Perspective on Schwann Cells Derived from Induced Pluripotent Stem Cells in Peripheral Nerve Tissue Engineering

 , and
, and
Abstract
1. Introduction
2. Schwann Cells in the Injured Peripheral Nerve
3. Purification and Culture of Primary Human Schwann Cells
4. Alternative Sources of Schwann Cells
5. Induced Pluripotent Stem Cells as a Source for Schwann Cells
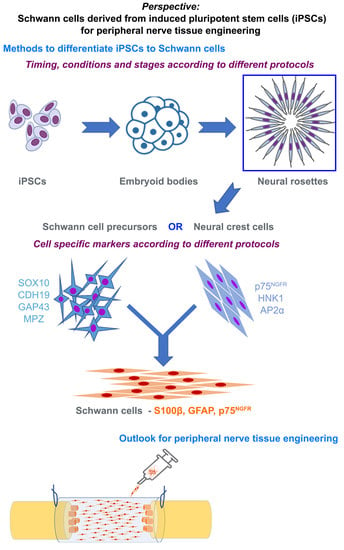
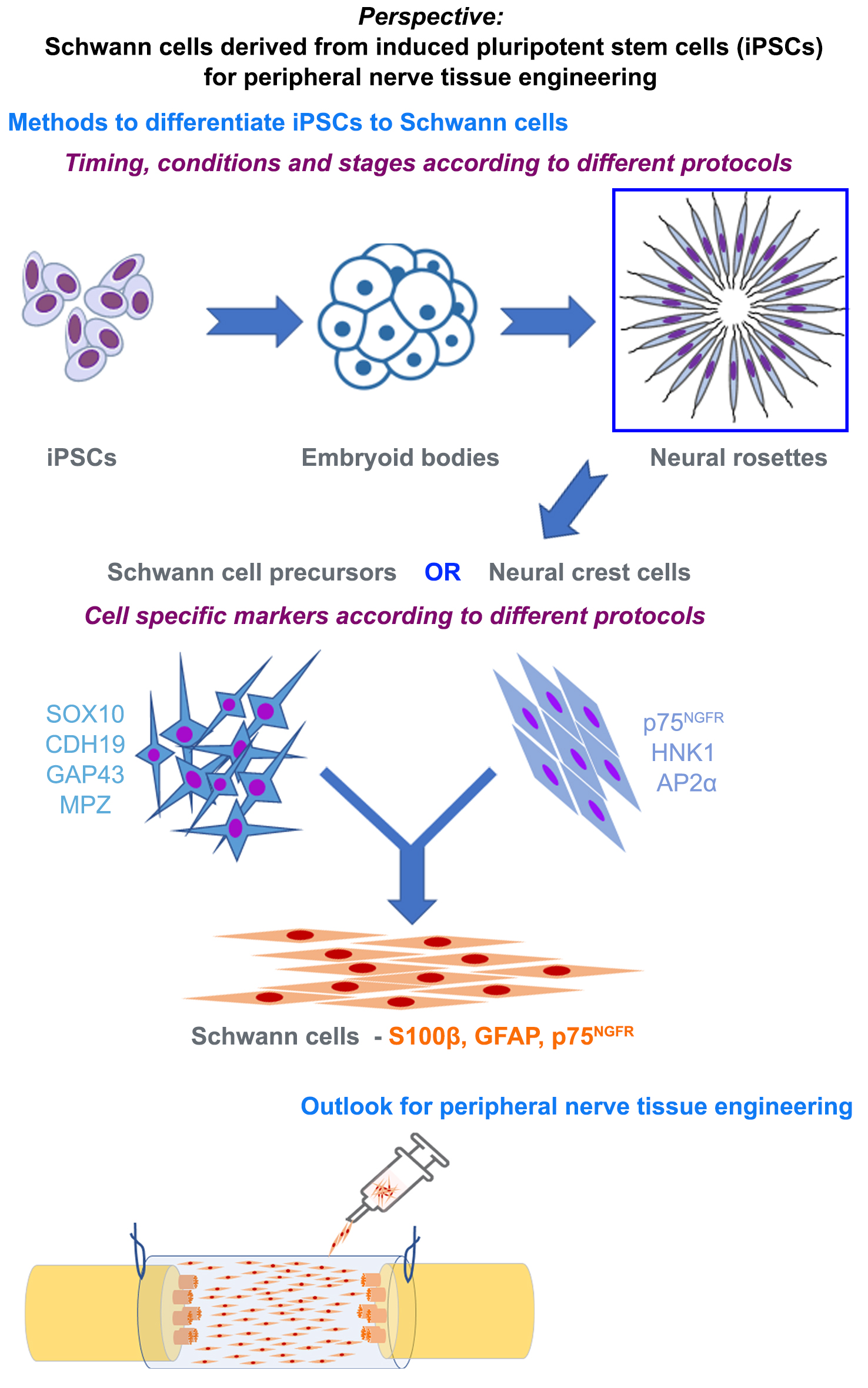
6. Methods to Differentiate iPSCs to Schwann Cells
7. Characterizing Schwann Cells Differentiated from iPSCs
8. Phenotype of Differentiated Schwann Cells—In Vitro and In Vivo
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Catala, M.; Kubis, N. Gross anatomy and development of the peripheral nervous system. Handb. Clin. Neurol. 2013, 115, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, S.; Odaci, E.; Unal, B.; Sahin, B.; Fornaro, M. Chapter 2: Development of the peripheral nerve. Int. Rev. Neurobiol. 2009, 87, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005, 6, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Deumens, R.; Bozkurt, A.; Meek, M.F.; Marcus, M.A.; Joosten, E.A.; Weis, J.; Brook, G.A. Repairing injured peripheral nerves: Bridging the gap. Prog. Neurobiol. 2010, 92, 245–276. [Google Scholar] [CrossRef]
- Navarro, X.; Vivo, M.; Valero-Cabre, A. Neural plasticity after peripheral nerve injury and regeneration. Prog. Neurobiol. 2007, 82, 163–201. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; et al. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 2012, 75, 633–647. [Google Scholar] [CrossRef]
- Jessen, K.R.; Arthur-Farraj, P. Repair Schwann cell update: Adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia 2019, 67, 421–437. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef]
- Pan, D.; Mackinnon, S.E.; Wood, M.D. Advances in the repair of segmental nerve injuries and trends in reconstruction. Muscle Nerve 2020, 61, 726–739. [Google Scholar] [CrossRef]
- Cattin, A.L.; Burden, J.J.; Van Emmenis, L.; Mackenzie, F.E.; Hoving, J.J.; Garcia Calavia, N.; Guo, Y.; McLaughlin, M.; Rosenberg, L.H.; Quereda, V.; et al. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 2015, 162, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Dun, X.P.; Carr, L.; Woodley, P.K.; Barry, R.W.; Drake, L.K.; Mindos, T.; Roberts, S.L.; Lloyd, A.C.; Parkinson, D.B. Macrophage-Derived Slit3 Controls Cell Migration and Axon Pathfinding in the Peripheral Nerve Bridge. Cell Rep. 2019, 26, 1458–1472.e1454. [Google Scholar] [CrossRef] [PubMed]
- Nocera, G.; Jacob, C. Mechanisms of Schwann cell plasticity involved in peripheral nerve repair after injury. Cell Mol. Life Sci. 2020, 77, 3977–3989. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.L.; Dun, X.P.; Doddrell, R.D.S.; Mindos, T.; Drake, L.K.; Onaitis, M.W.; Florio, F.; Quattrini, A.; Lloyd, A.C.; D’Antonio, M.; et al. Sox2 expression in Schwann cells inhibits myelination in vivo and induces influx of macrophages to the nerve. Development 2017, 144, 3114–3125. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, R.E.; Echevarria, F.D. Macrophage biology in the peripheral nervous system after injury. Prog. Neurobiol. 2019, 173, 102–121. [Google Scholar] [CrossRef] [PubMed]
