
Mechanisms by Which Different Functional States of Mitochondria Define Yeast Longevity


Abstract
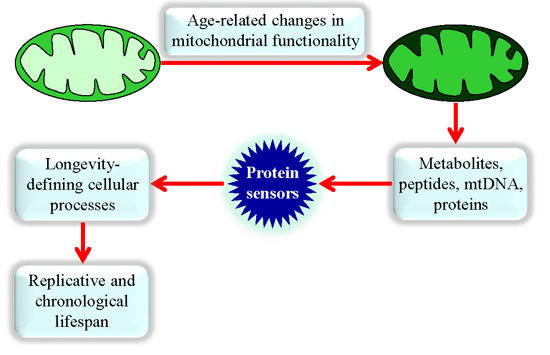
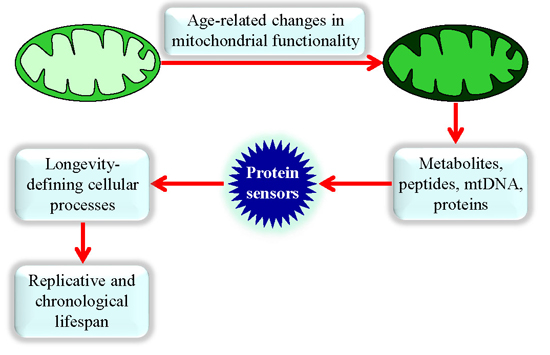
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Yeast S. cerevisiae Is a Beneficial Model Organism for Uncovering Mechanisms of Cellular Aging in Multicellular Eukaryotes
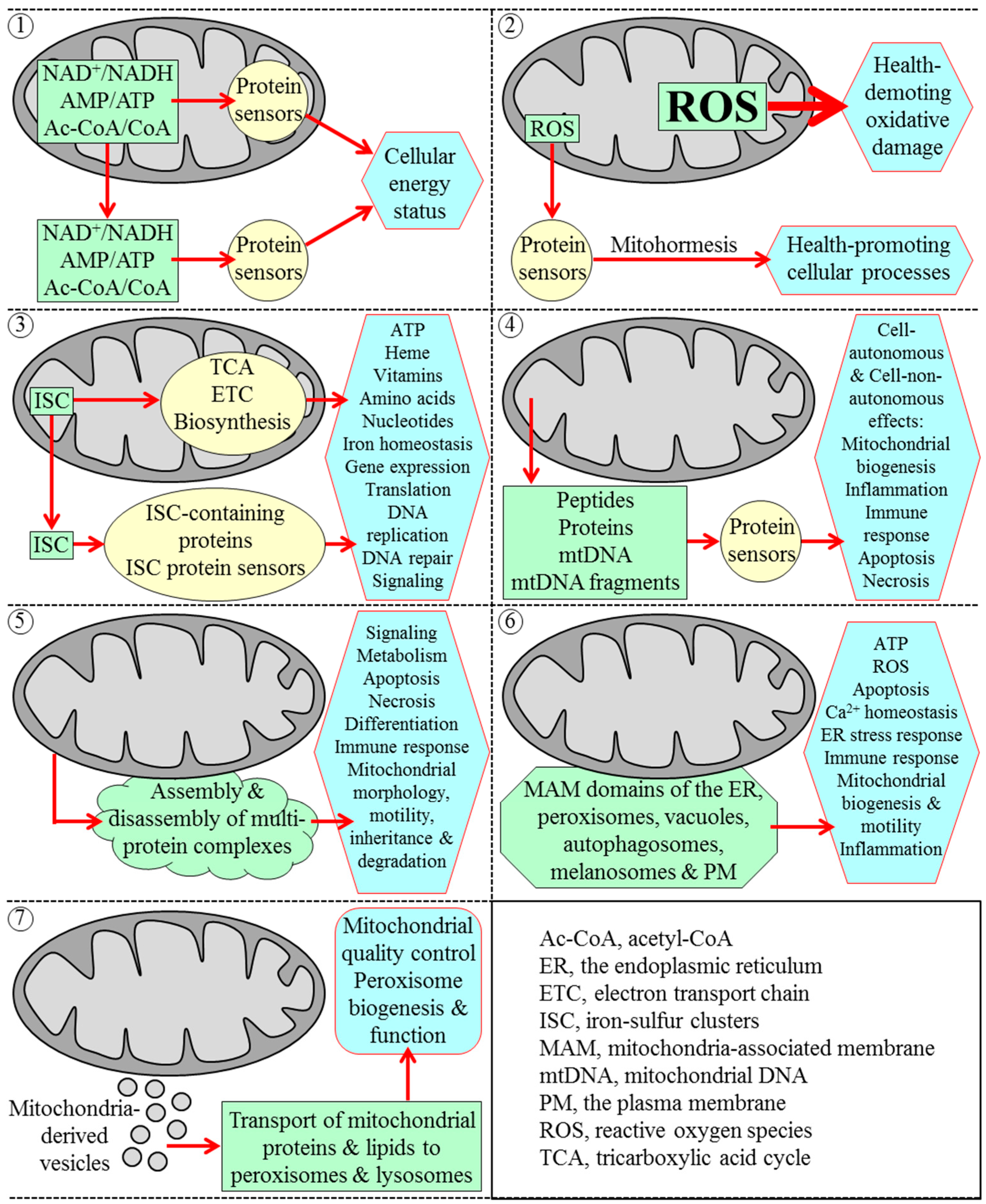
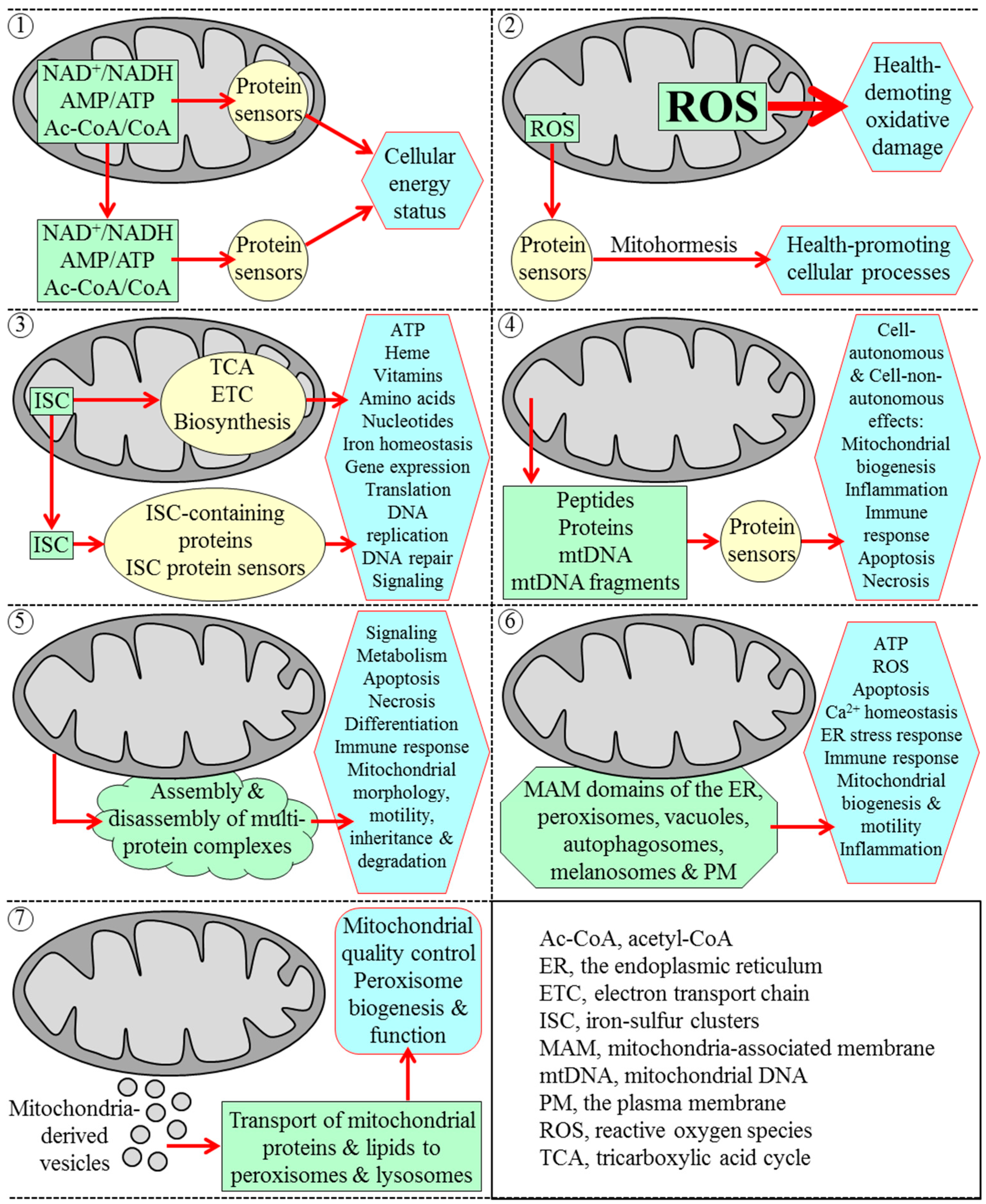
3. Mitochondria Are Signaling Organelles that Establish the Rate of Cellular Aging in Yeast by Orchestrating Many Processes Outside of Mitochondria
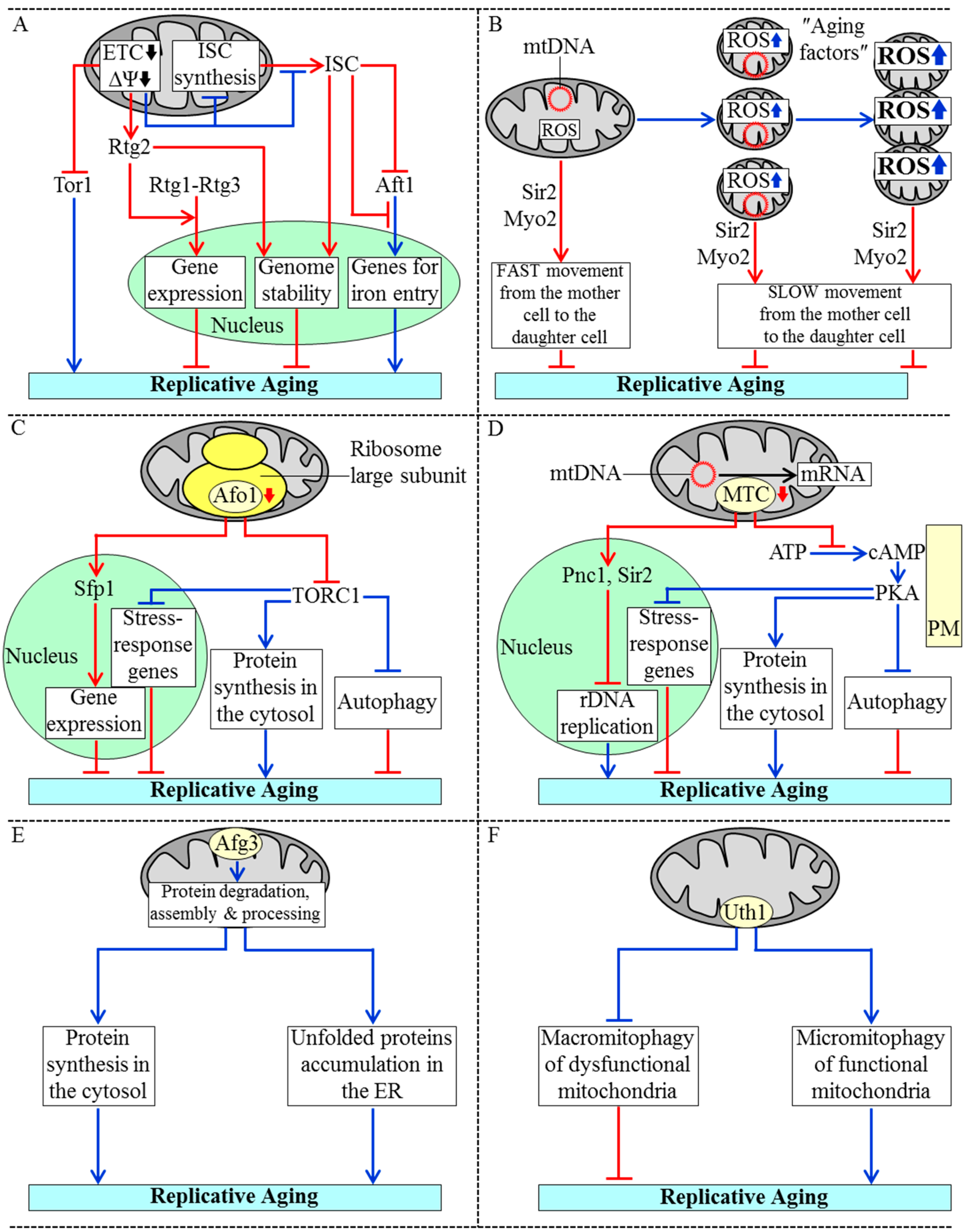
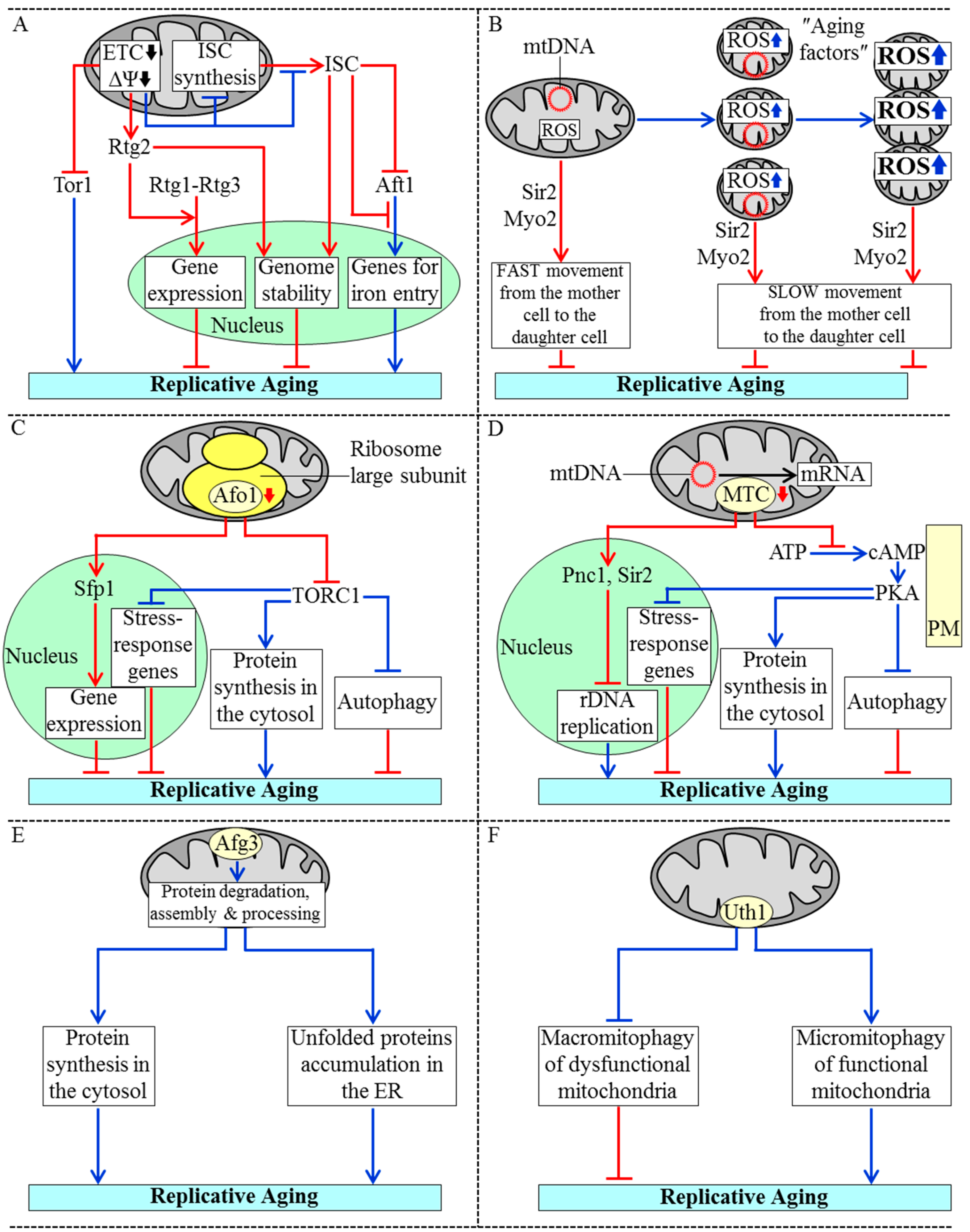
3.1. Mechanisms Underlying the Essential Role of Mitochondria in Yeast Replicative Aging
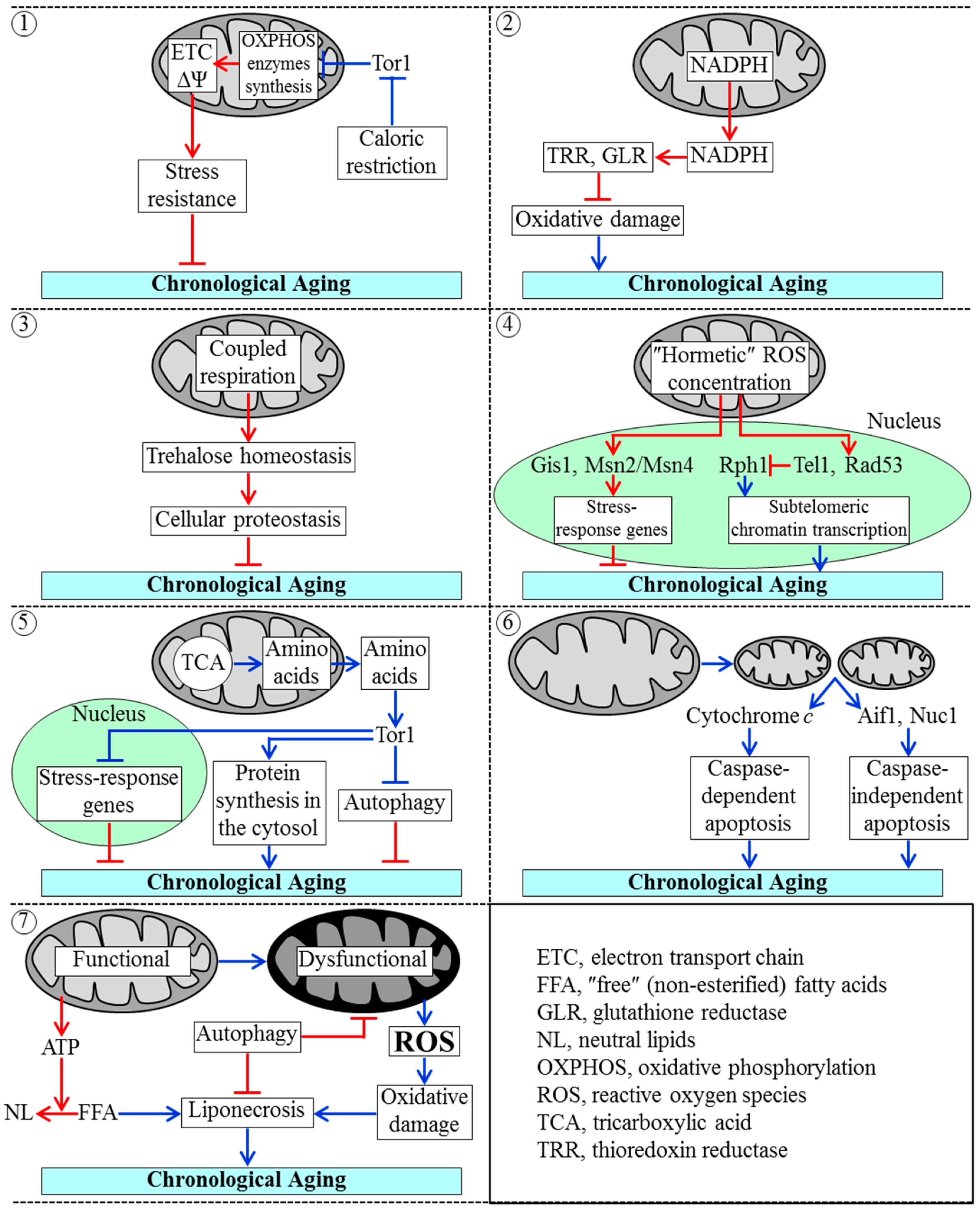
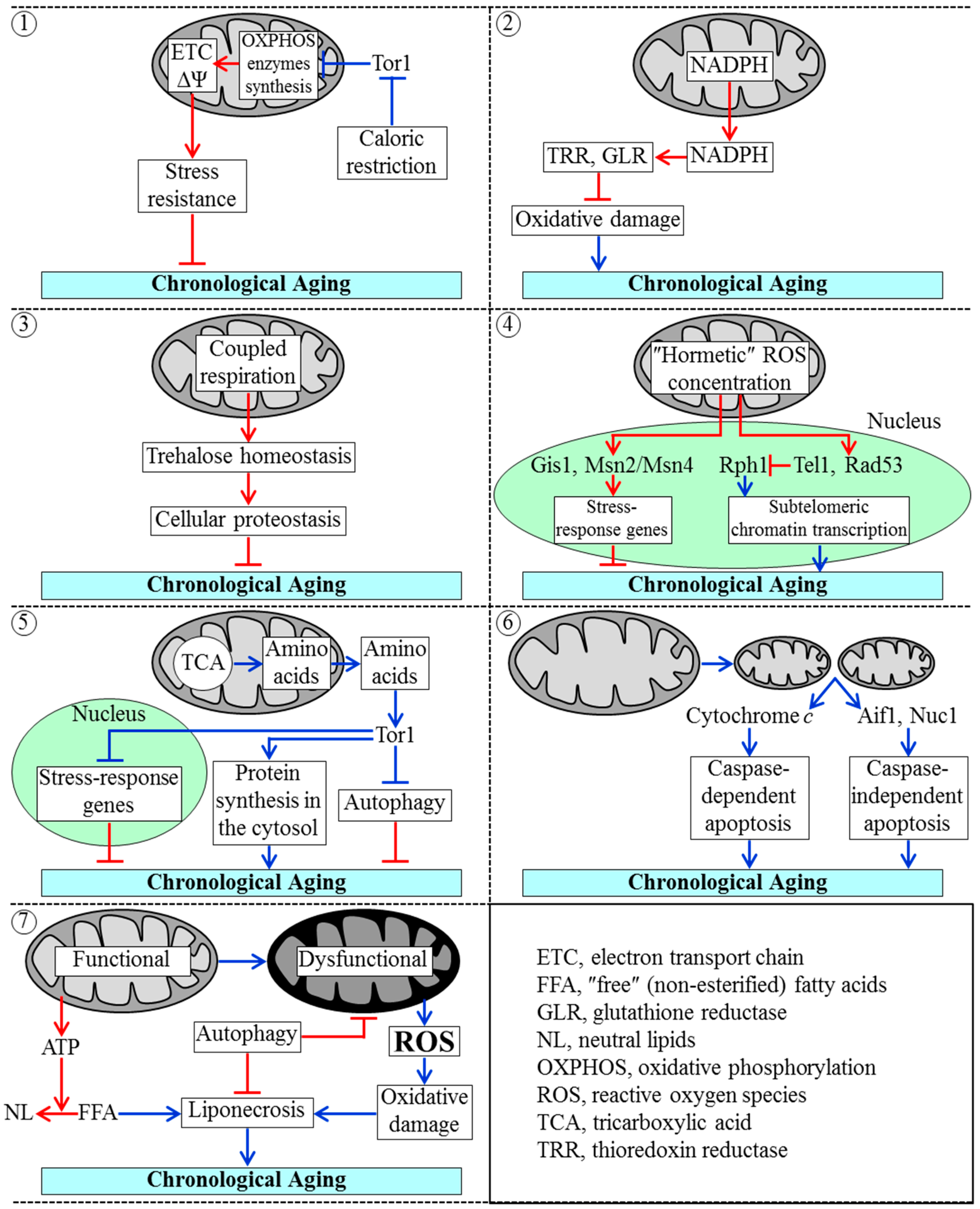
3.2. Mechanisms by Which Mitochondrial Functionality Impacts Chronological Aging of Yeast
4. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Breitenbach, M.; Laun, P.; Dickinson, J.R.; Klocker, A.; Rinnerthaler, M.; Dawes, I.W.; Aung-Htut, M.T.; Breitenbach-Koller, L.; Caballero, A.; Nyström, T.; et al. The role of mitochondria in the aging processes of yeast. Subcell. Biochem. 2012, 57, 55–78. [Google Scholar] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Rutter, J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013, 27, 2615–2627. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Power, Sex, Suicide: Mitochondria and the Meaning of Life; Oxford University Press: New York, NY, USA, 2006; p. 368. [Google Scholar]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.R., Jr.; Ballinger, S.; Dell’Italia, L.; Jianhua, Z.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Frohman, M.A. Lipid signaling on the mitochondrial surface. Biochim. Biophys. Acta 2009, 1791, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cochemé, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep. 2012, 13, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Frohman, M.A. Roles for the lipid-signaling enzyme MitoPLD in mitochondrial dynamics, piRNA biogenesis, and spermatogenesis. BMB Rep. 2012, 45, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.L.; Kornbluth, S. The tangled circuitry of metabolism and apoptosis. Mol. Cell 2013, 49, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Leonov, A.; Titorenko, V.I. A network of interorganellar communications underlies cellular aging. IUBMB Life 2013, 65, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Arlia-Ciommo, A.; Leonov, A.; Piano, A.; Svistkova, V.; Titorenko, V.I. Cell-autonomous mechanisms of chronological aging in the yeast Saccharomyces cerevisiae. Microb. Cell 2014, 1, 164–178. [Google Scholar] [CrossRef]
- Arlia-Ciommo, A.; Piano, A.; Leonov, A.; Svistkova, V.; Titorenko, V.I. Quasi-programmed aging of budding yeast: A trade-off between programmed processes of cell proliferation, differentiation, stress response, survival and death defines yeast lifespan. Cell Cycle 2014, 13, 3336–3349. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345. [Google Scholar] [CrossRef]
- Kasahara, A.; Scorrano, L. Mitochondria: From cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014, 24, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J.; Malhotra, J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1843, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Labbé, K.; Murley, A.; Nunnari, J. Determinants and functions of mitochondrial behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 357–391. [Google Scholar] [CrossRef] [PubMed]
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta 2014, 1843, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Haigis, M.C. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res. Rev. 2009, 8, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Hirschey, M.D. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012, 52, 23–35. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Newman, J.C.; Wang, M.Z.; Ho, L.; Verdin, E. Mitochondrial sirtuins: Regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 2012, 23, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Finkel, T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Wellen, K.E.; Thompson, C.B. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 270–276. [Google Scholar] [PubMed]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Burkewitz, K.; Zhang, Y.; Mair, W.B. AMPK at the nexus of energetics and aging. Cell Metab. 2014, 20, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD+-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Schroeder, S.; Andryushkova, A.; Pendl, T.; Küttner, V.; Bhukel, A.; Mariño, G.; Pietrocola, F.; Harger, A.; Zimmermann, A.; et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme A stimulates autophagy and prolongs lifespan. Cell Metab. 2014, 19, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3. [Google Scholar] [CrossRef]
