p38 MAPK in Glucose Metabolism of Skeletal Muscle: Beneficial or Harmful?

{kind=link}
Abstract
1. Introduction
1.1. Skeletal Muscle Energy Metabolism
1.1.1. Glucose Uptake
1.1.2. Fiber Type and Glucose Metabolism
1.2. P38 MAPK Is Involved in Various Aspects of Whole-Body Energy Metabolism
1.2.1. PPAR Gamma Activator-1 Alpha (PGC1α)
1.2.2. Activating Transcription Factor 2 (ATF2)
1.2.3. CCAAT/Enhancer-Binding Protein α (C/EBPα)
1.2.4. cAMP Response Element-Binding Protein (CREB)
1.2.5. Glycogen Synthase (GS)
1.2.6. Peroxisome Proliferator-Activated Receptor α (PPARα)
1.2.7. X-Box Binding Protein 1 (Xbp1)
2. Regulation of Glucose Metabolism by p38 MAPK in the Adaptation of Skeletal Muscle to Exercise
2.1. Glucose Uptake
2.2. Mitochondrial Activities
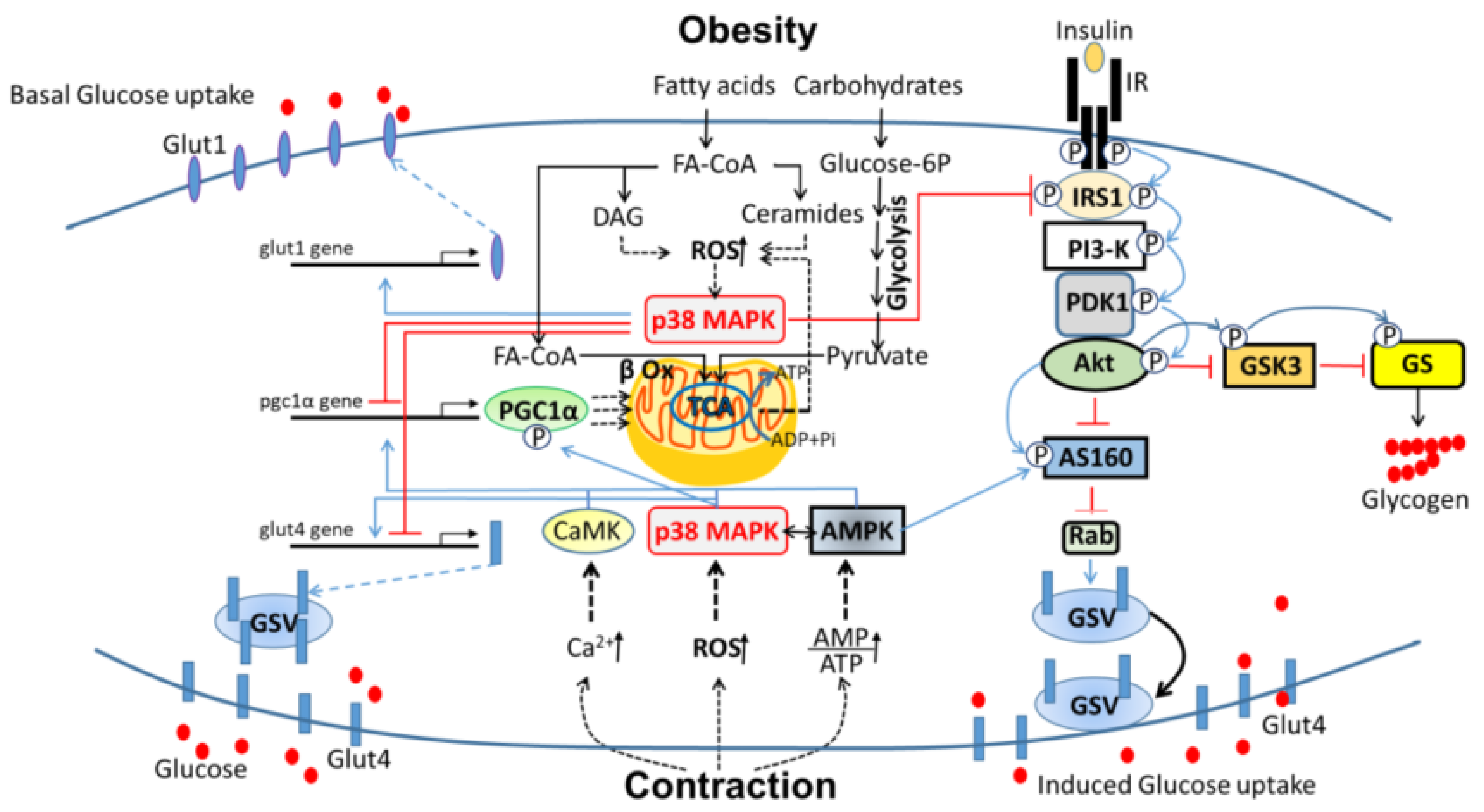
3. P38 MAPK in the Development of Insulin Resistance and Type 2 Diabetes (T2D)
3.1. Insulin Resistance
3.2. P38 MAPK in Skeletal Muscle Insulin Resistance
3.3. P38 MAPK as a Potential Target for Prevention of Obesity-Induced T2DM
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| MAPK | Mitogen-Activated Protein Kinase |
| ROS | reactive oxygen species |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; |
| SAPK | Stress-activated protein kinase |
| T2D | Type 2 Diabetes |
| AS160 | Akt substrate of 160 kDa |
| IRS | Insulin receptor substrate |
| Glut | Glucose transporter |
References
- Zurlo, F.; Larson, K.; Bogardus, C.; Ravussin, E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Investig. 1990, 86, 1423–1427. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Sato, Y.; Felig, P.; Wahren, J. Synergistic interaction between exercise and insulin on peripheral glucose uptake. J. Clin. Investig. 1981, 68, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Kriketos, A.D.; Baur, L.A.; O’Connor, J.; Carey, D.; King, S.; Caterson, I.D.; Storlien, L.H. Muscle fibre type composition in infant and adult populations and relationships with obesity. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1997, 21, 796–801. [Google Scholar] [CrossRef]
- Thayer, R.; Collins, J.; Noble, E.G.; Taylor, A.W. A decade of aerobic endurance training: Histological evidence for fibre type transformation. J. Sports Med. Phys. Fit. 2000, 40, 284–289. [Google Scholar]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef]
- Fan, M.; Rhee, J.; St-Pierre, J.; Handschin, C.; Puigserver, P.; Lin, J.; Jaeger, S.; Erdjument-Bromage, H.; Tempst, P.; Spiegelman, B.M. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: Modulation by p38 MAPK. Genes Dev. 2004, 18, 278–289. [Google Scholar] [CrossRef]
- Gupta, S.; Campbell, D.; Derijard, B.; Davis, R.J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 1995, 267, 389–393. [Google Scholar] [CrossRef]
- Cao, W.; Collins, Q.F.; Becker, T.C.; Robidoux, J.; Lupo, E.G., Jr.; Xiong, Y.; Daniel, K.W.; Floering, L.; Collins, S. p38 Mitogen-activated protein kinase plays a stimulatory role in hepatic gluconeogenesis. J. Biol. Chem. 2005, 280, 42731–42737. [Google Scholar] [CrossRef]