- Lindborg, J.A.; Mack, M.; Zigmond, R.E. Neutrophils Are Critical for Myelin Removal in a Peripheral Nerve Injury Model of Wallerian Degeneration. J. Neurosci. 2017, 37, 10258–10277. [Google Scholar] [CrossRef] [PubMed]
- Stratton, J.A.; Holmes, A.; Rosin, N.L.; Sinha, S.; Vohra, M.; Burma, N.E.; Trang, T.; Midha, R.; Biernaskie, J. Macrophages Regulate Schwann Cell Maturation after Nerve Injury. Cell Rep. 2018, 24, 2561–2572.e2566. [Google Scholar] [CrossRef]
- Parrinello, S.; Napoli, I.; Ribeiro, S.; Wingfield Digby, P.; Fedorova, M.; Parkinson, D.B.; Doddrell, R.D.; Nakayama, M.; Adams, R.H.; Lloyd, A.C. EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell 2010, 143, 145–155. [Google Scholar] [CrossRef]
- Klein, D.; Martini, R. Myelin and macrophages in the PNS: An intimate relationship in trauma and disease. Brain Res. 2016, 1641, 130–138. [Google Scholar] [CrossRef]
- Groh, J.; Klein, I.; Hollmann, C.; Wettmarshausen, J.; Klein, D.; Martini, R. CSF-1-activated macrophages are target-directed and essential mediators of Schwann cell dedifferentiation and dysfunction in Cx32-deficient mice. Glia 2015, 63, 977–986. [Google Scholar] [CrossRef]
- Shibuya, Y.; Mizoguchi, A.; Takeichi, M.; Shimada, K.; Ide, C. Localization of N-cadherin in the normal and regenerating nerve fibers of the chicken peripheral nervous system. Neuroscience 1995, 67, 253–261. [Google Scholar] [CrossRef]
- Chen, Y.Y.; McDonald, D.; Cheng, C.; Magnowski, B.; Durand, J.; Zochodne, D.W. Axon and Schwann cell partnership during nerve regrowth. J. Neuropathol. Exp. Neurol. 2005, 64, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Muscella, A.; Vetrugno, C.; Cossa, L.G.; Marsigliante, S. TGF-β1 activates RSC96 Schwann cells migration and invasion through MMP-2 and MMP-9 activities. J. Neurochem. 2020, 153, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.P.; Byrne, E.; Camarillo Guerrero, L.F.; Cattin, A.L.; Zakka, L.; Ashraf, A.; Burden, J.J.; Khadayate, S.; Lloyd, A.C.; Marguerat, S.; et al. The Wound Microenvironment Reprograms Schwann Cells to Invasive Mesenchymal-like Cells to Drive Peripheral Nerve Regeneration. Neuron 2017, 96, 98–114.e117. [Google Scholar] [CrossRef] [PubMed]
- Painter, M.W. Aging Schwann cells: Mechanisms, implications, future directions. Curr. Opin. Neurobiol. 2017, 47, 203–208. [Google Scholar] [CrossRef]
- Painter, M.W.; Brosius Lutz, A.; Cheng, Y.C.; Latremoliere, A.; Duong, K.; Miller, C.M.; Posada, S.; Cobos, E.J.; Zhang, A.X.; Wagers, A.J.; et al. Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron 2014, 83, 331–343. [Google Scholar] [CrossRef]
- Gonçalves, N.P.; Mohseni, S.; El Soury, M.; Ulrichsen, M.; Richner, M.; Xiao, J.; Wood, R.J.; Andersen, O.M.; Coulson, E.J.; Raimondo, S.; et al. Peripheral Nerve Regeneration Is Independent From Schwann Cell p75(NTR) Expression. Front. Cell Neurosci. 2019, 13, 235. [Google Scholar] [CrossRef]
