
Vimentin Fragmentation and Its Role in Amyloid-Beta Plaque Deposition in Alzheimer’s Disease

 ,
,  , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
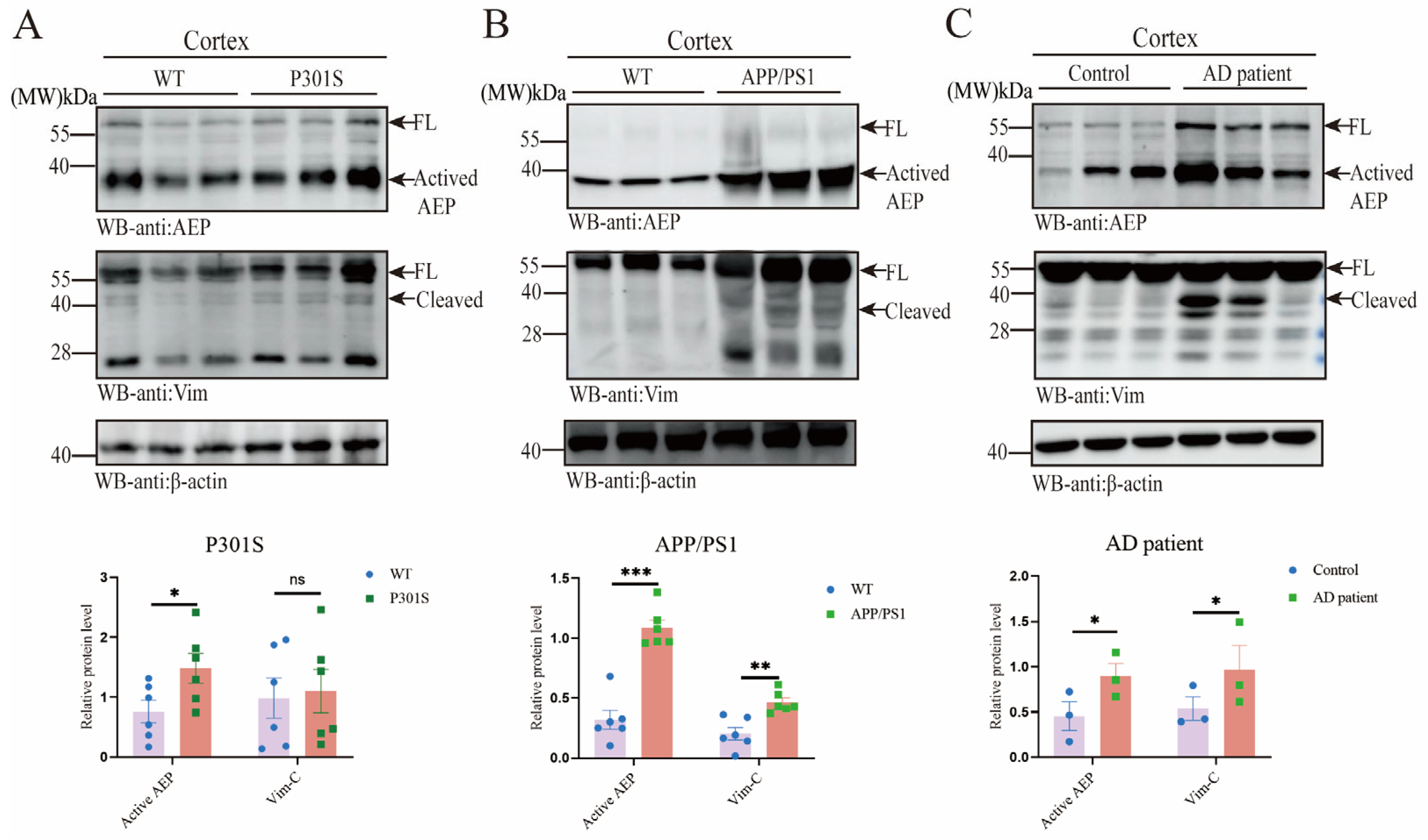
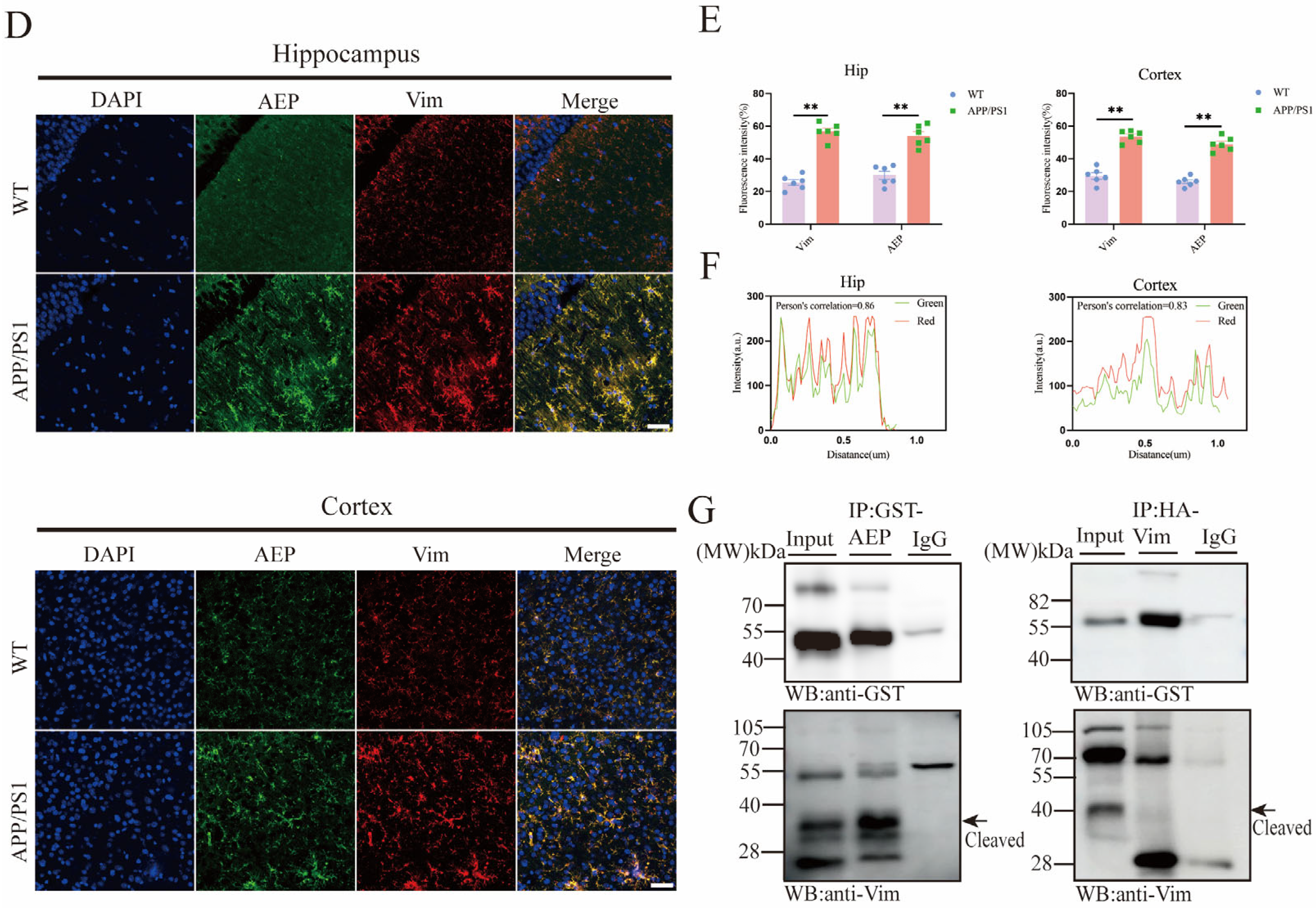
2.1. AEP and Vim Escalate and Interact with Each Other in AD Brain
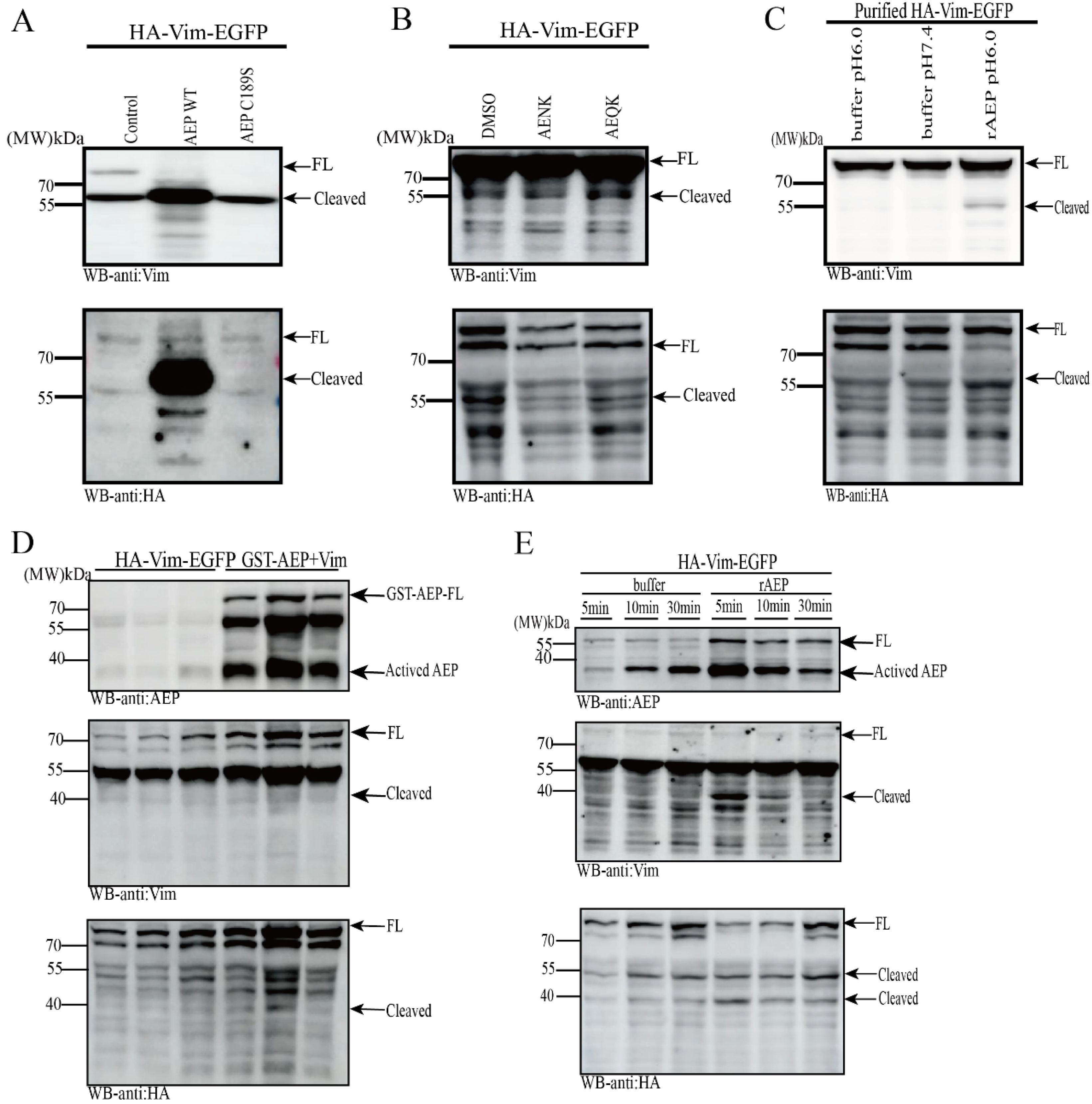
2.2. Vim Is a Substrate of AEP, Cleaved at N283 Residue
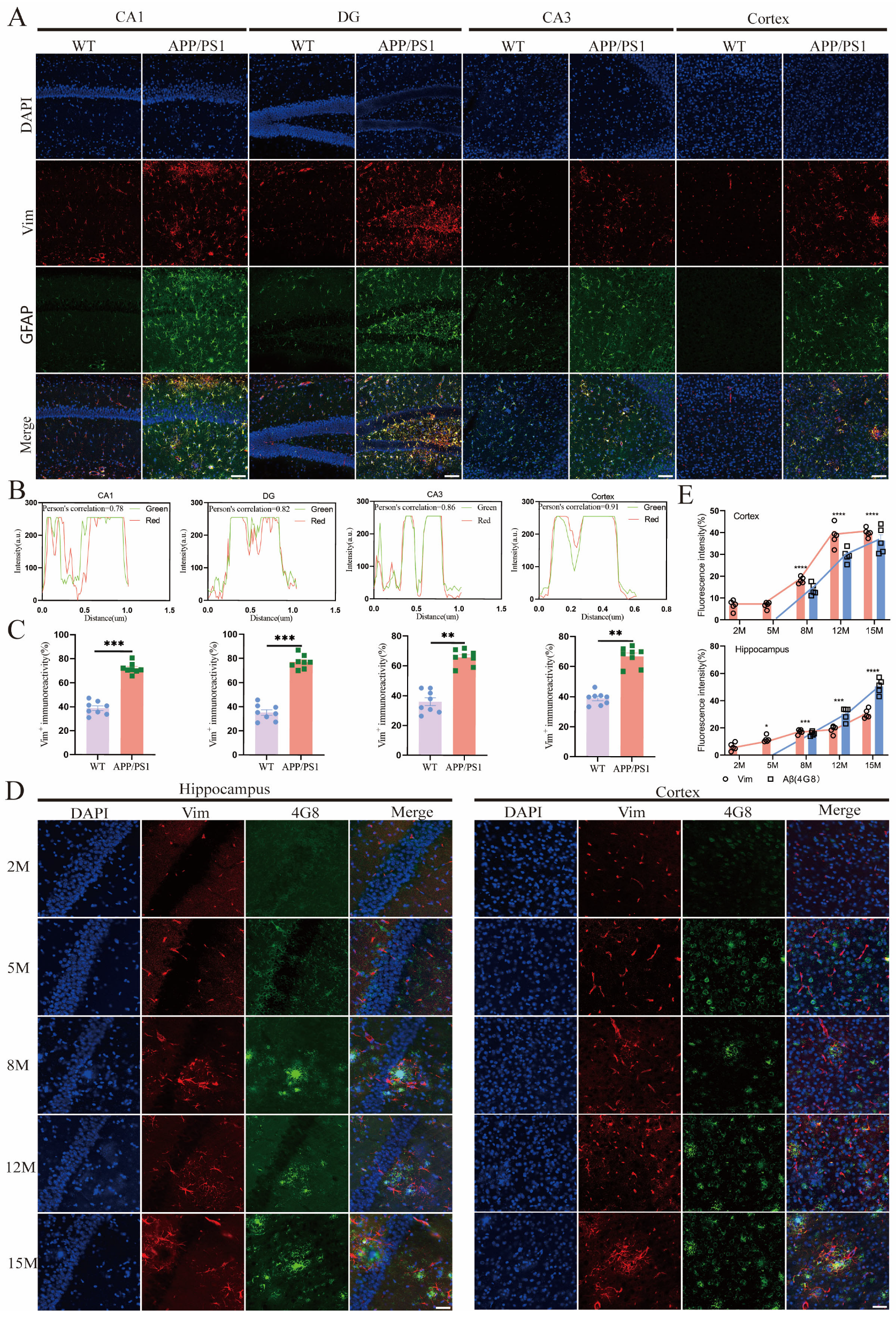
2.3. Vim Mainly Localized in APP/PS1 Mice Hippocampus Astrocytes, and Its Increase Precedes the Development of Aβ Pathology
2.4. Vim-Positive Astrocytes in APP/PS1 Mice Were More Likely to Cluster Around Aβ Plaques
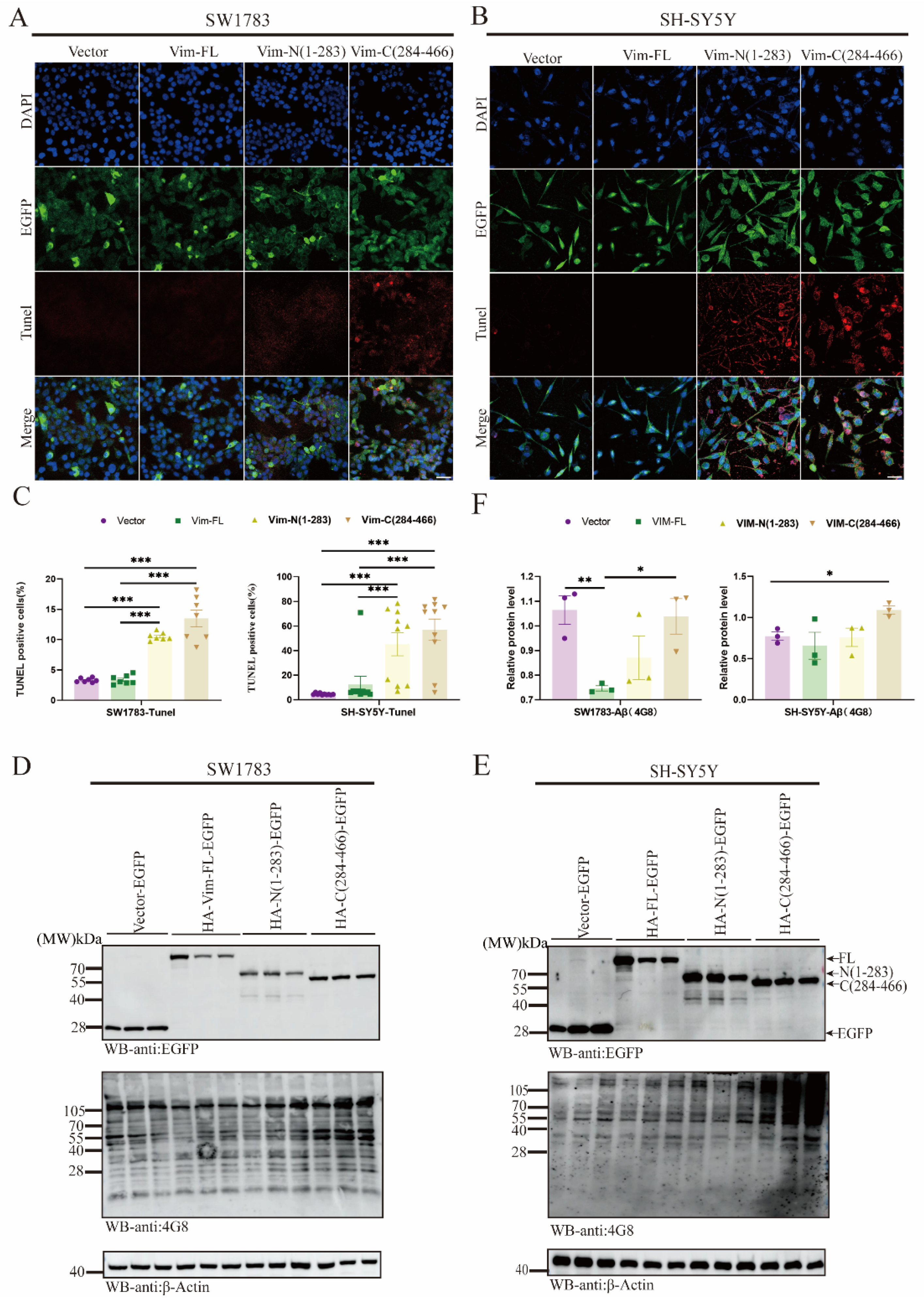
2.5. Vim Fragmentations Promote Cell Apoptosis and Aβ Aggregation via ApoE Downregulation
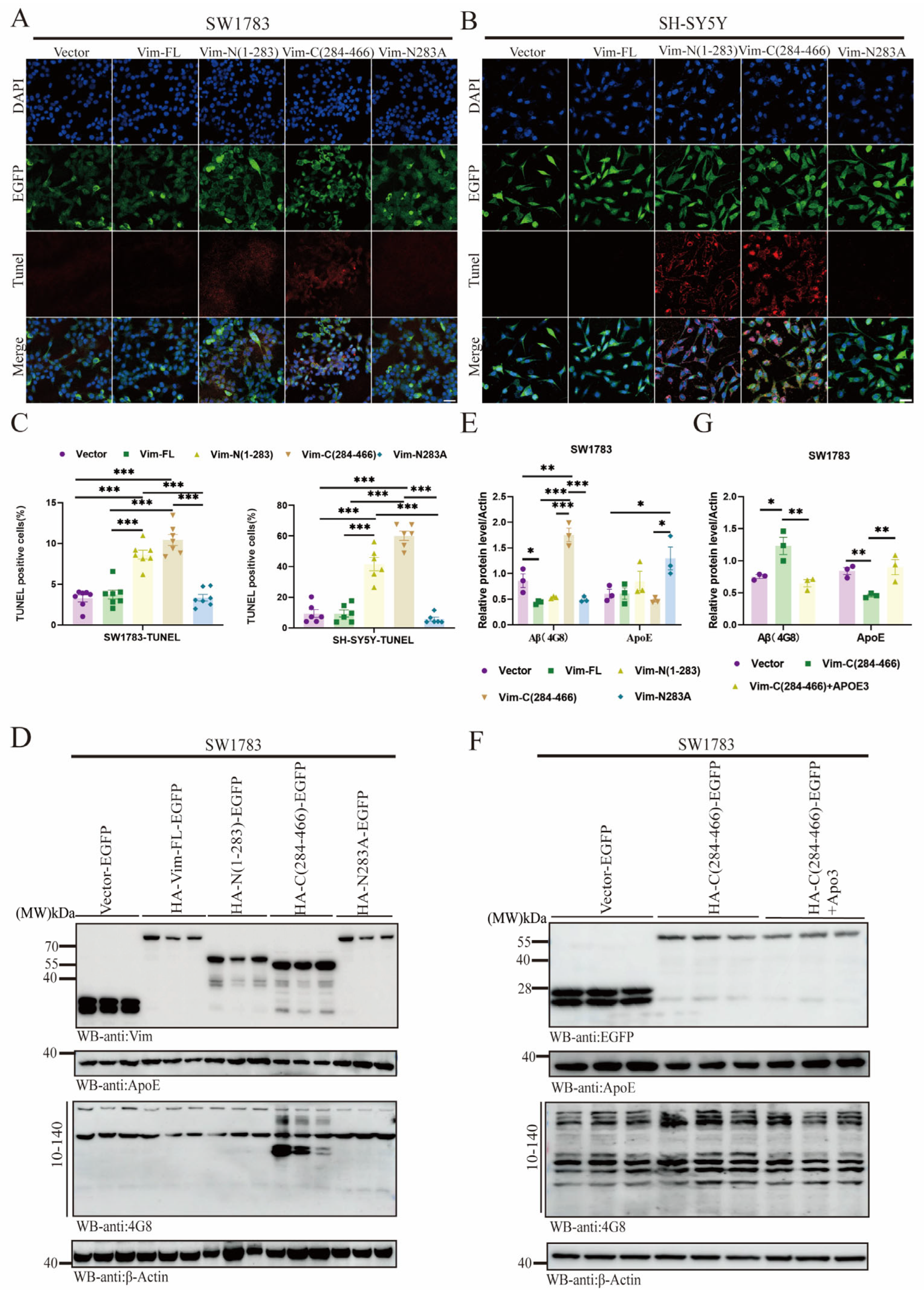
2.6. Vim-N283A Mutation Reverses Cell Apoptosis and Prevents Aβ Aggregation by Restoring ApoE Levels
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Western Blot Analysis
4.3. Immunofluorescence
4.4. Cell Culture and Transduction
4.5. Human Tissue Sample
4.6. In Vitro Vim Cleavage Assay
4.7. Co-IP Assay
4.8. Vim Fragmentation Plasmid Construction
4.9. Mass Spectrometry Analysis
4.10. Antibodies and Reagents
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| EOAD | Early onset Alzheimer’s disease |
| PD | Parkinson’s disease |
| sCJD | Creutzfeldt–Jakob disease |
| Vim | Intermediate filament protein vimentin |
| Aβ | Amyloid-β |
| AEP | Asparagine endopeptidase |
| GFAP | Glial fibrillary acidic protein |
| MAO | Monoamine oxidase |
| ApoE | Apolipoprotein E |
| LDLR | Low-density lipoprotein receptor |
References
- Graff-Radford, J.; Yong, K.X.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; Rabinovici, G.D.; Schott, J.M.; Jones, D.T.; Murray, M.E. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021, 20, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Tulbă, D.; Cozma, L.; Popescu, B.O.; Davidescu, E.I. Dysautonomia in Alzheimer’s Disease. Medicina 2020, 56, 337. [Google Scholar] [CrossRef] [PubMed]