- Maekawa, T.; Jin, W.; Ishii, S. The role of ATF-2 family transcription factors in adipocyte differentiation: Antiobesity effects of p38 inhibitors. Mol. Cell Biol. 2010, 30, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, L.E.; Orho-Melander, M.; William-Olsson, L.; Sjoholm, K.; Sjostrom, L.; Groop, L.; Carlsson, B.; Carlsson, L.M.; Olsson, B. CCAAT/enhancer binding protein alpha (C/EBPalpha) in adipose tissue regulates genes in lipid and glucose metabolism and a genetic variation in C/EBPalpha is associated with serum levels of triglycerides. J. Clin. Endocrinol. Metab. 2008, 93, 4880–4886. [Google Scholar] [CrossRef]
- Yang, J.; Croniger, C.M.; Lekstrom-Himes, J.; Zhang, P.; Fenyus, M.; Tenen, D.G.; Darlington, G.J.; Hanson, R.W. Metabolic response of mice to a postnatal ablation of CCAAT/enhancer-binding protein alpha. J. Biol. Chem. 2005, 280, 38689–38699. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; MacDougald, O.A.; Shao, J. CCAAT/enhancer-binding protein alpha mediates induction of hepatic phosphoenolpyruvate carboxykinase by p38 mitogen-activated protein kinase. J. Biol. Chem. 2006, 281, 24390–24397. [Google Scholar] [CrossRef]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Collins, Q.F.; Xiong, Y.; Lupo, E.G., Jr.; Liu, H.Y.; Cao, W. p38 Mitogen-activated protein kinase mediates free fatty acid-induced gluconeogenesis in hepatocytes. J. Biol. Chem. 2006, 281, 24336–24344. [Google Scholar] [CrossRef]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef]
- Kuma, Y.; Campbell, D.G.; Cuenda, A. Identification of glycogen synthase as a new substrate for stress-activated protein kinase 2b/p38beta. Biochem. J. 2004, 379, 133–139. [Google Scholar] [CrossRef]
- Lawrence, J.C., Jr.; Roach, P.J. New insights into the role and mechanism of glycogen synthase activation by insulin. Diabetes 1997, 46, 541–547. [Google Scholar] [CrossRef]
- Barger, P.M.; Browning, A.C.; Garner, A.N.; Kelly, D.P. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: A potential role in the cardiac metabolic stress response. J. Biol. Chem. 2001, 276, 44495–44501. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Roth, R.J.; Anderson, E.J.; Hong, E.G.; Lee, M.K.; Choi, C.S.; Neufer, P.D.; Shulman, G.I.; Kim, J.K.; Bennett, A.M. Mice lacking MAP kinase phosphatase-1 have enhanced MAP kinase activity and resistance to diet-induced obesity. Cell Metab. 2006, 4, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Collins, Q.F.; An, J.; Lupo, E., Jr.; Liu, H.Y.; Liu, D.; Robidoux, J.; Liu, Z.; Cao, W. p38 mitogen-activated protein kinase plays an inhibitory role in hepatic lipogenesis. J. Biol. Chem. 2007, 282, 4975–4982. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lee, J.; Reno, C.M.; Sun, C.; Park, S.W.; Chung, J.; Lee, J.; Fisher, S.J.; White, M.F.; Biddinger, S.B.; et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat. Med. 2011, 17, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef]
- Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [CrossRef]
- Borst, S.E. Interventions for sarcopenia and muscle weakness in older people. Age Ageing 2004, 33, 548–555. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef]
- Sigal, R.J.; Kenny, G.P.; Boule, N.G.; Wells, G.A.; Prud’homme, D.; Fortier, M.; Reid, R.D.; Tulloch, H.; Coyle, D.; Phillips, P.; et al. Effects of aerobic training, resistance training, or both on glycemic control in type 2 diabetes: A randomized trial. Ann. Intern. Med. 2007, 147, 357–369. [Google Scholar] [CrossRef]
