

Role of Coxsackievirus B3-Induced Immune Responses in the Transition from Myocarditis to Dilated Cardiomyopathy and Heart Failure


Abstract
1. Introduction
2. Immune-Associated Pathogenicity of Coxsackievirus B3 (CVB3)
2.1. CVB3 Proteases 2A and 3C Cleave Proteins Involved in Immune Responses
2.2. CVB3 Indirectly Impairs Cardiac Function by Inducing Inflammation That Results in Cardiomyocyte Necrosis and Fibrosis
3. Inflammation-Associated Metabolic Remodeling during CVB3-Induced Myocarditis
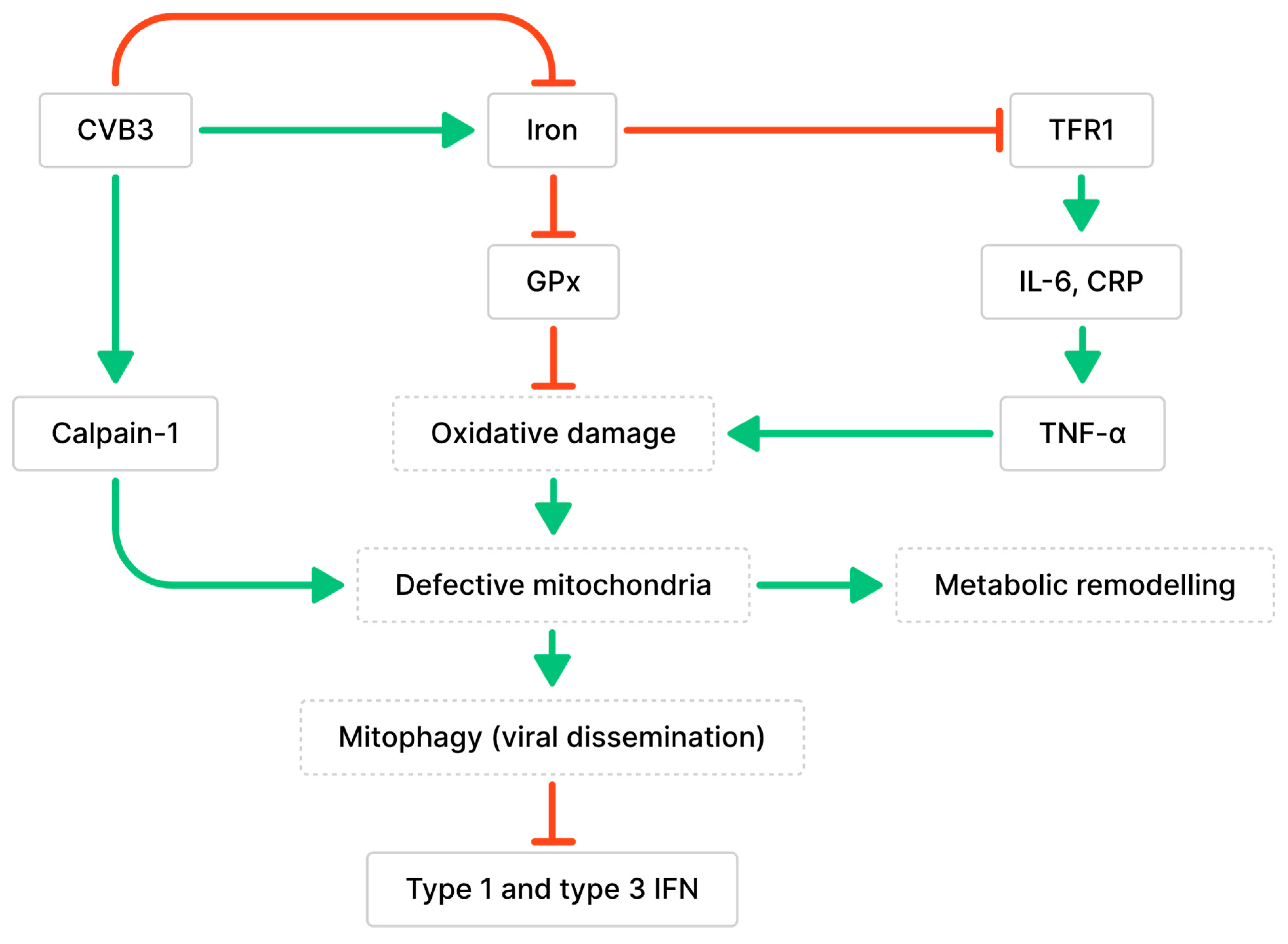
3.1. Impaired Mitochondrial Functions and Ferroptosis Caused by CVB3 Infection and Altered Iron Metabolism
3.2. Impaired Lipid and Glucose Metabolism Mediated by CVB3-Induced Inflammation
4. Immune-Associated Cells and Genes That Influence Cardiac Remodeling and Dilated Cardiomyopathy (DCM) Development
4.1. Monocytes
4.2. Macrophages
4.3. Neutrophils
4.4. T Cells
4.5. B Cells
4.6. Fibroblasts
5. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rezkalla, S.H.; Kloner, R.A. Viral myocarditis: 1917–2020: From the Influenza A to the COVID-19 pandemics. Trends Cardiovasc. Med. 2021, 31, 163–169. [Google Scholar] [CrossRef]
- Mele, D.; Flamigni, F.; Rapezzi, C.; Ferrari, R. Myocarditis in COVID-19 patients: Current problems. Intern. Emerg. Med. 2021, 16, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.-T.; Klingel, K.; Kandolf, R. Human Parvovirus B19–Associated Myocarditis. N. Engl. J. Med. 2010, 362, 1248–1249. [Google Scholar] [CrossRef]
- Bowles, N.E.; Ni, J.; Kearney, D.L.; Pauschinger, M.; Schultheiss, H.P.; McCarthy, R.; Hare, J.; Bricker, J.T.; Bowles, K.R.; Towbin, J.A. Detection of viruses in myocardial tissues by polymerase chain reaction: Evidence of adenovirus as a common cause of myocarditis in children and adults. J. Am. Coll. Cardiol. 2003, 42, 466–472. [Google Scholar] [CrossRef]
- Lasrado, N.; Reddy, J. An overview of the immune mechanisms of viral myocarditis. Rev. Med. Virol. 2020, 30, e2131. [Google Scholar] [CrossRef]
- Kühl, U.; Pauschinger, M.; Seeberg, B.; Lassner, D.; Noutsias, M.; Poller, W.; Schultheiss, H.-P. Viral Persistence in the Myocardium Is Associated with Progressive Cardiac Dysfunction. Circulation 2005, 112, 1965–1970. [Google Scholar] [CrossRef]
- Kearney, M.; Cotton, J.; Richardson, P.; Shah, A. Viral myocarditis and dilated cardiomyopathy: Mechanisms, manifestations, and management. Postgrad. Med. J. 2001, 77, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Leonard, E.G. Viral Myocarditis, Pediatr. Infect. Dis. J. 2004, 23, 665–666. [Google Scholar] [CrossRef]
- Knowlton, K.U. Dilated Cardiomyopathy. Circulation 2019, 139, 2339–2341. [Google Scholar] [CrossRef]
- Chau, D.H.W.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2007, 12, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Ye, X.; Zhang, H.M.; Hanson, P.; Zhao, G.; Tong, L.; Xie, R.; Yang, D. Cleavage of osmosensitive transcriptional factor NFAT5 by Coxsackieviral protease 2A promotes viral replication. PLoS Pathog. 2017, 13, e1006744. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Zhang, H.M.; Qiu, Y.; Ye, X.; Yang, D. Cleavage of Desmosomal Cadherins Promotes γ-Catenin Degradation and Benefits Wnt Signaling in Coxsackievirus B3-Induced Destruction of Cardiomyocytes. Front. Microbiol. 2020, 11, 767. [Google Scholar] [CrossRef] [PubMed]
- Mohamud, Y.; Xue, Y.C.; Liu, H.; Ng, C.S.; Bahreyni, A.; Luo, H. Autophagy Receptor Protein Tax1-Binding Protein 1/TRAF6-Binding Protein Is a Cellular Substrate of Enteroviral Proteinase. Front. Microbiol. 2021, 12, 647410. [Google Scholar] [CrossRef]
- Hanson, P.J.; Ye, X.; Qiu, Y.; Zhang, H.M.; Hemida, M.G.; Wang, F.; Lim, T.; Gu, A.; Cho, B.; Kim, H.; et al. Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes. Cell Death Differ. 2016, 23, 828–840. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The Coxsackievirus B 3Cpro Protease Cleaves MAVS and TRIF to Attenuate Host Type I Interferon and Apoptotic Signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef]
- Deonarain, R.; Cerullo, D.; Fuse, K.; Liu, P.P.; Fish, E.N. Protective Role for Interferon-β in Coxsackievirus B3 Infection. Circulation 2004, 110, 3540–3543. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Marques, J.T.; Yamashita, M.; Peters, K.L.; Smith, K.; Desai, A.; Williams, B.R.; Sen, G.C. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010, 29, 1762–1773. [Google Scholar] [CrossRef]
- Lei, Y.; Moore, C.B.; Liesman, R.M.; O’Connor, B.P.; Bergstralh, D.T.; Chen, Z.J.; Pickles, R.J.; Ting, J.P.-Y. MAVS-Mediated Apoptosis and Its Inhibition by Viral Proteins. PLoS ONE 2009, 4, e5466. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Offermann, M.K. Apoptosis Induced by the Toll-Like Receptor Adaptor TRIF Is Dependent on Its Receptor Interacting Protein Homotypic Interaction Motif. J. Immunol. 2005, 174, 4942–4952. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.I.; Doolittle, A.C.; Snuggs, J.W.; Shapiro, I.M.; Le Maitre, C.L.; Risbud, M.V. TNF-α promotes nuclear enrichment of the transcription factor TonEBP/NFAT5 to selectively control inflammatory but not osmoregulatory responses in nucleus pulposus cells. J. Biol. Chem. 2017, 292, 17561–17575. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wong, J.; Piesik, P.; Fung, G.; Zhang, J.; Jagdeo, J.; Li, X.; Jan, E.; Luo, H. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy 2013, 9, 1591–1603. [Google Scholar] [CrossRef]
