
Trophoblast Fusion in Hypertensive Disorders of Pregnancy and Preeclampsia



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Placental Materno-Fetal Interface
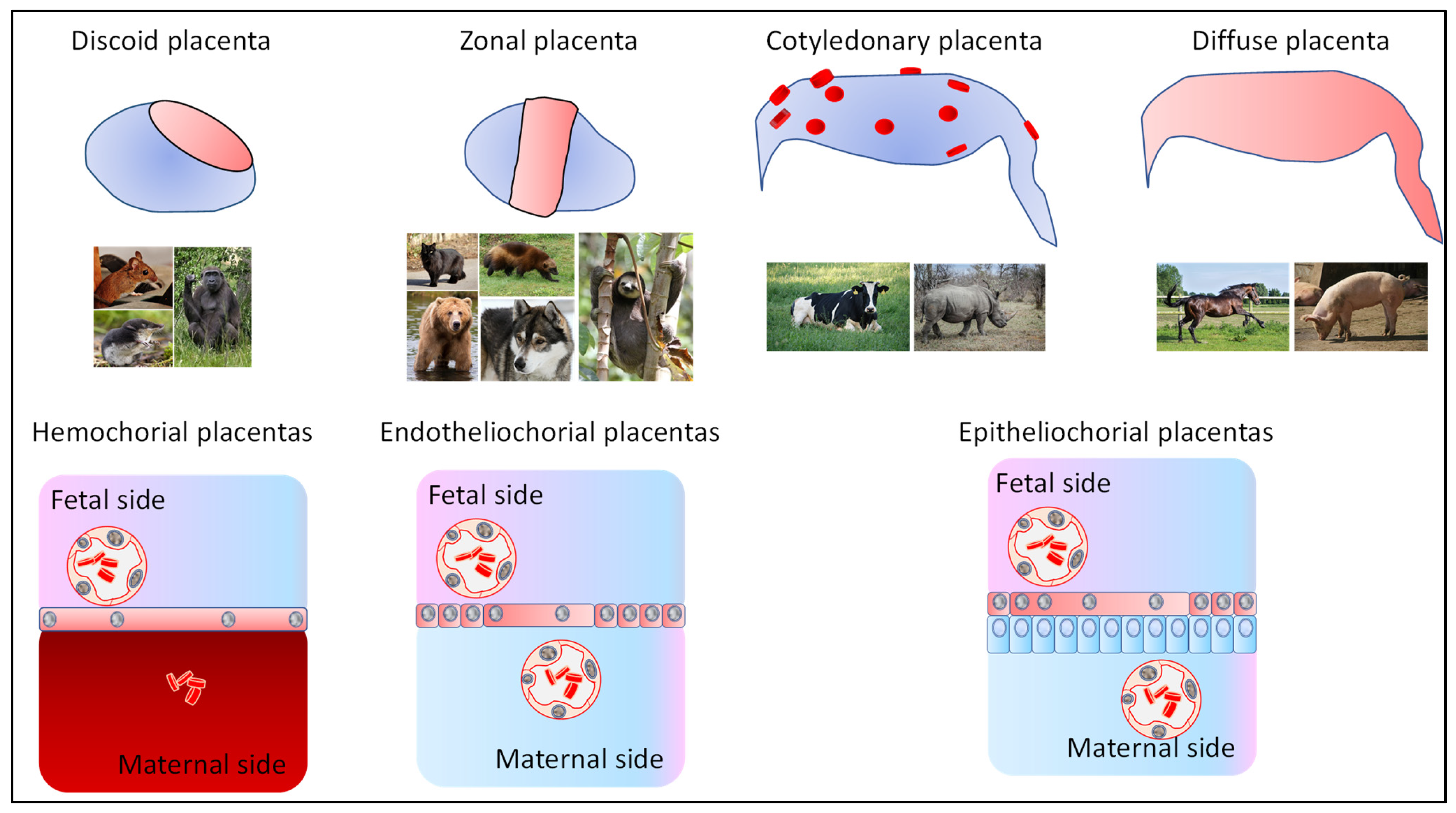
1.1. Various Mammalian Placentas
1.2. Placental Development
1.3. Syncytialization Is Systematically Present in the Various Placentas Throughout Evolution
1.4. How Do We Study the Syncytialization Process?
1.5. SCT Heterogeneity and Life Cycle
2. Trophoblast Fusion in Diseases of Placental Origin
2.1. Hypoxia and Fusion
2.2. Apoptosis and Fusion
2.3. Epigenetic Regulation of Fusion in the Context of Placental Disease
2.4. Direct Effects of Fusion
3. Estimating the Importance of the Balance of Trophoblast Renewal/Fusion in Placental Diseases Is a Challenge
4. Conclusions
Funding
Conflicts of Interest
References
- Zimmermann, W.; Kammerer, R. The immune-modulating pregnancy-specific glycoproteins evolve rapidly and their presence correlates with hemochorial placentation in primates. BMC Genom. 2021, 22, 128. [Google Scholar]
- Motomura, K.; Hara, M.; Ito, I.; Morita, H.; Matsumoto, K. Roles of human trophoblasts’ pattern recognition receptors in host defense and pregnancy complications. J. Reprod. Immunol. 2023, 156, 103811. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.H.; Murphy, H.A.; Chapman, H.; George, E.M. Syncytialization alters the extracellular matrix and barrier function of placental trophoblasts. Am. J. Physiol. Cell Physiol. 2021, 321, C694–C703. [Google Scholar] [PubMed]
- Turco, M.Y.; Moffett, A. Development of the human placenta. Development 2019, 146, dev163428. [Google Scholar]
- Stewart, J.R.; Blackburn, D.G. Reptilian Placentation: Structural Diversity and Terminology. Copeia 1988, 4, 839–852. [Google Scholar]
- Cornelis, G.; Funk, M.; Vernochet, C.; Leal, F.; Tarazona, O.A.; Meurice, G.; Heidmann, O.; Dupressoir, A.; Miralles, A.; Ramirez-Pinilla, M.P.; et al. An endogenous retroviral envelope syncytin and its cognate receptor identified in the viviparous placental Mabuya lizard. Proc. Natl. Acad. Sci. USA 2017, 114, E10991–E11000. [Google Scholar]
- Foster, C.S.P.; Van Dyke, J.U.; Thompson, M.B.; Smith, N.M.A.; Simpfendorfer, C.A.; Murphy, C.R.; Whittington, C.M. Different Genes are Recruited During Convergent Evolution of Pregnancy and the Placenta. Mol. Biol. Evol. 2022, 39, msac077. [Google Scholar]
- Blaise, S.; de Parseval, N.; Benit, L.; Heidmann, T. Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13013–13018. [Google Scholar] [CrossRef]
- Dupressoir, A.; Marceau, G.; Vernochet, C.; Benit, L.; Kanellopoulos, C.; Sapin, V.; Heidmann, T. Syncytin-A and syncytin-B, two fusogenic placenta-specific murine envelope genes of retroviral origin conserved in Muridae. Proc. Natl. Acad. Sci. USA 2005, 102, 725–730. [Google Scholar]
- Cornelis, G.; Heidmann, O.; Bernard-Stoecklin, S.; Reynaud, K.; Veron, G.; Mulot, B.; Dupressoir, A.; Heidmann, T. Ancestral capture of syncytin-Car1, a fusogenic endogenous retroviral envelope gene involved in placentation and conserved in Carnivora. Proc. Natl. Acad. Sci. USA 2012, 109, E432–E441. [Google Scholar]
- Cornelis, G.; Heidmann, O.; Degrelle, S.A.; Vernochet, C.; Lavialle, C.; Letzelter, C.; Bernard-Stoecklin, S.; Hassanin, A.; Mulot, B.; Guillomot, M.; et al. Captured retroviral envelope syncytin gene associated with the unique placental structure of higher ruminants. Proc. Natl. Acad. Sci. USA 2013, 110, E828–E837. [Google Scholar] [CrossRef]
- Cornelis, G.; Vernochet, C.; Malicorne, S.; Souquere, S.; Tzika, A.C.; Goodman, S.M.; Catzeflis, F.; Robinson, T.J.; Milinkovitch, M.C.; Pierron, G.; et al. Retroviral envelope syncytin capture in an ancestrally diverged mammalian clade for placentation in the primitive Afrotherian tenrecs. Proc. Natl. Acad. Sci. USA 2014, 111, E4332–E4341. [Google Scholar] [CrossRef]