- Stuart, J.A.; Maddalena, L.A.; Merilovich, M.; Robb, E.L. A midlife crisis for the mitochondrial free radical theory of aging. Longev. Healthspan 2014, 3. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, H.; Finkel, T. Unraveling the truth about antioxidants: ROS and disease: Finding the right balance. Nat. Med. 2014, 20, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.A.; Shadel, G.S. Alternative mitochondrial fuel extends life span. Cell Metab. 2012, 15, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Hwang, A.B.; Ryu, E.A.; Artan, M.; Chang, H.W.; Kabir, M.H.; Nam, H.J.; Lee, D.; Yang, J.S.; Kim, S.; Mair, W.B.; et al. Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2014, 111, E4458–E4467. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. TOR signaling couples oxygen sensing to lifespan in C. elegans. Cell Rep. 2014, 9, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chisholm, A.D. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev. Cell 2014, 31, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.; Yang, W.; Hekimi, S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 2014, 157, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Mühlenhoff, U. Maturation of iron-sulfur proteins in eukaryotes: Mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 2008, 77, 669–700. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.; Stehling, O.; Lill, R. Iron-sulfur proteins in health and disease. Trends Endocrinol. Metab. 2010, 21, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.M.; Møller, S.G. Iron-sulfur clusters: Biogenesis, molecular mechanisms, and their functional significance. Antioxid. Redox Signal. 2011, 15, 271–307. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Mühlenhoff, U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 2012, 1823, 1491–1508. [Google Scholar] [CrossRef] [PubMed]
- Beilschmidt, L.K.; Puccio, H.M. Mammalian Fe-S cluster biogenesis and its implication in disease. Biochimie 2014, 100, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Stehling, O.; Wilbrecht, C.; Lill, R. Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie 2014, 100, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Veatch, J.R.; McMurray, M.A.; Nelson, Z.W.; Gottschling, D.E. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 2009, 137, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Gari, K.; León Ortiz, A.M.; Borel, V.; Flynn, H.; Skehel, J.M.; Boulton, S.J. MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science 2012, 337, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Gottschling, D.E. Molecular biology. Fragile delivery to the genome. Science 2012, 337, 160–161. [Google Scholar] [CrossRef] [PubMed]
- Stehling, O.; Vashisht, A.A.; Mascarenhas, J.; Jonsson, Z.O.; Sharma, T.; Netz, D.J.; Pierik, A.J.; Wohlschlegel, J.A.; Lill, R. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 2012, 337, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—Protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Kirstein-Miles, J.; Morimoto, R.I. Peptides signal mitochondrial stress. Cell Metab. 2010, 11, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Haynes, C.M. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 2011, 36, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Vögtle, F.N.; Meisinger, C. Sensing mitochondrial homeostasis: The protein import machinery takes control. Dev. Cell 2012, 23, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Fiorese, C.J.; Lin, Y.F. Evaluating and responding to mitochondrial dysfunction: The mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 2013, 23, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.; van Remmen, H. Mitochondrial stress signaling in longevity: A new role for mitochondrial function in aging. Redox Biol. 2014, 2, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Mottis, A.; Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response in mammalian physiology. Mamm. Genome 2014, 25, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Aβ. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef] [PubMed]