- Rahnefeld, A.; Klingel, K.; Schuermann, A.; Diny, N.L.; Althof, N.; Lindner, A.; Bleienheuft, P.; Savvatis, K.; Respondek, D.; Opitz, E.; et al. Ubiquitin-Like Protein ISG15 (Interferon-Stimulated Gene of 15 kDa) in Host Defense Against Heart Failure in a Mouse Model of Virus-Induced Cardiomyopathy. Circulation 2014, 130, 1589–1600. [Google Scholar] [CrossRef]
- Nadkarni, R.; Chu, W.C.; Lee, C.Q.E.; Mohamud, Y.; Yap, L.; Toh, G.A.; Beh, S.; Lim, R.; Fan, Y.M.; Zhang, Y.L.; et al. Viral proteases activate the CARD8 inflammasome in the human cardiovascular system. J. Exp. Med. 2022, 219, e20212117. [Google Scholar] [CrossRef]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.F.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; et al. A Role for Toll-like Receptor 3 Variants in Host Susceptibility to Enteroviral Myocarditis and Dilated Cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Jensen, S.; Thomsen, A.R. Sensing of RNA Viruses: A Review of Innate Immune Receptors Involved in Recognizing RNA Virus Invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.E.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef]
- Kumagai, Y.; Takeuchi, O.; Akira, S. TLR9 as a key receptor for the recognition of DNA. Adv. Drug Deliv. Rev. 2008, 60, 795–804. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Michael Lavigne, G.; Russell, H.; Sherry, B.; Ke, R. Autocrine and paracrine interferon signalling as ‘ring vaccination’ and ‘contact tracing’ strategies to suppress virus infection in a host. Proc. R. Soc. B Biol. Sci. 2021, 288, 20203002. [Google Scholar] [CrossRef]
- Smolgovsky, S.; Ibeh, U.; Tamayo, T.P.; Alcaide, P. Adding insult to injury—Inflammation at the heart of cardiac fibrosis. Cell. Signal. 2021, 77, 109828. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef]
- Xu, D.; Wang, P.; Yang, J.; Qian, Q.; Li, M.; Wei, L.; Xu, W. Gr-1+ Cells Other Than Ly6G+ Neutrophils Limit Virus Replication and Promote Myocardial Inflammation and Fibrosis Following Coxsackievirus B3 Infection of Mice. Front. Cell. Infect. Microbiol. 2018, 8, 157. [Google Scholar] [CrossRef]
- Hammond, M.D.; Ai, Y.; Sansing, L.H. Gr1+ Macrophages and Dendritic Cells Dominate the Inflammatory Infiltrate 12 h After Experimental Intracerebral Hemorrhage. Transl. Stroke Res. 2012, 3, 125–131. [Google Scholar] [CrossRef]
- Niedermeier, M.; Reich, B.; Gomez, M.R.; Denzel, A.; Schmidbauer, K.; Göbel, N.; Talke, Y.; Schweda, F.; Mack, M. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 17892–17897. [Google Scholar] [CrossRef] [PubMed]
- Beinert, H.; Holm, R.H.; Münck, E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Tsujino, T.; Matsumoto, M.; Sakoda, T.; Ohyanagi, M.; Masuyama, T. Adaptive response of the heart to long-term anemia induced by iron deficiency. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H585–H593. [Google Scholar] [CrossRef]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef]
- Kobak, K.A.; Franczuk, P.; Schubert, J.; Dzięgała, M.; Kasztura, M.; Tkaczyszyn, M.; Drozd, M.; Kosiorek, A.; Kiczak, L.; Bania, J.; et al. Primary Human Cardiomyocytes and Cardiofibroblasts Treated with Sera from Myocarditis Patients Exhibit an Increased Iron Demand and Complex Changes in the Gene Expression. Cells 2021, 10, 818. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, E.; Testa, U.; Louache, F.; Thomopoulos, P.; Salvo, G.; Samoggia, P.; Peschle, C. Expression of transferrin receptors in phytohemagglutinin-stimulated human T-lymphocytes. Evidence for a three-step model. J. Biol. Chem. 1986, 261, 3036–3042. [Google Scholar] [CrossRef] [PubMed]
- Lanser, L.; Nemati, N.; Seifert, M.; Fuchs, D.; Weiss, G.; Pölzl, G.; Kurz, K. Inflammation, iron and vitamin D metabolism in different cardiomyopathy aetiologies. Pteridines 2020, 31, 28–37. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Yi, L.; Hu, Y.; Wu, Z.; Li, Y.; Kong, M.; Kang, Z.; Zuoyuan, B.; Yang, Z. TFRC upregulation promotes ferroptosis in CVB3 infection via nucleus recruitment of Sp1. Cell Death Dis. 2022, 13, 592. [Google Scholar] [CrossRef]
- Hong, M.; Rong, J.; Tao, X.; Xu, Y. The Emerging Role of Ferroptosis in Cardiovascular Diseases. Front. Pharmacol. 2022, 13, 822083. [Google Scholar] [CrossRef]
- Ursu, O.N.; Sauter, M.; Ettischer, N.; Kandolf, R.; Klingel, K. Heme Oxygenase-1 Mediates Oxidative Stress and Apoptosis in Coxsackievirus B3-Induced Myocarditis. Cell. Physiol. Biochem. 2014, 33, 52–66. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Chen, Z.; Yu, Y.; Shi, H.; Yu, Y.; Wang, Y.; Chen, R.; Ge, J. Mitochondrial calpain-1 activates NLRP3 inflammasome by cleaving ATP5A1 and inducing mitochondrial ROS in CVB3-induced myocarditis. Basic Res. Cardiol. 2022, 117, 40. [Google Scholar] [CrossRef]
