Extracellular ATP: A Feasible Target for Cancer Therapy



Abstract
:1. Introduction
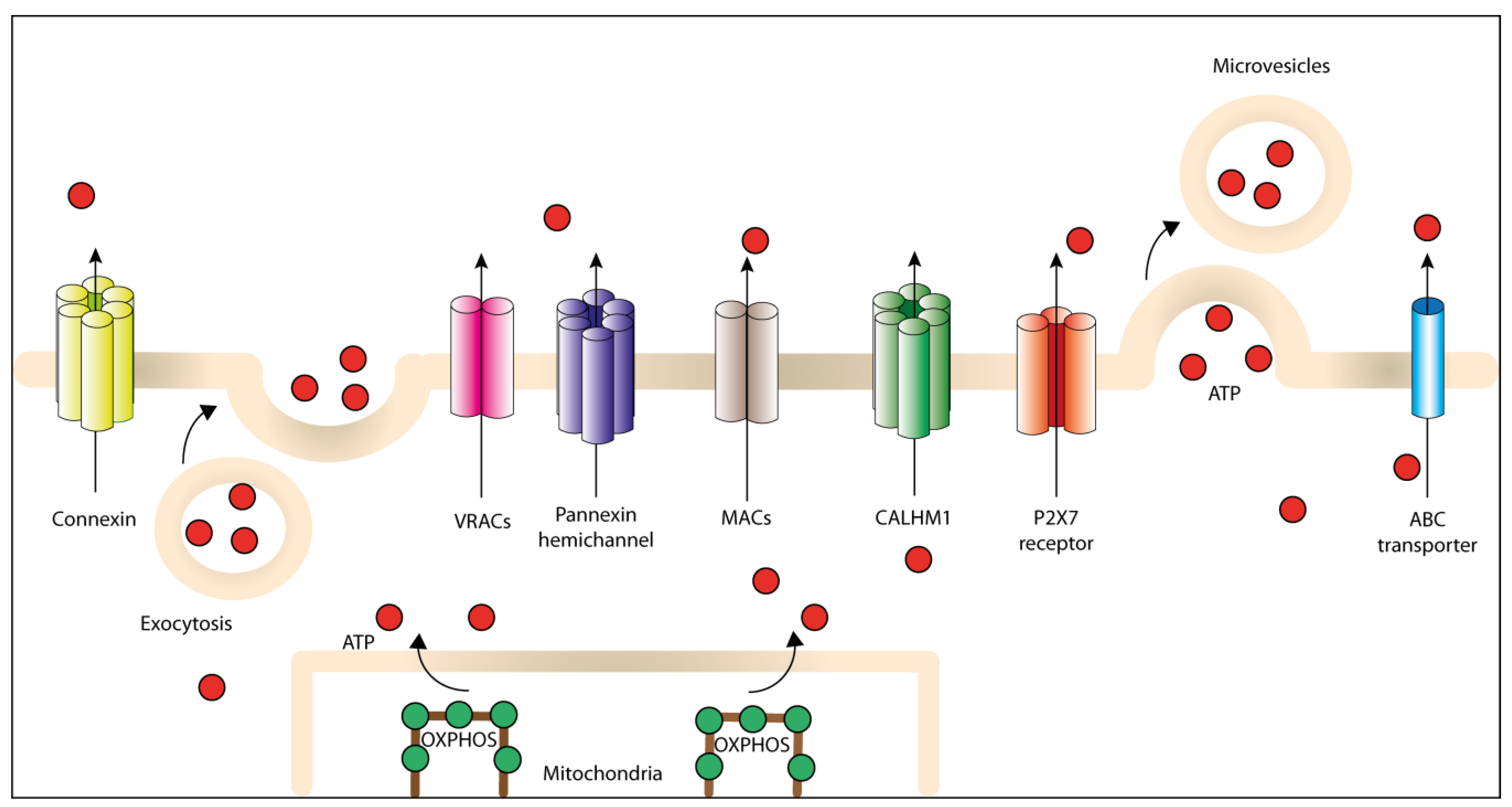
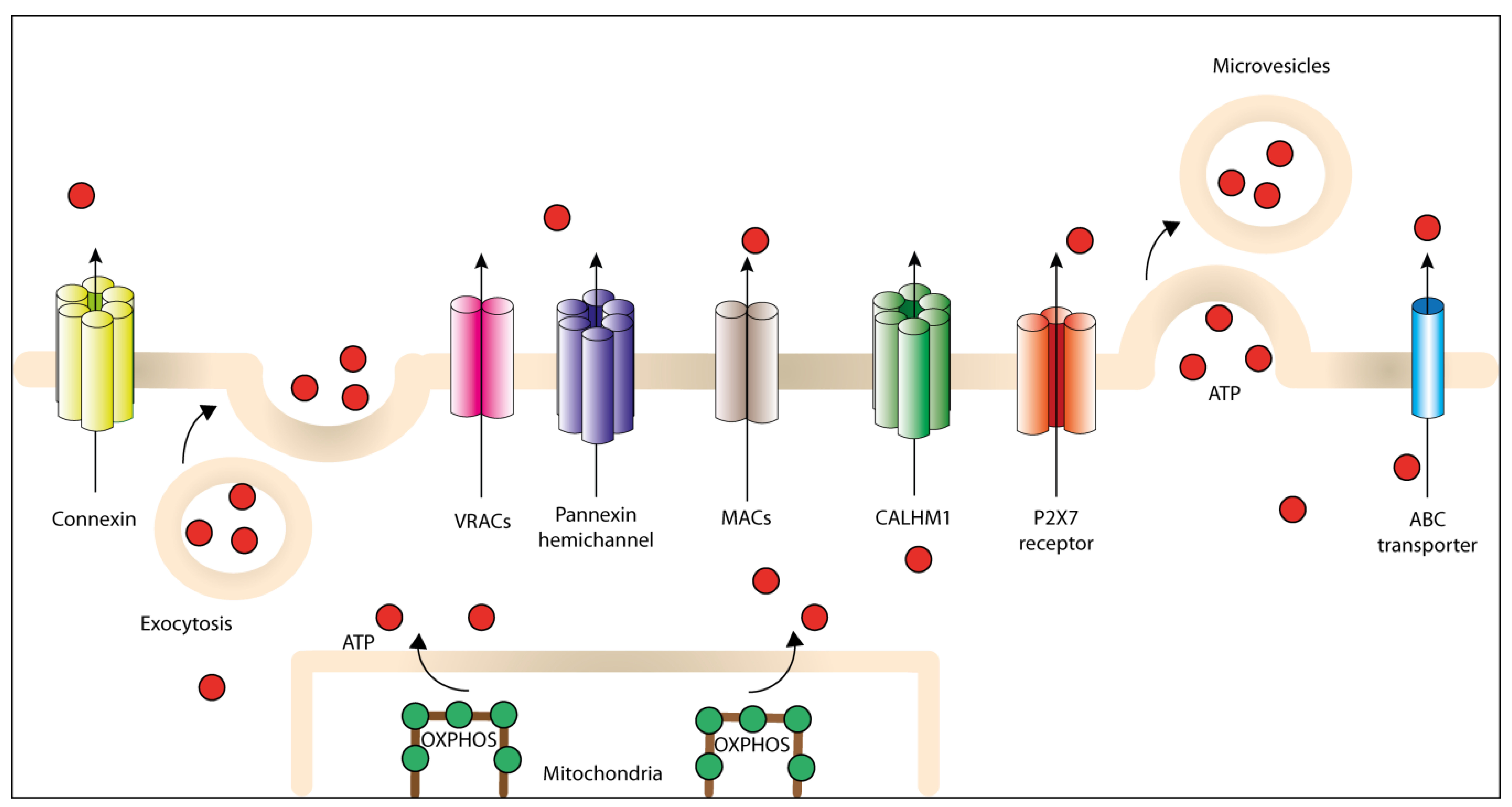
2. Mechanisms of ATP Release
3. Detection of Extracellular ATP in the TME
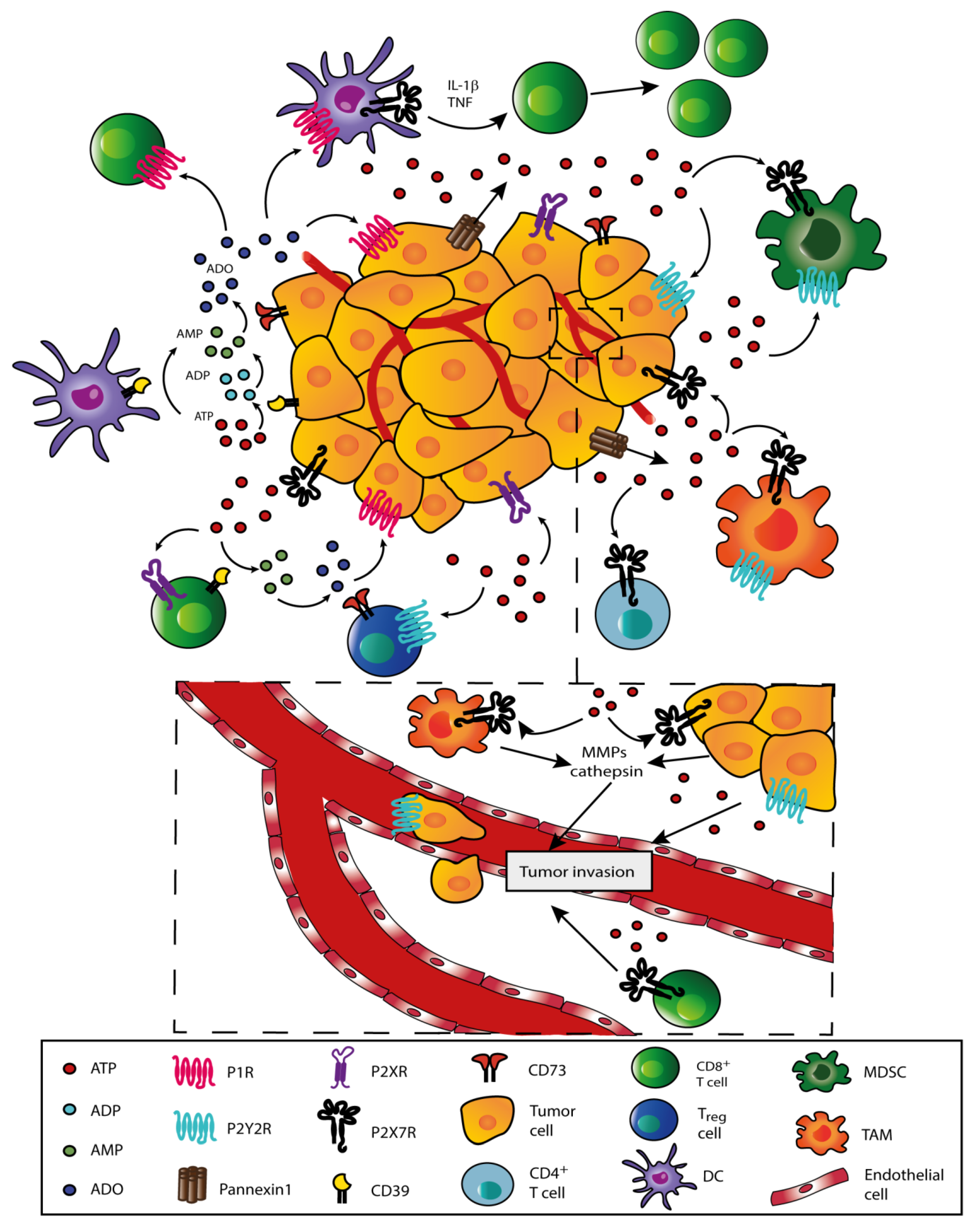
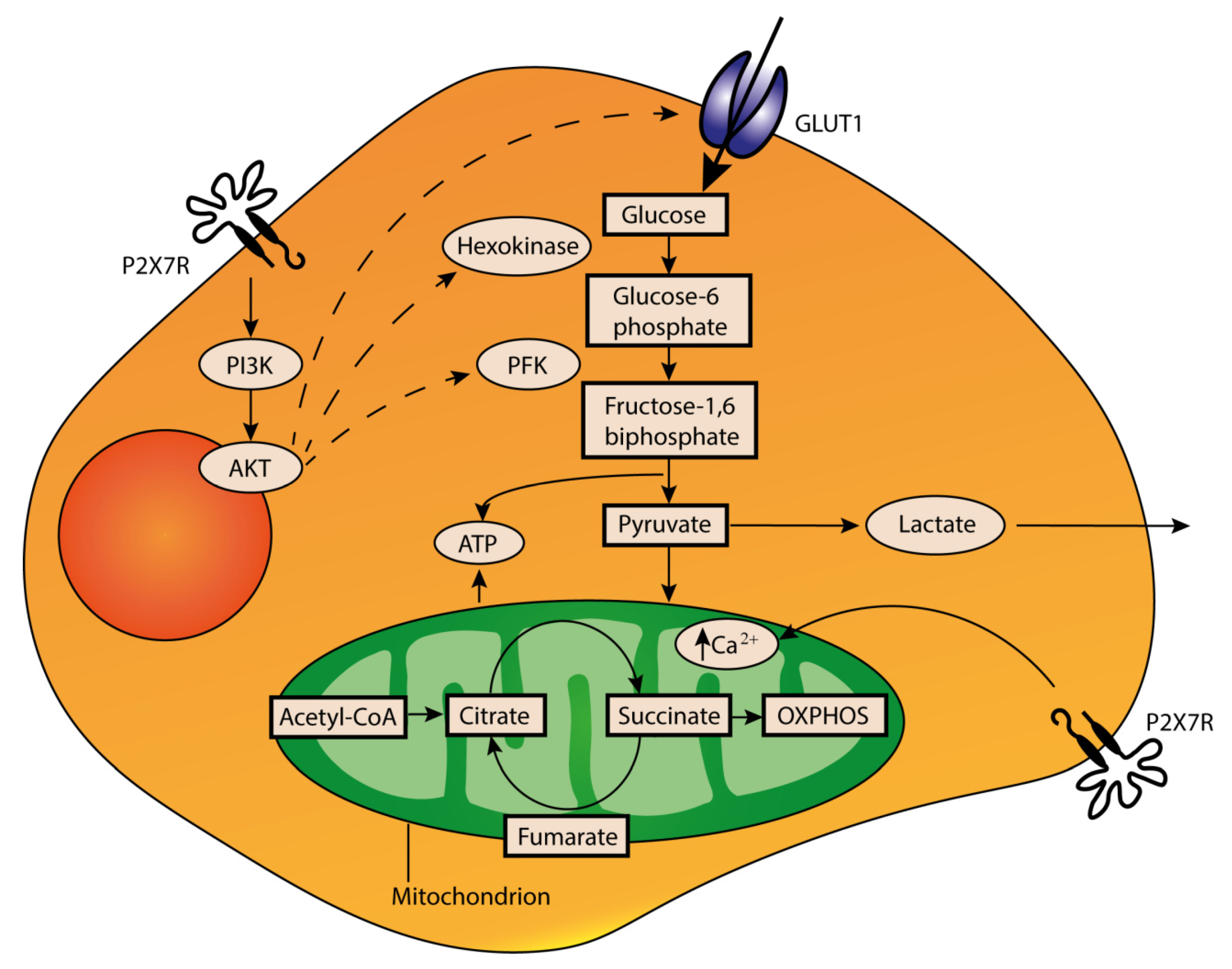
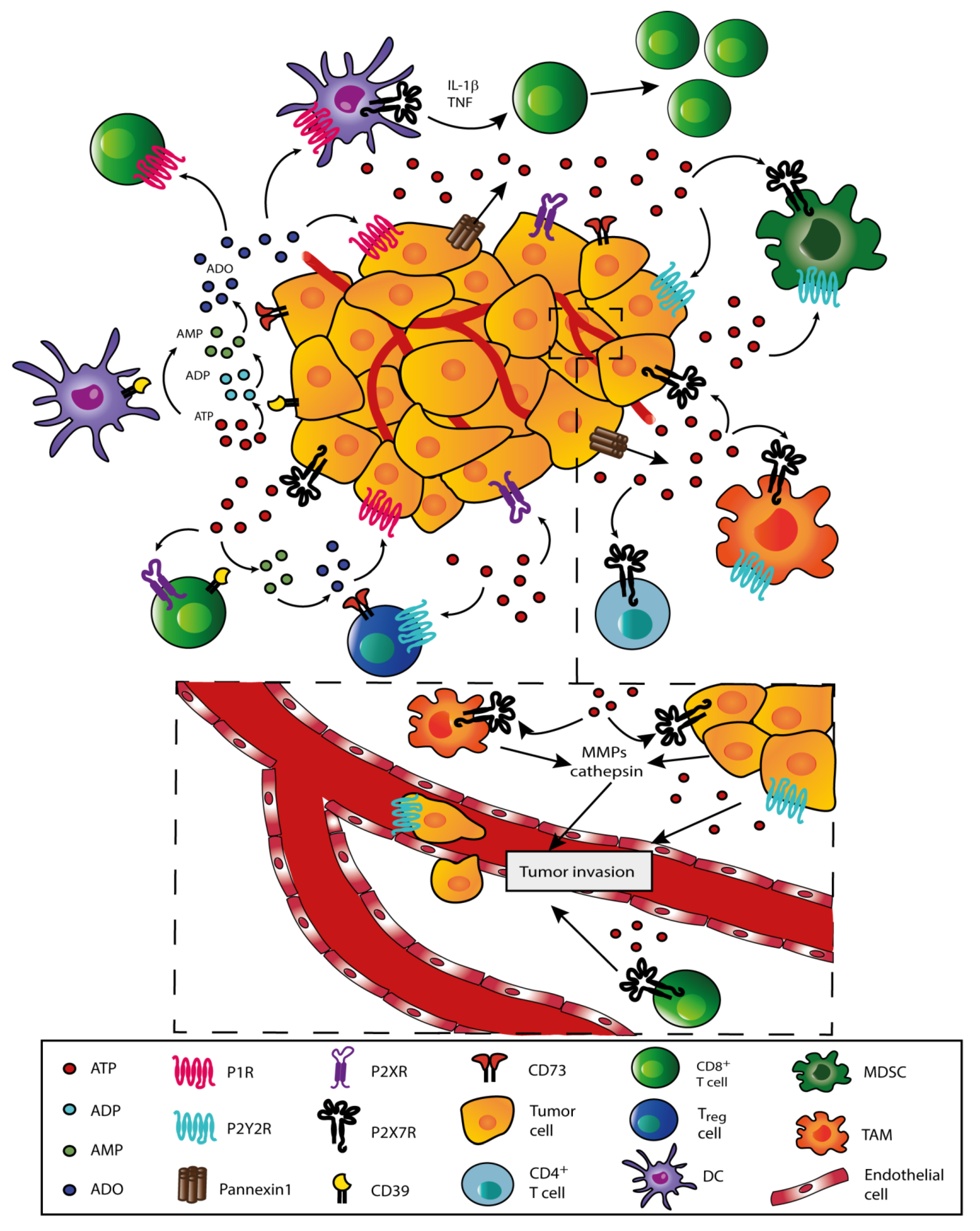
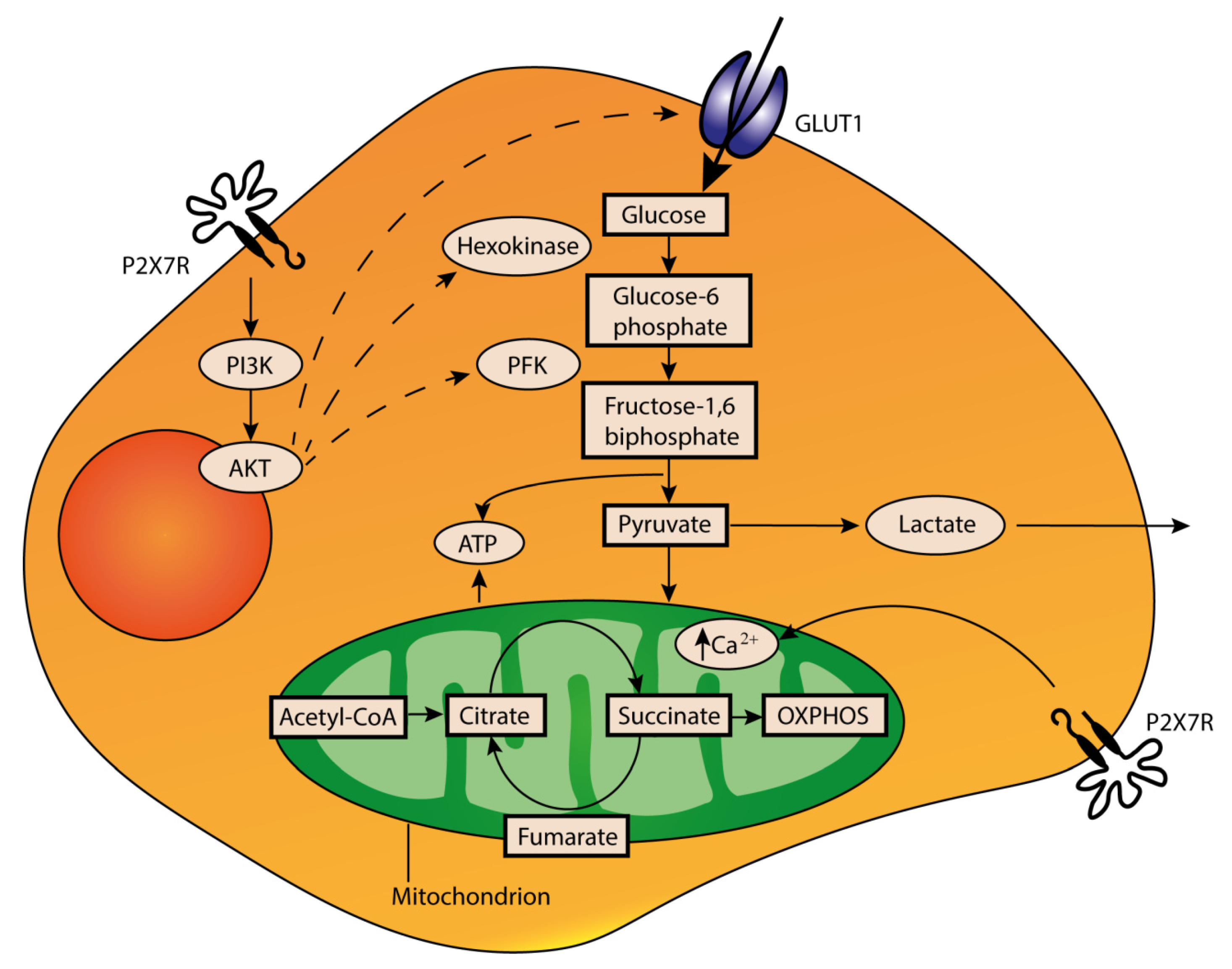
4. Role of Extracellular ATP in the TME
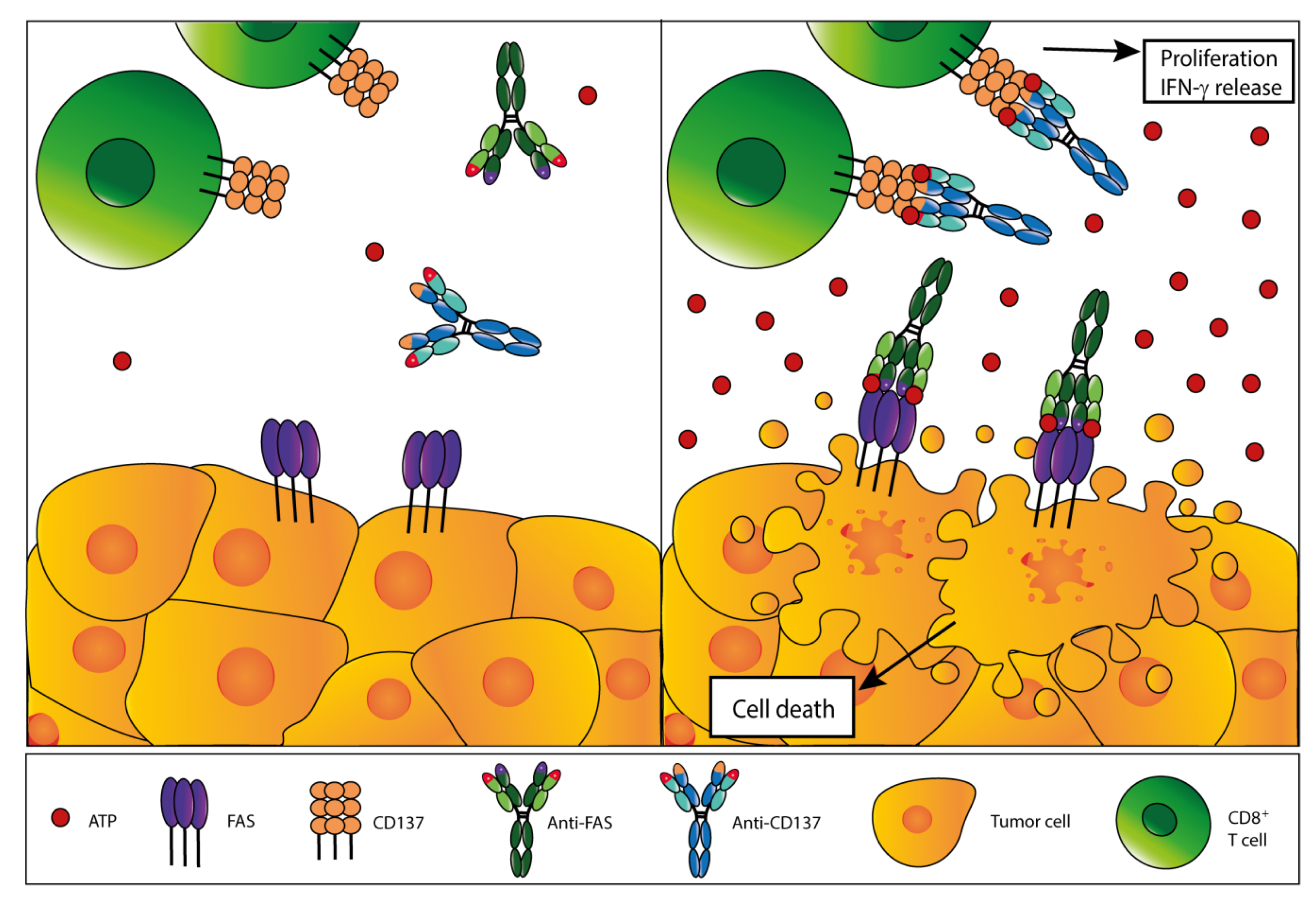
5. Extracellular ATP as a Target for Cancer Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Langen, P.; Hucho, F. Karl Lohmann and the discovery of ATP. Angew Chem. Int. Ed. Engl 2008, 47, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling: From discovery to current developments. Exp. Physiol. 2014, 99, 16–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G.; Campbell, G.; Satchell, D.; Smythe, A. Evidence that adenosine triphosphate or a related nucleotide is the transmitter substance released by non-adrenergic inhibitory nerves in the gut. Br. J. Pharmacol. 1970, 40, 668–688. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic nerves. Pharmacol. Rev. 1972, 24, 509–581. [Google Scholar] [PubMed]
- Webb, T.E.; Simon, J.; Krishek, B.J.; Bateson, A.N.; Smart, T.G.; King, B.F.; Burnstock, G.; Barnard, E.A. Cloning and functional expression of a brain G-protein-coupled ATP receptor. FEBS Lett. 1993, 324, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Valera, S.; Hussy, N.; Evans, R.J.; Adami, N.; North, R.A.; Surprenant, A.; Buell, G. A new class of ligand-gated ion channel defined by P2x receptor for extracellular ATP. Nature 1994, 371, 516–519. [Google Scholar] [CrossRef]
- Cockcroft, S.; Gomperts, B.D. The ATP4- receptor of rat mast cells. Biochem. J. 1980, 188, 789–798. [Google Scholar] [CrossRef] [Green Version]
- Rapaport, E.; Fishman, R.F.; Gercel, C. Growth inhibition of human tumor cells in soft-agar cultures by treatment with low levels of adenosine 5′-triphosphate. Cancer Res. 1983, 43, 4402–4406. [Google Scholar]
- Kepp, O.; Loos, F.; Liu, P.; Kroemer, G. Extracellular nucleosides and nucleotides as immunomodulators. Immunol. Rev. 2017, 280, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Giuliani, A.L.; Sarti, A.C.; Di Virgilio, F. Extracellular nucleotides and nucleosides as signalling molecules. Immunol. Lett. 2019, 205, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Chen, Y.F.; Cowan, P.J.; Chen, X.P. Extracellular ATP signaling and clinical relevance. Clin. Immunol. 2018, 188, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M. ATP: A ubiquitous gliotransmitter integrating neuron-glial networks. Semin. Cell Dev. Biol. 2011, 22, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Gu, J.G. P2X purinoceptors and sensory transmission. Pflugers Arch. 2006, 452, 598–607. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic signalling in endocrine organs. Purinergic Signal. 2014, 10, 189–231. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G. Purinergic signalling and disorders of the central nervous system. Nat. Rev. Drug Discov. 2008, 7, 575–590. [Google Scholar] [CrossRef]
- Erlinge, D.; Burnstock, G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. 2008, 4, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F. P2X receptors and inflammation. Curr. Med. Chem. 2015, 22, 866–877. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Falzoni, S.; Giuliani, A.L.; Adinolfi, E. P2 receptors in cancer progression and metastatic spreading. Curr. Opin. Pharmacol. 2016, 29, 17–25. [Google Scholar] [CrossRef]
- Forrester, T.; Williams, C.A. Release of adenosine triphosphate from isolated adult heart cells in response to hypoxia. J. Physiol. 1977, 268, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martins, I.; Ma, Y.; Kepp, O.; Galluzzi, L.; Kroemer, G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy 2013, 9, 1624–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follo, C.; Cheng, Y.; Richards, W.G.; Bueno, R.; Broaddus, V.C. Autophagy facilitates the release of immunogenic signals following chemotherapy in 3D models of mesothelioma. Mol. Carcinog. 2019, 58, 1754–1769. [Google Scholar] [CrossRef]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad Sci. USA 2009, 106, 20388–20393. [Google Scholar] [CrossRef] [Green Version]
- Dosch, M.; Gerber, J.; Jebbawi, F.; Beldi, G. Mechanisms of ATP Release by Inflammatory Cells. Int. J. Mol. Sci. 2018, 19, 1222. [Google Scholar] [CrossRef] [Green Version]
- Bodin, P.; Burnstock, G. Evidence that release of adenosine triphosphate from endothelial cells during increased shear stress is vesicular. J. Cardiovasc. Pharmacol. 2001, 38, 900–908. [Google Scholar] [CrossRef]
- Moriyama, Y.; Hiasa, M.; Sakamoto, S.; Omote, H.; Nomura, M. Vesicular nucleotide transporter (VNUT): Appearance of an actress on the stage of purinergic signaling. Purinergic Signal. 2017, 13, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Imura, Y.; Morizawa, Y.; Komatsu, R.; Shibata, K.; Shinozaki, Y.; Kasai, H.; Moriishi, K.; Moriyama, Y.; Koizumi, S. Microglia release ATP by exocytosis. Glia 2013, 61, 1320–1330. [Google Scholar] [CrossRef]