- Cornelis, G.; Vernochet, C.; Carradec, Q.; Souquere, S.; Mulot, B.; Catzeflis, F.; Nilsson, M.A.; Menzies, B.R.; Renfree, M.B.; Pierron, G.; et al. Retroviral envelope gene captures and syncytin exaptation for placentation in marsupials. Proc. Natl. Acad. Sci. USA 2015, 112, E487–E496. [Google Scholar] [CrossRef]
- Shao, H.; Gao, S.; Ying, X.; Zhu, X.; Hua, Y. Expression and Regulation of Aquaporins in Pregnancy Complications and Reproductive Dysfunctions. DNA Cell Biol. 2021, 40, 116–125. [Google Scholar] [CrossRef]
- Chung, J.J.; Huber, T.B.; Godel, M.; Jarad, G.; Hartleben, B.; Kwoh, C.; Keil, A.; Karpitskiy, A.; Hu, J.; Huh, C.J.; et al. Albumin-associated free fatty acids induce macropinocytosis in podocytes. J. Clin. Investig. 2015, 125, 2307–2316. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Buffat, C.; Mondon, F.; Rigourd, V.; Boubred, F.; Bessieres, B.; Fayol, L.; Feuerstein, J.M.; Gamerre, M.; Jammes, H.; Rebourcet, R.; et al. A hierarchical analysis of transcriptome alterations in intrauterine growth restriction (IUGR) reveals common pathophysiological pathways in mammals. J. Pathol. 2007, 213, 337–346. [Google Scholar] [CrossRef]
- Mparmpakas, D.; Zachariades, E.; Foster, H.; Kara, A.; Harvey, A.; Goumenou, A.; Karteris, E. Expression of mTOR and downstream signalling components in the JEG-3 and BeWo human placental choriocarcinoma cell lines. Int. J. Mol. Med. 2010, 25, 65–69. [Google Scholar]
- Yoshikawa, K.; Umekawa, T.; Maki, S.; Kubo, M.; Nii, M.; Tanaka, K.; Tanaka, H.; Osato, K.; Kamimoto, Y.; Kondo, E.; et al. Tadalafil Improves L-NG-Nitroarginine Methyl Ester-Induced Preeclampsia With Fetal Growth Restriction-Like Symptoms in Pregnant Mice. Am. J. Hypertens. 2017, 31, 89–96. [Google Scholar] [CrossRef]
- Tanaka, K.; Tanaka, H.; Tachibana, R.; Yoshikawa, K.; Kawamura, T.; Takakura, S.; Takeuchi, H.; Ikeda, T. Tadalafil Treatment of Mice with Fetal Growth Restriction and Preeclampsia Improves Placental mTOR Signaling. Int. J. Mol. Sci. 2022, 23, 1474. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.H.; Wang, Y.X.; Liu, T.H. A comprehensive review of human trophoblast fusion models: Recent developments and challenges. Cell Death Discov. 2023, 9, 372. [Google Scholar]
- Wice, B.; Menton, D.; Geuze, H.; Schwartz, A.L. Modulators of cyclic AMP metabolism induce syncytiotrophoblast formation in vitro. Exp. Cell Res. 1990, 186, 306–316. [Google Scholar] [CrossRef]
- Borges, M.; Bose, P.; Frank, H.G.; Kaufmann, P.; Potgens, A.J. A two-colour fluorescence assay for the measurement of syncytial fusion between trophoblast-derived cell lines. Placenta 2003, 24, 959–964. [Google Scholar] [CrossRef]
- Rothbauer, M.; Patel, N.; Gondola, H.; Siwetz, M.; Huppertz, B.; Ertl, P. A comparative study of five physiological key parameters between four different human trophoblast-derived cell lines. Sci. Rep. 2017, 7, 5892. [Google Scholar] [CrossRef]
- Jaremek, A.; Jeyarajah, M.J.; Jaju Bhattad, G.; Renaud, S.J. Omics Approaches to Study Formation and Function of Human Placental Syncytiotrophoblast. Front. Cell Dev. Biol. 2021, 9, 674162. [Google Scholar] [CrossRef]
- Ounadjela, J.R.; Zhang, K.; Kobayashi-Kirschvink, K.J.; Jin, K.; Russell, A.J.C.; Lackner, A.I.; Callahan, C.; Viggiani, F.; Dey, K.K.; Jagadeesh, K.; et al. Spatial multiomic landscape of the human placenta at molecular resolution. Nat. Med. 2024, 30, 3495–3508. [Google Scholar]
- Garcia-Flores, V.; Miller, D.; Galaz, J.; Gomez-Lopez, N. Dissociation of Placental Tissues for Single-Cell Techniques. Methods Mol. Biol. 2024, 2781, 143–154. [Google Scholar]
- Tsang, J.C.H.; Vong, J.S.L.; Ji, L.; Poon, L.C.Y.; Jiang, P.; Lui, K.O.; Ni, Y.B.; To, K.F.; Cheng, Y.K.Y.; Chiu, R.W.K.; et al. Integrative single-cell and cell-free plasma RNA transcriptomics elucidates placental cellular dynamics. Proc. Natl. Acad. Sci. USA 2017, 114, E7786–E7795. [Google Scholar]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polanski, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar]