- Shi, H.; Yu, Y.; Liu, X.; Yu, Y.; Li, M.; Wang, Y.; Zou, Y.; Chen, R.; Ge, J. Inhibition of calpain reduces cell apoptosis by suppressing mitochondrial fission in acute viral myocarditis. Cell Biol. Toxicol. 2022, 38, 487–504. [Google Scholar] [CrossRef]
- Oh, S.-J.; Lim, B.-K.; Yun, J.; Shin, O.S. CVB3-Mediated Mitophagy Plays an Important Role in Viral Replication via Abrogation of Interferon Pathways. Front. Cell. Infect. Microbiol. 2021, 11, 704494. [Google Scholar] [CrossRef] [PubMed]
- Sin, J.; McIntyre, L.; Stotland, A.; Feuer, R.; Gottlieb, R.A. Coxsackievirus B Escapes the Infected Cell in Ejected Mitophagosomes. J. Virol. 2017, 91, e01347-17. [Google Scholar] [CrossRef]
- Bu, L.; Wang, H.; Hou, P.; Guo, S.; He, M.; Xiao, J.; Li, P.; Zhong, Y.; Jia, P.; Cao, Y.; et al. The Ubiquitin E3 Ligase Parkin Inhibits Innate Antiviral Immunity Through K48-Linked Polyubiquitination of RIG-I and MDA5. Front. Immunol. 2020, 11, 1926. [Google Scholar] [CrossRef] [PubMed]
- Remels, A.H.V.; Derks, W.J.A.; Cillero-Pastor, B.; Verhees, K.J.P.; Kelders, M.C.; Heggermont, W.; Carai, P.; Summer, G.; Ellis, S.R.; de Theije, C.C.; et al. NF-κB-mediated metabolic remodelling in the inflamed heart in acute viral myocarditis. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P.; Clayton, D.A. Purification and characterization of human mitochondrial transcription factor 1. Mol. Cell. Biol. 1988, 8, 3496–3509. [Google Scholar] [CrossRef]
- Larsson, N.-G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Herrera, K.N.G.; Wikman-Kocher, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Chinen, L.T.D.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Dassanayaka, S.; Jones, S.P. O-GlcNAc and the cardiovascular system. Pharmacol. Ther. 2014, 142, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. WJG 2014, 20, 14205–14218. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Bailey, P.J.; Goldenberg, M.M.; Ford-Hutchinson, A.W. The Role of Arachidonic Acid Oxygenation Products in Pain and Inflammation. Annu. Rev. Immunol. 1984, 2, 335–357. [Google Scholar] [CrossRef]
- Fang, H.; Judd, R.L. Adiponectin regulation and function. In Comprehensive Physiology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 1031–1063. [Google Scholar] [CrossRef]
- Skurk, C.; Wittchen, F.; Suckau, L.; Witt, H.; Noutsias, M.; Fechner, H.; Schultheiss, H.-P.; Poller, W. Description of a local cardiac adiponectin system and its deregulation in dilated cardiomyopathy. Eur. Heart J. 2008, 29, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Jenke, A.; Holzhauser, L.; Löbel, M.; Savvatis, K.; Wilk, S.; Weithäuser, A.; Pinkert, S.; Tschöpe, C.; Klingel, K.; Poller, W.; et al. Adiponectin promotes coxsackievirus B3 myocarditis by suppression of acute anti-viral immune responses. Basic Res. Cardiol. 2014, 109, 408. [Google Scholar] [CrossRef] [PubMed]
- Frisancho-Kiss, S.; Nyland, J.F.; Davis, S.E.; Augusto Frisancho, J.; Barrett, M.A.; Rose, N.R.; Fairweather, D. Sex differences in coxsackievirus B3-induced myocarditis: IL-12Rβ1 signaling and IFN-γ increase inflammation in males independent from STAT4. Brain Res. 2006, 1126, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Yusung, S.; Frisancho, S.; Barrett, M.; Gatewood, S.; Steele, R.; Rose, N.R. IL-12 Receptor β1 and Toll-Like Receptor 4 Increase IL-1β- and IL-18-Associated Myocarditis and Coxsackievirus Replication. J. Immunol. 2003, 170, 4731–4737. [Google Scholar] [CrossRef] [PubMed]
- Cihakova, D.; Barin, J.G.; Afanasyeva, M.; Kimura, M.; Fairweather, D.; Berg, M.; Talor, M.V.; Baldeviano, G.C.; Frisancho, S.; Gabrielson, K.; et al. Interleukin-13 Protects Against Experimental Autoimmune Myocarditis by Regulating Macrophage Differentiation. Am. J. Pathol. 2008, 172, 1195–1208. [Google Scholar] [CrossRef]
- Choi, J.-M.; Bothwell, A.L.M. The Nuclear Receptor PPARs as Important Regulators of T-Cell Functions and Autoimmune Diseases. Mol. Cells 2012, 33, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Zhao, F.; Xie, X.; Liao, Y.; Song, Y.; Liu, C.; Wu, Y.; Wang, Y.; Liu, D.; Wang, Y.; et al. PPARα suppresses Th17 cell differentiation through IL-6/STAT3/RORγt pathway in experimental autoimmune myocarditis. Exp. Cell Res. 2019, 375, 22–30. [Google Scholar] [CrossRef]
- Zhou, L.; Lopes, J.E.; Chong, M.M.W.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits Th17 cell differentiation by antagonizing RORγt function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef]
- Tan, H.W.S.; Anjum, B.; Shen, H.-M.; Ghosh, S.; Yen, P.M.; Sinha, R.A. Lysosomal inhibition attenuates peroxisomal gene transcription via suppression of PPARA and PPARGC1A levels. Autophagy 2019, 15, 1455–1459. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor γ Coactivator 1 Coactivators, Energy Homeostasis, and Metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef]