- Woodward, H.N.; Anwar, A.; Riddle, S.; Taraseviciene-Stewart, L.; Fragoso, M.; Stenmark, K.R.; Gerasimovskaya, E.V. PI3K, Rho, and ROCK play a key role in hypoxia-induced ATP release and ATP-stimulated angiogenic responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L954–L964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, T.; Oike, M.; Ito, Y. Involvement of Rho-kinase and tyrosine kinase in hypotonic stress-induced ATP release in bovine aortic endothelial cells. J. Physiol. 2001, 532, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Historical review: ATP as a neurotransmitter. Trends Pharmacol. Sci. 2006, 27, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, H.; Zarnegar, B.; Westin, A.; Persson, V.; Peuckert, C.; Jonsson, J.; Hallgren, J.; Kullander, K. SLC10A4 regulates IgE-mediated mast cell degranulation in vitro and mast cell-mediated reactions in vivo. Sci. Rep. 2017, 7, 1085. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, A.; Tsukimoto, M.; Harada, H.; Moriyama, Y.; Kojima, S. Involvement of SLC17A9-dependent vesicular exocytosis in the mechanism of ATP release during T cell activation. J. Biol Chem 2010, 285, 17406–17416. [Google Scholar] [CrossRef] [Green Version]
- Schwiebert, E.M. ABC transporter-facilitated ATP conductive transport. Am. J. Physiol. 1999, 276, C1–C8. [Google Scholar] [CrossRef]
- Hyde, S.C.; Emsley, P.; Hartshorn, M.J.; Mimmack, M.M.; Gileadi, U.; Pearce, S.R.; Gallagher, M.P.; Gill, D.R.; Hubbard, R.E.; Higgins, C.F. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature 1990, 346, 362–365. [Google Scholar] [CrossRef]
- Abraham, E.H.; Prat, A.G.; Gerweck, L.; Seneveratne, T.; Arceci, R.J.; Kramer, R.; Guidotti, G.; Cantiello, H.F. The multidrug resistance (mdr1) gene product functions as an ATP channel. Proc. Natl. Acad. Sci. USA 1993, 90, 312–316. [Google Scholar] [CrossRef] [Green Version]
- Reisin, I.L.; Prat, A.G.; Abraham, E.H.; Amara, J.F.; Gregory, R.J.; Ausiello, D.A.; Cantiello, H.F. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J. Biol. Chem. 1994, 269, 20584–20591. [Google Scholar]
- Grygorczyk, R.; Tabcharani, J.A.; Hanrahan, J.W. CFTR channels expressed in CHO cells do not have detectable ATP conductance. J. Membr. Biol. 1996, 151, 139–148. [Google Scholar] [CrossRef]
- Roman, R.M.; Lomri, N.; Braunstein, G.; Feranchak, A.P.; Simeoni, L.A.; Davison, A.K.; Mechetner, E.; Schwiebert, E.M.; Fitz, J.G. Evidence for multidrug resistance-1 P-glycoprotein-dependent regulation of cellular ATP permeability. J. Membr. Biol. 2001, 183, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Nejime, N.; Kagota, S.; Tada, Y.; Nakamura, K.; Hashimoto, M.; Kunitomo, M.; Shinozuka, K. Possible participation of chloride ion channels in ATP release from cancer cells in suspension. Clin. Exp. Pharmacol. Physiol. 2009, 36, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.F.; O’Neal, W.K.; Huang, P.; Nicholas, R.A.; Ostrowski, L.E.; Craigen, W.J.; Lazarowski, E.R.; Boucher, R.C. Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J. Gen. Physiol. 2004, 124, 513–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taruno, A. ATP Release Channels. Int. J. Mol. Sci 2018, 19, 808. [Google Scholar] [CrossRef] [Green Version]
- Scemes, E.; Spray, D.C.; Meda, P. Connexins, pannexins, innexins: Novel roles of “hemi-channels”. Pflugers Arch. 2009, 457, 1207–1226. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 2008, 28, 4702–4711. [Google Scholar] [CrossRef]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004, 572, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Sosinsky, G.E.; Boassa, D.; Dermietzel, R.; Duffy, H.S.; Laird, D.W.; MacVicar, B.; Naus, C.C.; Penuela, S.; Scemes, E.; Spray, D.C.; et al. Pannexin channels are not gap junction hemichannels. Channels (Austin) 2011, 5, 193–197. [Google Scholar] [CrossRef]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Begandt, D.; Good, M.E.; Keller, A.S.; DeLalio, L.J.; Rowley, C.; Isakson, B.E.; Figueroa, X.F. Pannexin channel and connexin hemichannel expression in vascular function and inflammation. BMC Cell Biol. 2017, 18, 2. [Google Scholar] [CrossRef] [Green Version]
- Cotrina, M.L.; Lin, J.H.; Alves-Rodrigues, A.; Liu, S.; Li, J.; Azmi-Ghadimi, H.; Kang, J.; Naus, C.C.; Nedergaard, M. Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. USA 1998, 95, 15735–15740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Vinken, M.; Rogiers, V.; Bukauskas, F.F.; Bultynck, G.; Leybaert, L. Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta 2013, 1828, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Faigle, M.; Seessle, J.; Zug, S.; El Kasmi, K.C.; Eltzschig, H.K. ATP release from vascular endothelia occurs across Cx43 hemichannels and is attenuated during hypoxia. PLoS ONE 2008, 3, e2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Qin, W.; Xu, X.; Xiong, Y.; Zhang, Y.; Zhang, H.; Sun, B. Endotoxin-induced autocrine ATP signaling inhibits neutrophil chemotaxis through enhancing myosin light chain phosphorylation. Proc. Natl. Acad. Sci. USA 2017, 114, 4483–4488. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Eckle, T.; Mager, A.; Kuper, N.; Karcher, C.; Weissmuller, T.; Boengler, K.; Schulz, R.; Robson, S.C.; Colgan, S.P. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 2006, 99, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Penuela, S.; Gehi, R.; Laird, D.W. The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 2013, 1828, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Dahl, G. ATP release through pannexon channels. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2015, 370. [Google Scholar] [CrossRef]