- Parameshwar, P.K.; Sagrillo-Fagundes, L.; Fournier, C.; Girard, S.; Vaillancourt, C.; Moraes, C. Disease-specific extracellular matrix composition regulates placental trophoblast fusion efficiency. Biomater. Sci. 2021, 9, 7247–7256. [Google Scholar]
- Zhang, Y.; Le, T.; Grabau, R.; Mohseni, Z.; Kim, H.; Natale, D.R.; Feng, L.; Pan, H.; Yang, H. TMEM16F phospholipid scramblase mediates trophoblast fusion and placental development. Sci. Adv. 2020, 6, eaba0310. [Google Scholar]
- Ochiai, Y.; Suzuki, C.; Segawa, K.; Uchiyama, Y.; Nagata, S. Inefficient development of syncytiotrophoblasts in the Atp11a-deficient mouse placenta. Proc. Natl. Acad. Sci. USA 2022, 119, e2200582119. [Google Scholar]
- Carmeille, R.; Degrelle, S.A.; Plawinski, L.; Bouvet, F.; Gounou, C.; Evain-Brion, D.; Brisson, A.R.; Bouter, A. Annexin-A5 promotes membrane resealing in human trophoblasts. Biochim. Biophys. Acta 2015, 1853, 2033–2044. [Google Scholar]
- Ducat, A.; Couderc, B.; Bouter, A.; Biquard, L.; Aouache, R.; Passet, B.; Doridot, L.; Cohen, M.B.; Ribaux, P.; Apicella, C.; et al. Molecular Mechanisms of Trophoblast Dysfunction Mediated by Imbalance between STOX1 Isoforms. iScience 2020, 23, 101086. [Google Scholar]
- Mansilla, M.; Wang, Y.; Lim, R.; Palmer, K.; Nie, G. HtrA4 is up-regulated during trophoblast syncytialization and BeWo cells fail to syncytialize without HtrA4. Sci. Rep. 2021, 11, 14363. [Google Scholar]
- Pidoux, G.; Gerbaud, P.; Gnidehou, S.; Grynberg, M.; Geneau, G.; Guibourdenche, J.; Carette, D.; Cronier, L.; Evain-Brion, D.; Malassine, A.; et al. ZO-1 is involved in trophoblastic cell differentiation in human placenta. Am. J. Physiol. Cell Physiol. 2010, 298, C1517–C1526. [Google Scholar] [CrossRef]
- Sivasubramaniyam, T.; Garcia, J.; Tagliaferro, A.; Melland-Smith, M.; Chauvin, S.; Post, M.; Todros, T.; Caniggia, I. Where polarity meets fusion: Role of Par6 in trophoblast differentiation during placental development and preeclampsia. Endocrinology 2013, 154, 1296–1309. [Google Scholar]
- Frendo, J.L.; Cronier, L.; Bertin, G.; Guibourdenche, J.; Vidaud, M.; Evain-Brion, D.; Malassine, A. Involvement of connexin 43 in human trophoblast cell fusion and differentiation. J. Cell Sci. 2003, 116 Pt 16, 3413–3421. [Google Scholar]
- Ng, Y.H.; Zhu, H.; Leung, P.C. Twist modulates human trophoblastic cell invasion via regulation of N-cadherin. Endocrinology 2012, 153, 925–936. [Google Scholar]
- Yamada, T.; Kojima, T.; Akaishi, R.; Ishikawa, S.; Takeda, M.; Kawaguchi, S.; Nishida, R.; Morikawa, M.; Yamada, T.; Minakami, H. Problems in methods for the detection of significant proteinuria in pregnancy. J. Obstet. Gynaecol. Res. 2014, 40, 161–166. [Google Scholar]
- Iwahashi, N.; Ikezaki, M.; Matsuzaki, I.; Yamamoto, M.; Toujima, S.; Murata, S.I.; Ihara, Y.; Ino, K. Calreticulin Regulates Syncytialization Through Control of the Synthesis and Transportation of E-Cadherin in BeWo Cells. Endocrinology 2019, 160, 359–374. [Google Scholar] [CrossRef]
- Aghababaei, M.; Beristain, A.G. The Elsevier Trophoblast Research Award Lecture: Importance of metzincin proteases in trophoblast biology and placental development: A focus on ADAM12. Placenta 2015, 36 (Suppl. 1), S11–S19. [Google Scholar] [CrossRef]
- Ajuzieogu, O.V.; Ezike, H.A.; Amucheazi, A.O.; Enwereji, J. A retrospective study of the outcome of cesarean section for women with severe pre-eclampsia in a third world setting. Saudi J. Anaesth. 2011, 5, 15–18. [Google Scholar] [CrossRef]
- Weber, M.; Gohner, C.; San Martin, S.; Vattai, A.; Hutter, S.; Parraga, M.; Jeschke, U.; Schleussner, E.; Markert, U.R.; Fitzgerald, J.S. Unique trophoblast stem cell- and pluripotency marker staining patterns depending on gestational age and placenta-associated pregnancy complications. Cell Adh. Migr. 2016, 10, 56–65. [Google Scholar] [CrossRef]
- Lu, X.; Wang, R.; Zhu, C.; Wang, H.; Lin, H.Y.; Gu, Y.; Cross, J.C.; Wang, H. Fine-Tuned and Cell-Cycle-Restricted Expression of Fusogenic Protein Syncytin-2 Maintains Functional Placental Syncytia. Cell Rep. 2017, 21, 1150–1159. [Google Scholar] [CrossRef]