- Riehle, C.; Wende, A.R.; Zaha, V.G.; Pires, K.M.; Wayment, B.; Olsen, C.; Bugger, H.; Buchanan, J.; Wang, X.; Moreira, A.B.; et al. PGC-1β Deficiency Accelerates the Transition to Heart Failure in Pressure Overload Hypertrophy. Circ. Res. 2011, 109, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-γ coactivator 1α. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef]
- Jackson, S.M.; Wilson, P.C.; James, J.A.; Capra, J.D. Human B cell subsets. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2008; Chapter 5; pp. 151–224. [Google Scholar] [CrossRef]
- MacKenna, D.; Summerour, S.R.; Villarreal, F.J. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc. Res. 2000, 46, 257–263. [Google Scholar] [CrossRef]
- Yang, X.; Yue, Y.; Xiong, S. Dpep2 Emerging as a Modulator of Macrophage Inflammation Confers Protection Against CVB3-Induced Viral Myocarditis. Front. Cell. Infect. Microbiol. 2019, 9, 57. [Google Scholar] [CrossRef]
- Carai, P.; Ruozi, G.; Paye, A.; Debing, Y.; Bortolotti, F.; Lecomte, J.; Zentilin, L.; Jones, E.A.V.; Giacca, M.; Heymans, S. AAV9-mediated functional screening for cardioprotective cytokines in Coxsackievirus-B3-induced myocarditis. Sci. Rep. 2022, 12, 7304. [Google Scholar] [CrossRef]
- Yu, M.; Long, Q.; Li, H.-H.; Liang, W.; Liao, Y.-H.; Yuan, J.; Cheng, X. IL-9 Inhibits Viral Replication in Coxsackievirus B3-Induced Myocarditis. Front. Immunol. 2016, 7, 409. [Google Scholar] [CrossRef]
- Dhingra, S.; Sharma, A.K.; Arora, R.C.; Slezak, J.; Singal, P.K. IL-10 attenuates TNF-α-induced NFκB pathway activation and cardiomyocyte apoptosis. Cardiovasc. Res. 2009, 82, 59–66. [Google Scholar] [CrossRef]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef] [PubMed]
- YlÖstalo, J.H.; Bartosh, T.J.; Coble, K.; Prockop, D.J. Human Mesenchymal Stem/Stromal Cells Cultured as Spheroids are Self-activated to Produce Prostaglandin E2 that Directs Stimulated Macrophages into an Anti-inflammatory Phenotype. Stem Cells. 2012, 30, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Loynes, C.A.; Lee, J.A.; Robertson, A.L.; Steel, M.J.G.; Ellett, F.; Feng, Y.; Levy, B.D.; Whyte, M.K.B.; Renshaw, S.A. PGE2 production at sites of tissue injury promotes an anti-inflammatory neutrophil phenotype and determines the outcome of inflammation resolution in vivo. Sci. Adv. 2018, 4, eaar8320. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Cao, Y.; Tian, Y.; Gu, Y.; Lu, H.; Zhang, S.; Xu, H.; Su, Z. PGE2 ameliorated viral myocarditis development and promoted IL-10-producing regulatory B cell expansion via MAPKs/AKT-AP1 axis or AhR signaling. Cell. Immunol. 2020, 347, 104025. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, L.; Xiao, L.; Shangguan, J. Progranulin ameliorates coxsackievirus-B3-induced viral myocarditis by downregulating Th1 and Th17 cells. Exp. Cell Res. 2018, 367, 241–250. [Google Scholar] [CrossRef]
- Carai, P.; Papageorgiou, A.P.; Van Linthout, S.; Deckx, S.; Velthuis, S.; Lutgens, E.; Wijnands, E.; Tschöpe, C.; Schmuttermaier, C.; Kzhyshkowska, J.; et al. Stabilin-1 mediates beneficial monocyte recruitment and tolerogenic macrophage programming during CVB3-induced viral myocarditis. J. Mol. Cell. Cardiol. 2022, 165, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte Conversion During Inflammation and Injury. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef]
- Leuschner, F.; Courties, G.; Dutta, P.; Mortensen, L.J.; Gorbatov, R.; Sena, B.; Novobrantseva, T.I.; Borodovsky, A.; Fitzgerald, K.; Koteliansky, V.; et al. Silencing of CCR2 in myocarditis. Eur. Heart J. 2015, 36, 1478–1488. [Google Scholar] [CrossRef]
- Tsou, C.-L.; Peters, W.; Si, Y.; Slaymaker, S.; Aslanian, A.M.; Weisberg, S.P.; Mack, M.; Charo, I.F. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 2007, 117, 902–909. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Y.; Wei, B.; Wu, W.; Gao, X. CD80 Regulates Th17 Cell Differentiation in Coxsackie Virus B3-Induced Acute Myocarditis. Inflammation 2018, 41, 232–239. [Google Scholar] [CrossRef]
- De Giusti, C.J.; Ure, A.E.; Rivadeneyra, L.; Schattner, M.; Gomez, R.M. Macrophages and galectin 3 play critical roles in CVB3-induced murine acute myocarditis and chronic fibrosis. J. Mol. Cell. Cardiol. 2015, 85, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.M.; Cooper, L.T.; Kem, D.C.; Stavrakis, S.; Kosanke, S.D.; Shevach, E.M.; Fairweather, D.; Stoner, J.A.; Cox, C.J.; Cunningham, M.W. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight 2016, 1, e85851. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J. GM-CSF: The secret weapon in the TH17 arsenal. Nat. Immunol. 2011, 12, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Baldeviano, G.C.; Barin, J.G.; Talor, M.V.; Srinivasan, S.; Bedja, D.; Zheng, D.; Gabrielson, K.; Iwakura, Y.; Rose, N.R.; Cihakova, D. Interleukin-17A Is Dispensable for Myocarditis but Essential for the Progression to Dilated Cardiomyopathy. Circ. Res. 2010, 106, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Kraft, L.; Erdenesukh, T.; Sauter, M.; Tschöpe, C.; Klingel, K. Blocking the IL-1β signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res. Cardiol. 2019, 114, 11. [Google Scholar] [CrossRef] [PubMed]