- Retamal, M.A. Connexin and Pannexin hemichannels are regulated by redox potential. Front. Physiol. 2014, 5, 80. [Google Scholar] [CrossRef] [Green Version]
- Pelegrin, P.; Surprenant, A. The P2X(7) receptor-pannexin connection to dye uptake and IL-1beta release. Purinergic Signal. 2009, 5, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ambrosi, C.; Qiu, F.; Jackson, D.G.; Sosinsky, G.; Dahl, G. The membrane protein Pannexin1 forms two open-channel conformations depending on the mode of activation. Sci. Signal. 2014, 7, ra69. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.H.; Jin, X.; Medina, C.B.; Leonhardt, S.A.; Kiessling, V.; Bennett, B.C.; Shu, S.; Tamm, L.K.; Yeager, M.; Ravichandran, K.S.; et al. A quantized mechanism for activation of pannexin channels. Nat. Commun. 2017, 8, 14324. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Siebert, A.P.; Cheung, K.H.; Lee, R.J.; Johnson, B.; Cohen, A.S.; Vingtdeux, V.; Marambaud, P.; Foskett, J.K. Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc. Natl. Acad. Sci. USA 2012, E109, E1963–E1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebert, A.P.; Ma, Z.; Grevet, J.D.; Demuro, A.; Parker, I.; Foskett, J.K. Structural and functional similarities of calcium homeostasis modulator 1 (CALHM1) ion channel with connexins, pannexins, and innexins. J. Biol. Chem. 2013, 288, 6140–6153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, A.D.; Carey, R.M.; Chen, B.; Saunders, C.J.; Marambaud, P.; Mitchell, C.H.; Tordoff, M.G.; Lee, R.J.; Cohen, N.A. CALHM1-Mediated ATP Release and Ciliary Beat Frequency Modulation in Nasal Epithelial Cells. Sci. Rep. 2017, 7, 6687. [Google Scholar] [CrossRef]
- Taruno, A.; Vingtdeux, V.; Ohmoto, M.; Ma, Z.; Dvoryanchikov, G.; Li, A.; Adrien, L.; Zhao, H.; Leung, S.; Abernethy, M.; et al. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 2013, 495, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Eggermont, J.; Voets, T.; Buyse, G.; Manolopoulos, V.; Droogmans, G. Properties of volume-regulated anion channels in mammalian cells. Prog. Biophys. Mol. Biol. 1997, 68, 69–119. [Google Scholar] [CrossRef]
- Voets, T.; Droogmans, G.; Raskin, G.; Eggermont, J.; Nilius, B. Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proc. Natl. Acad. Sci. USA 1999, 96, 5298–5303. [Google Scholar] [CrossRef] [Green Version]
- Akita, T.; Fedorovich, S.V.; Okada, Y. Ca2+ nanodomain-mediated component of swelling-induced volume-sensitive outwardly rectifying anion current triggered by autocrine action of ATP in mouse astrocytes. Cell Physiol. Biochem. 2011, 28, 1181–1190. [Google Scholar] [CrossRef]
- Hazama, A.; Shimizu, T.; Ando-Akatsuka, Y.; Hayashi, S.; Tanaka, S.; Maeno, E.; Okada, Y. Swelling-induced, CFTR-independent ATP release from a human epithelial cell line: Lack of correlation with volume-sensitive cl(-) channels. J. Gen. Physiol. 1999, 114, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Braunstein, G.M.; Zsembery, A.; Tucker, T.A.; Schwiebert, E.M. Purinergic signaling underlies CFTR control of human airway epithelial cell volume. J. Cyst. Fibros. 2004, 3, 99–117. [Google Scholar] [CrossRef] [Green Version]
- Gaitan-Penas, H.; Gradogna, A.; Laparra-Cuervo, L.; Solsona, C.; Fernandez-Duenas, V.; Barrallo-Gimeno, A.; Ciruela, F.; Lakadamyali, M.; Pusch, M.; Estevez, R. Investigation of LRRC8-Mediated Volume-Regulated Anion Currents in Xenopus Oocytes. Biophys. J. 2016, 111, 1429–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Okada, T.; Islam, M.R.; Sabirov, R.Z. Molecular Identities and ATP Release Activities of Two Types of Volume-Regulatory Anion Channels, VSOR and Maxi-Cl. Curr. Top. Membr. 2018, 81, 125–176. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.K.; Sabirov, R.Z.; Uramoto, H.; Okada, Y. Role of ATP-conductive anion channel in ATP release from neonatal rat cardiomyocytes in ischaemic or hypoxic conditions. J. Physiol. 2004, 559, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Best, L. Study of a glucose-activated anion-selective channel in rat pancreatic beta-cells. Pflugers Arch. 2002, 445, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sabirov, R.Z.; Dutta, A.K.; Okada, Y. Volume-dependent ATP-conductive large-conductance anion channel as a pathway for swelling-induced ATP release. J. Gen. Physiol 2001, 118, 251–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabirov, R.Z.; Okada, Y. Wide nanoscopic pore of maxi-anion channel suits its function as an ATP-conductive pathway. Biophys. J. 2004, 87, 1672–1685. [Google Scholar] [CrossRef] [Green Version]