- Scheib, J.L.; Höke, A. An attenuated immune response by Schwann cells and macrophages inhibits nerve regeneration in aged rats. Neurobiol. Aging 2016, 45, 1–9. [Google Scholar] [CrossRef]
- Büttner, R.; Schulz, A.; Reuter, M.; Akula, A.K.; Mindos, T.; Carlstedt, A.; Riecken, L.B.; Baader, S.L.; Bauer, R.; Morrison, H. Inflammaging impairs peripheral nerve maintenance and regeneration. Aging Cell 2018, 17, e12833. [Google Scholar] [CrossRef]
- Poppler, L.H.; Ee, X.; Schellhardt, L.; Hoben, G.M.; Pan, D.; Hunter, D.A.; Yan, Y.; Moore, A.M.; Snyder-Warwick, A.K.; Stewart, S.A.; et al. Axonal Growth Arrests After an Increased Accumulation of Schwann Cells Expressing Senescence Markers and Stromal Cells in Acellular Nerve Allografts. Tissue Eng. Part. A 2016, 22, 949–961. [Google Scholar] [CrossRef]
- Heathman, T.R.; Nienow, A.W.; McCall, M.J.; Coopman, K.; Kara, B.; Hewitt, C.J. The translation of cell-based therapies: Clinical landscape and manufacturing challenges. Regen. Med. 2015, 10, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Monje, P.V. The properties of human Schwann cells: Lessons from in vitro culture and transplantation studies. Glia 2020, 68, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Monje, P.V. Schwann Cell Cultures: Biology, Technology and Therapeutics. Cells 2020, 9, 1848. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Martinez, D.; Garavito, Z.; Spinel, C.; Hurtado, H. Schwann cell-enriched cultures from adult human peripheral nerve: A technique combining short enzymatic dissociation and treatment with cytosine arabinoside (Ara-C). J. Neurosci. Methods 2002, 114, 1–8. [Google Scholar] [CrossRef]
- Weiss, T.; Taschner-Mandl, S.; Ambros, P.F.; Ambros, I.M. Detailed Protocols for the Isolation, Culture, Enrichment and Immunostaining of Primary Human Schwann Cells. Methods Mol. Biol. (Clifton N. J.) 2018, 1739, 67–86. [Google Scholar] [CrossRef]
- Jirsova, K.; Sodaar, P.; Mandys, V.; Bar, P.R. Cold jet: A method to obtain pure Schwann cell cultures without the need for cytotoxic, apoptosis-inducing drug treatment. J. Neurosci. Methods 1997, 78, 133–137. [Google Scholar] [CrossRef]
- Haastert, K.; Mauritz, C.; Chaturvedi, S.; Grothe, C. Human and rat adult Schwann cell cultures: Fast and efficient enrichment and highly effective non-viral transfection protocol. Nat. Protoc. 2007, 2, 99–104. [Google Scholar] [CrossRef]
- Haastert-Talini, K. Culture and proliferation of highly purified adult Schwann cells from rat, dog, and man. Methods Mol. Biol. (Clifton N. J.) 2012, 846, 189–200. [Google Scholar] [CrossRef]
- Dilwali, S.; Patel, P.B.; Roberts, D.S.; Basinsky, G.M.; Harris, G.J.; Emerick, K.S.; Stankovic, K.M. Primary culture of human Schwann and schwannoma cells: Improved and simplified protocol. Hear. Res. 2014, 315, 25–33. [Google Scholar] [CrossRef]
- Casella, G.T.; Bunge, R.P.; Wood, P.M. Improved method for harvesting human Schwann cells from mature peripheral nerve and expansion in vitro. Glia 1996, 17, 327–338. [Google Scholar] [CrossRef]
