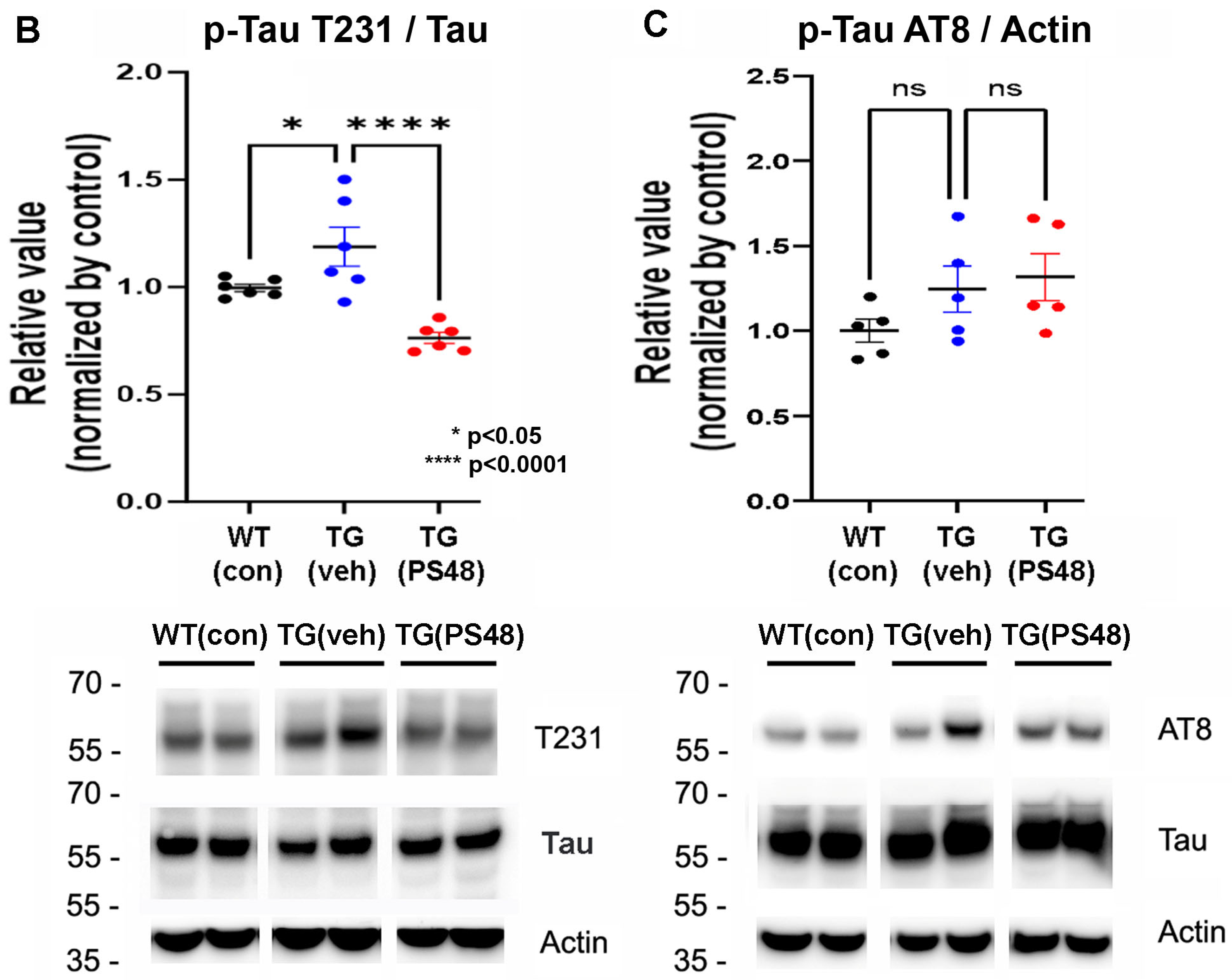
2.1. Immunoprecipitations: Western Blot and Kinase Activity Analysis
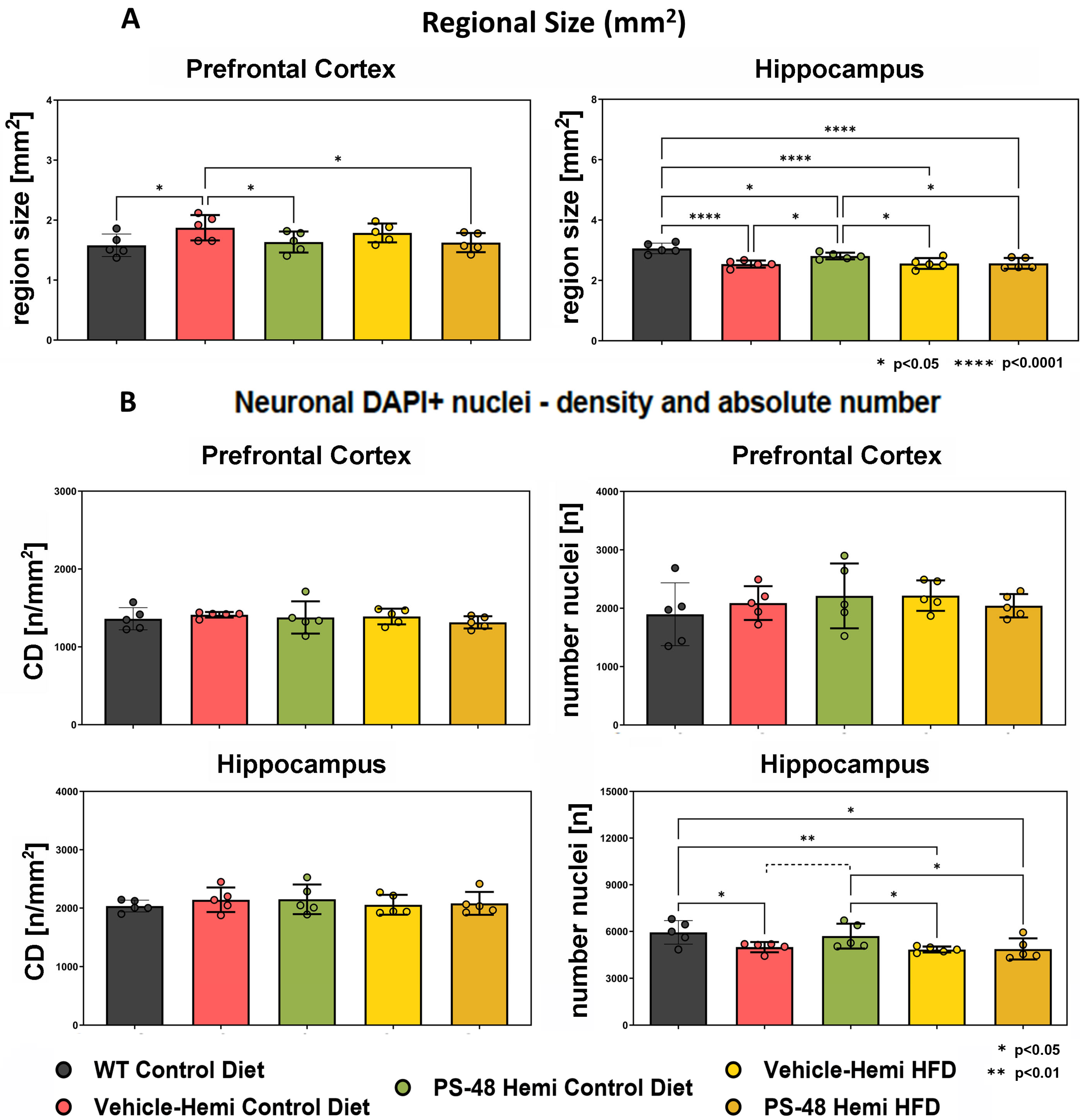
2.2. Immunohistochemistry: Regional Size and Neuron Number
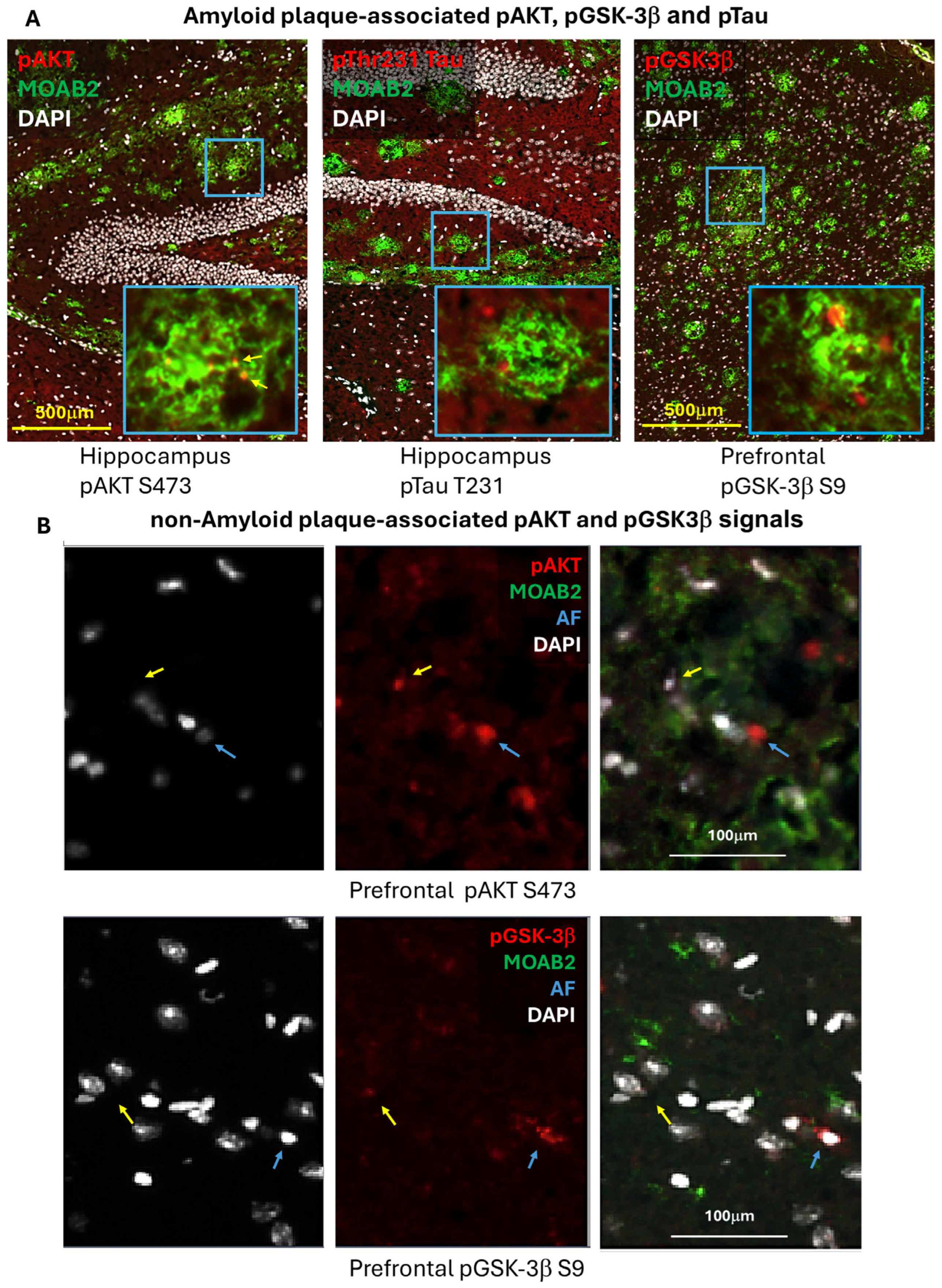
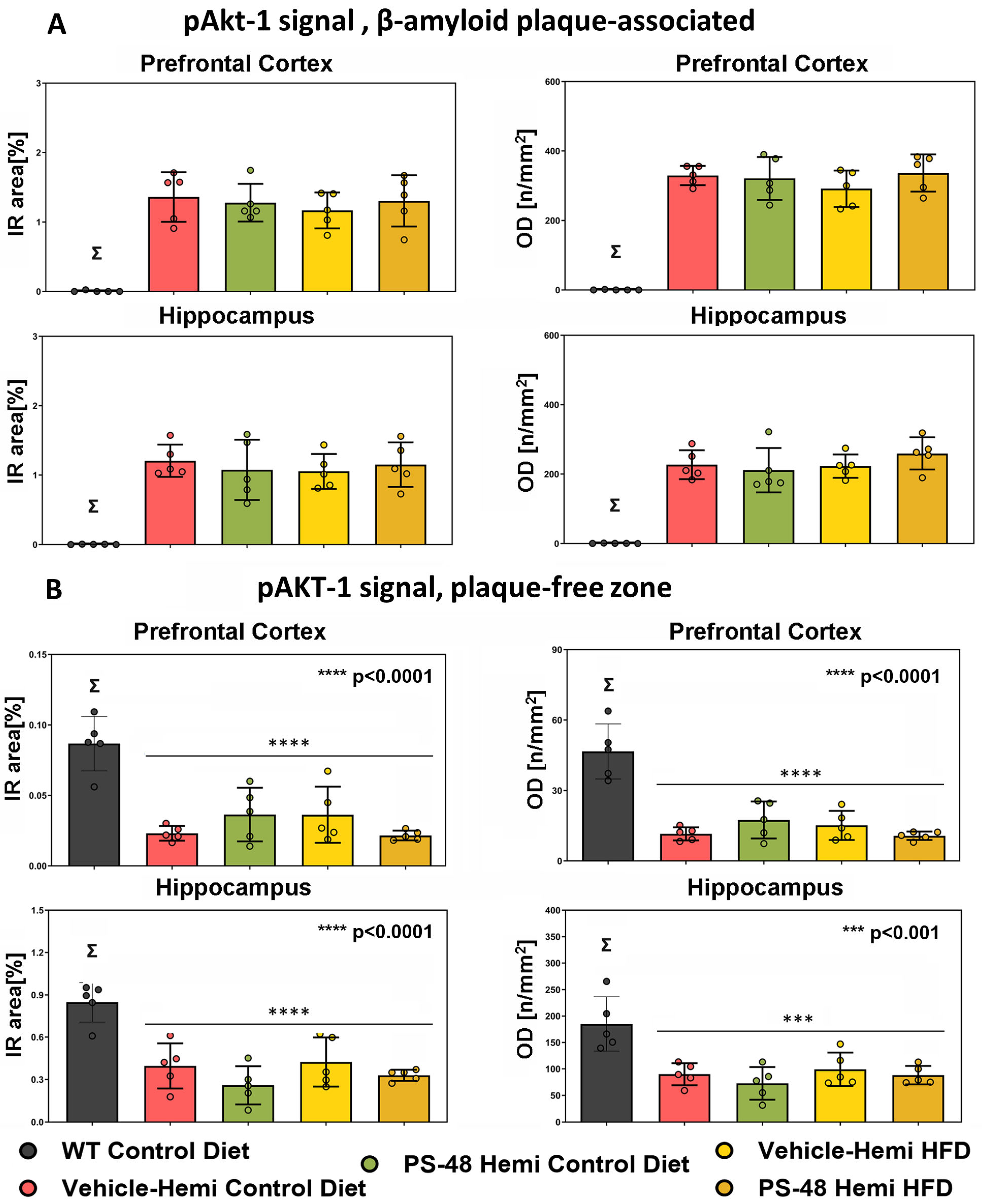
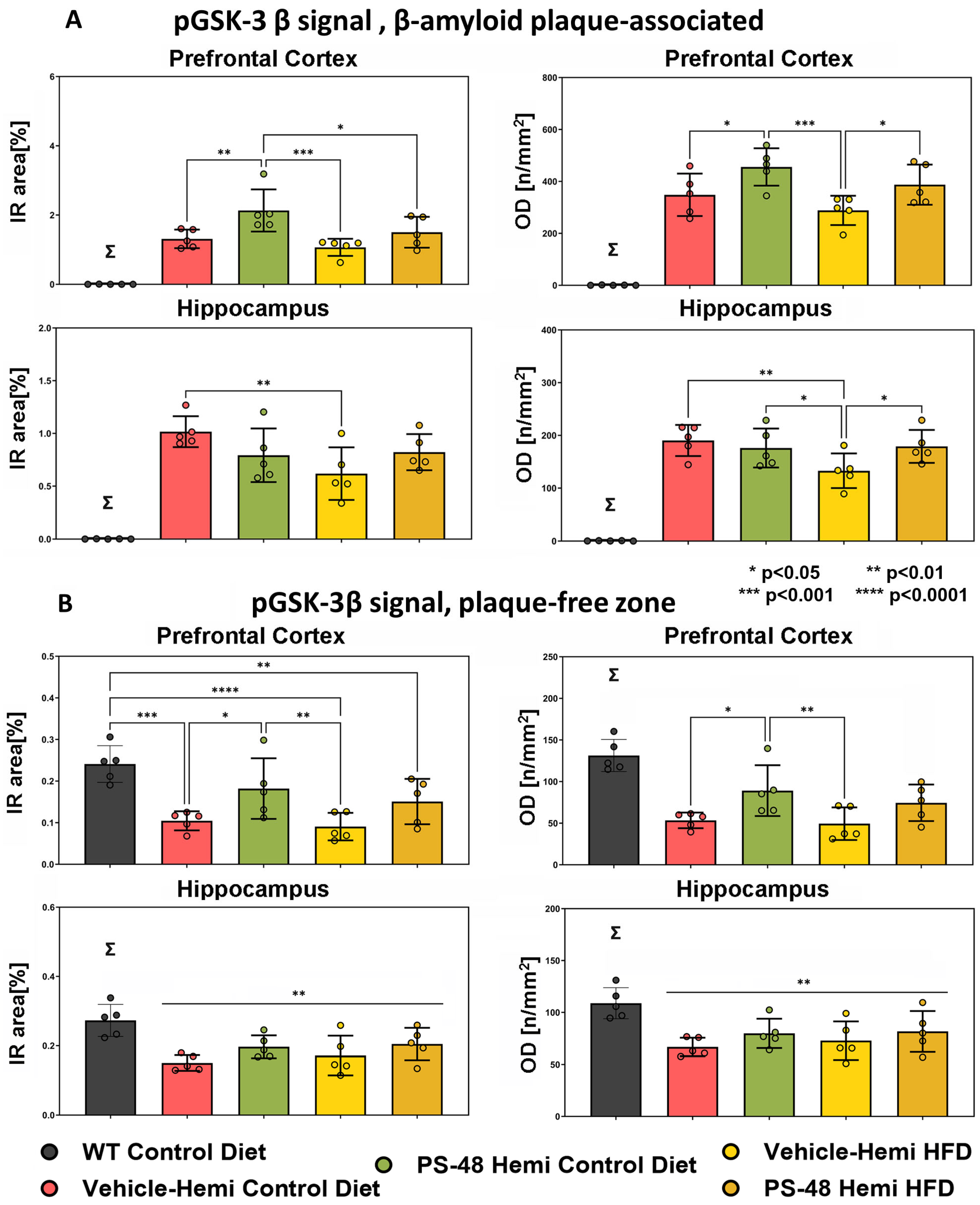
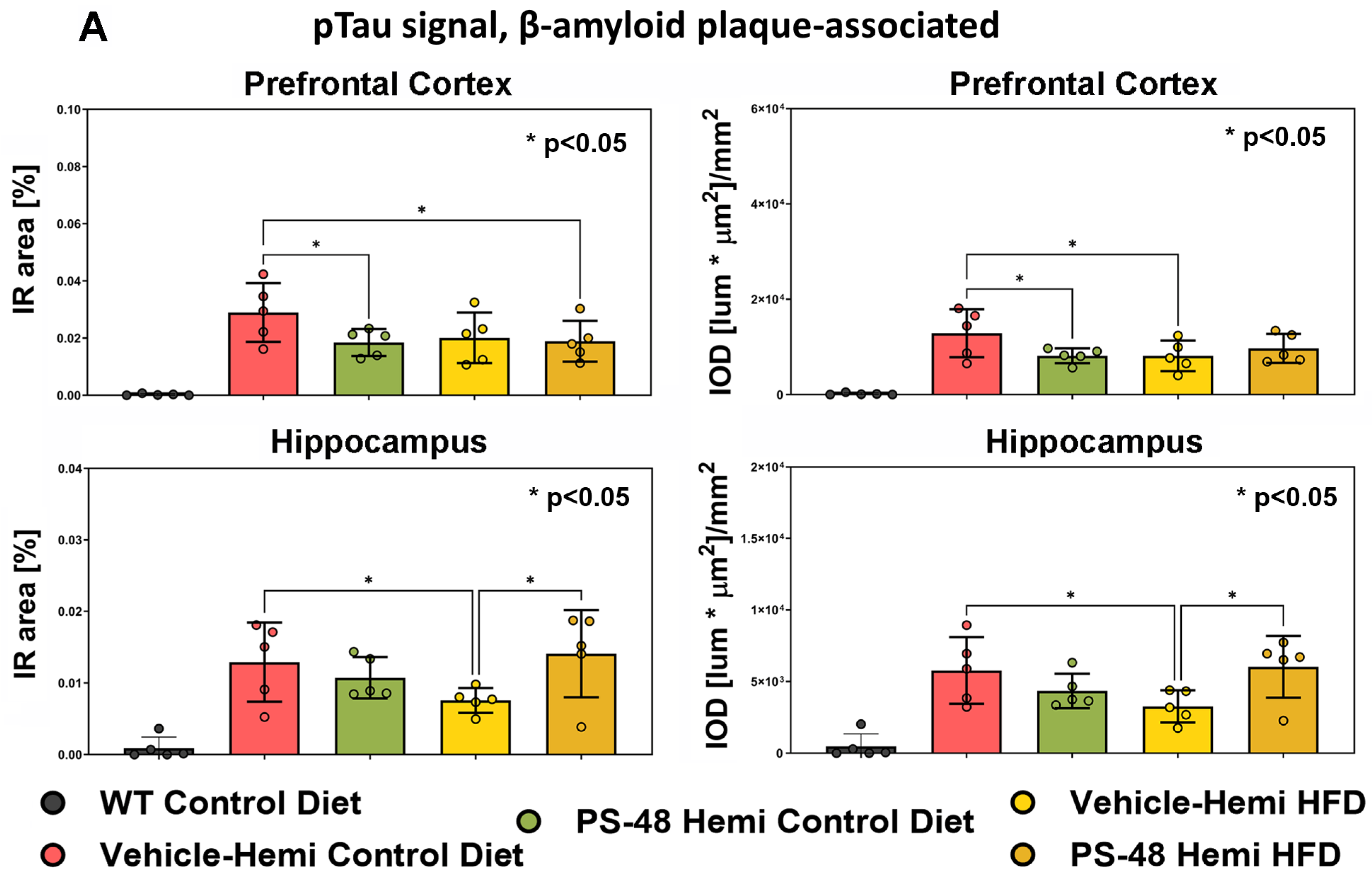
2.3. Immunohistochemistry: Phospho-Akt, -GSK-3β, and -Tau
2.4. β-Amyloid and PS48 Level Quantification
3. Discussion
4. Methods
4.1. Animals and Oral Drug Dosing
4.2. Brain Lysates
4.3. Western Blot Analysis
4.4. Akt1 Activity Assay
4.5. Histology and Immunofluorescence
4.6. Quantification
- Mean signal (lumens);
- Immunoreactive surface area (IR, noted as % of ROI size);
- Object density (OD, noted as number of objects per mm2);
- Relative integrated optical density (IOD, noted as lumen × μm2/mm2).
4.7. β-Amyloid Levels (ELISA)
4.8. PS48 Levels (MS-MS)
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Hyperinsulinemia and risk of Alzheimer disease. Neurology 2004, 63, 1187–1192. [Google Scholar] [PubMed]
- Patterson, C.; Feightner, J.; Garcia, A.; MacKnight, C. Primary prevention of dementia. Alzheimers Dement. 2007, 3, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a risk factor for dementia and mild cognitive impairment: A meta-analysis of longitudinal studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Ashford, J.W. APOE genotype effects on Alzheimer’s disease onset and epidemiology. J. Mol. Neurosci. 2004, 23, 157–165. [Google Scholar] [CrossRef]
- Schipper, H.M. Apolipoprotein E: Implications for AD neurobiology, epidemiology and risk assessment. Neurobiol. Aging 2011, 32, 778–790. [Google Scholar] [CrossRef]
- Peila, R.; Rodriguez, B.L.; Launer, L.J.; Honolulu-Asia Aging, S. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef]
- Ronnemaa, E.; Zethelius, B.; Sundelof, J.; Sundstrom, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008, 71, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Wang, H.Y.; Capuano, A.W.; Khan, A.; Taib, B.; Anokye-Danso, F.; Schneider, J.A.; Bennett, D.A.; Ahima, R.S.; Arnold, S.E. Brain Insulin Signaling, Alzheimer Disease Pathology, and Cognitive Function. Ann. Neurol. 2020, 88, 513–525. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Liao, Y.; Hung, M.C. Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2010, 2, 19–42. [Google Scholar]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Di, L.J. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 2022, 42, 946–982. [Google Scholar] [CrossRef]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Sayas, C.L.; Avila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Moller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Braendgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Meloni, A.R.; DeYoung, M.B.; Lowe, C.; Parkes, D.G. GLP-1 receptor activated insulin secretion from pancreatic beta-cells: Mechanism and glucose dependence. Diabetes Obes. Metab. 2013, 15, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Hummel, J.; Wagner, R.; Dannecker, C.; Vosseler, A.; Fritsche, L.; Veit, R.; Kantartzis, K.; Machann, J.; Birkenfeld, A.L.; et al. Empagliflozin Improves Insulin Sensitivity of the Hypothalamus in Humans With Prediabetes: A Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial. Diabetes Care 2022, 45, 398–406. [Google Scholar] [CrossRef]
- Heneka, M.T.; Fink, A.; Doblhammer, G. Effect of pioglitazone medication on the incidence of dementia. Ann. Neurol. 2015, 78, 284–294. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef]
- Cukierman-Yaffe, T.; Gerstein, H.C.; Colhoun, H.M.; Diaz, R.; Garcia-Perez, L.E.; Lakshmanan, M.; Bethel, A.; Xavier, D.; Probstfield, J.; Riddle, M.C.; et al. Effect of dulaglutide on cognitive impairment in type 2 diabetes: An exploratory analysis of the REWIND trial. Lancet Neurol. 2020, 19, 582–590. [Google Scholar] [CrossRef]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar]
- O’ Neill, C. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 2013, 48, 647–653. [Google Scholar] [CrossRef]
- Bhaskar, K.; Miller, M.; Chludzinski, A.; Herrup, K.; Zagorski, M.; Lamb, B.T. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol. Neurodegener. 2009, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.J.; Khatoon, S.; An, W.L.; Nordlinder, M.; Tanaka, T.; Braak, H.; Tsujio, I.; Takeda, M.; Alafuzoff, I.; Winblad, B.; et al. Role of protein kinase B in Alzheimer’s neurofibrillary pathology. Acta Neuropathol. 2003, 105, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Hoeffer, C.A.; Capetillo-Zarate, E.; Yu, F.; Wong, H.; Lin, M.T.; Tampellini, D.; Klann, E.; Blitzer, R.D.; Gouras, G.K. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e12845. [Google Scholar] [CrossRef]