- Renaud, S.J.; Chakraborty, D.; Mason, C.W.; Rumi, M.A.; Vivian, J.L.; Soares, M.J. OVO-like 1 regulates progenitor cell fate in human trophoblast development. Proc. Natl. Acad. Sci. USA 2015, 112, E6175–E6184. [Google Scholar] [CrossRef]
- Meinhardt, G.; Haider, S.; Kunihs, V.; Saleh, L.; Pollheimer, J.; Fiala, C.; Hetey, S.; Feher, Z.; Szilagyi, A.; Than, N.G.; et al. Pivotal role of the transcriptional co-activator YAP in trophoblast stemness of the developing human placenta. Proc. Natl. Acad. Sci. USA 2020, 117, 13562–13570. [Google Scholar] [CrossRef]
- Ray, S.; Saha, A.; Ghosh, A.; Roy, N.; Kumar, R.P.; Meinhardt, G.; Mukerjee, A.; Gunewardena, S.; Kumar, R.; Knofler, M.; et al. Hippo signaling cofactor, WWTR1, at the crossroads of human trophoblast progenitor self-renewal and differentiation. Proc. Natl. Acad. Sci. USA 2022, 119, e2204069119. [Google Scholar] [CrossRef]
- Hornbachner, R.; Lackner, A.; Papuchova, H.; Haider, S.; Knofler, M.; Mechtler, K.; Latos, P.A. MSX2 safeguards syncytiotrophoblast fate of human trophoblast stem cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2105130118. [Google Scholar] [CrossRef]
- Song, H.L.; Liu, T.H.; Wang, Y.H.; Li, F.F.; Ruan, L.L.; Adu-Gyamfi, E.A.; Hu, S.C.; Chen, X.M.; Ding, Y.B.; Fu, L.J. Appropriate expression of P57kip2 drives trophoblast fusion via cell cycle arrest. Reproduction 2021, 161, 633–644. [Google Scholar] [CrossRef]
- Jasmer, B.; Muschol-Steinmetz, C.; Kreis, N.N.; Friemel, A.; Kielland-Kaisen, U.; Bruggmann, D.; Jennewein, L.; Allert, R.; Solbach, C.; Yuan, J.; et al. Involvement of the oncogene B-cell lymphoma 6 in the fusion and differentiation process of trophoblastic cells of the placenta. Oncotarget 2017, 8, 108643–108654. [Google Scholar] [CrossRef]
- Schaiff, W.T.; Carlson, M.G.; Smith, S.D.; Levy, R.; Nelson, D.M.; Sadovsky, Y. Peroxisome proliferator-activated receptor-gamma modulates differentiation of human trophoblast in a ligand-specific manner. J. Clin. Endocrinol. Metab. 2000, 85, 3874–3881. [Google Scholar]
- Morasso, M.I.; Grinberg, A.; Robinson, G.; Sargent, T.D.; Mahon, K.A. Placental failure in mice lacking the homeobox gene Dlx3. Proc. Natl. Acad. Sci. USA 1999, 96, 162–167. [Google Scholar] [CrossRef]
- Roberson, M.S.; Meermann, S.; Morasso, M.I.; Mulvaney-Musa, J.M.; Zhang, T. A role for the homeobox protein Distal-less 3 in the activation of the glycoprotein hormone alpha subunit gene in choriocarcinoma cells. J. Biol. Chem. 2001, 276, 10016–10024. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Aronow, B.J.; Hossain, S.; Trapnell, B.; Kong, S.; Handwerger, S. Critical role for transcription factor AP-2alpha in human trophoblast differentiation. Physiol. Genom. 2004, 18, 99–107. [Google Scholar] [CrossRef]
- Hubert, M.A.; Sherritt, S.L.; Bachurski, C.J.; Handwerger, S. Involvement of transcription factor NR2F2 in human trophoblast differentiation. PLoS ONE 2010, 5, e9417. [Google Scholar] [CrossRef]
- Yi, C.; Song, H.; Liang, H.; Ran, Y.; Tang, J.; Chen, E.; Li, F.; Fu, L.; Wang, Y.; Chen, F.; et al. TBX3 reciprocally controls key trophoblast lineage decisions in villi during human placenta development in the first trimester. Int. J. Biol. Macromol. 2024, 263 Pt 1, 130220. [Google Scholar] [CrossRef]
- Cesana, M.; Tufano, G.; Panariello, F.; Zampelli, N.; Soldati, C.; Mutarelli, M.; Montefusco, S.; Grieco, G.; Sepe, L.V.; Rossi, B.; et al. TFEB controls syncytiotrophoblast formation and hormone production in placenta. Cell Death Differ. 2024, 31, 1439–1451. [Google Scholar] [CrossRef]
- Baczyk, D.; Drewlo, S.; Proctor, L.; Dunk, C.; Lye, S.; Kingdom, J. Glial cell missing-1 transcription factor is required for the differentiation of the human trophoblast. Cell Death Differ. 2009, 16, 719–727. [Google Scholar] [CrossRef]