- Casella, G.T.; Wieser, R.; Bunge, R.P.; Margitich, I.S.; Katz, J.; Olson, L.; Wood, P.M. Density dependent regulation of human Schwann cell proliferation. Glia 2000, 30, 165–177. [Google Scholar] [CrossRef]
- Bhangra, K.S.; Busuttil, F.; Phillips, J.B.; Rahim, A.A. Using Stem Cells to Grow Artificial Tissue for Peripheral Nerve Repair. Stem Cells Int. 2016, 2016, 7502178. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.; Gregory, H.; Powell, R.; Quick, T.J.; Phillips, J.B. Strategies for Peripheral Nerve Repair. Curr. Tissue Microenviron. Rep. 2020, 1, 49–59. [Google Scholar] [CrossRef]
- Kubiak, C.A.; Grochmal, J.; Kung, T.A.; Cederna, P.S.; Midha, R.; Kemp, S.W.P. Stem-cell-based therapies to enhance peripheral nerve regeneration. Muscle Nerve 2020, 61, 449–459. [Google Scholar] [CrossRef]
- Fairbairn, N.G.; Meppelink, A.M.; Ng-Glazier, J.; Randolph, M.A.; Winograd, J.M. Augmenting peripheral nerve regeneration using stem cells: A review of current opinion. World J. Stem Cells 2015, 7, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Wakao, S.; Matsuse, D.; Dezawa, M. Mesenchymal stem cells as a source of Schwann cells: Their anticipated use in peripheral nerve regeneration. Cells Tissues Organs 2014, 200, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, L.; Grigoryan, S.; Yang, I.H.; Thakor, N.V.; Goldstein, R.S. Efficient generation of schwann cells from human embryonic stem cell-derived neurospheres. Stem Cell Rev. Rep. 2011, 7, 394–403. [Google Scholar] [CrossRef]
- Zakrzewski, W.; Dobrzynski, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell. Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Rippon, H.J.; Bishop, A.E. Embryonic stem cells. Cell Prolif. 2004, 37, 23–34. [Google Scholar] [CrossRef]
- Martin, R.M.; Fowler, J.L.; Cromer, M.K.; Lesch, B.J.; Ponce, E.; Uchida, N.; Nishimura, T.; Porteus, M.H.; Loh, K.M. Improving the safety of human pluripotent stem cell therapies using genome-edited orthogonal safeguards. Nat. Commun. 2020, 11, 2713. [Google Scholar] [CrossRef]
- Cai, S.; Tsui, Y.P.; Tam, K.W.; Shea, G.K.; Chang, R.S.; Ao, Q.; Shum, D.K.; Chan, Y.S. Directed Differentiation of Human Bone Marrow Stromal Cells to Fate-Committed Schwann Cells. Stem Cell Rep. 2017, 9, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Kingham, P.J.; Kalbermatten, D.F.; Mahay, D.; Armstrong, S.J.; Wiberg, M.; Terenghi, G. Adipose-derived stem cells differentiate into a Schwann cell phenotype and promote neurite outgrowth in vitro. Exp. Neurol. 2007, 207, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.; Park, S.; Choi, Y.; Park, J.W.; Hong, Y.B.; Park, H.H.; Yu, Y.; Kwak, G.; Kim, H.S.; Ryu, K.H.; et al. Tonsil-Derived Mesenchymal Stem Cells Differentiate into a Schwann Cell Phenotype and Promote Peripheral Nerve Regeneration. Int J. Mol. Sci 2016, 17, 1867. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wang, Y.; Zhang, L.; Zhao, B.; Zhao, Z.; Chen, J.; Guo, Q.; Liu, S.; Sui, X.; Xu, W.; et al. Human umbilical cord Wharton’s jelly-derived mesenchymal stem cells differentiate into a Schwann-cell phenotype and promote neurite outgrowth in vitro. Brain Res. Bull. 2011, 84, 235–243. [Google Scholar] [CrossRef]