- Ahmad, F.; Singh, K.; Das, D.; Gowaikar, R.; Shaw, E.; Ramachandran, A.; Rupanagudi, K.V.; Kommaddi, R.P.; Bennett, D.A.; Ravindranath, V. Reactive Oxygen Species-Mediated Loss of Synaptic Akt1 Signaling Leads to Deficient Activity-Dependent Protein Translation Early in Alzheimer’s Disease. Antioxid. Redox Signal. 2017, 27, 1269–1280. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar]
- Lee, H.K.; Kumar, P.; Fu, Q.; Rosen, K.M.; Querfurth, H.W. The insulin/Akt signaling pathway is targeted by intracellular beta-amyloid. Mol. Biol. Cell 2009, 20, 1533–1544. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef]
- Hindie, V.; Stroba, A.; Zhang, H.; Lopez-Garcia, L.A.; Idrissova, L.; Zeuzem, S.; Hirschberg, D.; Schaeffer, F.; Jorgensen, T.J.; Engel, M.; et al. Structure and allosteric effects of low-molecular-weight activators on the protein kinase PDK1. Nat. Chem. Biol. 2009, 5, 758–764. [Google Scholar] [CrossRef]
- Leroux, A.E.; Schulze, J.O.; Biondi, R.M. AGC kinases, mechanisms of regulation and innovative drug development. Semin. Cancer Biol. 2018, 48, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, V.; Ouyang, W.; Wei, H.; Soto, N.; Lazorchak, A.; Gould, C.; Lowry, C.; Newton, A.C.; Mao, Y.; Miao, R.Q.; et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008, 27, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.; Marshall, J.; Parang, K.; Rioult-Pedotti, M.S.; Tiwari, R.; Kwon, B.; Reisinger, S.; Lee, H.K. A PDK-1 allosteric agonist neutralizes insulin signaling derangements and beta-amyloid toxicity in neuronal cells and in vitro. PLoS ONE 2022, 17, e0261696. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.; Slitt, A.; DiCamillo, A.; Surles, N.; DeBoef, B.; Lee, H.K. A PDK-1 allosteric agonist improves spatial learning and memory in a betaAPP/PS-1 transgenic mouse-high fat diet intervention model of Alzheimer’s disease. Behav. Brain Res. 2023, 438, 114183. [Google Scholar] [CrossRef]
- Kimura, T.; Sharma, G.; Ishiguro, K.; Hisanaga, S.I. Phospho-Tau Bar Code: Analysis of Phosphoisotypes of Tau and Its Application to Tauopathy. Front. Neurosci. 2018, 12, 44. [Google Scholar] [CrossRef]