- Tajima, H.; Niikura, T.; Hashimoto, Y.; Ito, Y.; Kita, Y.; Terashita, K.; Yamazaki, K.; Koto, A.; Aiso, S.; Nishimoto, I. Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer’s disease-related insults. Neurosci. Lett. 2002, 324, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Zhai, D.; Cabezas, E.; Welsh, K.; Nouraini, S.; Satterthwait, A.C.; Reed, J.C. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 2003, 423, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, M.; Liu, B.; Hashimoto, Y.; Ma, L.; Lee, K.W.; Niikura, T.; Nishimoto, I.; Cohen, P. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 13042–13047. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Habata, Y.; Hosoya, M.; Nishi, K.; Fujii, R.; Kobayashi, M.; Hinuma, S. N-formylated humanin activates both formyl peptide receptor-like 1 and 2. Biochem. Biophys. Res. Commun. 2004, 324, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.; Luciano, F.; Zhu, X.; Guo, B.; Satterthwait, A.C.; Reed, J.C. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J. Biol. Chem. 2005, 280, 15815–15824. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yen, K.; Cohen, P. Humanin: A harbinger of mitochondrial-derived peptides? Trends Endocrinol. Metab. 2013, 24, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Lee, C.; Mehta, H.; Cohen, P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J. Mol. Endocrinol. 2013, 50, R11–R19. [Google Scholar] [CrossRef] [PubMed]
- Rabiet, M.J.; Huet, E.; Boulay, F. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur. J. Immunol. 2005, 35, 2486–2495. [Google Scholar] [CrossRef] [PubMed]
- Hauser, C.J.; Sursal, T.; Rodriguez, E.K.; Appleton, P.T.; Zhang, Q.; Itagaki, K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. J. Orthop. Trauma 2010, 24, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Raoof, M.; Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. J. Trauma 2010, 68, 1328–1332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J. Mitochondria: Sovereign of inflammation? Eur. J. Immunol. 2011, 41, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Lindsley, T.A.; Sheridan, A.; Bhoiwala, D.L.; Hushmendy, S.F.; Yager, E.J.; Ruggiero, E.A.; Crawford, D.R. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J. Alzheimers Dis. 2012, 30, 617–627. [Google Scholar] [PubMed]
- Sun, S.; Sursal, T.; Adibnia, Y.; Zhao, C.; Zheng, Y.; Li, H.; Otterbein, L.E.; Hauser, C.J.; Itagaki, K. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One 2013, 8, e59989. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Berendzen, K.M.; Dillin, A. Systemic stress signalling: Understanding the cell non-autonomous control of proteostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Esseltine, J.L.; Scott, J.D. AKAP signaling complexes: Pointing towards the next generation of therapeutic targets? Trends Pharmacol. Sci. 2013, 34, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T. Mitochondrial-mediated antiviral immunity. Biochim. Biophys. Acta 2013, 1833, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Zhang, P.; Steichen, J.M.; Keshwani, M.M.; Kornev, A.P. PKA: Lessons learned after twenty years. Biochim. Biophys. Acta 2013, 1834, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Cassina, A.; Garcia-Haro, L.; Choi, C.S.; Osundiji, M.A.; Lane, E.A.; Huang, H.; Yildirim, M.A.; Szlyk, B.; Fisher, J.K.; Polak, K.; et al. Regulation of hepatic energy metabolism and gluconeogenesis by BAD. Cell Metab. 2014, 19, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Pourcelot, M.; Arnoult, D. Mitochondrial dynamics and the innate antiviral immune response. FEBS J. 2014, 281, 3791–3802. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial inheritance in yeast. Biochim. Biophys. Acta 2014, 1837, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Helle, S.C.; Kanfer, G.; Kolar, K.; Lang, A.; Michel, A.H.; Kornmann, B. Organization and function of membrane contact sites. Biochim. Biophys. Acta 2013, 1833, 2526–2541. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B. The molecular hug between the ER and the mitochondria. Curr. Opin. Cell Biol. 2013, 25, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Daniele, T.; Schiaffino, M.V. Organelle biogenesis and interorganellar connections: Better in contact than in isolation. Commun. Integr. Biol. 2014, 7. [Google Scholar] [CrossRef]