- Sabirov, R.Z.; Merzlyak, P.G.; Okada, T.; Islam, M.R.; Uramoto, H.; Mori, T.; Makino, Y.; Matsuura, H.; Xie, Y.; Okada, Y. The organic anion transporter SLCO2A1 constitutes the core component of the Maxi-Cl channel. EMBO J. 2017, 36, 3309–3324. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Schmalzing, G.; Markwardt, F. The Elusive P2X7 Macropore. Trends Cell. Biol. 2018, 28, 392–404. [Google Scholar] [CrossRef]
- Pellegatti, P.; Falzoni, S.; Pinton, P.; Rizzuto, R.; Di Virgilio, F. A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol. Biol. Cell 2005, 16, 3659–3665. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, B.; Kaschubowski, K.E.; Nader, S.; Schneider, E.; Nicola, J.A.; Fliegert, R.; Wolf, I.M.A.; Guse, A.H.; Nikolaev, V.O.; Koch-Nolte, F.; et al. P2X7-mediated ATP secretion is accompanied by depletion of cytosolic ATP. Purinergic Signal. 2019, 15, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Brandao-Burch, A.; Key, M.L.; Patel, J.J.; Arnett, T.R.; Orriss, I.R. The P2X7 Receptor is an Important Regulator of Extracellular ATP Levels. Front. Endocrinol. (Lausanne) 2012, 3, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, Y.; Tsukimoto, M.; Takenouchi, T.; Harada, H.; Suzuki, A.; Sato, M.; Kitani, H.; Kojima, S. gamma-Irradiation induces P2X(7) receptor-dependent ATP release from B16 melanoma cells. Biochim. Biophys. Acta 2010, 1800, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Fabre, A.C.; Vantourout, P.; Champagne, E.; Terce, F.; Rolland, C.; Perret, B.; Collet, X.; Barbaras, R.; Martinez, L.O. Cell surface adenylate kinase activity regulates the F(1)-ATPase/P2Y (13)-mediated HDL endocytosis pathway on human hepatocytes. Cell Mol. Life Sci. 2006, 63, 2829–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yegutkin, G.G.; Henttinen, T.; Samburski, S.S.; Spychala, J.; Jalkanen, S. The evidence for two opposite, ATP-generating and ATP-consuming, extracellular pathways on endothelial and lymphoid cells. Biochem. J. 2002, 367, 121–128. [Google Scholar] [CrossRef]
- Zeiner, J.; Loukovaara, S.; Losenkova, K.; Zuccarini, M.; Korhonen, A.M.; Lehti, K.; Kauppinen, A.; Kaarniranta, K.; Muller, C.E.; Jalkanen, S.; et al. Soluble and membrane-bound adenylate kinase and nucleotidases augment ATP-mediated inflammation in diabetic retinopathy eyes with vitreous hemorrhage. J. Mol. Med. (Berl) 2019, 97, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Agren, G.; Ponten, J.; Ronquist, G.; Westermark, B. Nucleoside diphosphate kinase at the cell surface of neoplastic human cells in culture. J. Cell Physiol. 1974, 83, 91–101. [Google Scholar] [CrossRef]
- Boison, D.; Yegutkin, G.G. Adenosine Metabolism: Emerging Concepts for Cancer Therapy. Cancer Cell 2019, 36, 582–596. [Google Scholar] [CrossRef]
- Chi, S.L.; Pizzo, S.V. Angiostatin is directly cytotoxic to tumor cells at low extracellular pH: A mechanism dependent on cell surface-associated ATP synthase. Cancer Res. 2006, 66, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Kita, T.; Arakaki, N. Contribution of extracellular ATP on the cell-surface F1F0-ATP synthase-mediated intracellular triacylglycerol accumulation. Biomed. Res. 2015, 36, 115–120. [Google Scholar] [CrossRef] [Green Version]
- McAllister, S.S.; Weinberg, R.A. Tumor-host interactions: A far-reaching relationship. J. Clin. Oncol. 2010, 28, 4022–4028. [Google Scholar] [CrossRef]
- Rajendran, M.; Dane, E.; Conley, J.; Tantama, M. Imaging Adenosine Triphosphate (ATP). Biol. Bull. 2016, 231, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigi, R.; Kobatake, E.; Aizawa, M.; Dubyak, G.R. Detection of local ATP release from activated platelets using cell surface-attached firefly luciferase. Am. J. Physiol 1999, 276, C267–C278. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.W.; Egan, M.E.; Jena, B.P.; Guggino, W.B.; Oberleithner, H.; Geibel, J.P. Continuous detection of extracellular ATP on living cells by using atomic force microscopy. Proc. Natl. Acad. Sci. USA 1999, 96, 12180–12185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corriden, R.; Insel, P.A.; Junger, W.G. A novel method using fluorescence microscopy for real-time assessment of ATP release from individual cells. Am. J. Physiol. Cell Physiol. 2007, 293, C1420–C1425. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, G.; Yang, L.; Gajewski, C.D.; Mattiazzi, M. Measurements of ATP in mammalian cells. Methods 2002, 26, 317–326. [Google Scholar] [CrossRef]
- Hayashi, S.; Hazama, A.; Dutta, A.K.; Sabirov, R.Z.; Okada, Y. Detecting ATP release by a biosensor method. Sci. STKE 2004, 2004, pl14. [Google Scholar] [CrossRef]
- Llaudet, E.; Hatz, S.; Droniou, M.; Dale, N. Microelectrode biosensor for real-time measurement of ATP in biological tissue. Anal. Chem. 2005, 77, 3267–3273. [Google Scholar] [CrossRef]