- Whitwell, J.L. Alzheimer’s disease neuroimaging. Curr. Opin. Neurol. 2018, 31, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Petrucelli, L.; Lewis, J. Targeting Abeta and tau in Alzheimer’s disease, an early interim report. Exp. Neurol. 2010, 223, 252–266. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Abeta and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. [Google Scholar] [CrossRef]
- Quax, W.; Egberts, W.V.; Hendriks, W.; Quax-Jeuken, Y.; Bloemendal, H. The structure of the vimentin gene. Cell 1983, 35, 215–223. [Google Scholar] [CrossRef]
- Dodemont, H.J.; Soriano, P.; Quax, W.J.; Ramaekers, F.; Lenstra, J.A.; Groenen, M.A.; Bernardi, G.; Bloemendal, H. The genes coding for the cytoskeletal proteins actin and vimentin in warm-blooded vertebrates. EMBO J. 1982, 1, 167–171. [Google Scholar] [CrossRef]
- Ridge, K.M.; Eriksson, J.E.; Pekny, M.; Goldman, R.D. Roles of vimentin in health and disease. Genes. Dev. 2022, 36, 391–407. [Google Scholar] [CrossRef]
- Antfolk, D.; Sjöqvist, M.; Cheng, F.; Isoniemi, K.; Duran, C.L.; Rivero-Muller, A.; Antila, C.; Niemi, R.; Landor, S.; Bouten, C.V.C.; et al. Selective regulation of Notch ligands during angiogenesis is mediated by vimentin. Proc. Natl. Acad. Sci. USA 2017, 114, E4574–E4581. [Google Scholar] [CrossRef]
- Chen, K.Z.; Liu, S.X.; Li, Y.W.; He, T.; Zhao, J.; Wang, T.; Qiu, X.X.; Wu, H.F. Vimentin as a potential target for diverse nervous system diseases. Neural Regen. Res. 2023, 18, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsson, U.; Pozo-Rodrigalvarez, A.; Kalm, M.; de Pablo, Y.; Widestrand, Å.; Pekna, M.; Pekny, M. The role of GFAP and vimentin in learning and memory. Biol. Chem. 2019, 400, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Aimone, J.B.; Gage, F.H. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010, 11, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Garber, C.; Vasek, M.J.; Vollmer, L.L.; Sun, T.; Jiang, X.; Klein, R.S. Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nat. Immunol. 2018, 19, 151–161. [Google Scholar] [CrossRef]
- Kamphuis, W.; Kooijman, L.; Orre, M.; Stassen, O.; Pekny, M.; Hol, E.M. GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer’s disease. Glia 2015, 63, 1036–1056. [Google Scholar] [CrossRef]