- Stroba, A.; Schaeffer, F.; Hindie, V.; Lopez-Garcia, L.; Adrian, I.; Frohner, W.; Hartmann, R.W.; Biondi, R.M.; Engel, M. 3,5-Diphenylpent-2-enoic acids as allosteric activators of the protein kinase PDK1: Structure-activity relationships and thermodynamic characterization of binding as paradigms for PIF-binding pocket-targeting compounds. J. Med. Chem. 2009, 52, 4683–4693. [Google Scholar] [CrossRef]
- Biondi, R.M.; Kieloch, A.; Currie, R.A.; Deak, M.; Alessi, D.R. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001, 20, 4380–4390. [Google Scholar] [CrossRef]
- Biondi, R.M.; Cheung, P.C.; Casamayor, A.; Deak, M.; Currie, R.A.; Alessi, D.R. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 2000, 19, 979–988. [Google Scholar] [CrossRef]
- Alessi, D.R. Discovery of PDK1, one of the missing links in insulin signal transduction. Colworth Medal Lecture. Biochem. Soc. Trans. 2001, 29, 1–14. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Zhou, H.; Wei, W.; Ambasudhan, R.; Lin, T.; Kim, J.; Zhang, K.; Ding, S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 2010, 7, 651–655. [Google Scholar] [CrossRef]
- Cuesto, G.; Enriquez-Barreto, L.; Carames, C.; Cantarero, M.; Gasull, X.; Sandi, C.; Ferrus, A.; Acebes, A.; Morales, M. Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J. Neurosci. 2011, 31, 2721–2733. [Google Scholar] [CrossRef] [PubMed]
- Joshi, H.; Bharath, S.; Balachandar, R.; Sadanand, S.; Vishwakarma, H.V.; Aiyappan, S.; Saini, J.; Kumar, K.J.; John, J.P.; Varghese, M. Differentiation of Early Alzheimer’s Disease, Mild Cognitive Impairment, and Cognitively Healthy Elderly Samples Using Multimodal Neuroimaging Indices. Brain Connect. 2019, 9, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lu, H.; Metmer, H.; Li, W.X.Y.; Lu, J. Evaluating functional connectivity of executive control network and frontoparietal network in Alzheimer’s disease. Brain Res. 2018, 1678, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Mercado-Gomez, O.; Hernandez-Fonseca, K.; Villavicencio-Queijeiro, A.; Massieu, L.; Chimal-Monroy, J.; Arias, C. Inhibition of Wnt and PI3K signaling modulates GSK-3beta activity and induces morphological changes in cortical neurons: Role of tau phosphorylation. Neurochem. Res. 2008, 33, 1599–1609. [Google Scholar] [CrossRef]
- Lee, C.W.; Lau, K.F.; Miller, C.C.; Shaw, P.C. Glycogen synthase kinase-3 beta-mediated tau phosphorylation in cultured cell lines. Neuroreport 2003, 14, 257–260. [Google Scholar] [CrossRef]