- Elbaz-Alon, Y.; Rosenfeld-Gur, E.; Shinder, V.; Futerman, A.H.; Geiger, T.; Schuldiner, M. A dynamic interface between vacuoles and mitochondria in yeast. Dev. Cell 2014, 30, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Hönscher, C.; Mari, M.; Auffarth, K.; Bohnert, M.; Griffith, J.; Geerts, W.; van der Laan, M.; Cabrera, M.; Reggiori, F.; Ungermann, C. Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev. Cell 2014, 30, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Klecker, T.; Böckler, S.; Westermann, B. Making connections: Interorganelle contacts orchestrate mitochondrial behavior. Trends Cell Biol. 2014, 24, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Prinz, WA. Bridging the gap: Membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 2014, 205, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Van der Laan, M.; Bohnert, M.; Wiedemann, N.; Pfanner, N. Role of MINOS in mitochondrial membrane architecture and biogenesis. Trends Cell Biol. 2012, 22, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Böckler, S.; Westermann, B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev. Cell 2014, 28, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Chao, J.T.; Tavassoli, S.; Wong, A.K.; Choudhary, V.; Young, B.P.; Loewen, C.J.; Prinz, W.A. A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 2014, 12, e1001969. [Google Scholar] [CrossRef] [PubMed]
- Schlattner, U.; Tokarska-Schlattner, M.; Rousseau, D.; Boissan, M.; Mannella, C.; Epand, R.; Lacombe, M.L. Mitochondrial cardiolipin/phospholipid trafficking: The role of membrane contact site complexes and lipid transfer proteins. Chem. Phys. Lipids 2014, 179, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Sesaki, H.; Endo, T. Phospholipid transport via mitochondria. Traffic 2014, 15, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Scharwey, M.; Langer, T. Mitochondrial lipid trafficking. Trends Cell Biol. 2014, 24, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Andrade-Navarro, M.A.; Rachubinski, R.A.; McBride, H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 2010, 20, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 2012, 7, e52830. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.; McBride, H.M. Emerging roles of mitochondria in the evolution, biogenesis, and function of peroxisomes. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Schiff, M.; Bénit, P.; Jacobs, H.T.; Vockley, J.; Rustin, P. Therapies in inborn errors of oxidative metabolism. Trends Endocrinol. Metab. 2012, 23, 488–495. [Google Scholar] [PubMed]
- Ylikallio, E.; Suomalainen, A. Mechanisms of mitochondrial diseases. Ann. Med. 2012, 44, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Scorrano, L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J. 2012, 31, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, A.; Szewczyk, A. Mitochondria as a pharmacological target: Magnum overview. IUBMB Life 2013, 65, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.; da Cunha, F.M.; Oliveira, G.A.; Tahara, E.B.; Kowaltowski, A.J. Yeast as a model to study mitochondrial mechanisms in ageing. Mech. Ageing Dev. 2010, 131, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y. Mitochondria, reactive oxygen species, and chronological aging: A message from yeast. Exp. Gerontol. 2011, 46, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, S.M. The retrograde response and other pathways of interorganelle communication in yeast replicative aging. Subcell. Biochem. 2012, 57, 79–100. [Google Scholar] [PubMed]