- Chen, Y.; Corriden, R.; Inoue, Y.; Yip, L.; Hashiguchi, N.; Zinkernagel, A.; Nizet, V.; Insel, P.A.; Junger, W.G. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006, 314, 1792–1795. [Google Scholar] [CrossRef] [Green Version]
- Vancraenenbroeck, R.; Webb, M.R. A Fluorescent, Reagentless Biosensor for ATP, Based on Malonyl-Coenzyme A Synthetase. ACS Chem. Biol. 2015, 10, 2650–2657. [Google Scholar] [CrossRef]
- Conley, J.M.; Radhakrishnan, S.; Valentino, S.A.; Tantama, M. Imaging extracellular ATP with a genetically-encoded, ratiometric fluorescent sensor. PLoS ONE 2017, 12, e0187481. [Google Scholar] [CrossRef] [Green Version]
- Trull, K.J.; Miller, P.; Tat, K.; Varney, S.A.; Conley, J.M.; Tantama, M. Detection of Osmotic Shock-Induced Extracellular Nucleotide Release with a Genetically Encoded Fluorescent Sensor of ADP and ATP. Sensors 2019, 19, 3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef] [PubMed]
- Lecciso, M.; Ocadlikova, D.; Sangaletti, S.; Trabanelli, S.; De Marchi, E.; Orioli, E.; Pegoraro, A.; Portararo, P.; Jandus, C.; Bontadini, A.; et al. ATP Release from Chemotherapy-Treated Dying Leukemia Cells Elicits an Immune Suppressive Effect by Increasing Regulatory T Cells and Tolerogenic Dendritic Cells. Front. Immunol. 2017, 8, 1918. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Sarti, A.C.; Marchi, S.; Missiroli, S.; Falzoni, S.; Raffaghello, L.; Pistoia, V.; Giorgi, C.; Di Virgilio, F.; Pinton, P. Use of luciferase probes to measure ATP in living cells and animals. Nat. Protoc. 2017, 12, 1542–1562. [Google Scholar] [CrossRef]
- De Marchi, E.; Orioli, E.; Pegoraro, A.; Adinolfi, E.; Di Virgilio, F. Detection of Extracellular ATP in the Tumor Microenvironment, Using the pmeLUC Biosensor. Methods Mol. Biol. 2020, 2041, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Vuerich, M.; Pellegatti, P.; Marimpietri, D.; Emionite, L.; Marigo, I.; Bronte, V.; Di Virgilio, F.; Pistoia, V.; Raffaghello, L. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. 2014, 5, e1135. [Google Scholar] [CrossRef] [Green Version]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Rao, S.; Enot, D.P.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Jacquelot, N.; Yamazaki, T.; et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 2016, 30, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Investig. 2002, 110, 993–1002. [Google Scholar] [CrossRef]
- Dwyer, K.M.; Deaglio, S.; Gao, W.; Friedman, D.; Strom, T.B.; Robson, S.C. CD39 and control of cellular immune responses. Purinergic Signal. 2007, 3, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, K.M.; Kishore, B.K.; Robson, S.C. Conversion of extracellular ATP into adenosine: A master switch in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Yegutkin, G.G.; Pacher, P.; Blandizzi, C.; Hasko, G. Anti-CD73 in cancer immunotherapy: Awakening new opportunities. Trends Cancer 2016, 2, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, M.R.; Fernandez, D.; Flores-Santibanez, F.; Rosemblatt, M.; Sauma, D. CD73 and CD39 ectonucleotidases in T cell differentiation: Beyond immunosuppression. FEBS Lett. 2015, 589, 3454–3460. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Kunzli, B.M.; YI, A.R.; Sevigny, J.; Berberat, P.O.; Enjyoji, K.; Csizmadia, E.; Friess, H.; Robson, S.C. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16788–16793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Mirandola, P.; Milani, D.; Varani, K.; Gessi, S.; Klotz, K.N.; Leung, E.; Baraldi, P.G.; Borea, P.A. Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J. Investig. Dermatol 2002, 119, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.G.; Jacobson, K.A. A2B Adenosine Receptor and Cancer. Int. J. Mol. Sci. 2019, 20, 5139. [Google Scholar] [CrossRef] [Green Version]
- Gessi, S.; Sacchetto, V.; Fogli, E.; Merighi, S.; Varani, K.; Baraldi, P.G.; Tabrizi, M.A.; Leung, E.; Maclennan, S.; Borea, P.A. Modulation of metalloproteinase-9 in U87MG glioblastoma cells by A3 adenosine receptors. Biochem. Pharmacol. 2010, 79, 1483–1495. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Menetrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [Green Version]
- Linden, J.; Cekic, C. Regulation of lymphocyte function by adenosine. Arterioscler Thromb. Vasc. Biol. 2012, 32, 2097–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G. Purine and pyrimidine receptors. Cell Mol. Life Sci. 2007, 64, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Di Virgilio, F. Purinergic signalling and cancer. Purinergic Signal. 2013, 9, 491–540. [Google Scholar] [CrossRef] [PubMed]
- Schulien, I.; Hockenjos, B.; van Marck, V.; Ayata, C.K.; Follo, M.; Thimme, R.; Hasselblatt, P. Extracellular ATP and Purinergic P2Y2 Receptor Signaling Promote Liver Tumorigenesis in Mice by Exacerbating DNA Damage. Cancer Res. 2020, 80, 699–708. [Google Scholar] [CrossRef]