- Brenner, M. Role of GFAP in CNS injuries. Neurosci. Lett. 2014, 565, 7–13. [Google Scholar] [CrossRef]
- Ding, M.; Eliasson, C.; Betsholtz, C.; Hamberger, A.; Pekny, M. Altered taurine release following hypotonic stress in astrocytes from mice deficient for GFAP and vimentin. Brain Res. Mol. Brain Res. 1998, 62, 77–81. [Google Scholar] [CrossRef]
- Dall, E.; Brandstetter, H. Structure and function of legumain in health and disease. Biochimie 2016, 122, 126–150. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Kang, S.S.; Kwon, I.S.; Duong, D.M.; Seyfried, N.T.; Hu, W.T.; Liu, Z.; Wang, J.Z.; et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 2014, 20, 1254–1262. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Su Kang, S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping Yu, S.; et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 2015, 6, 8762. [Google Scholar] [CrossRef]
- Xiong, J.; Zhang, Z.; Ye, K. C/EBPβ/AEP Signaling Drives Alzheimer’s Disease Pathogenesis. Neurosci. Bull. 2023, 39, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wang, Z.H.; Zhang, Z.; Liu, X.; Yu, S.P.; Wang, J.Z.; Wang, X.C.; Ye, K. Delta- and beta- secretases crosstalk amplifies the amyloidogenic pathway in Alzheimer’s disease. Prog. Progress. Neurobiol. 2021, 204, 102113. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Tang, L.; Dai, L.; Lei, C.; Xiong, M.; Zhang, X.; He, M.; Tian, Y.; Xiong, J.; Ke, W.; et al. Cofilin promotes tau pathology in Alzheimer’s disease. Cell Rep. 2023, 42, 112138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kang, S.S.; Liu, X.; Ahn, E.H.; Zhang, Z.; He, L.; Iuvone, P.M.; Duong, D.M.; Seyfried, N.T.; Benskey, M.J.; et al. Asparagine endopeptidase cleaves α-synuclein and mediates pathologic activities in Parkinson’s disease. Nat. Struct. Mol. Biol. 2017, 24, 632–642. [Google Scholar] [CrossRef]
- Ramaekers, F.C.; Poels, L.G.; Jap, P.H.; Bloemendal, H. Simultaneous demonstration of microfilaments and intermediate-sized filaments in the lens by double immunofluorescence. Exp. Eye Res. 1982, 35, 363–369. [Google Scholar] [CrossRef]