- Knerr, I.; Schubert, S.W.; Wich, C.; Amann, K.; Aigner, T.; Vogler, T.; Jung, R.; Dotsch, J.; Rascher, W.; Hashemolhosseini, S. Stimulation of GCMa and syncytin via cAMP mediated PKA signaling in human trophoblastic cells under normoxic and hypoxic conditions. FEBS Lett. 2005, 579, 3991–3998. [Google Scholar] [CrossRef]
- Yang, M.; Lei, Z.M.; Rao Ch, V. The central role of human chorionic gonadotropin in the formation of human placental syncytium. Endocrinology 2003, 144, 1108–1120. [Google Scholar] [CrossRef]
- Morrish, D.W.; Bhardwaj, D.; Paras, M.T. Transforming growth factor beta 1 inhibits placental differentiation and human chorionic gonadotropin and human placental lactogen secretion. Endocrinology 1991, 129, 22–26. [Google Scholar] [CrossRef]
- Pidoux, G.; Gerbaud, P.; Dompierre, J.; Lygren, B.; Solstad, T.; Evain-Brion, D.; Tasken, K. A PKA-ezrin-Cx43 signaling complex controls gap junction communication and thereby trophoblast cell fusion. J. Cell Sci. 2014, 127 Pt 19, 4172–4185. [Google Scholar]
- Multhaup, A.; Huppertz, B.; Gohner, C.; Bohringer, M.; Mai, M.; Markert, U.; Schleussner, E.; Groten, T. N-cadherin knockdown leads to disruption of trophoblastic and endothelial cell interaction in a 3D cell culture model—New insights in trophoblast invasion failure. Cell Adh. Migr. 2018, 12, 259–270. [Google Scholar]
- Getsios, S.; MacCalman, C.D. Cadherin-11 modulates the terminal differentiation and fusion of human trophoblastic cells in vitro. Dev. Biol. 2003, 257, 41–54. [Google Scholar]
- Hart, N.R. Paradoxes: Cholesterol and Hypoxia in Preeclampsia. Biomolecules 2024, 14, 691. [Google Scholar] [CrossRef]
- Sethuraman, V.; Pu, Y.; Gingrich, J.; Jing, J.; Long, R.; Olomu, I.N.; Veiga-Lopez, A. Expression of ABC transporters during syncytialization in preeclampsia. Pregnancy Hypertens. 2022, 27, 181–188. [Google Scholar] [CrossRef]
- Renaud, S.J.; Jeyarajah, M.J. How trophoblasts fuse: An in-depth look into placental syncytiotrophoblast formation. Cell. Mol. Life Sci. 2022, 79, 433. [Google Scholar] [CrossRef]
- Wang, M.; Liu, Y.; Sun, R.; Liu, F.; Li, J.; Yan, L.; Zhang, J.; Xie, X.; Li, D.; Wang, Y.; et al. Single-nucleus multi-omic profiling of human placental syncytiotrophoblasts identifies cellular trajectories during pregnancy. Nat. Genet. 2024, 56, 294–305. [Google Scholar]
- Burton, G.J.; Jones, C.J. Syncytial knots, sprouts, apoptosis, and trophoblast deportation from the human placenta. Taiwan J. Obstet. Gynecol. 2009, 48, 28–37. [Google Scholar]
- Roland, C.S.; Hu, J.; Ren, C.E.; Chen, H.; Li, J.; Varvoutis, M.S.; Leaphart, L.W.; Byck, D.B.; Zhu, X.; Jiang, S.W. Morphological changes of placental syncytium and their implications for the pathogenesis of preeclampsia. Cell. Mol. Life Sci. 2016, 73, 365–376. [Google Scholar]
- Schmidt, M.; Hoffmann, B.; Beelen, D.; Gellhaus, A.; Winterhager, E.; Kimmig, R.; Kasimir-Bauer, S. Detection of circulating trophoblast particles in peripheral maternal blood in preeclampsia complicated pregnancies. Hypertens. Pregnancy 2008, 27, 131–142. [Google Scholar]
- Heazell, A.E.; Moll, S.J.; Jones, C.J.; Baker, P.N.; Crocker, I.P. Formation of syncytial knots is increased by hyperoxia, hypoxia and reactive oxygen species. Placenta 2007, 28 (Suppl. A), S33–S40. [Google Scholar]
- Nonn, O.; Debnath, O.; Valdes, D.S.; Sallinger, K.; Secener, A.K.; Fischer, C.; Tiesmeyer, S.; Nimo, J.; Kuenzer, T.; Ulrich, J.; et al. Senescent Syncytiotrophoblast Secretion During Early Onset Preeclampsia. Hypertension 2025, online ahead of print. [Google Scholar]
- Cinkornpumin, J.K.; Kwon, S.Y.; Prandstetter, A.M.; Maxian, T.; Sirois, J.; Goldberg, J.; Zhang, J.; Saini, D.; Dasgupta, P.; Jeyarajah, M.J.; et al. Hypoxia and loss of GCM1 expression prevents differentiation and contact inhibition in human trophoblast stem cells. bioRxiv 2024. [Google Scholar]
- Alsat, E.; Wyplosz, P.; Malassine, A.; Guibourdenche, J.; Porquet, D.; Nessmann, C.; Evain-Brion, D. Hypoxia impairs cell fusion and differentiation process in human cytotrophoblast, in vitro. J. Cell. Physiol. 1996, 168, 346–353. [Google Scholar]