- Longo, V.D.; Shadel, G.S.; Kaeberlein, M.; Kennedy, B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012, 16, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, S.M. The retrograde response: When mitochondrial quality control is not enough. Biochim. Biophys. Acta 2013, 1833, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.T.; Titorenko, V.I. A mitochondrially targeted compound delays aging in yeast through a mechanism linking mitochondrial membrane lipid metabolism to mitochondrial redox biology. Redox Biol. 2014, 2, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Mirisola, M.G.; Longo, V.D. A radical signal activates the epigenetic regulation of longevity. Cell Metab. 2013, 17, 812–813. [Google Scholar] [CrossRef] [PubMed]
- Steinkraus, K.A.; Kaeberlein, M.; Kennedy, B.K. Replicative aging in yeast: The means to the end. Annu. Rev. Cell Dev. Biol. 2008, 24, 29–54. [Google Scholar] [CrossRef] [PubMed]
- Steffen, K.K.; Kennedy, B.K.; Kaeberlein, M. Measuring replicative life span in the budding yeast. J. Vis. Exp. 2009, 28. [Google Scholar] [CrossRef]
- Kaeberlein, M. Lessons on longevity from budding yeast. Nature 2010, 464, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Denoth Lippuner, A.; Julou, T.; Barral, Y. Budding yeast as a model organism to study the effects of age. FEMS Microbiol. Rev. 2014, 38, 300–325. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.A.; Bourque, S.D.; Kyryakov, P.; Gregg, C.; Boukh-Viner, T.; Beach, A.; Burstein, M.T.; Machkalyan, G.; Richard, V.; Rampersad, S.; et al. Effect of calorie restriction on the metabolic history of chronologically aging yeast. Exp. Gerontol. 2009, 44, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Fabrizio, P. Chronological aging in Saccharomyces cerevisiae. Subcell. Biochem. 2012, 57, 101–121. [Google Scholar] [PubMed]
- Piper, P.W. Maximising the yeast chronological lifespan. Subcell. Biochem. 2012, 57, 145–159. [Google Scholar] [PubMed]
- Weissman, J.; Guthrie, C.; Fink, G.R. Guide to Yeast Genetics: Functional Genomics, Proteomics, and Other Systems Analysis, 2nd ed.; Academic Press: Burlington, CO, USA, 2010; p. 892. [Google Scholar]
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for 21st Century biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Sutphin, G.L.; Olsen, B.A.; Kennedy, B.K.; Kaeberlein, M. Genome-wide analysis of yeast aging. Subcell. Biochem. 2012, 57, 251–289. [Google Scholar] [PubMed]
- Richard, V.R.; Bourque, S.D.; Titorenko, V.I. Metabolomic and lipidomic analyses of chronologically aging yeast. Methods Mol. Biol. 2014, 1205, 359–373. [Google Scholar] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.A.; Bourque, S.D.; Kyryakov, P.; Boukh-Viner, T.; Gregg, C.; Beach, A.; Burstein, M.T.; Machkalyan, G.; Richard, V.; Rampersad, S.; et al. A novel function of lipid droplets in regulating longevity. Biochem. Soc. Trans. 2009, 37, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.A.; Richard, V.R.; Kyryakov, P.; Bourque, S.D.; Beach, A.; Burstein, M.T.; Glebov, A.; Koupaki, O.; Boukh-Viner, T.; Gregg, C.; et al. Chemical genetic screen identifies lithocholic acid as an anti-aging compound that extends yeast chronological life span in a TOR-independent manner, by modulating housekeeping longevity assurance processes. Aging 2010, 2, 393–414. [Google Scholar] [PubMed]
- Kapahi, P; Chen, D.; Rogers, A.N.; Katewa, S.D.; Li, P.W.; Thomas, E.L.; Kockel, L. With TOR, less is more: A key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010, 11, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Titorenko, V.I.; Terlecky, S.R. Peroxisome metabolism and cellular aging. Traffic 2011, 12, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Beach, A.; Burstein, M.T.; Richard, V.R.; Leonov, A.; Levy, S.; Titorenko, V.I. Integration of peroxisomes into an endomembrane system that governs cellular aging. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef]
- Beach, A.; Titorenko, V.I. Essential roles of peroxisomally produced and metabolized biomolecules in regulating yeast longevity. Subcell. Biochem. 2013, 69, 153–167. [Google Scholar] [PubMed]
- De Cabo, R.; Carmona-Gutierrez, D.; Bernier, M.; Hall, M.N.; Madeo, F. The search for antiaging interventions: From elixirs to fasting regimens. Cell 2014, 157, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Nyström, T.; Liu, B. Protein quality control in time and space—Links to cellular aging. FEMS Yeast Res. 2014, 14, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.A.; Guarente, L. Small-molecule allosteric activators of sirtuins. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Gottschling, D.E. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature 2012, 492, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, S.M.; Kriete, A. The yeast retrograde response as a model of intracellular signaling of mitochondrial dysfunction. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef]