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef]
- Boppart, M.D.; Hirshman, M.F.; Sakamoto, K.; Fielding, R.A.; Goodyear, L.J. Static stretch increases c-Jun NH2-terminal kinase activity and p38 phosphorylation in rat skeletal muscle. Am. J. Physiol. Cell Physiol. 2001, 280, C352–C358. [Google Scholar] [CrossRef] [PubMed]
- Coffey, V.G.; Zhong, Z.; Shield, A.; Canny, B.J.; Chibalin, A.V.; Zierath, J.R.; Hawley, J.A. Early signaling responses to divergent exercise stimuli in skeletal muscle from well-trained humans. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Stepto, N.K.; Chibalin, A.V.; Fryer, L.G.; Carling, D.; Krook, A.; Hawley, J.A.; Zierath, J.R. Metabolic and mitogenic signal transduction in human skeletal muscle after intense cycling exercise. J. Physiol. 2003, 546, 327–335. [Google Scholar] [CrossRef]
- Mounier, R.; Theret, M.; Lantier, L.; Foretz, M.; Viollet, B. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol. Metab. TEM 2015, 26, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.A.; Moylan, J.S.; Smith, J.D.; Goodyear, L.J.; Reid, M.B. Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. J. Physiol. 2009, 587, 3363–3373. [Google Scholar] [CrossRef] [PubMed]
- Combes, A.; Dekerle, J.; Webborn, N.; Watt, P.; Bougault, V.; Daussin, F.N. Exercise-induced metabolic fluctuations influence AMPK, p38-MAPK and CaMKII phosphorylation in human skeletal muscle. Physiol. Rep. 2015, 3, e12462. [Google Scholar] [CrossRef]
- Kim, N.; Lee, J.O.; Lee, H.J.; Lee, Y.W.; Kim, H.I.; Kim, S.J.; Park, S.H.; Lee, C.S.; Ryoo, S.W.; Hwang, G.S.; et al. AMPK, a metabolic sensor, is involved in isoeugenol-induced glucose uptake in muscle cells. J. Endocrinol. 2016, 228, 105–114. [Google Scholar] [CrossRef]
- Lemieux, K.; Konrad, D.; Klip, A.; Marette, A. The AMP-activated protein kinase activator AICAR does not induce GLUT4 translocation to transverse tubules but stimulates glucose uptake and p38 mitogen-activated protein kinases alpha and beta in skeletal muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1658–1665. [Google Scholar] [CrossRef]
- Zetser, A.; Gredinger, E.; Bengal, E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J. Biol. Chem. 1999, 274, 5193–5200. [Google Scholar] [CrossRef]
- Zhao, M.; New, L.; Kravchenko, V.V.; Kato, Y.; Gram, H.; di Padova, F.; Olson, E.N.; Ulevitch, R.J.; Han, J. Regulation of the MEF2 family of transcription factors by p38. Mol. Cell Biol. 1999, 19, 21–30. [Google Scholar] [CrossRef]
- McGee, S.L.; Hargreaves, M. Exercise and myocyte enhancer factor 2 regulation in human skeletal muscle. Diabetes 2004, 53, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Somwar, R.; Perreault, M.; Kapur, S.; Taha, C.; Sweeney, G.; Ramlal, T.; Kim, D.Y.; Keen, J.; Cote, C.H.; Klip, A.; et al. Activation of p38 mitogen-activated protein kinase alpha and beta by insulin and contraction in rat skeletal muscle: Potential role in the stimulation of glucose transport. Diabetes 2000, 49, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.N.; Huang, C.; Niu, W.; Liu, Z.; Eyers, P.A.; Heidenreich, K.A.; Bilan, P.J.; Klip, A. Reduction of insulin-stimulated glucose uptake in L6 myotubes by the protein kinase inhibitor SB203580 is independent of p38MAPK activity. Endocrinology 2005, 146, 3773–3781. [Google Scholar] [CrossRef]