- Al-Kofahi, M.; Omura, S.; Tsunoda, I.; Sato, F.; Becker, F.; Gavins, F.N.; Woolard, M.D.; Pattillo, C.; Zawieja, D.; Muthuchamy, M.; et al. IL-1β reduces cardiac lymphatic muscle contraction via COX-2 and PGE2 induction: Potential role in myocarditis. Biomed. Pharmacother. 2018, 107, 1591–1600. [Google Scholar] [CrossRef]
- Savvatis, K.; Müller, I.; Fröhlich, M.; Pappritz, K.; Zietsch, C.; Hamdani, N.; Grote, K.; Schieffer, B.; Klingel, K.; Van Linthout, S.; et al. Interleukin-6 receptor inhibition modulates the immune reaction and restores titin phosphorylation in experimental myocarditis. Basic Res. Cardiol. 2014, 109, 449. [Google Scholar] [CrossRef]
- Valaperti, A.; Nishii, M.; Liu, Y.; Naito, K.; Chan, M.; Zhang, L.; Skurk, C.; Schultheiss, H.-P.; Wells, G.A.; Eriksson, U.; et al. Innate Immune Interleukin-1 Receptor–Associated Kinase 4 Exacerbates Viral Myocarditis by Reducing CCR5+CD11b+ Monocyte Migration and Impairing Interferon Production. Circulation 2013, 128, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- López, B.; González, A.; Lindner, D.; Westermann, D.; Ravassa, S.; Beaumont, J.; Gallego, I.; Zudaire, A.; Brugnolaro, C.; Querejeta, R.; et al. Osteopontin-mediated myocardial fibrosis in heart failure: A role for lysyl oxidase? Cardiovasc. Res. 2013, 99, 111–120. [Google Scholar] [CrossRef]
- Antoniak, S.; Owens, A.P., III; Baunacke, M.; Williams, J.C.; Lee, R.D.; Weithäuser, A.; Sheridan, P.A.; Malz, R.; Luyendyk, J.P.; Esserman, D.A.; et al. PAR-1 contributes to the innate immune response during viral infection. J. Clin. Investig. 2013, 123, 1310–1322. [Google Scholar] [CrossRef]
- Weithauser, A.; Bobbert, P.; Antoniak, S.; Böhm, A.; Rauch, B.H.; Klingel, K.; Savvatis, K.; Kroemer, H.K.; Tschope, C.; Stroux, A.; et al. Protease-Activated Receptor-2 Regulates the Innate Immune Response to Viral Infection in a Coxsackievirus B3–Induced Myocarditis. J. Am. Coll. Cardiol. 2013, 62, 1737–1745. [Google Scholar] [CrossRef]
- Müller, I.; Vogl, T.; Pappritz, K.; Miteva, K.; Savvatis, K.; Rohde, D.; Most, P.; Lassner, D.; Pieske, B.; Kühl, U.; et al. Pathogenic Role of the Damage-Associated Molecular Patterns S100A8 and S100A9 in Coxsackievirus B3–Induced Myocarditis. Circ. Heart Fail. 2017, 10, e004125. [Google Scholar] [CrossRef]
- Li, Y.; Chen, B.; Yang, X.; Zhang, C.; Jiao, Y.; Li, P.; Liu, Y.; Li, Z.; Qiao, B.; Bond Lau, W.; et al. S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocyte Death in Response to Ischemic/Reperfusion Injury. Circulation 2019, 140, 751–764. [Google Scholar] [CrossRef]
- Xue, Y.-L.; Zhang, S.-X.; Zheng, C.-F.; Li, Y.-F.; Zhang, L.-H.; Hao, Y.-F.; Wang, S.; Li, X.-W. Silencing of STAT4 Protects Against Autoimmune Myocarditis by Regulating Th1/Th2 Immune Response via Inactivation of the NF-κB Pathway in Rats. Inflammation 2019, 42, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Wang, W.; Morales-Nebreda, L.; Feng, G.; Wu, M.; Zhou, X.; Lafyatis, R.; Lee, J.; Hinchcliff, M.; Feghali-Bostwick, C.; et al. Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 2016, 7, 11703. [Google Scholar] [CrossRef]
- Juni, R.P.; Kuster, D.W.D.; Goebel, M.; Helmes, M.; Musters, R.J.P.; van der Velden, J.; Koolwijk, P.; Paulus, W.J.; van Hinsbergh, V.W.M. Cardiac Microvascular Endothelial Enhancement of Cardiomyocyte Function Is Impaired by Inflammation and Restored by Empagliflozin. JACC Basic Transl. Sci. 2019, 4, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chen, M.; Dawood, F.; Zurawska, U.; Li, J.Y.; Parker, T.; Kassiri, Z.; Kirshenbaum, L.A.; Arnold, M.; Khokha, R.; et al. Tumor Necrosis Factor-α Mediates Cardiac Remodeling and Ventricular Dysfunction After Pressure Overload State. Circulation 2007, 115, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A.; Sartini, D. Roles of Tumor Necrosis Factor Alpha (TNF-α) and the p55 TNF Receptor in CD1d Induction and Coxsackievirus B3-Induced Myocarditis. J. Virol. 2005, 79, 2659–2665. [Google Scholar] [CrossRef]
- Suematsu, N.; Tsutsui, H.; Wen, J.; Kang, D.; Ikeuchi, M.; Ide, T.; Hayashidani, S.; Shiomi, T.; Kubota, T.; Hamasaki, N.; et al. Oxidative Stress Mediates Tumor Necrosis Factor-α–Induced Mitochondrial DNA Damage and Dysfunction in Cardiac Myocytes. Circulation 2003, 107, 1418–1423. [Google Scholar] [CrossRef]
- Fang, M.; Zhang, A.; Du, Y.; Lu, W.; Wang, J.; Minze, L.J.; Cox, T.C.; Li, X.C.; Xing, J.; Zhang, Z. TRIM18 is a critical regulator of viral myocarditis and organ inflammation. J. Biomed. Sci. 2022, 29, 55. [Google Scholar] [CrossRef]