- Ksiezak-Reding, H.; Pyo, H.K.; Feinstein, B.; Pasinetti, G.M. Akt/PKB kinase phosphorylates separately Thr212 and Ser214 of tau protein in vitro. Biochim. Biophys. Acta 2003, 1639, 159–168. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Ortiz-Barajas, O.; Gamero-Carrasco, C.; de la Rosa, P.R.; Infante-Garcia, C.; Zopeque-Garcia, N.; Lechuga-Sancho, A.M.; Garcia-Alloza, M. Prediabetes-induced vascular alterations exacerbate central pathology in APPswe/PS1dE9 mice. Psychoneuroendocrinology 2014, 48, 123–135. [Google Scholar] [CrossRef]
- Walker, J.M.; Dixit, S.; Saulsberry, A.C.; May, J.M.; Harrison, F.E. Reversal of high fat diet-induced obesity improves glucose tolerance, inflammatory response, beta-amyloid accumulation and cognitive decline in the APP/PSEN1 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2017, 100, 87–98. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.; Shi, J.; Yin, D.; El-Akkad, E.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. PKA modulates GSK-3beta- and cdk5-catalyzed phosphorylation of tau in site- and kinase-specific manners. FEBS Lett. 2006, 580, 6269–6274. [Google Scholar] [CrossRef]
- Sengupta, A.; Kabat, J.; Novak, M.; Wu, Q.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998, 357, 299–309. [Google Scholar] [CrossRef]
- Cho, J.H.; Johnson, G.V. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J. Biol. Chem. 2003, 278, 187–193. [Google Scholar] [CrossRef]
- Liu, F.; Li, B.; Tung, E.J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 2007, 26, 3429–3436. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Johnson, G.V. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J. Neurochem. 2004, 88, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, M.; Kadavath, H.; Biernat, J.; Ozenne, V.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Structural Impact of Tau Phosphorylation at Threonine 231. Structure 2015, 23, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 52. [Google Scholar] [CrossRef]
- Quartey, M.O.; Nyarko, J.N.K.; Maley, J.M.; Barnes, J.R.; Bolanos, M.A.C.; Heistad, R.M.; Knudsen, K.J.; Pennington, P.R.; Buttigieg, J.; De Carvalho, C.E.; et al. The Abeta(1-38) peptide is a negative regulator of the Abeta(1-42) peptide implicated in Alzheimer disease progression. Sci. Rep. 2021, 11, 431. [Google Scholar] [CrossRef]