- Ribe, D.; Yang, J.; Patel, S.; Koumanov, F.; Cushman, S.W.; Holman, G.D. Endofacial competitive inhibition of glucose transporter-4 intrinsic activity by the mitogen-activated protein kinase inhibitor SB203580. Endocrinology 2005, 146, 1713–1717. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Somwar, R.; Koterski, S.; Sweeney, G.; Sciotti, R.; Djuric, S.; Berg, C.; Trevillyan, J.; Scherer, P.E.; Rondinone, C.M.; Klip, A. A dominant-negative p38 MAPK mutant and novel selective inhibitors of p38 MAPK reduce insulin-stimulated glucose uptake in 3T3-L1 adipocytes without affecting GLUT4 translocation. J. Biol. Chem. 2002, 277, 50386–50395. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, M.; Gotoh, Y.; Katagiri, H.; Sakoda, H.; Ogihara, T.; Anai, M.; Onishi, Y.; Ono, H.; Funaki, M.; Inukai, K.; et al. MKK6/3 and p38 MAPK pathway activation is not necessary for insulin-induced glucose uptake but regulates glucose transporter expression. J. Biol. Chem. 2001, 276, 19800–19806. [Google Scholar] [CrossRef]
- Geiger, P.C.; Wright, D.C.; Han, D.H.; Holloszy, J.O. Activation of p38 MAP kinase enhances sensitivity of muscle glucose transport to insulin. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E782–E788. [Google Scholar] [CrossRef][Green Version]
- Kim, J.H.; Lee, J.O.; Lee, S.K.; Moon, J.W.; You, G.Y.; Kim, S.J.; Park, S.H.; Park, J.M.; Lim, S.Y.; Suh, P.G.; et al. The glutamate agonist homocysteine sulfinic acid stimulates glucose uptake through the calcium-dependent AMPK-p38 MAPK-protein kinase C zeta pathway in skeletal muscle cells. J. Biol. Chem. 2011, 286, 7567–7576. [Google Scholar] [CrossRef]
- Sylow, L.; Nielsen, I.L.; Kleinert, M.; Moller, L.L.; Ploug, T.; Schjerling, P.; Bilan, P.J.; Klip, A.; Jensen, T.E.; Richter, E.A. Rac1 governs exercise-stimulated glucose uptake in skeletal muscle through regulation of GLUT4 translocation in mice. J. Physiol. 2016, 594, 4997–5008. [Google Scholar] [CrossRef]
- Henriquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun. 2019, 10, 4623. [Google Scholar] [CrossRef]
- Ho, R.C.; Alcazar, O.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. p38gamma MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R342–R349. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Baar, K.; Wende, A.R.; Jones, T.E.; Marison, M.; Nolte, L.A.; Chen, M.; Kelly, D.P.; Holloszy, J.O. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Calvo, J.A.; Daniels, T.G.; Wang, X.; Paul, A.; Lin, J.; Spiegelman, B.M.; Stevenson, S.C.; Rangwala, S.M. Muscle-specific expression of PPARgamma coactivator-1alpha improves exercise performance and increases peak oxygen uptake. J. Appl. Physiol. 2008, 104, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Kang, C. Role of PGC-1alpha in sarcopenia: Etiology and potential intervention—A mini-review. Gerontology 2015, 61, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef]