- Lasrado, N.; Borcherding, N.; Arumugam, R.; Starr, T.K.; Reddy, J. Dissecting the cellular landscape and transcriptome network in viral myocarditis by single-cell RNA sequencing. IScience 2022, 25, 103865. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Field, A.E.; Pitha-Rowe, P.M. Virus-induced Heterodimer Formation betweenIRF-5 and IRF-7 Modulates Assembly of theIFNA Enhanceosome in Vivo and Transcriptional Activity of IFNA Genes. J. Biol. Chem. 2003, 278, 16630–16641. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5060–5065. [Google Scholar] [CrossRef]
- Sharma, U.; Rhaleb, N.-E.; Pokharel, S.; Harding, P.; Rasoul, S.; Peng, H.; Carretero, O.A. Novel anti-inflammatory mechanisms of N-Acetyl-Ser-Asp-Lys-Pro in hypertension-induced target organ damage. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1226–H1232. [Google Scholar] [CrossRef]
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef]
- Rider, P.; Voronov, E.; Dinarello, C.A.; Apte, R.N.; Cohen, I. Alarmins: Feel the Stress. J. Immunol. 2017, 198, 1395–1402. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Averill, M.M.; Kerkhoff, C.; Bornfeldt, K.E. S100A8 and S100A9 in Cardiovascular Biology and Disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 223–229. [Google Scholar] [CrossRef]
- Yang, J.; Anholts, J.; Kolbe, U.; Stegehuis-Kamp, J.A.; Claas, F.H.J.; Eikmans, M. Calcium-Binding Proteins S100A8 and S100A9: Investigation of Their Immune Regulatory Effect in Myeloid Cells. Int. J. Mol. Sci. 2018, 19, 1833. [Google Scholar] [CrossRef]
- Crowe, L.A.N.; McLean, M.; Kitson, S.M.; Melchor, E.G.; Patommel, K.; Cao, H.M.; Reilly, J.H.; Leach, W.J.; Rooney, B.P.; Spencer, S.J.; et al. S100A8 & S100A9: Alarmin mediated inflammation in tendinopathy. Sci. Rep. 2019, 9, 1463. [Google Scholar] [CrossRef]
- Guo, Q.; Zhao, Y.; Li, J.; Liu, J.; Yang, X.; Guo, X.; Kuang, M.; Xia, H.; Zhang, Z.; Cao, L.; et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Microbe 2021, 29, 222–235.e4. [Google Scholar] [CrossRef] [PubMed]
- Szalay, G.; Sauter, M.; Hald, J.; Weinzierl, A.; Kandolf, R.; Klingel, K. Sustained Nitric Oxide Synthesis Contributes to Immunopathology in Ongoing Myocarditis Attributable to Interleukin-10 Disorders. Am. J. Pathol. 2006, 169, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.A.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Stertz, S.; Kochs, G. The Mx GTPase family of interferon-induced antiviral proteins. Microbes Infect. 2007, 9, 1636–1643. [Google Scholar] [CrossRef]
- Hwang, S.-M.; Sharma, G.; Verma, R.; Byun, S.; Rudra, D.; Im, S.-H. Inflammation-induced Id2 promotes plasticity in regulatory T cells. Nat. Commun. 2018, 9, 4736. [Google Scholar] [CrossRef]
- Gassner, F.J.; Zaborsky, N.; Catakovic, K.; Rebhandl, S.; Huemer, M.; Egle, A.; Hartmann, T.N.; Greil, R.; Geisberger, R. Chronic lymphocytic leukaemia induces an exhausted T cell phenotype in the TCL1 transgenic mouse model. Br. J. Haematol. 2015, 170, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.S.; De Labastida Rivera, F.; Yan, J.; Corvino, D.; Das, I.; Zhang, P.; Kuns, R.; Chauhan, S.B.; Hou, J.; Li, X.-Y.; et al. The NK cell granule protein NKG7 regulates cytotoxic granule exocytosis and inflammation. Nat. Immunol. 2020, 21, 1205–1218. [Google Scholar] [CrossRef]
- Basavalingappa, R.H.; Arumugam, R.; Lasrado, N.; Yalaka, B.; Massilamany, C.; Gangaplara, A.; Riethoven, J.-J.; Xiang, S.-H.; Steffen, D.; Reddy, J. Viral myocarditis involves the generation of autoreactive T cells with multiple antigen specificities that localize in lymphoid and non-lymphoid organs in the mouse model of CVB3 infection. Mol. Immunol. 2020, 124, 218–228. [Google Scholar] [CrossRef]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.E.; McKenzie, D.R.; Bastow, C.R.; Gregor, C.E.; Fenix, K.A.; Ogunniyi, A.D.; Paton, J.C.; Mack, M.; Pombal, D.R.; Seillet, C.; et al. CCR2 defines in vivo development and homing of IL-23-driven GM-CSF-producing Th17 cells. Nat. Commun. 2015, 6, 8644. [Google Scholar] [CrossRef] [PubMed]
- Deswal, A.; Petersen, N.J.; Feldman, A.M.; Young, J.B.; White, B.G.; Mann, D.L. Cytokines and Cytokine Receptors in Advanced Heart Failure. Circulation 2001, 103, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Satoh, M.; Nakamura, M.; Saitoh, H.; Satoh, H.; Maesawa, C.; Segawa, I.; Tashiro, A.; Hiramori, K. Tumor Necrosis Factor-α–Converting Enzyme and Tumor Necrosis Factor-α in Human Dilated Cardiomyopathy. Circulation 1999, 99, 3260–3265. [Google Scholar] [CrossRef]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Medeiros, N.I.; Gomes, J.A.S.; Fiuza, J.A.; Sousa, G.R.; Almeida, E.F.; Novaes, R.O.; Rocha, V.L.S.; Chaves, A.T.; Dutra, W.O.; Rocha, M.O.C.; et al. MMP-2 and MMP-9 plasma levels are potential biomarkers for indeterminate and cardiac clinical forms progression in chronic Chagas disease. Sci. Rep. 2019, 9, 14170. [Google Scholar] [CrossRef]
- Westermann, D.; Savvatis, K.; Schultheiss, H.P.; Tschöpe, C. Immunomodulation and matrix metalloproteinases in viral myocarditis. J. Mol. Cell. Cardiol. 2010, 48, 468–473. [Google Scholar] [CrossRef]