- Kudo, Y.; Boyd, C.A.; Sargent, I.L.; Redman, C.W. Hypoxia alters expression and function of syncytin and its receptor during trophoblast cell fusion of human placental BeWo cells: Implications for impaired trophoblast syncytialisation in pre-eclampsia. Biochim. Biophys. Acta 2003, 1638, 63–71. [Google Scholar]
- Colson, A.; Depoix, C.L.; Baldin, P.; Hubinont, C.; Sonveaux, P.; Debieve, F. Hypoxia-inducible factor 2 alpha impairs human cytotrophoblast syncytialization: New insights into placental dysfunction and fetal growth restriction. FASEB J. 2020, 34, 15222–15235. [Google Scholar]
- Sugimoto, J.; Schust, D.J.; Sugimoto, M.; Jinno, Y.; Kudo, Y. Controlling Trophoblast Cell Fusion in the Human Placenta-Transcriptional Regulation of Suppressyn, an Endogenous Inhibitor of Syncytin-1. Biomolecules 2023, 13, 1627. [Google Scholar] [CrossRef]
- Jaremek, A.; Shaha, S.; Jeyarajah, M.J.; Jaju Bhattad, G.; Chowdhury, D.; Riddell, M.; Renaud, S.J. Genome-Wide Analysis of Hypoxia-Inducible Factor Binding Reveals Targets Implicated in Impaired Human Placental Syncytiotrophoblast Formation under Low Oxygen. Am. J. Pathol. 2023, 193, 846–865. [Google Scholar]
- Singh, H.; Zhao, M.; Chen, Q.; Wang, Y.; Li, Y.; Kaitu’u-Lino, T.J.; Tong, S.; Nie, G. Human HtrA4 Expression Is Restricted to the Placenta, Is Significantly Up-Regulated in Early-Onset Preeclampsia, and High Levels of HtrA4 Cause Endothelial Dysfunction. J. Clin. Endocrinol. Metab. 2015, 100, E936–E945. [Google Scholar] [PubMed]
- Yuan, X.; Liu, X.; Zhu, F.; Huang, B.; Lin, L.; Huang, J.; Wen, L.; Kilby, M.D.; Baker, P.N.; Fu, Y.; et al. Endoplasmic reticulum stress impairs trophoblast syncytialization through upregulation of HtrA4 and causes early-onset preeclampsia. J. Hypertens. 2023, 41, 2095–2106. [Google Scholar] [PubMed]
- Melland-Smith, M.; Ermini, L.; Chauvin, S.; Craig-Barnes, H.; Tagliaferro, A.; Todros, T.; Post, M.; Caniggia, I. Disruption of sphingolipid metabolism augments ceramide-induced autophagy in preeclampsia. Autophagy 2015, 11, 653–669. [Google Scholar] [PubMed]
- Bailey, L.J.; Alahari, S.; Tagliaferro, A.; Post, M.; Caniggia, I. Augmented trophoblast cell death in preeclampsia can proceed via ceramide-mediated necroptosis. Cell Death Dis. 2017, 8, e2590. [Google Scholar]
- Chen, Y.X.; Allars, M.; Maiti, K.; Angeli, G.L.; Abou-Seif, C.; Smith, R.; Nicholson, R.C. Factors affecting cytotrophoblast cell viability and differentiation: Evidence of a link between syncytialisation and apoptosis. Int. J. Biochem. Cell Biol. 2011, 43, 821–828. [Google Scholar]
- Lestari, B.; Fukushima, T.; Utomo, R.Y.; Wahyuningsih, M.S.H. Apoptotic and non-apoptotic roles of caspases in placenta physiology and pathology. Placenta 2024, 151, 37–47. [Google Scholar]
- Kasture, V.; Sundrani, D.; Randhir, K.; Wagh, G.; Joshi, S. Placental apoptotic markers are associated with placental morphometry. Placenta 2021, 115, 1–11. [Google Scholar]
- Chen, C.P.; Wang, L.K.; Chen, C.Y.; Chen, C.Y.; Kuo, Y.H.; Wu, Y.H. Decreased junctional adhesion molecule 3 expression induces reactive oxygen species production and apoptosis in trophoblastsdagger. Biol. Reprod. 2022, 107, 1264–1278. [Google Scholar]
- Wang, Y.N.; Chen, X.L.; Yang, J.; Gong, X.X.; Zhang, H.F.; Zhang, Y.M.; Zeng, D.F.; Chen, P.S.; Chen, H.B. Reduced syncytin-1 regulates trophoblast invasion and apoptosis in preeclampsia. Placenta 2024, 155, 32–41. [Google Scholar]
- Leavey, K.; Benton, S.J.; Grynspan, D.; Kingdom, J.C.; Bainbridge, S.A.; Cox, B.J. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension 2016, 68, 137–147. [Google Scholar]