- Perez-Schindler, J.; Summermatter, S.; Santos, G.; Zorzato, F.; Handschin, C. The transcriptional coactivator PGC-1alpha is dispensable for chronic overload-induced skeletal muscle hypertrophy and metabolic remodeling. Proc. Natl. Acad. Sci. USA 2013, 110, 20314–20319. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. USA 2000, 97, 14400–14405. [Google Scholar] [CrossRef]
- Wu, H.; Rothermel, B.; Kanatous, S.; Rosenberg, P.; Naya, F.J.; Shelton, J.M.; Hutcheson, K.A.; DiMaio, J.M.; Olson, E.N.; Bassel-Duby, R.; et al. Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO J. 2001, 20, 6414–6423. [Google Scholar] [CrossRef]
- Puigserver, P.; Adelmant, G.; Wu, Z.; Fan, M.; Xu, J.; O‘Malley, B.; Spiegelman, B.M. Activation of PPARgamma coactivator-1 through transcription factor docking. Science 1999, 286, 1368–1371. [Google Scholar] [CrossRef]
- Knutti, D.; Kressler, D.; Kralli, A. Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proc. Natl. Acad. Sci. USA 2001, 98, 9713–9718. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef]
- Pogozelski, A.R.; Geng, T.; Li, P.; Yin, X.; Lira, V.A.; Zhang, M.; Chi, J.T.; Yan, Z. p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS ONE 2009, 4, e7934. [Google Scholar] [CrossRef]
- Foster, W.H.; Tidball, J.G.; Wang, Y. p38gamma activity is required for maintenance of slow skeletal muscle size. Muscle Nerve 2012, 45, 266–273. [Google Scholar] [CrossRef]
- Meyer, C.; Dostou, J.M.; Welle, S.L.; Gerich, J.E. Role of human liver, kidney, and skeletal muscle in postprandial glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E419–E427. [Google Scholar] [CrossRef]
- Bonen, A.; Parolin, M.L.; Steinberg, G.R.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Heigenhauser, G.J.; Dyck, D.J. Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 1144–1146. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Neufer, P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. TEM 2012, 23, 142–153. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef]
- Gehart, H.; Kumpf, S.; Ittner, A.; Ricci, R. MAPK signalling in cellular metabolism: Stress or wellness? EMBO Rep. 2010, 11, 834–840. [Google Scholar] [CrossRef]
- de Alvaro, C.; Teruel, T.; Hernandez, R.; Lorenzo, M. Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. J. Biol. Chem. 2004, 279, 17070–17078. [Google Scholar] [CrossRef]
- Kim, S.J.; Roy, R.R.; Kim, J.A.; Zhong, H.; Haddad, F.; Baldwin, K.M.; Edgerton, V.R. Gene expression during inactivity-induced muscle atrophy: Effects of brief bouts of a forceful contraction countermeasure. J. Appl. Physiol. 2008, 105, 1246–1254. [Google Scholar] [CrossRef]
- Dokken, B.B.; Saengsirisuwan, V.; Kim, J.S.; Teachey, M.K.; Henriksen, E.J. Oxidative stress-induced insulin resistance in rat skeletal muscle: Role of glycogen synthase kinase-3. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E615–E621. [Google Scholar] [CrossRef]
- Koistinen, H.A.; Chibalin, A.V.; Zierath, J.R. Aberrant p38 mitogen-activated protein kinase signalling in skeletal muscle from Type 2 diabetic patients. Diabetologia 2003, 46, 1324–1328. [Google Scholar] [CrossRef]
- Archuleta, T.L.; Lemieux, A.M.; Saengsirisuwan, V.; Teachey, M.K.; Lindborg, K.A.; Kim, J.S.; Henriksen, E.J. Oxidant stress-induced loss of IRS-1 and IRS-2 proteins in rat skeletal muscle: Role of p38 MAPK. Free Radic. Biol. Med. 2009, 47, 1486–1493. [Google Scholar] [CrossRef]