- Cheung, C.; Marchant, D.; Walker, E.K.-Y.; Luo, Z.; Zhang, J.; Yanagawa, B.; Rahmani, M.; Cox, J.; Overall, C.; Senior, R.M.; et al. Ablation of Matrix Metalloproteinase-9 Increases Severity of Viral Myocarditis in Mice. Circulation 2008, 117, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Diny, N.L.; Hou, X.; Barin, J.G.; Chen, G.; Talor, M.V.; Schaub, J.; Russell, S.D.; Klingel, K.; Rose, N.R.; Čiháková, D. Macrophages and cardiac fibroblasts are the main producers of eotaxins and regulate eosinophil trafficking to the heart. Eur. J. Immunol. 2016, 46, 2749–2760. [Google Scholar] [CrossRef]
- Daniel, R.; He, Z.; Carmichael, K.P.; Halper, J.; Bateman, A. Cellular Localization of Gene Expression for Progranulin. J. Histochem. Cytochem. 2000, 48, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dienz, O.; Roberts, B.; Moussawi, M.; Rincon, M.; Huber, S.A. IL-21R expression on CD8+ T cells promotes CD8+ T cell activation in coxsackievirus B3 induced myocarditis. Exp. Mol. Pathol. 2012, 92, 327–333. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Lin, C.-Y.; Evans, S.; Williams, J.; Dun, H.; Li, W.; Mpoy, C.; Andhey, P.S.; Rogers, B.E.; et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight 2020, 5, e134700. [Google Scholar] [CrossRef] [PubMed]
- Cen, Z.; Li, Y.; Wei, B.; Wu, W.; Huang, Y.; Lu, J. The Role of B Cells in Regulation of Th Cell Differentiation in Coxsackievirus B3–Induced Acute Myocarditis. Inflammation 2021, 44, 1949–1960. [Google Scholar] [CrossRef]
- Wei, B.; Deng, Y.; Huang, Y.; Gao, X.; Wu, W. IL-10-producing B cells attenuate cardiac inflammation by regulating Th1 and Th17 cells in acute viral myocarditis induced by coxsackie virus B3. Life Sci. 2019, 235, 116838. [Google Scholar] [CrossRef]
- Yu, M.; Wen, S.; Wang, M.; Liang, W.; Li, H.-H.; Long, Q.; Guo, H.P.; Liao, Y.-H.; Yuan, J. TNF-α-Secreting B Cells Contribute to Myocardial Fibrosis in Dilated Cardiomyopathy. J. Clin. Immunol. 2013, 33, 1002–1008. [Google Scholar] [CrossRef]
- Stoian, I.; Oros, A.; Moldoveanu, E. Apoptosis and Free Radicals. Biochem. Mol. Med. 1996, 59, 93–97. [Google Scholar] [CrossRef]
- Tsutamoto, T.; Wada, A.; Matsumoto, T.; Maeda, K.; Mabuchi, N.; Hayashi, M.; Tsutsui, T.; Ohnishi, M.; Sawaki, M.; Fujii, M.; et al. Relationship between tumor necrosis factor-alpha production and oxidative stress in the failing hearts of patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2001, 37, 2086–2092. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, H.; Yu, J.; Wei, L.; Li, M.; Xu, W. Stem cell factor/mast cell/CCL2/monocyte/macrophage axis promotes Coxsackievirus B3 myocarditis and cardiac fibrosis by increasing Ly6Chigh monocyte influx and fibrogenic mediators production. Immunology 2022, 167, 590–605. [Google Scholar] [CrossRef]
- Hamdani, N.; Herwig, M.; Linke, W.A. Tampering with springs: Phosphorylation of titin affecting the mechanical function of cardiomyocytes. Biophys. Rev. 2017, 9, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Kötter, S.; Kazmierowska, M.; Andresen, C.; Bottermann, K.; Grandoch, M.; Gorressen, S.; Heinen, A.; Moll, J.M.; Scheller, J.; Gödecke, A.; et al. Titin-Based Cardiac Myocyte Stiffening Contributes to Early Adaptive Ventricular Remodeling After Myocardial Infarction. Circ. Res. 2016, 119, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Kötter, S.; Gout, L.; Von Frieling-Salewsky, M.; Müller, A.E.; Helling, S.; Marcus, K.; Dos Remedios, C.; Linke, W.A.; Krüger, M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc. Res. 2013, 99, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Therapeutic Targeting of the Interleukin-6 Receptor. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Sébert, M.; Sola-Tapias, N.; Mas, E.; Barreau, F.; Ferrand, A. Protease-Activated Receptors in the Intestine: Focus on Inflammation and Cancer. Front. Endocrinol. 2019, 10, 717. [Google Scholar] [CrossRef]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 4. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, Z.; Lim, T.; Zhang, H.; He, J.; Walker, E.; Shier, C.; Wang, Y.; Su, Y.; Sall, A.; et al. CXCL10 Inhibits Viral Replication Through Recruitment of Natural Killer Cells in Coxsackievirus B3-Induced Myocarditis. Circ. Res. 2009, 104, 628–638. [Google Scholar] [CrossRef]
- Bode, M.F.; Schmedes, C.M.; Egnatz, G.J.; Bharathi, V.; Hisada, Y.M.; Martinez, D.; Kawano, T.; Weithauser, A.; Rosenfeldt, L.; Rauch, U.; et al. Cell type-specific roles of PAR1 in Coxsackievirus B3 infection. Sci. Rep. 2021, 11, 14264. [Google Scholar] [CrossRef] [PubMed]
- Mohamud, Y.; Shi, J.; Qu, J.; Poon, T.; Xue, Y.C.; Deng, H.; Zhang, J.; Luo, H. Enteroviral Infection Inhibits Autophagic Flux via Disruption of the SNARE Complex to Enhance Viral Replication. Cell Rep. 2018, 22, 3292–3303. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Protein | Myocarditis/ Cardiomyopathy | Function | Effects during CVB3 Infection | Reference |