- Cullen, N.; Janelidze, S.; Palmqvist, S.; Stomrud, E.; Mattsson-Carlgren, N.; Hansson, O.; Alzheimer’s Disease Neuroimaging, I. Association of CSF Abeta(38) Levels With Risk of Alzheimer Disease-Related Decline. Neurology 2022, 98, e958–e967. [Google Scholar] [CrossRef]
- Theriault, P.; ElAli, A.; Rivest, S. High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar] [CrossRef]
- Elhaik Goldman, S.; Goez, D.; Last, D.; Naor, S.; Liraz Zaltsman, S.; Sharvit-Ginon, I.; Atrakchi-Baranes, D.; Shemesh, C.; Twitto-Greenberg, R.; Tsach, S.; et al. High-fat diet protects the blood-brain barrier in an Alzheimer’s disease mouse model. Aging Cell 2018, 17, e12818. [Google Scholar] [CrossRef]
- Malm, T.M.; Iivonen, H.; Goldsteins, G.; Keksa-Goldsteine, V.; Ahtoniemi, T.; Kanninen, K.; Salminen, A.; Auriola, S.; Van Groen, T.; Tanila, H.; et al. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. J. Neurosci. 2007, 27, 3712–3721. [Google Scholar] [CrossRef]
- Yi, J.H.; Baek, S.J.; Heo, S.; Park, H.J.; Kwon, H.; Lee, S.; Jung, J.; Park, S.J.; Kim, B.C.; Lee, Y.C.; et al. Direct pharmacological Akt activation rescues Alzheimer’s disease like memory impairments and aberrant synaptic plasticity. Neuropharmacology 2018, 128, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Mondal, S.; Tan, D.; Nagata, E.; Takizawa, S.; Sharma, A.K.; Hou, Q.; Shanmugasundaram, K.; Prasad, A.; Tung, J.K.; et al. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc. Natl. Acad. Sci. USA 2012, 109, 10581–10586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Y.; Li, H.; Xiong, R.; Zhao, Z.; Chu, X.; Li, Q.; Sun, S.; Chen, S. Neuroprotective effects of salidroside through PI3K/Akt pathway activation in Alzheimer’s disease models. Drug Des. Devel Ther. 2016, 10, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Chen, L.; Wang, Z.; He, Y.; Ruganzu, J.B.; Guo, H.; Zhang, X.; Ji, S.; Zheng, L.; Yang, W. Tanshinone IIA regulates glycogen synthase kinase-3beta-related signaling pathway and ameliorates memory impairment in APP/PS1 transgenic mice. Eur. J. Pharmacol. 2022, 918, 174772. [Google Scholar] [CrossRef]
- Giunti, E.; Collu, R.; Daley, S.; Querfurth, H.; Morin, P.; Killick, R.; Melamed, R.D.; Xia, W. Reduction of Phosphorylated Tau in Alzheimer’s Disease Induced Pluripotent Stem Cell-Derived Neuro-Spheroids by Rho-Associated Coiled-Coil Kinase Inhibitor Fasudil. J. Alzheimers Dis. 2023, 96, 1695–1709. [Google Scholar] [CrossRef]