- Crunkhorn, S.; Dearie, F.; Mantzoros, C.; Gami, H.; da Silva, W.S.; Espinoza, D.; Faucette, R.; Barry, K.; Bianco, A.C.; Patti, M.E. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: Potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 2007, 282, 15439–15450. [Google Scholar] [CrossRef]
- Kawamoto, E.; Koshinaka, K.; Yoshimura, T.; Masuda, H.; Kawanaka, K. Immobilization rapidly induces muscle insulin resistance together with the activation of MAPKs (JNK and p38) and impairment of AS160 phosphorylation. Physiol. Rep. 2016, 4, e12876. [Google Scholar] [CrossRef]
- Brown, A.E.; Palsgaard, J.; Borup, R.; Avery, P.; Gunn, D.A.; De Meyts, P.; Yeaman, S.J.; Walker, M. p38 MAPK activation upregulates proinflammatory pathways in skeletal muscle cells from insulin-resistant type 2 diabetic patients. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E63–E70. [Google Scholar] [CrossRef]
- Talbot, N.A.; Wheeler-Jones, C.P.; Cleasby, M.E. Palmitoleic acid prevents palmitic acid-induced macrophage activation and consequent p38 MAPK-mediated skeletal muscle insulin resistance. Mol. Cell. Endocrinol. 2014, 393, 129–142. [Google Scholar] [CrossRef]
- Daugaard, J.R.; Nielsen, J.N.; Kristiansen, S.; Andersen, J.L.; Hargreaves, M.; Richter, E.A. Fiber type-specific expression of GLUT4 in human skeletal muscle: Influence of exercise training. Diabetes 2000, 49, 1092–1095. [Google Scholar] [CrossRef]
- Lawan, A.; Min, K.; Zhang, L.; Canfran-Duque, A.; Jurczak, M.J.; Camporez, J.P.G.; Nie, Y.; Gavin, T.P.; Shulman, G.I.; Fernandez-Hernando, C.; et al. Skeletal Muscle-Specific Deletion of MKP-1 Reveals a p38 MAPK/JNK/Akt Signaling Node That Regulates Obesity-Induced Insulin Resistance. Diabetes 2018, 67, 624–635. [Google Scholar] [CrossRef]
- Roth, R.J.; Le, A.M.; Zhang, L.; Kahn, M.; Samuel, V.T.; Shulman, G.I.; Bennett, A.M. MAPK phosphatase-1 facilitates the loss of oxidative myofibers associated with obesity in mice. J. Clin. Investig. 2009, 119, 3817–3829. [Google Scholar] [CrossRef]
- Bost, F.; Aouadi, M.; Caron, L.; Even, P.; Belmonte, N.; Prot, M.; Dani, C.; Hofman, P.; Pages, G.; Pouyssegur, J.; et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes 2005, 54, 402–411. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef]
- Yoon, M.J.; Lee, G.Y.; Chung, J.J.; Ahn, Y.H.; Hong, S.H.; Kim, J.B. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes 2006, 55, 2562–2570. [Google Scholar] [CrossRef]
- Mao, X.; Kikani, C.K.; Riojas, R.A.; Langlais, P.; Wang, L.; Ramos, F.J.; Fang, Q.; Christ-Roberts, C.Y.; Hong, J.Y.; Kim, R.Y.; et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. 2006, 8, 516–523. [Google Scholar] [CrossRef]
- Sayeed, M.; Gautam, S.; Verma, D.P.; Afshan, T.; Kumari, T.; Srivastava, A.K.; Ghosh, J.K. A collagen domain-derived short adiponectin peptide activates APPL1 and AMPK signaling pathways and improves glucose and fatty acid metabolisms. J. Biol. Chem. 2018, 293, 13509–13523. [Google Scholar] [CrossRef]
- Parker, L.; Stepto, N.K.; Shaw, C.S.; Serpiello, F.R.; Anderson, M.; Hare, D.L.; Levinger, I. Acute High-Intensity Interval Exercise-Induced Redox Signaling Is Associated with Enhanced Insulin Sensitivity in Obese Middle-Aged Men. Front. Physiol. 2016, 7, 411. [Google Scholar] [CrossRef]