- Razavi, S.; Ahmadi, N.; Kazemi, M.; Mardani, M.; Esfandiari, E. Efficient transdifferentiation of human adipose-derived stem cells into Schwann-like cells: A promise for treatment of demyelinating diseases. Adv. Biomed. Res. 2012, 1, 12. [Google Scholar] [CrossRef]
- Xie, S.; Lu, F.; Han, J.; Tao, K.; Wang, H.; Simental, A.; Hu, D.; Yang, H. Efficient generation of functional Schwann cells from adipose-derived stem cells in defined conditions. Cell Cycle 2017, 16, 841–851. [Google Scholar] [CrossRef]
- Matsuse, D.; Kitada, M.; Kohama, M.; Nishikawa, K.; Makinoshima, H.; Wakao, S.; Fujiyoshi, Y.; Heike, T.; Nakahata, T.; Akutsu, H.; et al. Human umbilical cord-derived mesenchymal stromal cells differentiate into functional Schwann cells that sustain peripheral nerve regeneration. J. Neuropathol. Exp. Neurol. 2010, 69, 973–985. [Google Scholar] [CrossRef]
- McKenzie, I.A.; Biernaskie, J.; Toma, J.G.; Midha, R.; Miller, F.D. Skin-derived precursors generate myelinating Schwann cells for the injured and dysmyelinated nervous system. J. Neurosci. 2006, 26, 6651–6660. [Google Scholar] [CrossRef]
- Khuong, H.T.; Kumar, R.; Senjaya, F.; Grochmal, J.; Ivanovic, A.; Shakhbazau, A.; Forden, J.; Webb, A.; Biernaskie, J.; Midha, R. Skin derived precursor Schwann cells improve behavioral recovery for acute and delayed nerve repair. Exp. Neurol. 2014, 254, 168–179. [Google Scholar] [CrossRef]
- Sakaue, M.; Sieber-Blum, M. Human epidermal neural crest stem cells as a source of Schwann cells. Development 2015, 142, 3188–3197. [Google Scholar] [CrossRef]
- Martens, W.; Sanen, K.; Georgiou, M.; Struys, T.; Bronckaers, A.; Ameloot, M.; Phillips, J.; Lambrichts, I. Human dental pulp stem cells can differentiate into Schwann cells and promote and guide neurite outgrowth in an aligned tissue-engineered collagen construct in vitro. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Sanen, K.; Martens, W.; Georgiou, M.; Ameloot, M.; Lambrichts, I.; Phillips, J. Engineered neural tissue with Schwann cell differentiated human dental pulp stem cells: Potential for peripheral nerve repair? J. Tissue Eng. Regen. Med. 2017, 11, 3362–3372. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Liu, C.L.; Ting, C.Y.; Chiu, Y.T.; Cheng, Y.C.; Nicholson, M.W.; Hsieh, P.C.H. Human iPSC banking: Barriers and opportunities. J. Biomed. Sci. 2019, 26, 87. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Martín-Ibáñez, R.; Sareen, D. Manufacturing of human iPSC-derived cell therapies: Road to the clinic. Cell Gene Ther. Insights 2020, 6, 177–191. [Google Scholar] [CrossRef]
- Turner, M.; Leslie, S.; Martin, N.G.; Peschanski, M.; Rao, M.; Taylor, C.J.; Trounson, A.; Turner, D.; Yamanaka, S.; Wilmut, I. Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell 2013, 13, 382–384. [Google Scholar] [CrossRef]