- Long, H.Z.; Cheng, Y.; Zhou, Z.W.; Luo, H.Y.; Wen, D.D.; Gao, L.C. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer’s Disease and Parkinson’s Disease. Front. Pharmacol. 2021, 12, 648636. [Google Scholar] [CrossRef]
- Oomura, Y.; Hori, N.; Shiraishi, T.; Fukunaga, K.; Takeda, H.; Tsuji, M.; Matsumiya, T.; Ishibashi, M.; Aou, S.; Li, X.L.; et al. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides 2006, 27, 2738–2749. [Google Scholar] [CrossRef]
- Nakagami, Y. Inhibitors beta-amyloid-induced toxicity by modulating the Akt signaling pathway. Drug News Perspect. 2004, 17, 655–660. [Google Scholar] [CrossRef]
- Kumar, H.B.; Manandhar, S.; Rathi, E.; Kabekkodu, S.P.; Mehta, C.H.; Nayak, U.Y.; Kini, S.G.; Pai, K.S.R. Identification of potential Akt activators: A ligand and structure-based computational approach. Mol. Divers. 2024, 28, 1485–1503. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Dietary regulation of PI3K/AKT/GSK-3beta pathway in Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 35. [Google Scholar] [CrossRef]
- Mordhorst, B.R.; Kerns, K.C.; Schauflinger, M.; Zigo, M.; Murphy, S.L.; Ross, R.M.; Wells, K.D.; Green, J.A.; Sutovsky, P.; Prather, R.S. Pharmacologic treatment with CPI-613 and PS48 decreases mitochondrial membrane potential and increases quantity of autolysosomes in porcine fibroblasts. Sci. Rep. 2019, 9, 9417. [Google Scholar] [CrossRef]
- Spate, L.D.; Brown, A.; Redel, B.K.; Whitworth, K.M.; Prather, R.S. PS48 can replace bovine serum albumin in pig embryo culture medium, and improve in vitro embryo development by phosphorylating AKT. Mol. Reprod. Dev. 2015, 82, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Apostolova, L.; Rabinovici, G.D.; Atri, A.; Aisen, P.; Greenberg, S.; Hendrix, S.; Selkoe, D.; Weiner, M.; Petersen, R.C.; et al. Lecanemab: Appropriate Use Recommendations. J. Prev. Alzheimers Dis. 2023, 10, 362–377. [Google Scholar] [CrossRef]