- McFaline-Figueroa, J.R.; Vevea, J.; Swayne, T.C.; Zhou, C.; Liu, C.; Leung, G.; Boldogh, I.R.; Pon, L.A. Mitochondrial quality control during inheritance is associated with lifespan and mother-daughter age asymmetry in budding yeast. Aging Cell 2011, 10, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, S.; Paoletti, C.; Goulev, Y.; Ungureanu, A.; Aguilaniu, H.; Charvin, G. Aging yeast cells undergo a sharp entry into senescence unrelated to the loss of mitochondrial membrane potential. Cell Rep. 2013, 5, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Knorre, D.A.; Popadin, K.Y.; Sokolov, S.S.; Severin, F.F. Roles of mitochondrial dynamics under stressful and normal conditions in yeast cells. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef]
- Sorokin, M.I.; Knorre, D.A.; Fedor, F.; Severin, F.F. Early manifestations of replicative aging in the yeast Saccharomyces cerevisiae. Microbial Cell 2014, 1, 37–42. [Google Scholar] [CrossRef]
- Higuchi, R.; Vevea, J.D.; Swayne, T.C.; Chojnowski, R.; Hill, V.; Boldogh, I.R.; Pon, L.A. Actin dynamics affect mitochondrial quality control and aging in budding yeast. Curr. Biol. 2013, 23, 2417–2422. [Google Scholar] [CrossRef] [PubMed]
- Nyström, T.; Liu, B. The mystery of aging and rejuvenation—A budding topic. Curr. Opin. Microbiol. 2014, 18, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Vevea, J.D.; Swayne, T.C.; Boldogh, I.R.; Pon, L.A. Inheritance of the fittest mitochondria in yeast. Trends Cell Biol. 2014, 24, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Heeren, G.; Rinnerthaler, M.; Laun, P.; von Seyerl, P.; Kössler, S.; Klinger, H.; Hager, M.; Bogengruber, E.; Jarolim, S.; Simon-Nobbe, B.; et al. The mitochondrial ribosomal protein of the large subunit, Afo1p, determines cellular longevity through mitochondrial back-signaling via TOR1. Aging 2009, 1, 622–636. [Google Scholar] [PubMed]
- Chen, X.J. The search for nonconventional mitochondrial determinants of aging. Mol. Cell 2011, 42, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Jensen, L.J.; Gagneur, J.; Ahting, U.; von Mering, C.; Bork, P.; Prokisch, H.; Steinmetz, L.M. Assessing systems properties of yeast mitochondria through an interaction map of the organelle. PLoS Genet. 2006, 2, e170. [Google Scholar] [CrossRef] [PubMed]
- Merz, S.; Westermann, B. Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol. 2009, 10. [Google Scholar] [CrossRef]
- Caballero, A.; Ugidos, A.; Liu, B.; Öling, D.; Kvint, K.; Hao, X.; Mignat, C.; Nachin, L.; Molin, M.; Nyström, T. Absence of mitochondrial translation control proteins extends life span by activating sirtuin-dependent silencing. Mol. Cell 2011, 42, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Kim, J.; Park, S.; Jeon, J.; Shin, Y.E.; Kim, S. Spatial and functional organization of mitochondrial protein network. Sci. Rep. 2013, 3. [Google Scholar] [PubMed]
- Koppen, M.; Langer, T. Protein degradation within mitochondria: Versatile activities of AAA proteases and other peptidases. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Delaney, J.R.; Ahmed, U.; Chou, A.; Sim, S.; Carr, D.; Murakami, C.J.; Schleit, J.; Sutphin, G.L.; An, E.H.; Castanza, A.; et al. Stress profiling of longevity mutants identifies Afg3 as a mitochondrial determinant of cytoplasmic mRNA translation and aging. Aging Cell 2013, 12, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Kissová, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [Google Scholar] [CrossRef] [PubMed]
- Bhatia-Kiššová, I.; Camougrand, N. Mitophagy in yeast: Actors and physiological roles. FEMS Yeast Res. 2010, 10, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Welter, E.; Montino, M.; Reinhold, R.; Schlotterhose, P.; Krick, R.; Dudek, J.; Rehling, P.; Thumm, M. Uth1 is a mitochondrial inner membrane protein dispensable for post-log-phase and rapamycin-induced mitophagy. FEBS J. 2013, 280, 4970–4982. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.T.; Kyryakov, P.; Beach, A.; Richard, V.R.; Koupaki, O.; Gomez-Perez, A.; Leonov, A.; Levy, S.; Noohi, F.; Titorenko, V.I. Lithocholic acid extends longevity of chronologically aging yeast only if added at certain critical periods of their lifespan. Cell Cycle 2012, 11, 3443–3462. [Google Scholar] [CrossRef] [PubMed]