- Takai, E.; Tsukimoto, M.; Harada, H.; Kojima, S. Autocrine signaling via release of ATP and activation of P2X7 receptor influences motile activity of human lung cancer cells. Purinergic Signal. 2014, 10, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Yu, X.; Tang, L.; Li, G.; He, T. P2X7 receptor stimulates breast cancer cell invasion and migration via the AKT pathway. Oncol. Rep. 2015, 34, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.L.; Liu, Y.; Yang, H.; Zhang, H.Q.; Tian, X.X.; Fang, W.G. ATP-P2Y2-beta-catenin axis promotes cell invasion in breast cancer cells. Cancer Sci. 2017, 108, 1318–1327. [Google Scholar] [CrossRef]
- Adinolfi, E.; Melchiorri, L.; Falzoni, S.; Chiozzi, P.; Morelli, A.; Tieghi, A.; Cuneo, A.; Castoldi, G.; Di Virgilio, F.; Baricordi, O.R. P2X7 receptor expression in evolutive and indolent forms of chronic B lymphocytic leukemia. Blood 2002, 99, 706–708. [Google Scholar] [CrossRef]
- Raffaghello, L.; Chiozzi, P.; Falzoni, S.; Di Virgilio, F.; Pistoia, V. The P2X7 receptor sustains the growth of human neuroblastoma cells through a substance P-dependent mechanism. Cancer Res. 2006, 66, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, F.; Capece, M.; Rotondo, A.; Cangelosi, D.; Ferracin, M.; Franceschini, A.; Raffaghello, L.; Pistoia, V.; Varesio, L.; Adinolfi, E. The P2X7 receptor is a key modulator of the PI3K/GSK3beta/VEGF signaling network: Evidence in experimental neuroblastoma. Oncogene 2015, 34, 5240–5251. [Google Scholar] [CrossRef]
- Robinson, L.E.; Shridar, M.; Smith, P.; Murrell-Lagnado, R.D. Plasma membrane cholesterol as a regulator of human and rodent P2X7 receptor activation and sensitization. J. Biol. Chem 2014, 289, 31983–31994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, J.P.; Lee, J.S.; Sohn, B.H.; Yu, X.; Lopez-Terrada, D.; Finegold, M.J.; Goss, J.A.; Thevananther, S. P2X3 purinergic receptor overexpression is associated with poor recurrence-free survival in hepatocellular carcinoma patients. Oncotarget 2015, 6, 41162–41179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, A.V.; Linge, C.; Healy, V.; Lim, P.; Clayton, E.; Rustin, M.H.; McGrouther, D.A.; Burnstock, G. Expression of purinergic receptors in non-melanoma skin cancers and their functional roles in A431 cells. J. Invest. Dermatol. 2003, 121, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Ledderose, C.; Woehrle, T.; Ledderose, S.; Strasser, K.; Seist, R.; Bao, Y.; Zhang, J.; Junger, W.G. Cutting off the power: Inhibition of leukemia cell growth by pausing basal ATP release and P2X receptor signaling? Purinergic Signal. 2016, 12, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Lapel, M.; Weston, P.; Strassheim, D.; Karoor, V.; Burns, N.; Lyubchenko, T.; Paucek, P.; Stenmark, K.R.; Gerasimovskaya, E.V. Glycolysis and oxidative phosphorylation are essential for purinergic receptor-mediated angiogenic responses in vasa vasorum endothelial cells. Am. J. Physiol. Cell Physiol. 2017, 312, C56–C70. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, E.; Callegari, M.G.; Ferrari, D.; Bolognesi, C.; Minelli, M.; Wieckowski, M.R.; Pinton, P.; Rizzuto, R.; Di Virgilio, F. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol. Biol. Cell 2005, 16, 3260–3272. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, F.; Falzoni, S.; Adinolfi, E.; Ferrari, D.; Di Virgilio, F. The P2X7 receptor is a key modulator of aerobic glycolysis. Cell Death Dis. 2012, 3, e370. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 2016, 39, 1–6. [Google Scholar] [CrossRef]
- Junger, W.G. Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 2011, 11, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Aswad, F.; Kawamura, H.; Dennert, G. High sensitivity of CD4+CD25+ regulatory T cells to extracellular metabolites nicotinamide adenine dinucleotide and ATP: A role for P2X7 receptors. J. Immunol. 2005, 175, 3075–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, D.; La Sala, A.; Chiozzi, P.; Morelli, A.; Falzoni, S.; Girolomoni, G.; Idzko, M.; Dichmann, S.; Norgauer, J.; Di Virgilio, F. The P2 purinergic receptors of human dendritic cells: Identification and coupling to cytokine release. FASEB J. 2000, 14, 2466–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adinolfi, E.; De Marchi, E.; Orioli, E.; Pegoraro, A.; Di Virgilio, F. Role of the P2X7 receptor in tumor-associated inflammation. Curr. Opin. Pharmacol. 2019, 47, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E.; Heppel, L.A. A Specific effect of external ATP on the permeability of transformed 3T3 cells. Biochem. Biophys. Res. Commun. 1975, 67, 1581–1588. [Google Scholar] [CrossRef]
- Landry, Y.; Lehninger, A.L. Transport of calcium ions by Ehrlich ascites-tumour cells. Biochem J. 1976, 158, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Zambon, A.; Bronte, V.; Di Virgilio, F.; Hanau, S.; Steinberg, T.H.; Collavo, D.; Zanovello, P. Role of extracellular ATP in cell-mediated cytotoxicity: A study with ATP-sensitive and ATP-resistant macrophages. Cell Immunol. 1994, 156, 458–467. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Bronte, V.; Collavo, D.; Zanovello, P. Responses of mouse lymphocytes to extracellular adenosine 5′-triphosphate (ATP). Lymphocytes with cytotoxic activity are resistant to the permeabilizing effects of ATP. J. Immunol. 1989, 143, 1955–1960. [Google Scholar]