- Ebell, M.H.; Barry, H.C.; Baduni, K.; Grasso, G. Clinically Important Benefits and Harms of Monoclonal Antibodies Targeting Amyloid for the Treatment of Alzheimer Disease: A Systematic Review and Meta-Analysis. Ann. Fam. Med. 2024, 22, 50–62. [Google Scholar] [CrossRef]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Fu, Q.; Ni, D.; Zhang, J.; Li, X.; Lu, S. The chemical diversity and structure-based discovery of allosteric modulators for the PIF-pocket of protein kinase PDK1. J. Enzym. Inhib. Med. Chem. 2019, 34, 361–374. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef]
- Onos, K.D.; Sukoff Rizzo, S.J.; Howell, G.R.; Sasner, M. Toward more predictive genetic mouse models of Alzheimer’s disease. Brain Res. Bull. 2016, 122, 1–11. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group no. | Group Label (Genotype-Diet-Drug) | Final n | Diet Intervention at 2 mo. | Oral Drug or Vehicle at 10 mo. | Behavioral Testing 12 to 14 mo. | Study Terminated 14 mo. n for Westerns |
|---|---|---|---|---|---|---|
| 1 | WT-SD | 8 | standard (SD) | - | 5 | |
| 2 | TG-SD-V | 8 | standard (SD) | vehicle (V) | 5 | |
| 3 | TG-SD-PS48 | 8 | standard (SD) | PS48 | 5 | |
| 4 | TG-HFD-V | 6 | high fat (HFD) | vehicle (V) | 5 | |
| 5 | TG-HFD-PS48 | 6 | high fat (HFD) | PS48 | 5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Querfurth, H.W.; Lemere, C.; Ciola, J.; Havas, D.; Xia, W.; Lee, H.K. Target Validation Studies of PS48, a PDK-1 Allosteric Agonist, for the Treatment of Alzheimer’s Disease Phenotype in APP/PS1 Transgenic Mice. Int. J. Mol. Sci. 2025, 26, 3473. https://doi.org/10.3390/ijms26083473
Querfurth HW, Lemere C, Ciola J, Havas D, Xia W, Lee HK. Target Validation Studies of PS48, a PDK-1 Allosteric Agonist, for the Treatment of Alzheimer’s Disease Phenotype in APP/PS1 Transgenic Mice. International Journal of Molecular Sciences. 2025; 26(8):3473. https://doi.org/10.3390/ijms26083473
Chicago/Turabian StyleQuerfurth, Henry W., Cynthia Lemere, Jason Ciola, Daniel Havas, Weiming Xia, and Han Kyu Lee. 2025. "Target Validation Studies of PS48, a PDK-1 Allosteric Agonist, for the Treatment of Alzheimer’s Disease Phenotype in APP/PS1 Transgenic Mice" International Journal of Molecular Sciences 26, no. 8: 3473. https://doi.org/10.3390/ijms26083473
APA StyleQuerfurth, H. W., Lemere, C., Ciola, J., Havas, D., Xia, W., & Lee, H. K. (2025). Target Validation Studies of PS48, a PDK-1 Allosteric Agonist, for the Treatment of Alzheimer’s Disease Phenotype in APP/PS1 Transgenic Mice. International Journal of Molecular Sciences, 26(8), 3473. https://doi.org/10.3390/ijms26083473