- Bonawitz, N.D.; Chatenay-Lapointe, M.; Pan, Y.; Shadel, G.S. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007, 5, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Shadel, G.S. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging 2009, 1, 131–145. [Google Scholar] [PubMed]
- Pan, Y.; Schroeder, E.A.; Ocampo, A.; Barrientos, A.; Shadel, G.S. Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 2011, 13, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Liu, J.; Schroeder, E.A.; Shadel, G.S.; Barrientos, A. Mitochondrial respiratory thresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab. 2012, 16, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, D.G. Yeast Intermediary Metabolism; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2011; p. 434. [Google Scholar]
- Cai, L.; Tu, B.P. Driving the cell cycle through metabolism. Annu. Rev. Cell Dev. Biol. 2012, 28, 59–87. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Tienson, H.; Lindemann, A.; Vitvitsky, V.; Reichmann, D.; Banerjee, R.; Jakob, U. Time line of redox events in aging postmitotic cells. Elife 2013, 2. [Google Scholar] [CrossRef]
- Kyryakov, P.; Beach, A.; Richard, V.R.; Burstein, M.T.; Leonov, A.; Levy, S.; Titorenko, V.I. Caloric restriction extends yeast chronological lifespan by altering a pattern of age-related changes in trehalose concentration. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef]
- Beach, A.; Richard, V.R.; Leonov, A.; Burstein, M.T.; Bourque, S.D.; Koupaki, O.; Juneau, M.; Feldman, R.; Iouk, T.; Titorenko, V.I. Mitochondrial membrane lipidome defines yeast longevity. Aging 2013, 5, 551–574. [Google Scholar] [PubMed]
- Schroeder, E.A.; Raimundo, N.; Shadel, G.S. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 2013, 17, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.A.; Shadel, G.S. Crosstalk between mitochondrial stress signals regulates yeast chronological lifespan. Mech. Ageing Dev. 2014, 135, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Broach, J.R. Nutritional control of growth and development in yeast. Genetics 2012, 192, 73–105. [Google Scholar] [CrossRef] [PubMed]
- De Virgilio, C. The essence of yeast quiescence. FEMS Microbiol. Rev. 2012, 36, 306–339. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.L.; Powers, T.; Fowler, B.; Hall, M.N. The TOR-controlled transcription activators GLN3, RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc. Natl. Acad. Sci. USA 2002, 99, 6784–6789. [Google Scholar] [CrossRef] [PubMed]
- Powers, R.W.; Kaeberlein, M.; Caldwell, S.D.; Kennedy, B.K.; Fields, S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006, 20, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Schothorst, J.; Kankipati, H.N.; van Zeebroeck, G.; Rubio-Texeira, M.; Thevelein, J.M. Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2014, 38, 254–299. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, E.; Ghillebert, R.; Wilms, T.; Winderickx, J. Molecular mechanisms linking the evolutionary conserved TORC1-Sch9 nutrient signalling branch to lifespan regulation in Saccharomyces cerevisiae. FEMS Yeast Res. 2014, 14, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Wissing, S.; Ludovico, P.; Herker, E.; Büttner, S.; Engelhardt, S.M.; Decker, T.; Link, A.; Proksch, A.; Rodrigues, F.; Corte-Real, M.; et al. An AIF orthologue regulates apoptosis in yeast. J. Cell Biol. 2004, 166, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Büttner, S.; Eisenberg, T.; Carmona-Gutierrez, D.; Ruli, D.; Knauer, H.; Ruckenstuhl, C.; Sigrist, C.; Wissing, S.; Kollroser, M.; Fröhlich, K.U.; et al. Endonuclease G regulates budding yeast life and death. Mol. Cell 2007, 25, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Büttner, S.; Kroemer, G.; Madeo, F. The mitochondrial pathway in yeast apoptosis. Apoptosis 2007, 12, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Silva, R.D.; Saraiva, L.; Johansson, B.; Sousa, M.J.; Côrte-Real, M. Mitochondria-dependent apoptosis in yeast. Biochim. Biophys. Acta 2008, 1783, 1286–1302. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Carmona-Gutierrez, D.; Ring, J.; Büttner, S.; Eisenberg, T.; Kroemer, G. Caspase-dependent and caspase-independent cell death pathways in yeast. Biochem. Biophys. Res. Commun. 2009, 382, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; Eisenberg, T.; Büttner, S.; Meisinger, C.; Kroemer, G.; Madeo, F. Apoptosis in yeast: Triggers, pathways, subroutines. Cell Death Differ. 2010, 17, 763–773. [Google Scholar] [CrossRef]