- Zanovello, P.; Bronte, V.; Rosato, A.; Pizzo, P.; Di Virgilio, F. Responses of mouse lymphocytes to extracellular ATP. II. Extracellular ATP causes cell type-dependent lysis and DNA fragmentation. J. Immunol. 1990, 145, 1545–1550. [Google Scholar]
- Pizzo, P.; Murgia, M.; Zambon, A.; Zanovello, P.; Bronte, V.; Pietrobon, D.; Di Virgilio, F. Role of P2z purinergic receptors in ATP-mediated killing of tumor necrosis factor (TNF)-sensitive and TNF-resistant L929 fibroblasts. J. Immunol. 1992, 149, 3372–3378. [Google Scholar]
- Rapaport, E. Treatment of human tumor cells with ADP or ATP yields arrest of growth in the S phase of the cell cycle. J. Cell Physiol 1983, 114, 279–283. [Google Scholar] [CrossRef]
- Rapaport, E. Experimental cancer therapy in mice by adenine nucleotides. Eur. J. Cancer Clin. Oncol 1988, 24, 1491–1497. [Google Scholar] [CrossRef]
- Shabbir, M.; Thompson, C.; Jarmulowiczc, M.; Mikhailidis, D.; Burnstock, G. Effect of extracellular ATP on the growth of hormone-refractory prostate cancer in vivo. BJU Int. 2008, 102, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Haskell, C.M.; Mendoza, E.; Pisters, K.M.; Fossella, F.V.; Figlin, R.A. Phase II study of intravenous adenosine 5′-triphosphate in patients with previously untreated stage IIIB and stage IV non-small cell lung cancer. Invest. New Drugs 1998, 16, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Agteresch, H.J.; Dagnelie, P.C.; Rietveld, T.; van den Berg, J.W.; Danser, A.H.; Wilson, J.H. Pharmacokinetics of intravenous ATP in cancer patients. Eur J. Clin. Pharmacol. 2000, 56, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Matty, M.A.; Knudsen, D.R.; Walton, E.M.; Beerman, R.W.; Cronan, M.R.; Pyle, C.J.; Hernandez, R.E.; Tobin, D.M. Potentiation of P2RX7 as a host-directed strategy for control of mycobacterial infection. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Pizzirani, C.; Gulinelli, S.; Callegari, G.; Chiozzi, P.; Idzko, M.; Panther, E.; Di Virgilio, F. Modulation of P2X7 receptor functions by polymyxin B: Crucial role of the hydrophobic tail of the antibiotic molecule. Br. J. Pharmacol. 2007, 150, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Bidula, S.M.; Cromer, B.A.; Walpole, S.; Angulo, J.; Stokes, L. Mapping a novel positive allosteric modulator binding site in the central vestibule region of human P2X7. Sci. Rep. 2019, 9, 3231. [Google Scholar] [CrossRef]
- de Andrade Mello, P.; Bian, S.; Savio, L.E.B.; Zhang, H.; Zhang, J.; Junger, W.; Wink, M.R.; Lenz, G.; Buffon, A.; Wu, Y.; et al. Hyperthermia and associated changes in membrane fluidity potentiate P2X7 activation to promote tumor cell death. Oncotarget 2017, 8, 67254–67268. [Google Scholar] [CrossRef]
- Munerati, M.; Cortesi, R.; Ferrari, D.; Di Virgilio, F.; Nastruzzi, C. Macrophages loaded with doxorubicin by ATP-mediated permeabilization: Potential carriers for antitumor therapy. Biochim. Biophys. Acta 1994, 1224, 269–276. [Google Scholar] [CrossRef]
- Modderman, W.E.; Weidema, A.F.; Vrijheid-Lammers, T.; Wassenaar, A.M.; Nijweide, P.J. Permeabilization of cells of hemopoietic origin by extracellular ATP4-: Elimination of osteoclasts, macrophages, and their precursors from isolated bone cell populations and fetal bone rudiments. Calcif. Tissue Int. 1994, 55, 141–150. [Google Scholar] [CrossRef]
- Qi, B.; Yu, T.; Wang, C.; Wang, T.; Yao, J.; Zhang, X.; Deng, P.; Xia, Y.; Junger, W.G.; Sun, D. Shock wave-induced ATP release from osteosarcoma U2OS cells promotes cellular uptake and cytotoxicity of methotrexate. J. Exp. Clin. Cancer Res. 2016, 35, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapaport, E.; Salikhova, A.; Abraham, E.H. Continuous intravenous infusion of ATP in humans yields large expansions of erythrocyte ATP pools but extracellular ATP pools are elevated only at the start followed by rapid declines. Purinergic Signal. 2015, 11, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Saldivar, P.; Huidobro-Toro, J.P. ATP-loaded biomimetic nanoparticles as controlled release system for extracellular drugs in cancer applications. Int. J. Nanomedicine 2019, 14, 2433–2447. [Google Scholar] [CrossRef] [Green Version]
- Feiz, M.S.; Meshkini, A. Targeted delivery of adenosine 5′-triphosphate using chitosan-coated mesoporous hydroxyapatite: A theranostic pH-sensitive nanoplatform with enhanced anti-cancer effect. Int J. Biol. Macromol. 2019, 129, 1090–1102. [Google Scholar] [CrossRef] [PubMed]
- Moesta, A.K.; Li, X.Y.; Smyth, M.J. Targeting CD39 in cancer. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Zhang, B. Targeting CD73 to augment cancer immunotherapy. Curr. Opin Pharmacol 2020, 53, 66–76. [Google Scholar] [CrossRef]
- Li, X.Y.; Moesta, A.K.; Xiao, C.; Nakamura, K.; Casey, M.; Zhang, H.; Madore, J.; Lepletier, A.; Aguilera, A.R.; Sundarrajan, A.; et al. Targeting CD39 in Cancer Reveals an Extracellular ATP- and Inflammasome-Driven Tumor Immunity. Cancer Discov. 2019, 9, 1754–1773. [Google Scholar] [CrossRef] [Green Version]