- Apicella, C.; Ruano, C.S.M.; Thilaganathan, B.; Khalil, A.; Giorgione, V.; Gascoin, G.; Marcellin, L.; Gaspar, C.; Jacques, S.; Murdoch, C.E.; et al. Pan-Genomic Regulation of Gene Expression in Normal and Pathological Human Placentas. Cells 2023, 12, 578. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Rousseaux, S.; Agier, L.; Giorgis-Allemand, L.; Tost, J.; Galineau, J.; Hulin, A.; Siroux, V.; Vaiman, D.; Charles, M.A.; et al. Pregnancy exposure to atmospheric pollution and meteorological conditions and placental DNA methylation. Environ. Int. 2018, 118, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, S.; Seyve, E.; Chuffart, F.; Bourova-Flin, E.; Benmerad, M.; Charles, M.A.; Forhan, A.; Heude, B.; Siroux, V.; Slama, R.; et al. Immediate and durable effects of maternal tobacco consumption alter placental DNA methylation in enhancer and imprinted gene-containing regions. BMC Med. 2020, 18, 306. [Google Scholar] [CrossRef] [PubMed]
- Broseus, L.; Guilbert, A.; Hough, I.; Kloog, I.; Chauvaud, A.; Seyve, E.; Vaiman, D.; Heude, B.; Chevrier, C.; Tost, J.; et al. Placental DNA methylation signatures of prenatal air pollution exposure and potential effects on birth outcomes: An analysis of three prospective cohorts. Lancet Planet. Health 2024, 8, e297–e308. [Google Scholar] [CrossRef]
- Mortillo, M.; Marsit, C.J. Select Early-Life Environmental Exposures and DNA Methylation in the Placenta. Curr. Environ. Health Rep. 2023, 10, 22–34. [Google Scholar] [CrossRef]
- Broseus, L.; Vaiman, D.; Tost, J.; Martin, C.R.S.; Jacobi, M.; Schwartz, J.D.; Beranger, R.; Slama, R.; Heude, B.; Lepeule, J. Maternal blood pressure associates with placental DNA methylation both directly and through alterations in cell-type composition. BMC Med. 2022, 20, 397. [Google Scholar] [CrossRef]
- Kulkarni, A.; Chavan-Gautam, P.; Mehendale, S.; Yadav, H.; Joshi, S. Global DNA methylation patterns in placenta and its association with maternal hypertension in pre-eclampsia. DNA Cell Biol. 2011, 30, 79–84. [Google Scholar] [CrossRef]
- Fan, W.; Mao, Y.; Wu, L.; Feng, P.; Zhang, X.; Hu, J.; Jin, Y.; Yang, X.; Li, H.; Liu, Q.; et al. Association between CORIN promoter methylation and hypertensive disorders of pregnancy—A nested case-control study. Placenta 2024, 148, 77–83. [Google Scholar] [CrossRef]
- Dave, K.; Kaur, L.; Sundrani, D.; Sharma, P.; Bayyana, S.; Mehendale, S.; Randhir, K.; Chandak, G.R.; Joshi, S. Association of placental fatty acid desaturase 2 (FADS2) methylation with maternal fatty acid levels in women with preeclampsia. Prostaglandins Leukot. Essent. Fatty Acids 2022, 184, 102472. [Google Scholar] [CrossRef]
- Rietze, A.H.; Conley, Y.P.; Ren, D.; Anderson, C.M.; Roberts, J.M.; Jeyabalan, A.; Hubel, C.A.; Schmella, M.J. DNA Methylation of Endoglin Pathway Genes in Pregnant Women With and Without Preeclampsia. Epigenet. Insights 2020, 13, 2516865720959682. [Google Scholar] [CrossRef]
- Zakeri, S.; Rahimi, Z.; Rezvani, N.; Vaisi-Raygani, A.; Alibakhshi, R.; Zakeri, S.; Yari, K. The influence of Nrf2 gene promoter methylation on gene expression and oxidative stress parameters in preeclampsia. BMC Med. Genom. 2024, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Torres-Salazar, Q.; Martinez-Lopez, Y.; Reyes-Romero, M.; Perez-Morales, R.; Sifuentes-Alvarez, A.; Salvador-Moysen, J. Differential Methylation in Promoter Regions of the Genes NR3C1 and HSP90AA1, Involved in the Regulation, and Bioavailability of Cortisol in Leukocytes of Women With Preeclampsia. Front. Med. 2020, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Kulandavelu, S.; Dulce, R.A.; Murray, C.I.; Bellio, M.A.; Fritsch, J.; Kanashiro-Takeuchi, R.; Arora, H.; Paulino, E.; Soetkamp, D.; Balkan, W.; et al. S-Nitrosoglutathione Reductase Deficiency Causes Aberrant Placental S-Nitrosylation and Preeclampsia. J. Am. Heart Assoc. 2022, 11, e024008. [Google Scholar] [CrossRef] [PubMed]
- Than, N.G.; Romero, R.; Xu, Y.; Erez, O.; Xu, Z.; Bhatti, G.; Leavitt, R.; Chung, T.H.; El-Azzamy, H.; LaJeunesse, C.; et al. Evolutionary origins of the placental expression of chromosome 19 cluster galectins and their complex dysregulation in preeclampsia. Placenta 2014, 35, 855–865. [Google Scholar] [CrossRef]