- Ilic, D. iPSC in the past decade: The Japanese dominance. Regen. Med. 2016, 11, 747–749. [Google Scholar] [CrossRef]
- Okano, T.; Sawa, Y.; Barber, E.; Umezawa, A. Regenerative therapy by fusion of medicine and engineering: First-in-human clinical trials with induced pluripotent stem cells and cell sheet technology: A report of the Symposium of Regenerative Medicine for Patients. Regen. Ther. 2015, 2, 2–5. [Google Scholar] [CrossRef]
- Deinsberger, J.; Reisinger, D.; Weber, B. Global trends in clinical trials involving pluripotent stem cells: A systematic multi-database analysis. NPJ Regen. Med. 2020, 5, 15. [Google Scholar] [CrossRef]
- Garber, K. RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 890–891. [Google Scholar] [CrossRef]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Winanto; Ng, S.Y. Replacing what’s lost: A new era of stem cell therapy for Parkinson’s disease. Transl. Neurodegener. 2020, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.-Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Stoddard-Bennett, T.; Reijo Pera, R. Treatment of Parkinson’s Disease through Personalized Medicine and Induced Pluripotent Stem Cells. Cells 2019, 8, 26. [Google Scholar] [CrossRef]
- Bartlett, R.D.; Burley, S.; Ip, M.; Phillips, J.B.; Choi, D. Cell Therapies for Spinal Cord Injury: Trends and Challenges of Current Clinical Trials. Neurosurgery 2020, 87, E456–E472. [Google Scholar] [CrossRef]
- Tsuji, O.; Sugai, K.; Yamaguchi, R.; Tashiro, S.; Nagoshi, N.; Kohyama, J.; Iida, T.; Ohkubo, T.; Itakura, G.; Isoda, M.; et al. Concise Review: Laying the Groundwork for a First-In-Human Study of an Induced Pluripotent Stem Cell-Based Intervention for Spinal Cord Injury. Stem Cells 2019, 37, 6–13. [Google Scholar] [CrossRef]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef]
- Jang, Y.; Choi, J.; Park, N.; Kang, J.; Kim, M.; Kim, Y.; Ju, J.H. Development of immunocompatible pluripotent stem cells via CRISPR-based human leukocyte antigen engineering. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef]
- Sekine, K.; Tsuzuki, S.; Yasui, R.; Kobayashi, T.; Ikeda, K.; Hamada, Y.; Kanai, E.; Camp, J.G.; Treutlein, B.; Ueno, Y.; et al. Robust detection of undifferentiated iPSC among differentiated cells. Sci. Rep. 2020, 10, 10293. [Google Scholar] [CrossRef]
- Ma, M.S.; Boddeke, E.; Copray, S. Pluripotent stem cells for Schwann cell engineering. Stem Cell Rev. 2015, 11, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Furlan, A.; Adameyko, I. Schwann cell precursor: A neural crest cell in disguise? Dev. Biol. 2018, 444 (Suppl. 1), S25–S35. [Google Scholar] [CrossRef] [PubMed]