- Schnitzer, J.; Franke, W.W.; Schachner, M. Immunocytochemical demonstration of vimentin in astrocytes and ependymal cells of developing and adult mouse nervous system. J. Cell Biol. 1981, 90, 435–447. [Google Scholar] [CrossRef]
- Prahlad, V.; Yoon, M.; Moir, R.D.; Vale, R.D.; Goldman, R.D. Rapid movements of vimentin on microtubule tracks: Kinesin-dependent assembly of intermediate filament networks. J. Cell Biol. 1998, 143, 159–170. [Google Scholar] [CrossRef]
- Galou, M.; Colucci-Guyon, E.; Ensergueix, D.; Ridet, J.L.; Gimenez y Ribotta, M.; Privat, A.; Babinet, C.; Dupouey, P. Disrupted glial fibrillary acidic protein network in astrocytes from vimentin knockout mice. J. Cell Biol. 1996, 133, 853–863. [Google Scholar] [CrossRef]
- Brown, M.J.; Hallam, J.A.; Colucci-Guyon, E.; Shaw, S. Rigidity of circulating lymphocytes is primarily conferred by vimentin intermediate filaments. J. Immunol. 2001, 166, 6640–6646. [Google Scholar] [CrossRef]
- Capote, C.; Maccioni, R.B. The association of tau-like proteins with vimentin filaments in cultured cells. Exp. Cell Res. 1998, 239, 202–213. [Google Scholar] [CrossRef]
- Vanderheyden, W.M.; Lim, M.M.; Musiek, E.S.; Gerstner, J.R. Alzheimer’s Disease and Sleep-Wake Disturbances: Amyloid, Astrocytes, and Animal Models. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 2901–2910. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.; Aguilar, X.; Sehlin, D.; Fang, X.T.; Antoni, G.; Erlandsson, A.; Syvänen, S. Astroglial Responses to Amyloid-Beta Progression in a Mouse Model of Alzheimer’s Disease. Mol. Imaging Biol. 2018, 20, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H.; Abney, E.R.; David, S.; Ffrench-Constant, C.; Lindsay, R.; Patel, R.; Stone, J.; Raff, M.C. Is reactive gliosis a property of a distinct subpopulation of astrocytes? J. Neurosci. Off. J. Soc. Neurosci. 1986, 6, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, Y.; Kohsaka, S.; Toya, S.; Otani, M.; Tsukada, Y. Immunohistochemical studies on the proliferation of reactive astrocytes and the expression of cytoskeletal proteins following brain injury in rats. Brain Res. 1988, 466, 201–210. [Google Scholar] [CrossRef]