- Yang, Y.F.; Li, Z.H.; Liu, T.H.; Li, C.; Xie, Y.L.; Ge, L.X.; Tang, J.; Li, F.F.; Luo, X.; Fu, L.J.; et al. Evaluating the role of DNA methyltransferases in trophoblast fusion and preeclampsia development: Insights from methylation-regulated genes. FASEB J. 2024, 38, e23706. [Google Scholar] [CrossRef]
- Apicella, C.; Ruano, C.S.M.; Jacques, S.; Gascoin, G.; Mehats, C.; Vaiman, D.; Miralles, F. Urothelial Cancer Associated 1 (UCA1) and miR-193 Are Two Non-coding RNAs Involved in Trophoblast Fusion and Placental Diseases. Front. Cell Dev. Biol. 2021, 9, 633937. [Google Scholar] [CrossRef]
- Kong, X.; Li, R.; Chen, M.; Zheng, R.; Wang, J.; Sun, C.; Qu, Y. Endogenous retrovirus HERVH-derived lncRNA UCA1 controls human trophoblast development. Proc. Natl. Acad. Sci. USA 2024, 121, e2318176121. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Gao, Y. Abnormal H3K27 histone methylation of RASA1 gene leads to unexplained recurrent spontaneous abortion by regulating Ras-MAPK pathway in trophoblast cells. Mol. Biol. Rep. 2021, 48, 5109–5119. [Google Scholar] [CrossRef]
- Hahn, L.; Meister, S.; Mannewitz, M.; Beyer, S.; Corradini, S.; Hasbargen, U.; Mahner, S.; Jeschke, U.; Kolben, T.; Burges, A. Gal-2 Increases H3K4me(3) and H3K9ac in Trophoblasts and Preeclampsia. Biomolecules 2022, 12, 707. [Google Scholar] [CrossRef]
- Chen, C.P.; Wang, K.G.; Chen, C.Y.; Yu, C.; Chuang, H.C.; Chen, H. Altered placental syncytin and its receptor ASCT2 expression in placental development and pre-eclampsia. BJOG Int. J. Obstet. Gynaecol. 2006, 113, 152–158. [Google Scholar] [CrossRef]
- Duan, F.M.; Fu, L.J.; Wang, Y.H.; Adu-Gyamfi, E.A.; Ruan, L.L.; Xu, Z.W.; Xiao, S.Q.; Chen, X.M.; Wang, Y.X.; Liu, T.H.; et al. THBS1 regulates trophoblast fusion through a CD36-dependent inhibition of cAMP, and its upregulation participates in preeclampsia. Genes Dis. 2021, 8, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, T.; Yoshizawa, T.; Sato, Y.; Ito, T.; Tsuyama, T.; Satoh, A.; Araki, S.; Tsujita, K.; Tamura, M.; Oike, Y.; et al. SIRT7 Deficiency Protects against Aging-Associated Glucose Intolerance and Extends Lifespan in Male Mice. Cells 2022, 11, 3609. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y.; et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Wu, Y.; He, Y.; Xiang, J.; Huang, J.; Lash, G.E.; Li, P. SIRT1 regulates trophoblast senescence in premature placental aging in preeclampsia. Placenta 2022, 122, 56–65. [Google Scholar]
- Yu, H.; Chen, L.; Du, P.; Liu, X.; Xia, Y. Effects of sirtuin 1 deficiency on trophoblasts and its implications in the pathogenesis of pre-eclampsia. J. Obstet. Gynaecol. 2023, 43, 2282103. [Google Scholar]
- Cox, L.S.; Redman, C. The role of cellular senescence in ageing of the placenta. Placenta 2017, 52, 139–145. [Google Scholar]
- Mayne, B.T.; Leemaqz, S.Y.; Smith, A.K.; Breen, J.; Roberts, C.T.; Bianco-Miotto, T. Accelerated placental aging in early onset preeclampsia pregnancies identified by DNA methylation. Epigenomics 2017, 9, 279–289. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benouda, I.; Vaiman, D.; Miralles, F. Trophoblast Fusion in Hypertensive Disorders of Pregnancy and Preeclampsia. Int. J. Mol. Sci. 2025, 26, 2859. https://doi.org/10.3390/ijms26072859
Benouda I, Vaiman D, Miralles F. Trophoblast Fusion in Hypertensive Disorders of Pregnancy and Preeclampsia. International Journal of Molecular Sciences. 2025; 26(7):2859. https://doi.org/10.3390/ijms26072859
Chicago/Turabian StyleBenouda, Ikram, Daniel Vaiman, and Francisco Miralles. 2025. "Trophoblast Fusion in Hypertensive Disorders of Pregnancy and Preeclampsia" International Journal of Molecular Sciences 26, no. 7: 2859. https://doi.org/10.3390/ijms26072859
APA StyleBenouda, I., Vaiman, D., & Miralles, F. (2025). Trophoblast Fusion in Hypertensive Disorders of Pregnancy and Preeclampsia. International Journal of Molecular Sciences, 26(7), 2859. https://doi.org/10.3390/ijms26072859