- Sheibani, S.; Richard, V.R.; Beach, A.; Leonov, A.; Feldman, R.; Mattie, S.; Khelghatybana, L.; Piano, A.; Greenwood, M.; Vali, H.; et al. Macromitophagy, neutral lipids synthesis, and peroxisomal fatty acid oxidation protect yeast from “liponecrosis”, a previously unknown form of programmed cell death. Cell Cycle 2014, 13, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Richard, V.R.; Beach, A.; Piano, A.; Leonov, A.; Feldman, R.; Burstein, M.T.; Kyryakov, P.; Gomez-Perez, A.; Arlia-Ciommo, A.; Baptista, S.; et al. Mechanism of liponecrosis, a distinct mode of programmed cell death. Cell Cycle 2014, 13, 3707–3726. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Wang, K.; Cao, Y.; Baba, M.; Klionsky, D.J. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 2009, 17, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kondo-Okamoto, N.; Ohsumi, Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 2009, 17, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Beach, A.; Titorenko, V.I. In search of housekeeping pathways that regulate longevity. Cell Cycle 2011, 10, 3042–3044. [Google Scholar] [CrossRef] [PubMed]
- Richard, V.R.; Leonov, A.; Beach, A.; Burstein, M.T.; Koupaki, O.; Gomez-Perez, A.; Levy, S.; Pluska, L.; Mattie, S.; et al. Macromitophagy is a longevity assurance process that in chronologically aging yeast limited in calorie supply sustains functional mitochondria and maintains cellular lipid homeostasis. Aging 2013, 5, 234–269. [Google Scholar] [PubMed]
- Cheng, X.; Ivessa, A.S. The migration of mitochondrial DNA fragments to the nucleus affects the chronological aging process of Saccharomyces cerevisiae. Aging Cell 2010, 9, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ivessa, A.S. Accumulation of linear mitochondrial DNA fragments in the nucleus shortens the chronological life span of yeast. Eur. J. Cell Biol. 2012, 91, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Arlia-Ciommo, A.; Piano, A.; Svistkova, V.; Mohtashami, S.; Titorenko, V.I. Mechanisms underlying the anti-aging and anti-tumor effects of lithocholic bile acid. Int. J. Mol. Sci. 2014, 15, 16522–16543. [Google Scholar] [CrossRef] [PubMed]
- Beach, A.; Richard, V.R.; Bourque, S.; Boukh-Viner, T.; Kyryakov, P.; Gomez-Perez, A.; Arlia-Ciommo, A.; Feldman, R.; Leonov, A.; Piano, A.; et al. Lithocholic bile acid accumulated in yeast mitochondria orchestrates a development of an anti-aging cellular pattern by causing age-related changes in cellular proteome. Cell Cycle 2015, in press. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beach, A.; Leonov, A.; Arlia-Ciommo, A.; Svistkova, V.; Lutchman, V.; Titorenko, V.I. Mechanisms by Which Different Functional States of Mitochondria Define Yeast Longevity. Int. J. Mol. Sci. 2015, 16, 5528-5554. https://doi.org/10.3390/ijms16035528
Beach A, Leonov A, Arlia-Ciommo A, Svistkova V, Lutchman V, Titorenko VI. Mechanisms by Which Different Functional States of Mitochondria Define Yeast Longevity. International Journal of Molecular Sciences. 2015; 16(3):5528-5554. https://doi.org/10.3390/ijms16035528
Chicago/Turabian StyleBeach, Adam, Anna Leonov, Anthony Arlia-Ciommo, Veronika Svistkova, Vicky Lutchman, and Vladimir I. Titorenko. 2015. "Mechanisms by Which Different Functional States of Mitochondria Define Yeast Longevity" International Journal of Molecular Sciences 16, no. 3: 5528-5554. https://doi.org/10.3390/ijms16035528
APA StyleBeach, A., Leonov, A., Arlia-Ciommo, A., Svistkova, V., Lutchman, V., & Titorenko, V. I. (2015). Mechanisms by Which Different Functional States of Mitochondria Define Yeast Longevity. International Journal of Molecular Sciences, 16(3), 5528-5554. https://doi.org/10.3390/ijms16035528