- Calvo, J.L.; Carbonell, A.L.; Boya, J. Co-expression of glial fibrillary acidic protein and vimentin in reactive astrocytes following brain injury in rats. Brain Res. 1991, 566, 333–336. [Google Scholar] [CrossRef]
- Franke, W.W.; Schmid, E.; Winter, S.; Osborn, M.; Weber, K. Widespread occurrence of intermediate-sized filaments of the vimentin-type in cultured cells from diverse vertebrates. Exp. Cell Res. 1979, 123, 25–46. [Google Scholar] [CrossRef]
- Jing, R.; Wilhelmsson, U.; Goodwill, W.; Li, L.; Pan, Y.; Pekny, M.; Skalli, O. Synemin is expressed in reactive astrocytes in neurotrauma and interacts differentially with vimentin and GFAP intermediate filament networks. J. Cell Sci. 2007, 120, 1267–1277. [Google Scholar] [CrossRef]
- Nakamura, S.; Kawamata, T.; Akiguchi, I.; Kameyama, M.; Nakamura, N.; Kimura, H. Expression of monoamine oxidase B activity in astrocytes of senile plaques. Acta Neuropathol. 1990, 80, 419–425. [Google Scholar] [CrossRef]
- Gulyás, B.; Pavlova, E.; Kása, P.; Gulya, K.; Bakota, L.; Várszegi, S.; Keller, E.; Horváth, M.C.; Nag, S.; Hermecz, I.; et al. Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-L-deprenyl using whole hemisphere autoradiography. Neurochem. Int. 2011, 58, 60–68. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Manzoor, S.; Hoda, N. A comprehensive review of monoamine oxidase inhibitors as Anti-Alzheimer’s disease agents: A review. Eur. J. Med. Chem. 2020, 206, 112787. [Google Scholar] [CrossRef] [PubMed]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Wang, N.; Chen, Y.; Inoue, Y.; Shue, F.; Ren, Y.; Wang, M.; Qiao, W.; Ikezu, T.C.; Li, Z.; et al. Cell-autonomous effects of APOE4 in restricting microglial response in brain homeostasis and Alzheimer’s disease. Nat. Immunol. 2023, 24, 1854–1866. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lundkvist, A.; Andersson, D.; Wilhelmsson, U.; Nagai, N.; Pardo, A.C.; Nodin, C.; Ståhlberg, A.; Aprico, K.; Larsson, K.; et al. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow. Metab. Off. J. Int. Soc. Cereb. Blood Flow. Metab. 2008, 28, 468–481. [Google Scholar] [CrossRef]