|---|---|---|---|---|
| Dpep2 | Alleviate | Inhibits macrophage NF-κB signaling | Reduced inflammation | [79] |
| IL-3, IL-4, IL-9, IL-13, IL-15 (synergy) | Alleviate | Recruits helper T cells | Reduced cardiomyocyte necrosis Reduced fibrosis Reduced LV anomalies Reduced viral load | [80] |
| IL-9 | Alleviate | Reduces cardiomyocyte CAR expression Inhibits Th17 differentiation | Reduced viral load | [80,81] |
| IL-10 | Alleviate | Inhibits TNF-α-induced apoptosis | Reduced cardiomyocyte apoptosis | [82] |
| PGE2 | Alleviate | Induces anti-inflammatory effects on neutrophils and macrophages Induces B10 expansion Suppresses Th17 differentiation | Reduced LV anomalies Reduced inflammation | [83,84,85,86] |
| PGRN | Alleviate | Inhibits Th1 differentiation Inhibits Th17 differentiation | Reduced inflammation Reduced cardiomyocyte necrosis Reduced viral load | [87] |
| PPAR-α | Alleviate | Inhibits NF-κB Inhibits Th17 differentiation | Reduced inflammation | [67,68] |
| Stabilin-1 | Alleviate | Increases monocyte recruitment Promotes monocyte differentiation into regulatory macrophages | Reduced inflammation Reduced cardiomyocyte necrosis Reduced mouse mortality | [88,89] |
| APN | Exacerbate | Decreases TLR signaling Increases cytokine production | Increased inflammation Increased viral load Increased LV anomalies | [63] |
| Calpain-1 | Exacerbate | Increases pyroptosis Promotes mitochondrial dysfunction | Increased cardiomyocyte necrosis Increased fibrosis | [49] |
| CCR2 | Exacerbate | Increases monocyte recruitment | Increased fibrosis Increased inflammation | [90,91] |
| CD80 | Exacerbate | Increases IL-17 production | Increased cardiomyocyte necrosis | [92] |
| Gal-3 | Exacerbate | Induces transition from fibroblasts to myofibroblasts Mediates the migration of macrophages towards fibroblasts | Increased fibrosis Increased inflammation | [93] |
| GM-CSF | Exacerbate | Increases Th17 differentiation | Increased inflammation | [94,95] |
| IL-17A | Exacerbate | Increases pro-fibrotic cytokines Increases pro-inflammatory cytokine expression Increases Th17 differentiation Recruits myeloid cells | Increased fibrosis Increased inflammation Increased LV anomalies | [96] |
| IL-1β | Exacerbate | Increases matricellular protein expression Increases Th17 differentiation | Increased fibrosis Increased inflammation Increased LV anomalies | [97,98] |
| IL-23 | Exacerbate | Increases GM-CSF secretion | Increased inflammation | [94] |
| IL-6 | Exacerbate | Induces titin phosphorylation Increases Th17 differentiation Mediates TNF-α expression Recruits T cells and macrophages | Increased cardiomyocyte necrosis Increased fibrosis Increased LV anomalies Increased viral load | [99] |
| IRAK4 | Exacerbate | Inhibits IFN production Inhibits migration of protective macrophages and monocytes | Increased LV anomalies Increased viral load | [100] |
| OPN | Exacerbate | Increases insoluble collagen | Increased fibrosis Increased LV anomalies | [97,101] |
| PAR1/2 | Exacerbate | Increases CAR and DAF expression Reduces autophagic flux | Increased inflammation Increased LV anomalies Increased viral load | [102,103] |
| S100A9 | Exacerbate | Increases ROS production Inhibits mitochondrial complex 1 | Increased inflammation Increased LV anomalies Increased viral load Mitochondrial respiratory dysfunction | [104,105] |
| STAT4 | Exacerbate | Upregulates NF-κB pathway | Increased cardiomyocyte necrosis Increased fibrosis Increased inflammation | [106] |
| TN-C | Exacerbate | Increases fibroblast migration | Increased fibrosis | [97,107] |
| TNF-α | Exacerbate | Increases ROS production Activates autoimmune CD8+ T cells | Increased cardiomyocyte apoptosis Increased fibrosis Increased inflammation Increased LV anomalies | [108,109,110,111] |
| TRIM18 | Exacerbate | Inhibits IFN production | Increased inflammation Increased LV anomalies | [112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yip, F.; Lai, B.; Yang, D. Role of Coxsackievirus B3-Induced Immune Responses in the Transition from Myocarditis to Dilated Cardiomyopathy and Heart Failure. Int. J. Mol. Sci. 2023, 24, 7717. https://doi.org/10.3390/ijms24097717
Yip F, Lai B, Yang D. Role of Coxsackievirus B3-Induced Immune Responses in the Transition from Myocarditis to Dilated Cardiomyopathy and Heart Failure. International Journal of Molecular Sciences. 2023; 24(9):7717. https://doi.org/10.3390/ijms24097717
Chicago/Turabian StyleYip, Fione, Brian Lai, and Decheng Yang. 2023. "Role of Coxsackievirus B3-Induced Immune Responses in the Transition from Myocarditis to Dilated Cardiomyopathy and Heart Failure" International Journal of Molecular Sciences 24, no. 9: 7717. https://doi.org/10.3390/ijms24097717
APA StyleYip, F., Lai, B., & Yang, D. (2023). Role of Coxsackievirus B3-Induced Immune Responses in the Transition from Myocarditis to Dilated Cardiomyopathy and Heart Failure. International Journal of Molecular Sciences, 24(9), 7717. https://doi.org/10.3390/ijms24097717