- Somwar, R.; Kim, D.Y.; Sweeney, G.; Huang, C.; Niu, W.; Lador, C.; Ramlal, T.; Klip, A. GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: Potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem. J. 2001, 359, 639–649. [Google Scholar] [CrossRef]
- Thong, F.S.; Derave, W.; Urso, B.; Kiens, B.; Richter, E.A. Prior exercise increases basal and insulin-induced p38 mitogen-activated protein kinase phosphorylation in human skeletal muscle. J. Appl. Physiol. 2003, 94, 2337–2341. [Google Scholar] [CrossRef]
- Ishiki, M.; Klip, A. Minireview: Recent developments in the regulation of glucose transporter-4 traffic: New signals, locations, and partners. Endocrinology 2005, 146, 5071–5078. [Google Scholar] [CrossRef]
- Klip, A. The many ways to regulate glucose transporter 4. Appl. Physiol. Nutr. Metab. 2009, 34, 481–487. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, G.; Sin, K.W.; Liu, Z.; Lin, R.K.; Li, M.; Li, Y.P. Activin A induces skeletal muscle catabolism via p38beta mitogen-activated protein kinase. J. Cachexia Sarcopenia Muscle 2017, 8, 202–212. [Google Scholar] [CrossRef]
- Paul, P.K.; Gupta, S.K.; Bhatnagar, S.; Panguluri, S.K.; Darnay, B.G.; Choi, Y.; Kumar, A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell Biol. 2010, 191, 1395–1411. [Google Scholar] [CrossRef]
- Qin, W.; Pan, J.; Wu, Y.; Bauman, W.A.; Cardozo, C. Protection against dexamethasone-induced muscle atrophy is related to modulation by testosterone of FOXO1 and PGC-1alpha. Biochem. Biophys. Res. Commun. 2010, 403, 473–478. [Google Scholar] [CrossRef]
- Zhang, G.; Jin, B.; Li, Y.P. C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011, 30, 4323–4335. [Google Scholar] [CrossRef]
- Zhang, G.; Li, Y.P. p38beta MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPbeta. Skelet. Muscle 2012, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Odeh, M.; Tamir-Livne, Y.; Haas, T.; Bengal, E. P38alpha MAPK coordinates the activities of several metabolic pathways that together induce atrophy of denervated muscles. FEBS J. 2020, 287, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Papaconstantinou, J.; Wang, C.Z.; Zhang, M.; Yang, S.; Deford, J.; Bulavin, D.V.; Ansari, N.H. Attenuation of p38alpha MAPK stress response signaling delays the in vivo aging of skeletal muscle myofibers and progenitor cells. Aging 2015, 7, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, K.; Okubo, K.; Yoda, M.; Otsu, K.; Ishii, Y.; Nakamura, M.; Itoh, Y.; Horiuchi, K. Targeted ablation of p38alpha MAPK suppresses denervation-induced muscle atrophy. Sci. Rep. 2018, 8, 9037. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.J.; Li, Y.Y. The role of p38 mitogen-activated protein kinase in the pathogenesis of inflammatory bowel disease. J. Dig. Dis. 2011, 12, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Liu, W.; Cao, H.; Zhang, D.; Yao, X.; Zhang, S.; Xia, H.; Li, D.; Wang, Y.C.; Yan, J.; et al. Hepatic p38alpha regulates gluconeogenesis by suppressing AMPK. J. Hepatol. 2015, 62, 1319–1327. [Google Scholar] [CrossRef]