- Kraft, A.W.; Hu, X.; Yoon, H.; Yan, P.; Xiao, Q.; Wang, Y.; Gil, S.C.; Brown, J.; Wilhelmsson, U.; Restivo, J.L.; et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 187–198. [Google Scholar] [CrossRef]
- Aswendt, M.; Wilhelmsson, U.; Wieters, F.; Stokowska, A.; Schmitt, F.J.; Pallast, N.; de Pablo, Y.; Mohammed, L.; Hoehn, M.; Pekna, M.; et al. Reactive astrocytes prevent maladaptive plasticity after ischemic stroke. Prog. Progress. Neurobiol. 2022, 209, 102199. [Google Scholar] [CrossRef]
- Thal, D.R. The role of astrocytes in amyloid β-protein toxicity and clearance. Exp. Neurol. 2012, 236, 1–5. [Google Scholar] [CrossRef]
- Mulder, S.D.; Veerhuis, R.; Blankenstein, M.A.; Nielsen, H.M. The effect of amyloid associated proteins on the expression of genes involved in amyloid-β clearance by adult human astrocytes. Exp. Neurol. 2012, 233, 373–379. [Google Scholar] [CrossRef]
- Sommers, C.L.; Walker-Jones, D.; Heckford, S.E.; Worland, P.; Valverius, E.; Clark, R.; McCormick, F.; Stampfer, M.; Abularach, S.; Gelmann, E.P. Vimentin rather than keratin expression in some hormone-independent breast cancer cell lines and in oncogene-transformed mammary epithelial cells. Cancer Res. 1989, 49, 4258–4263. [Google Scholar]
- Perreau, J.; Lilienbaum, A.; Vasseur, M.; Paulin, D. Nucleotide sequence of the human vimentin gene and regulation of its transcription in tissues and cultured cells. Gene 1988, 62, 7–16. [Google Scholar] [CrossRef]
- Smith, T.A.; Strelkov, S.V.; Burkhard, P.; Aebi, U.; Parry, D.A. Sequence comparisons of intermediate filament chains: Evidence of a unique functional/structural role for coiled-coil segment 1A and linker L1. J. Struct. Biol. 2002, 137, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.; Hess, J.F.; Budamagunta, M.S.; Voss, J.C.; Kuzin, A.P.; Huang, Y.J.; Xiao, R.; Montelione, G.T.; FitzGerald, P.G.; Hunt, J.F. The structure of vimentin linker 1 and rod 1B domains characterized by site-directed spin-labeling electron paramagnetic resonance (SDSL-EPR) and X-ray crystallography. J. Biol. Chem. 2012, 287, 28349–28361. [Google Scholar] [CrossRef] [PubMed]