- Le Douarin, N.M. Cell line segregation during peripheral nervous system ontogeny. Science 1986, 231, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Brennan, A.; Morgan, L.; Mirsky, R.; Kent, A.; Hashimoto, Y.; Gavrilovic, J. The Schwann cell precursor and its fate: A study of cell death and differentiation during gliogenesis in rat embryonic nerves. Neuron 1994, 12, 509–527. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, J.; Lee, D.Y.; Kim, Y.D.; Kim, J.Y.; Lim, H.J.; Lim, S.; Cho, Y.S. Schwann Cell Precursors from Human Pluripotent Stem Cells as a Potential Therapeutic Target for Myelin Repair. Stem Cell Rep. 2017, 8, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Okawa, T.; Kamiya, H.; Himeno, T.; Kato, J.; Seino, Y.; Fujiya, A.; Kondo, M.; Tsunekawa, S.; Naruse, K.; Hamada, Y.; et al. Transplantation of neural crest-like cells derived from induced pluripotent stem cells improves diabetic polyneuropathy in mice. Cell Transplant. 2013, 22, 1767–1783. [Google Scholar] [CrossRef]
- Wang, A.; Tang, Z.; Park, I.H.; Zhu, Y.; Patel, S.; Daley, G.Q.; Li, S. Induced pluripotent stem cells for neural tissue engineering. Biomaterials 2011, 32, 5023–5032. [Google Scholar] [CrossRef]
- Kreitzer, F.R.; Salomonis, N.; Sheehan, A.; Huang, M.; Park, J.S.; Spindler, M.J.; Lizarraga, P.; Weiss, W.A.; So, P.L.; Conklin, B.R. A robust method to derive functional neural crest cells from human pluripotent stem cells. Am. J. Stem Cells 2013, 2, 119–131. [Google Scholar]
- Liu, Q.; Spusta, S.C.; Mi, R.; Lassiter, R.N.; Stark, M.R.; Höke, A.; Rao, M.S.; Zeng, X. Human neural crest stem cells derived from human ESCs and induced pluripotent stem cells: Induction, maintenance, and differentiation into functional schwann cells. Stem Cells Transl. Med. 2012, 1, 266–278. [Google Scholar] [CrossRef]
- Huang, C.W.; Huang, W.C.; Qiu, X.; Fernandes Ferreira da Silva, F.; Wang, A.; Patel, S.; Nesti, L.J.; Poo, M.M.; Li, S. The Differentiation Stage of Transplanted Stem Cells Modulates Nerve Regeneration. Sci Rep. 2017, 7, 17401. [Google Scholar] [CrossRef]
- Cheng, Z.; Ito, S.; Nishio, N.; Xiao, H.; Zhang, R.; Suzuki, H.; Okawa, Y.; Murohara, T.; Isobe, K. Establishment of induced pluripotent stem cells from aged mice using bone marrow-derived myeloid cells. J. Mol. Cell Biol 2011, 3, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wu, M.; Peng, C.; Zhao, G.; Gu, R. GFAP expression in injured astrocytes in rats. Exp. Ther. Med. 2017, 14, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Haastert-Talini, K. Appropriate Animal Models for Translational Nerve Research. In Peripheral Nerve Tissue Engineering and Regeneration; Phillips, J., Hercher, D., Hausner, T., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–17. [Google Scholar] [CrossRef]
- Achilleos, A.; Trainor, P.A. Neural crest stem cells: Discovery, properties and potential for therapy. Cell Res. 2012, 22, 288–304. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Medium 1 | Medium 2 | Medium 3 | Medium 4 | Medium 5 | Medium 6 |
|---|---|---|---|---|---|
| 80% KO DMEM/F12, 20% KSR, 1% NEAA 1 mM L-Glu 0.1 mM 2-ME | KO DMEM/F12, 2% StemPro neural suppl., 20 ng/mL FGF-2, 20 ng/mL EGF | N2 M., 10 ng/mL CNTF, 10 ng/mL FGF-2, 1 mM dBcAMP, 20 ng/mL NRG1 | 50% (Neurobasal M., MEM-NEAA, GlutaMAX, B27, 20 ng/mL FGF-2) + 50% (SDIA condition M, 10 μM Y-27632, 200 μM AA) | MesenPRO M., 20 ng/mL NRG1 | SDIA condition M., 10% KSR, 0.1 mM NEAA, 1 mM pyruvate, 0.1 mM 2-ME |