- Hegarty, B.D.; Cooney, G.J.; Kraegen, E.W.; Furler, S.M. Increased efficiency of fatty acid uptake contributes to lipid accumulation in skeletal muscle of high fat-fed insulin-resistant rats. Diabetes 2002, 51, 1477–1484. [Google Scholar] [CrossRef]
- James, A.M.; Collins, Y.; Logan, A.; Murphy, M.P. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol. Metab. TEM 2012, 23, 429–434. [Google Scholar] [CrossRef]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef]
- Holloszy, J.O. “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 2013, 62, 1036–1040. [Google Scholar] [CrossRef]
- Koh, J.H.; Johnson, M.L.; Dasari, S.; LeBrasseur, N.K.; Vuckovic, I.; Henderson, G.C.; Cooper, S.A.; Manjunatha, S.; Ruegsegger, G.N.; Shulman, G.I.; et al. TFAM Enhances Fat Oxidation and Attenuates High-Fat Diet-Induced Insulin Resistance in Skeletal Muscle. Diabetes 2019, 68, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Choi, C.S.; Chin, S.; Kim, S.; Kawamori, D.; Kurpad, A.J.; Neubauer, N.; Hu, J.; Mootha, V.K.; Kim, Y.B.; et al. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J. Clin. Investig. 2007, 117, 3463–3474. [Google Scholar] [CrossRef]
- Izumiya, Y.; Hopkins, T.; Morris, C.; Sato, K.; Zeng, L.; Viereck, J.; Hamilton, J.A.; Ouchi, N.; LeBrasseur, N.K.; Walsh, K. Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008, 7, 159–172. [Google Scholar] [CrossRef] [PubMed]
- LeBrasseur, N.K.; Walsh, K.; Arany, Z. Metabolic benefits of resistance training and fast glycolytic skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E3–E10. [Google Scholar] [CrossRef] [PubMed]
- Pospisilik, J.A.; Knauf, C.; Joza, N.; Benit, P.; Orthofer, M.; Cani, P.D.; Ebersberger, I.; Nakashima, T.; Sarao, R.; Neely, G.; et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 2007, 131, 476–491. [Google Scholar] [CrossRef] [PubMed]
- Brunmair, B.; Staniek, K.; Gras, F.; Scharf, N.; Althaym, A.; Clara, R.; Roden, M.; Gnaiger, E.; Nohl, H.; Waldhausl, W.; et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: A common mechanism contributing to their antidiabetic actions? Diabetes 2004, 53, 1052–1059. [Google Scholar] [CrossRef]
- Wessels, B.; Ciapaite, J.; van den Broek, N.M.; Nicolay, K.; Prompers, J.J. Metformin impairs mitochondrial function in skeletal muscle of both lean and diabetic rats in a dose-dependent manner. PLoS ONE 2014, 9, e100525. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bengal, E.; Aviram, S.; Hayek, T. p38 MAPK in Glucose Metabolism of Skeletal Muscle: Beneficial or Harmful? Int. J. Mol. Sci. 2020, 21, 6480. https://doi.org/10.3390/ijms21186480
Bengal E, Aviram S, Hayek T. p38 MAPK in Glucose Metabolism of Skeletal Muscle: Beneficial or Harmful? International Journal of Molecular Sciences. 2020; 21(18):6480. https://doi.org/10.3390/ijms21186480
Chicago/Turabian StyleBengal, Eyal, Sharon Aviram, and Tony Hayek. 2020. "p38 MAPK in Glucose Metabolism of Skeletal Muscle: Beneficial or Harmful?" International Journal of Molecular Sciences 21, no. 18: 6480. https://doi.org/10.3390/ijms21186480
APA StyleBengal, E., Aviram, S., & Hayek, T. (2020). p38 MAPK in Glucose Metabolism of Skeletal Muscle: Beneficial or Harmful? International Journal of Molecular Sciences, 21(18), 6480. https://doi.org/10.3390/ijms21186480