- Chernyatina, A.A.; Nicolet, S.; Aebi, U.; Herrmann, H.; Strelkov, S.V. Atomic structure of the vimentin central α-helical domain and its implications for intermediate filament assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 13620–13625. [Google Scholar] [CrossRef] [PubMed]
- Premchandar, A.; Mücke, N.; Poznański, J.; Wedig, T.; Kaus-Drobek, M.; Herrmann, H.; Dadlez, M. Structural Dynamics of the Vimentin Coiled-coil Contact Regions Involved in Filament Assembly as Revealed by Hydrogen-Deuterium Exchange. J. Biol. Chem. 2016, 291, 24931–24950. [Google Scholar] [CrossRef]
- Blumenberg, M. Evolution of homologous domains of cytoplasmic intermediate filament proteins and lamins. Mol. Biol. Evol. 1989, 6, 53–65. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.C.; Qiao, W.; Bu, G. Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 347–357. [Google Scholar] [CrossRef]
- Chai, A.B.; Lam, H.H.J.; Kockx, M.; Gelissen, I.C. Apolipoprotein E isoform-dependent effects on the processing of Alzheimer’s amyloid-β. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2021, 1866, 158980. [Google Scholar] [CrossRef]
- Xia, Z.; Prescott, E.E.; Urbanek, A.; Wareing, H.E.; King, M.C.; Olerinyova, A.; Dakin, H.; Leah, T.; Barnes, K.A.; Matuszyk, M.M.; et al. Co-aggregation with Apolipoprotein E modulates the function of Amyloid-β in Alzheimer’s disease. Nat. Commun. 2024, 15, 4695. [Google Scholar] [CrossRef]
- Kaji, S.; Berghoff, S.A.; Spieth, L.; Schlaphoff, L.; Sasmita, A.O.; Vitale, S.; Büschgens, L.; Kedia, S.; Zirngibl, M.; Nazarenko, T.; et al. Apolipoprotein E aggregation in microglia initiates Alzheimer’s disease pathology by seeding β-amyloidosis. Immunity 2024, 57, 2651–2668.e2612. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, X.; Zou, H.; Guan, M.; Fu, C.; Wang, W.; Zhang, Z.R.; Geng, Y.; Chen, Y. Differential and substrate-specific inhibition of γ-secretase by the C-terminal region of ApoE2, ApoE3, and ApoE4. Neuron 2023, 111, 1898–1913.e1895. [Google Scholar] [CrossRef]