| Medium 7 | Medium 8 | Medium 9 | according to Kreitzer et al. [88] | ||
| KSR medium | N2 medium | ||||
| DMEM/F12, 20 ng/mL FGF-2, 1% N2, 2% B27, 0.05% BSA fraction V, 1% GlutMax, 1% MEM-NEAA, 110 µM 2-ME, 10 µM Y-27632 | Advanced DMEM/F12 + Neurobasal M. (1:1 mix), 1% N2, 2% B27, 0.005% BSA, 2 mM GlutaMax, 0.11 mM 2-ME, 3 mM CT99021, 20 mM SB-431542 | DMEM/low glucose, 1% FBS, 4 mM FK, 200 ng/mL NRG1, 100 nM all-trans RA, 10 ng/mL PDGF-BB | Knockout DMEM, 15%KSR, 1% MEM-NEAA, 1% GlutaMax, 55 µM 2-ME | DMEM/F12, 0.15% glucose, 1% N2, 20 µg/mL insulin, 5 mM HEPES | |
| Progenitor Cell Stages | Schwann Cell Stages | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study | Source iPSC | Culture Condition | Type | Duration (days) | Cell Markers | Induction Medium | Type of Induction | Duration (days) | Cell Markers | Functionality Analysis |
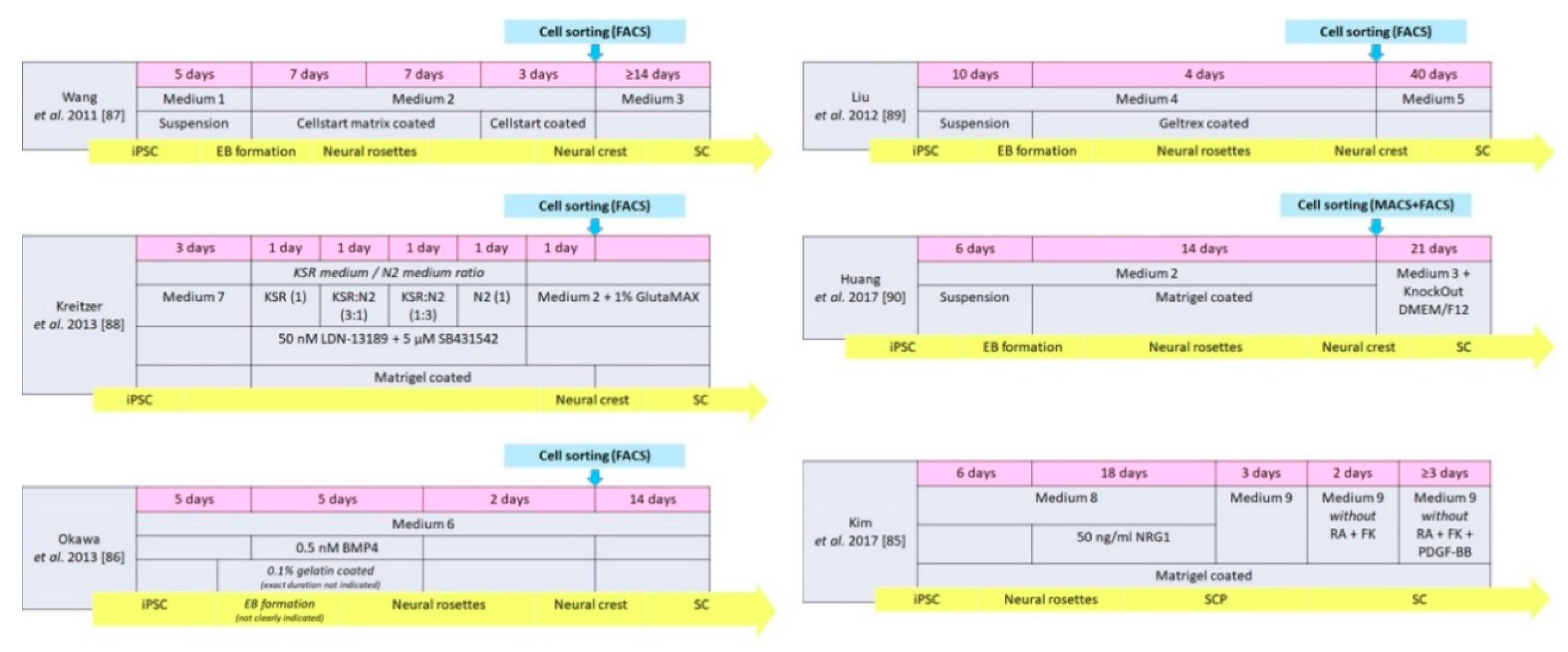
| Wang et al. 2011 [87] | Human | Medium 1 + 2 | Neural crest | 22 | p75NGFR, HNK1 Vimentin, Nestin Slug, AP2α | Medium 3 | Directly | ≥14 | S100β, GFAP | None |
| Liu et al. 2012 [89] | Human | Medium 4 | Neural crest | 14 | p75NGFR, HNK1, Sox9, Sox10, CD44 | Medium 5 | Directly | 40 | GFAP, S100, p75NGFR | In vitro |
| Kreitzer et al. 2013 [88] | Human | Medium 7, KSR medium, N2 medium, Medium 2, GlutaMAX | Neural crest | 8 | p75NGFR, HNK1 AP2α, Sox10 | Medium 2 GlutaMAX | Spontaneous in vitro | Not mentioned | GFAP | None |
| Huang et al. 2017 [90] | Human | Medium 2 | Neural crest | 20 | HNK1, AP2α, Sox10 | Medium 3 KnockOut DMEM/F12 | Directly | 21 | S100β, GFAP | In vivo |
| Kim et al. 2017 [85] | Human | Medium 8 | SC precursor | 24 | Sox 10, CDH19, MPZ, GAP43 | Medium 9 | Directly | ≥ 7 | NGFR, S100, EGR2, MPZ | In vitro and in vivo |
| Okawa et al. 2013 [86] | Mouse | Medium 6 BMP4 | Neural crest | 12 | p75NGFR, AP2α | Medium 6 | Spontaneous after implantation | 14 | S100β | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Powell, R.; Phillips, J.B.; Haastert-Talini, K. Perspective on Schwann Cells Derived from Induced Pluripotent Stem Cells in Peripheral Nerve Tissue Engineering. Cells 2020, 9, 2497. https://doi.org/10.3390/cells9112497
Huang Z, Powell R, Phillips JB, Haastert-Talini K. Perspective on Schwann Cells Derived from Induced Pluripotent Stem Cells in Peripheral Nerve Tissue Engineering. Cells. 2020; 9(11):2497. https://doi.org/10.3390/cells9112497
Chicago/Turabian StyleHuang, Zhong, Rebecca Powell, James B. Phillips, and Kirsten Haastert-Talini. 2020. "Perspective on Schwann Cells Derived from Induced Pluripotent Stem Cells in Peripheral Nerve Tissue Engineering" Cells 9, no. 11: 2497. https://doi.org/10.3390/cells9112497
APA StyleHuang, Z., Powell, R., Phillips, J. B., & Haastert-Talini, K. (2020). Perspective on Schwann Cells Derived from Induced Pluripotent Stem Cells in Peripheral Nerve Tissue Engineering. Cells, 9(11), 2497. https://doi.org/10.3390/cells9112497