- Festag, J.; Thelemann, T.; Schell, M.; Raith, S.; Michel, S.; Jaschinski, F.; Klar, R. Preventing ATP Degradation by ASO-Mediated Knockdown of CD39 and CD73 Results in A2aR-Independent Rescue of T Cell Proliferation. Mol. Ther. Nucleic Acids 2020, 21, 656–669. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Sato, H.; Okonogi, N.; Nakano, T. Rationale of combination of anti-PD-1/PD-L1 antibody therapy and radiotherapy for cancer treatment. Int. J. Clin. Oncol. 2020, 25, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Kamata-Sakurai, M.; Narita, Y.; Hori, Y.; Nemoto, T.; Uchikawa, R.; Honda, M.; Hironiwa, N.; Taniguchi, K.; Shida-Kawazoe, M.; Metsugi, S.; et al. Antibody to CD137 activated by extracellular adenosine triphosphate is tumor selective and broadly effective in vivo without systemic immune activation. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- Furlow, P.W.; Zhang, S.; Soong, T.D.; Halberg, N.; Goodarzi, H.; Mangrum, C.; Wu, Y.G.; Elemento, O.; Tavazoie, S.F. Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat. Cell Biol. 2015, 17, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, F.; Salaro, E.; Falzoni, S.; Chiozzi, P.; Giuliani, A.L.; Cavallesco, G.; Maniscalco, P.; Puozzo, A.; Bononi, I.; Martini, F.; et al. P2X7 targeting inhibits growth of human mesothelioma. Oncotarget 2016, 7, 49664–49676. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Shen, J.J.; Zhan, Y.C.; Li, H.Y.; Wang, Z. Ouabain impairs cancer metabolism and activates AMPK-Src signaling pathway in human cancer cell lines. Acta Pharmacol. Sin. 2020, 41, 110–118. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Wang, X.; Li, Y.; Cao, Y.; Chen, X. Extracellular ATP a New Player in Cancer Metabolism: NSCLC Cells Internalize ATP In Vitro and In Vivo Using Multiple Endocytic Mechanisms. Mol. Cancer Res. 2016, 14, 1087–1096. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, Y.; Qian, Y.; Cao, Y.; Shriwas, P.; Zhang, H.; Chen, X. Extracellular ATP, as an energy and phosphorylating molecule, induces different types of drug resistances in cancer cells through ATP internalization and intracellular ATP level increase. Oncotarget 2017, 8, 87860–87877. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, X.; Li, Y.; Evers, M.; Zhang, H.; Chen, X. Extracellular and macropinocytosis internalized ATP work together to induce epithelial-mesenchymal transition and other early metastatic activities in lung cancer. Cancer Cell Int. 2019, 19, 254. [Google Scholar] [CrossRef]
- Loo, J.M.; Scherl, A.; Nguyen, A.; Man, F.Y.; Weinberg, E.; Zeng, Z.; Saltz, L.; Paty, P.B.; Tavazoie, S.F. Extracellular metabolic energetics can promote cancer progression. Cell 2015, 160, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Geng, Y.H.; Wang, P.; Yang, H.; Zhou, Y.T.; Zhang, H.Q.; He, H.Y.; Fang, W.G.; Tian, X.X. Extracellular ATP promotes breast cancer invasion and chemoresistance via SOX9 signaling. Oncogene 2020, 39, 5795–5810. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Identifier Code | Target |
|---|---|---|
| Study of SRF617 in patients with advanced solid tumors | NCT04336098 | CD39 |
| TTX-030 in combination with immunotherapy and/or chemotherapy in subjects with advanced cancers | NCT04306900 NCT03884556 | CD39 |
| A study of the CD73 inhibitor LY3475070 alone or in combination with Pembrolizumab in participants with advanced cancer | NCT04148937 | CD73 |
| CPI-006 alone and in combination with ciforadenant and pembrolizumab for patients with advanced cancer | NCT03454451 | CD73 |
| Study of TJ004309 in combination with atezolizumab in patients with advanced or metastatic cancer | NCT03835949 | CD73 |
| A phase I/Ib study of NZV930 alone and in combination with PDR001 and/or NIR178 in patients with advanced malignancies | NCT03549000 | CD73 |
| A phase I, open-label study to assess the safety, tolerability, pharmacokinetics and antitumor activity of MED19447 (oleclumab) in Japanese patients with advanced solid malignancies | JapicCTI-184194 | CD73 |
| An investigational immune-therapy study of experimental medication BMS986179 given alone and in combination with nivolumab | NCT02754141 | CD73 |
| A phase Ia/Ib study of STA551 as a single agent and in combination with atezolizumab in patients with solid tumors | JapicCTI-205153 | CD137 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vultaggio-Poma, V.; Sarti, A.C.; Di Virgilio, F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells 2020, 9, 2496. https://doi.org/10.3390/cells9112496
Vultaggio-Poma V, Sarti AC, Di Virgilio F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells. 2020; 9(11):2496. https://doi.org/10.3390/cells9112496
Chicago/Turabian StyleVultaggio-Poma, Valentina, Alba Clara Sarti, and Francesco Di Virgilio. 2020. "Extracellular ATP: A Feasible Target for Cancer Therapy" Cells 9, no. 11: 2496. https://doi.org/10.3390/cells9112496
APA StyleVultaggio-Poma, V., Sarti, A. C., & Di Virgilio, F. (2020). Extracellular ATP: A Feasible Target for Cancer Therapy. Cells, 9(11), 2496. https://doi.org/10.3390/cells9112496