- Siciliano, R.A.; Mazzeo, M.F.; Ferretta, A.; Pacelli, C.; Rosato, A.; Papa, F.; Scacco, S.; Papa, S.; Cocco, T.; Lippolis, R. Decreased amount of vimentin N-terminal truncated proteolytic products in parkin-mutant skin fibroblasts. Biochem. Biophys. Res. Commun. 2020, 521, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.E.; He, T.; Trejo-Skalli, A.V.; Härmälä-Braskén, A.S.; Hellman, J.; Chou, Y.H.; Goldman, R.D. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 2004, 117, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.A.; Herrando-Grabulosa, M.; Velasco, R.; Blasco, I.; Povedano, M.; Navarro, X. TDP-43 Cytoplasmic Translocation in the Skin Fibroblasts of ALS Patients. Cells 2022, 11, 209. [Google Scholar] [CrossRef]
- Geng, H.; An, Q.; Zhang, Y.; Huang, Y.; Wang, L.; Wang, Y. Role of Peptidylarginine Deiminase 4 in Central Nervous System Diseases. Mol. Neurobiol. 2023, 60, 6748–6756. [Google Scholar] [CrossRef]
- Wang, L.; Chen, H.; Tang, J.; Guo, Z.; Wang, Y. Peptidylarginine Deiminase and Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 473–484. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Wang, J.; Yan, Y.; Xiang, L.; Zhai, X.; Cai, L.; Sun, Z.; Pi, M.; Xiong, Q.; Zhou, H.; et al. Vimentin Fragmentation and Its Role in Amyloid-Beta Plaque Deposition in Alzheimer’s Disease. Int. J. Mol. Sci. 2025, 26, 2857. https://doi.org/10.3390/ijms26072857
Zhang L, Wang J, Yan Y, Xiang L, Zhai X, Cai L, Sun Z, Pi M, Xiong Q, Zhou H, et al. Vimentin Fragmentation and Its Role in Amyloid-Beta Plaque Deposition in Alzheimer’s Disease. International Journal of Molecular Sciences. 2025; 26(7):2857. https://doi.org/10.3390/ijms26072857
Chicago/Turabian StyleZhang, Lan, Ji Wang, Yalong Yan, Lihong Xiang, Xinyue Zhai, Lianmei Cai, Zhuoran Sun, Mingshan Pi, Qi Xiong, Hongyan Zhou, and et al. 2025. "Vimentin Fragmentation and Its Role in Amyloid-Beta Plaque Deposition in Alzheimer’s Disease" International Journal of Molecular Sciences 26, no. 7: 2857. https://doi.org/10.3390/ijms26072857
APA StyleZhang, L., Wang, J., Yan, Y., Xiang, L., Zhai, X., Cai, L., Sun, Z., Pi, M., Xiong, Q., Zhou, H., Gui, Y., Wang, X., Shu, X., & Xia, Y. (2025). Vimentin Fragmentation and Its Role in Amyloid-Beta Plaque Deposition in Alzheimer’s Disease. International Journal of Molecular Sciences, 26(7), 2857. https://doi.org/10.3390/ijms26072857